

Abstract
Osteoclast motility is thought to depend on rapid podosome assembly and disassembly. Both μ-calpain and m-calpain, which promote the formation and disassembly of focal adhesions, were observed in the podosome belt of osteoclasts. Calpain inhibitors disrupted the podosome belt, blocked the constitutive cleavage of the calpain substrates filamin A, talin, and Pyk2, which are enriched in the podosome belt, induced osteoclast retraction, and reduced osteoclast motility and bone resorption. The motility and resorbing activity of μ-calpain−/− osteoclast-like cells were also reduced, indicating that μ-calpain is required for normal osteoclast activity. Histomorphometric analysis of tibias from μ-calpain−/− mice revealed increased osteoclast numbers and decreased trabecular bone volume that was apparent at 10 weeks but not at 5 weeks of age. In vitro studies suggested that the increased osteoclast number in the μ-calpain−/−bones resulted from increased osteoclast survival, not increased osteoclast formation. Calcitonin disrupted the podosome ring, induced osteoclast retraction, and reduced osteoclast motility and bone resorption in a manner similar to the effects of calpain inhibitors and had no further effect on these parameters when added to osteoclasts pretreated with calpain inhibitors. Calcitonin inhibited the constitutive cleavage of a fluorogenic calpain substrate and transiently blocked the constitutive cleavage of filamin A, talin, and Pyk2 by a protein kinase C-dependent mechanism, demonstrating that calcitonin induces the inhibition of calpain in osteoclasts. These results indicate that calpain activity is required for normal osteoclast activity and suggest that calcitonin inhibits osteoclast bone resorbing activity in part by down-regulating calpain activity.
Osteoclasts are large multinucleated cells of the monocyte-macrophage lineage that play a critical role in skeletal development and repair and in calcium homeostasis by resorbing mineralized cartilage and bone. They display a high degree of motility, which is required for normal bone resorbing activity. As in other highly motile cells, the integrin-based attachment complexes of osteoclasts are podosomes, which are structurally and functionally distinct from focal adhesions (1–6). Although many of the same proteins are present in both podosomes and focal adhesions, podosomes are notably more dynamic, undergoing assembly and disassembly within minutes (5–7). The rapid and cyclic podosome assembly and disassembly and their dynamic interaction with elements of the actin cytoskeleton are thought to be critical for the high motility of osteoclasts (7).
Recently, calpains have been found to play crucial roles in the regulation of motility in cells such as fibroblasts that form focal adhesions (8). Calpains are a family of cytosolic cysteine proteases, many of which are Ca2+-dependent, that catalyze the limited cleavage of specific proteins during regulatory signaling (9, 10). The most studied members of the calpain family are the ubiquitously expressed μ-calpain (calpain 1) and m-calpain (calpain 2). These calpains are heterodimers composed of a large 80-kDa catalytic subunit that is unique for each of the two enzymes and a common 30-kDa regulatory subunit, termed calpain 4 (11).
In these cells, calpains are thought to promote cell spreading and locomotion by modulating the assembly and disassembly of the focal adhesion complexes (8, 12). μ-Calpain promotes the formation of adhesion sites in the leading edge of the cell. Conversely, recent evidence suggests that m-calpain promotes the disassembly of focal adhesion structures (8), in part by cleaving talin (13). Calpains have been reported to cleave a number of attachment-related proteins that are enriched in the peripheral podosome belt of osteoclasts, including talin (14–17), vinculin (18), paxillin (15), β3 integrin (10, 19), Src (20), Pyk2 (21), and focal adhesion kinase (15, 22, 23). Therefore, it is likely that one or more calpains could contribute to the regulation of podosome formation and assembly and thereby help regulate osteoclast attachment and motility. Calpains could also affect the regulation of osteoclast bone resorption by cleaving caspase 3 (24–26), thereby promoting osteoclast apoptosis.
Calcitonin (CT)5 is a potent inhibitor of osteoclastic bone resorption that acts directly and rapidly on osteoclasts via the calcitonin receptor, a class B-type G protein-coupled receptor that couples to numerous signaling pathways via Gs, Gq/11, and Gi (27–38). Treatment of osteoclasts with CT reduces osteoclast motility, induces cell retraction, and disrupts the peripheral belt of podosomes that forms in osteoclasts plated on glass or dentin (27, 30, 39). This suggests that CT regulates at least some of the mechanisms responsible for podosome formation and function, but little is known of the specific mechanisms by which CT exerts its effects. The CT receptor (CTR) activates both cyclic AMP-dependent protein kinase (PKA) and protein kinase C (PKC), and it also induces transient increases in [Ca2+]i (29, 34, 36–38). All of these signaling mechanisms are known to modulate calpain activity, either directly or indirectly through the highly specific endogenous calpain inhibitor calpastatin (40–43), suggesting that CT could regulate calpain activity and thereby modulate the assembly/disassembly of podosomes and consequently osteoclast motility and bone resorption.
In this study, we investigated whether calpain might play a role in the regulation of osteoclast retraction, motility, and bone resorbing activity and whether the inhibitory effects of CT upon osteoclast motility and bone resorption might involve the modulation of calpain activity. We found that reducing calpain activity with chemical inhibitors or by deleting the μ-calpain gene impaired osteoclast spreading, motility, and bone resorption in vitro and that CT transiently inhibited calpain activity. At the same time, reducing calpain activity prolonged osteoclast survival in a manner similar to the effect of CT. The in vivo effect of deleting the μ-calpain gene was a marked increase in the numbers of osteoclasts with low intrinsic bone resorbing activity, resulting in late onset osteopenia.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Salmon calcitonin was purchased from Peninsula Laboratories, Inc. (Belmont, CA). Calphostin C, prostaglandin E2, minimum essential medium, α modification (α-MEM), Igepal CA-630, and fetal bovine serum (FBS) were from Sigma. Calpeptin, MDL28170, PD151746, (Rp)isomer of 8-bromo-adenosine 3′,5′-cyclic monophosphorothioate ((Rp)-8-Br-cAMPs), H-89, and anti-m-calpain antibody were purchased from Calbiochem. Anti-μ-calpain antibody was purchased from Biomol (Plymouth Meeting, PA). Anti-talin antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-filamin antibody was purchased from Chemicon International (Temecula, CA). Anti-Pyk2/CAK antibody was purchased from BD Biosciences. Anti-cleaved caspase-3 antibody was purchased from New England Biolabs, Inc. (Beverly, MA). Both anti-mouse and anti-rabbit horseradish peroxidase-conjugated secondary antibodies were obtained from Fisher. Boc-Leu-Met-7-amino-4-chloromethyl-coumarin (Boc-LM-CMAC) was purchased from Molecular Probes, Inc. (Eugene, OR).
Osteoclasts and Osteoclast-like Cells
Rabbit osteoclasts and osteoclast-like cells (OCLs) were generated as described elsewhere (44) with modifications. Briefly, the long bones and scapulae of neonatal New Zealand White rabbits (60–90 g) were dissected free of soft tissue and minced in α-MEM with 0.55 mg/liter sodium bicarbonate and 10 mm HEPES (pH 7.10). The bone fragments were then allowed to settle, the cell suspension was collected, and the cells were pelleted by centrifugation at 1500 × g for 5 min at 4 °C. To isolate mature osteoclasts, the cell pellets were resuspended in medium supplemented with 10% FBS, aliquoted (150 μl) onto coverslips in 6-well dishes, and cultured for 1 h at 37 °C in humidified air with 5% CO2. Then 2 ml of medium supplemented with 10% FBS was added to each well. To produce rabbit OCLs, the resuspended cells were cultured in medium supplemented with 10% FBS and 10−8 m 1,25-dihydroxyvitamin D3 (Proskelia, Romainville, France) for 9 or 10 days. The medium was changed every other day. Cells were cultured in medium with 0.1% FBS for 4 h before stimulation. Murine OCLs were produced by coculturing calvarial osteoblasts with bone marrow cells, as described elsewhere (45) and used immediately for Western blotting or immunofluorescence analysis, tartrate-resistant acid phosphatase (TRAP) staining or migration, retraction, and bone resorption assays as described below. All animal protocols were approved by the Yale University Institutional Animal Care and Use Committee.
Immunoprecipitation and Immunoblotting
Prior to treatment, rabbit and murine cocultures were incubated for 18 h in medium containing 0.5% FBS. The serum-starved OCLs were then purified by peeling away the layer of less adherent, mainly stromal cells. The OCLs were stimulated with CT or other agents in the absence of the cocultured mesenchymal cells at 37 °C. The OCLs were lysed in 1% Igepal CA-630, 150 mm NaCl, 50 mm Tris-HCl (pH 8.0), 5 mm EGTA, 1 mm phenyl-methanesulfonyl fluoride, 10 mm NaF, 0.4 mm Na2VO4, and 10 μg/ml leupeptin and aprotinin. The lysates were processed for immunoprecipitation and incubated with specific antibodies and protein A- or protein G-Sepharose, and the immune complexes were washed with mRIPA, containing 5 mm EGTA, 1 mm phenylmethanesulfonyl fluoride, 10 mm NaF, 0.4 mm Na2VO4, and 10 μg/ml each aprotinin and leupeptin. The samples were then processed for immunoblotting, as described elsewhere (46).
Calpain Activity Assay
Authentic osteoclasts were plated on glass coverslips for 24 h. They were then loaded with 30 μm Boc-LM-CMAC for 1 h at 37 °C. After loading, images of the cells were obtained before and after CT addition. Calpain-catalyzed cleavage of Boc-LM-CMAC creates a fluorescent product; fluorescence correlates with calpain activity (47–49).
Immunofluorescence Analysis
Authentic osteoclasts on coverslips were fixed in phosphate-buffered saline (PBS) containing 3.7% formaldehyde for 10 min at room temperature and washed in PBS. Coverslips for actin labeling were extracted in ice-cold acetone for 3–5 min and returned to PBS. All other coverslips were permeabilized in 0.05% saponin for 30 min. The coverslips were blocked in 5% normal goat serum for 30 min and then incubated in the appropriate primary antibody for 2 h, washed in PBS, incubated for 1 h in the appropriate fluorescein- or rhodamine-conjugated secondary antibody, and washed. Those cover-slips used for actin labeling were incubated in a 1:40 dilution (in PBS) of rhodamine phalloidin (Molecular Probes) for 20 min and washed. All coverslips were mounted in FluorSave. Cells were examined using a scanning laser confocal imaging system (MRC-600; Bio-Rad). Images were recorded, composite images were compiled, and image enhancements were performed using Adobe Photoshop 6.0.
Cell Retraction Assay
Authentic rabbit osteoclasts were plated on glass or dentine slices for 24 h and then incubated for 30 min with vehicle or calpain inhibitors as described under “Results.” Cells were then fixed (inhibitor only) or treated with CT for 20 min before fixing (CT only, inhibitor plus CT). The fixed cells were stained with toluidine blue or TRAP-stained (50, 51), and the area of at least 70 osteoclasts/ group was measured with a computerized system for histomorphometry (Osteometrics, Atlanta, GA).
Cell Migration Assay
Cell migration was analyzed as described elsewhere (52) with modifications. Authentic murine osteoclasts or OCLs were plated on semipermeable membranes coated with vitronectin in Boyden chambers (CHEMICON) and incubated for 18 h with and without calpain inhibitors as described under “Results” and then fixed and stained for TRAP. The TRAP-positive cells on the bottom side of the membrane were counted. Data values are expressed as a ratio relative to the values obtained from the untreated control chambers.
In Vitro Bone Resorption
Authentic murine or rabbit osteoclasts or murine OCLs were plated on dentine slices and cultured for 24 h. They were then incubated with or without CT or calpain inhibitors for an additional 16 h and then fixed and stained with toluidine blue. The pit area was measured with a computerized system for histomorphometry (Osteometrics, Inc., Atlanta, GA).
Histology and Histomorphometry
μ-Calpain−/− mice and genetically matched wild type mice were sacrificed by cervical dislocation at 5 and 10 weeks of age after being previously injected intraperitoneally with calcein (30 mg/kg), 7 and 2 days before sacrifice. Bone specimens were fixed in 4% formalin and then embedded in methylmethacrylate as described (52). 5-μm sections were cut and stained either with toluidine blue or by the Von Kossa method for staining calcified tissues (52). 10-μm sections were cut from the same samples and coverslipped unstained for dynamic measurements. Histomorphometric analysis was carried out with an Osteomeasure system (Osteometrics, Inc.), using standard procedures (53) and in blind fashion. Tibial sections were measured in the proximal metaphysis beginning 340 μm below the chondro-osseous junction. Tibial cortical thickness and periosteal mineral appositional rates were measured beginning 680 μm from the chondro-osseous junction on the anterofibular side. Serum levels of pyridinoline cross-links (collagen type I fragments resulting from proteolytic digestion), a marker of osteoclastic bone resorption, were measured using an ELISA kit from Nordic Biosciences (Copenhagen, Denmark). At least six animals per group were examined. Statistical analysis was performed using analysis of variance, with p values less than 0.05 accepted as significant; error bars represent ± S.D.
Osteoclast Survival
OCL survival was measured as reported previously (45). OCLs were generated by coculturing bone marrow cells from μ-calpain−/− mice and genetically matched wild type mice with calvarial osteoblasts from neonatal CD-1 mice. Once the OCLs were formed, the calvarial cell layer was removed by collagenase/dispase treatment. Some plates were immediately stained for TRAP. The remaining cells were cultured in μ-MEM and 10% FBS with vehicle, calpain inhibitors, or CT for various times up to 24 h. The OCLs were then fixed and TRAP-stained. The numbers of TRAP-positive multinucleated cells were determined and expressed as a percentage of the TRAP-positive multinucleated cells present at the time when the calvarial cell layer was removed.
RESULTS
Both μ-Calpain and m-calpain Are Expressed in Osteoclasts and Localize in Podosomes
Calpain is known to modulate spreading (54) and motility (8, 55) of cells that form focal adhesions by cleaving several focal adhesion proteins, cytoskeletal proteins, and downstream effectors, including talin, focal adhesion kinase, Pyk2, Src, Rac1, and RhoA, and thereby regulating focal complex assembly/disassembly (8, 56). We hypothesized that calpains might therefore regulate the turnover of the podosomes and consequently osteoclast motility and bone resorption. Western blotting of lysates of OCLs generated in vitro revealed the presence of both μ-calpain and m-calpain (Fig. 1, A and B). In addition, immunofluorescence analysis showed that high levels of both μ-calpain (Fig. 1, C and D) and m-calpain (Fig. 1, E and F) were associated with the F-actin-rich belt of podosomes in authentic osteoclasts from both rabbits (Fig. 1, C and E) and mice (Fig. 1, D and F). Calpain was enriched throughout the region between the individual podosomes (Fig. 2C).
FIGURE 1. Osteoclasts express both μ-calpain and m-calpain.
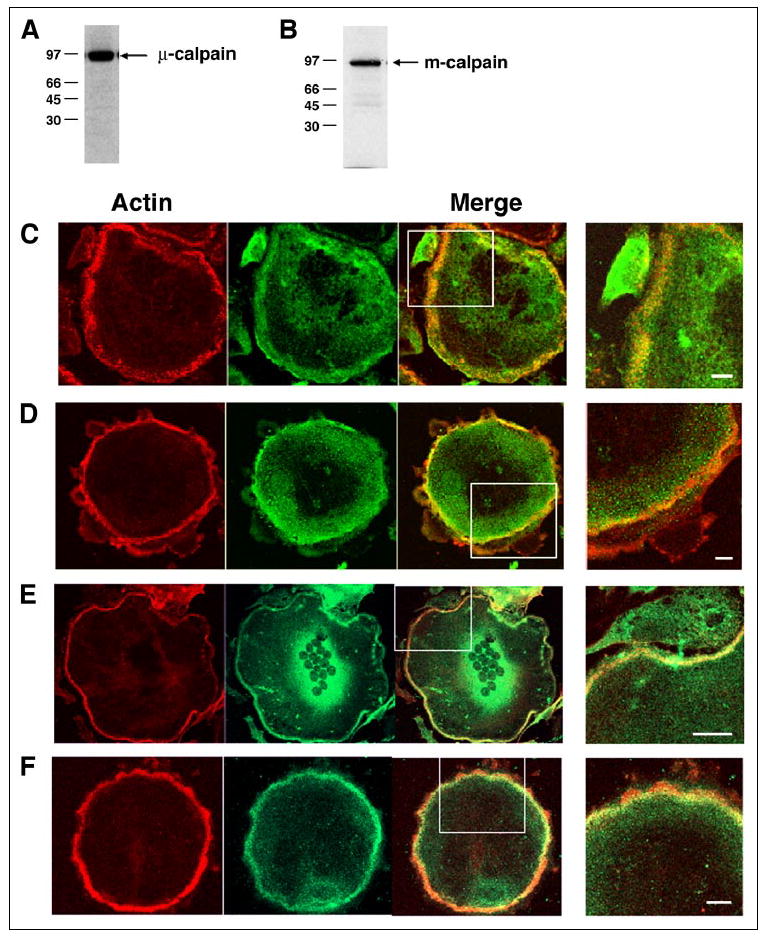
Western blotting of lysates of purified rabbit (A) or mouse (B) OCLs with antibodies against μ-calpain and m-calpain, respectively, identified immunoreactive bands at the predicted relative mobilities. Double immunofluorescence staining of authentic rabbit (C and E) or mouse (D and F) osteoclasts for F-actin (red) and μ-calpain (C and D) or m-calpain (E and F) (green) revealed that both calpains are present and distributed throughout the cell, with high levels seen in the peripheral F-actin-rich belt of podosomes. Scale bars, 5 μm.
FIGURE 2. Talin and filamin A are enriched in the osteoclast podosome belt.

Double immunofluorescence staining of actin (red) and talin (A), filamin A (B), or μ-calpain (C) (green) revealed that both proteins localize in the actin-rich podosome ring. Talin was primarily localized in a ring surrounding the F-actin-containing podosome core, as previously described (3), whereas filamin and μ-calpain were more evenly distributed throughout the region between the individual podosomes. Scale bars, 2 μm.
Talin, Filamin A, and Pyk2 Are Present in the Podosome Belt and Are Constitutively Cleaved by Calpain in Osteoclasts
The calpain substrates talin and filamin A are actin-binding proteins that associate with focal adhesion structures (14, 57–59), and talin has been shown to localize to podosomes in osteoclasts and other highly motile cells (60, 61). Immunofluorescence confirmed that talin is associated with podosomes (Fig. 2A) and showed that filamin A is also expressed in the osteoclasts and localized in and around podosomes (Fig. 2B) in a manner similar to calpain (Fig. 2C). Pyk2, another calpain substrate protein (21), is a nonreceptor tyrosine kinase related to focal adhesion kinase that is highly enriched in the podosomes of osteoclasts (39, 62) and plays a key role in the Src-dependent regulation of osteoclast adhesion and motility following the activation of αvβ3 integrin (46, 63, 64). Since podosomes form and disappear at a much higher rate than focal adhesions (5–7), we reasoned that if calpain-catalyzed proteolysis of these or other podosome-associated proteins contributes to the regulation of podosome turnover, and thus to the regulation of osteoclast attachment, motility, and spreading, it might be possible to detect calpain-generated cleavage products from these proteins in osteoclasts. Western blotting of OCL lysates with antibodies against talin (Fig. 3A), filamin A (Fig. 3B), or Pyk2 (Fig. 3C) detected all three proteins as well as less intense bands below the parent proteins of sizes consistent with reported cleavage products (15, 21, 65). Pretreatment of the OCLs with the calpain inhibitors calpeptin or MDL28170, which inhibit calpains by binding to the catalytic site (66), or PD151746, which inhibits calpains by binding to the calcium-binding site (67), reduced the amount of the lower bands in all three blots, strongly suggesting that one or more calpains are cleaving podosome-associated proteins in osteoclasts.
FIGURE 3. Calpain constitutively cleaves talin, filamin A, and Pyk2 in osteoclasts.
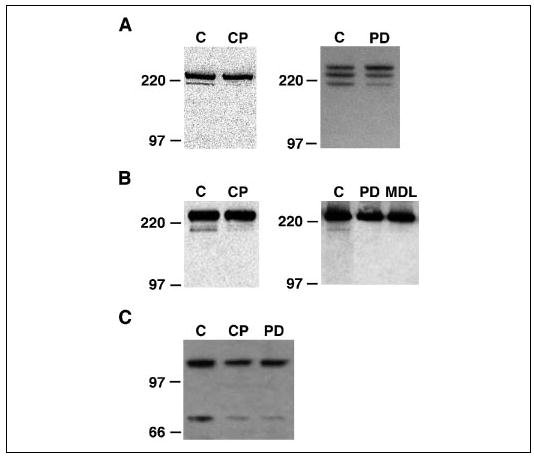
Western blotting of lysates of untreated rabbit (A and B, left panels) or mouse (A and B, right panels; C) OCL with antibodies against talin (A), filamin A (B), or Pyk2 (C) detected major bands at the predicted relative mobilities of the two proteins and less intense bands just below the parent protein (C lanes). Pretreatment of OCLs with membrane-permeable calpain inhibitors that block the catalytic site (calpeptin (CP; 40 μg/ml), MDL28170 (MDL; 50 μm)) or the calcium-binding site (PD151746 (PD); 100 μm)) largely eliminated the lower bands, indicating that calpain constitutively cleaves talin, filamin A, and Pyk2 in osteoclasts.
Calpain Is Required for Normal Osteoclast Spreading and Motility
Attachment, spreading, and motility of cells that form focal adhesions require the binding of attachment proteins, typically integrins, to the extracellular matrix and the subsequent engagement of the cytoskeleton by the integrin intracellular domain. The tension generated by these interactions leads to cell locomotion (68, 69). Since the calpain-catalyzed cleavage of cytoskeletal and attachment-related proteins regulates the assembly and disassembly of adhesion complexes (54) and cell motility (17, 55, 70, 71), we examined the effects of calpain inhibitors on osteoclast spreading and motility.
To determine if calpain promotes osteoclast spreading, authentic osteoclasts were isolated from rabbits or mice, cultured for 24 h, and then treated with calpain inhibitors or with CT as a positive control and fixed as described under “Experimental Procedures.” Both CT and the calpain inhibitors induced a prompt cellular retraction of rabbit (Fig. 4A) and mouse (not shown) osteoclasts plated on either glass (not shown) or dentine (Fig. 4A) to 30–50% of the area of untreated cells, suggesting that calpain activity is required for the normal spreading of osteoclasts. Similar results were obtained with three different calpain inhibitors (calpeptin, MDL28170, and PD151746). The addition of both CT and a calpain inhibitor together induced no more retraction than either agent alone.
FIGURE 4. Calpain promotes osteoclast spreading and migration.

A, rabbit osteoclasts were plated on dentine slices and cultured for 24 h and then treated with 10−8 m CT and/or with 40 μg/ml calpeptin (CP), 50 μm MDL28170 (MDL), or 100 μm PD151746 (PD), as indicated, for 20 min. The cells were prepared, and the areas of the cells were analyzed as described under “Experimental Procedures.” *, p < 0.01 relative to untreated control. B, to test the effects of calpain inhibitors on osteoclast migration, osteoclasts were cultured as described under “Experimental Procedures” for 16 h on semipermeable membranes in Boyden chambers in the presence of 10−9 m CT and/or the calpain inhibitors CP (40 μg/ml) and MDL (50 μm) as indicated. The membranes were stained for TRAP as described under “Experimental Procedures,” and the TRAP-positive cells on the bottom surface of the membranes were counted. *, p < 0.01 relative to untreated controls (C). C, the migration assay was performed with OCLs generated from bone marrow cells obtained from μ-calpain−/− mice and genetically matched wild type animals, as described under “Experimental Procedures,” in the presence or absence of 10−9 m CT. *, p < 0.01 relative to the untreated WT. #, p < 0.01 relative to the untreated μ-calpain−/− OCLs.
The effect of calpain inhibitors on osteoclast spreading suggested that calpain might also play a role in osteoclast motility. To address this question, osteoclasts were plated on vitronectin-coated semipermeable membranes in Boyden chambers, incubated with or without CT (positive control) or calpain inhibitors for 16 h, and analyzed for the amount of migration across the membrane (Fig. 4B) as described under “Experimental Procedures.” Data were normalized to untreated controls. Treatment with calpain inhibitors caused a reduction of cell motility that was comparable with that induced by CT and, as was true of cell spreading, there was no additive reduction of motility when CT and a calpain inhibitor were both present.
To examine the specific role of μ-calpain in osteoclast migration, we analyzed the migration of OCLs generated from precursors obtained from μ-calpain−/− mice (72) and genetically matched wild type animals. The μ-calpain−/− OCLs exhibited less motility than the wild type controls but more motility than the CT-treated wild type OCLs (Fig. 4C). However, in contrast to the inability of CT to further reduce the motility of osteoclasts treated with calpain inhibitors (Fig. 4B), treatment with CT further reduced the motility of the μ-calpain−/− OCLs. These results, together with the more complete inhibition obtained with different calpain inhibitors, suggest that osteoclast locomotion is also promoted by calpains other than μ-calpain, most likely m-calpain, and that CT down-regulates all of the calpains that are blocked by the chemical inhibitors.
Calpain Is Required for Normal Osteoclast Bone Resorbing Activity
Bone resorbing activity is dependent on osteoclast motility (73). Thus, reducing motility by inhibiting calpain might reduce bone resorption. To test this, authentic rabbit osteoclasts were isolated as described under “Experimental Procedures,” plated on dentine slices, and incubated for 16–18 h in the presence or absence of calpain inhibitors or CT as a positive control, as indicated (Fig. 5A). The calpain inhibitors reduced pit formation by 80–90%, at least as much as the CT-induced inhibition. There was no additive effect when the osteoclasts were treated with combinations of CT and calpain inhibitors. To examine the specific role of μ-calpain, we analyzed the resorbing activity of μ-calpain−/− OCLs plated on dentine slices for 16–18 h (Fig. 5B). Consistent with the results obtained with the rabbit osteoclasts, CT reduced the bone resorbing activity of the wild type murine OCLs by about 75%. The resorption by the μ-calpain−/− OCLs was about half of the resorption by the wild type OCLs, a significant reduction from the wild type activity but not as great a reduction as that achieved by treating the wild type OCLs with CT or by treating the rabbit osteoclasts with either CT or the calpain inhibitors. In contrast to the lack of an additional effect of CT on the bone resorbing activity of rabbit osteoclasts treated with calpain inhibitors, however, treatment of the μ-calpain−/−OCLs with CT induced a further reduction in the bone resorbing activity, suggesting again that at least one other effector that is inhibited by the calpain inhibitors and CT, most likely m-calpain, also contributes to osteoclast bone resorbing activity.
FIGURE 5. Bone resorption requires calpain activity.
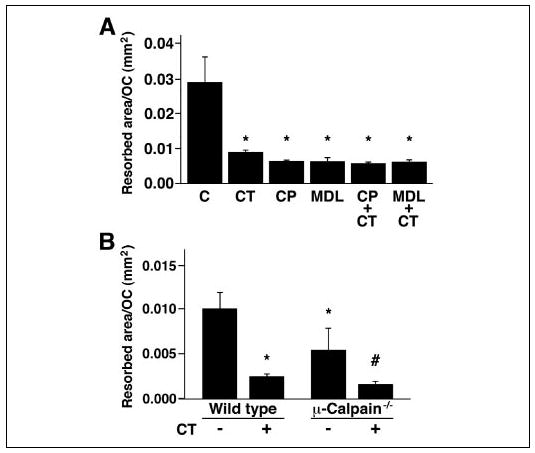
A, rabbit osteoclasts were plated on dentine slices and incubated for 18 h in the presence of calpain inhibitors (40 μg/ml CP, 50 μm MDL) and/or CT (10−9 m) as a positive control, as indicated. *, p < 0.05 relative to untreated cells. B, OCLs were generated from bone marrow cells obtained from μ-calpain−/− mice and genetically matched wild type animals as described under “Experimental Procedures.” Aliquots of OCL preparations were transferred onto dentine slices and cultured for an additional 12 h in the presence or absence of 10−9 m CT. *, p < 0.01 relative to untreated WT; #, p < 0.05 relative to untreated μ-calpain−/− OCLs.
Calcitonin Modulates Calpain Activity in Osteoclasts
The similar effects of CT and calpain inhibitors on osteoclast retraction, motility, and bone resorbing activity, together with the lack of any additive effect of the two agents on these functions, suggested that CT might act at least in part by inhibiting calpain. Calpain activity can be inhibited by either PKC or PKA (43), both of which are activated downstream of the CTR (29, 34, 36, 37). Thus, CT could inhibit calpain activity in osteoclasts and thereby modulate osteoclast attachment, motility, and bone resorbing activity. To determine whether CT affected calpain activity in osteoclasts, rabbit osteoclasts were incubated in the presence of 30 μm Boc-LM-CMAC, a membrane-permeable fluorogenic calpain substrate, for 1 h, during which time the fluorescence increased in the osteoclasts and contaminating cells as a consequence of constitutive calpain activity. Following the administration of CT, the fluorescence in the osteoclasts decreased, in contrast to the continued increased fluorescence in contaminating cells, particularly erythrocytes, that do not express CT receptors (Fig. 6A), consistent with the hypothesized down-regulation of osteoclast calpain activity by CT.
FIGURE 6. CT inhibits calpain activity in osteoclasts.
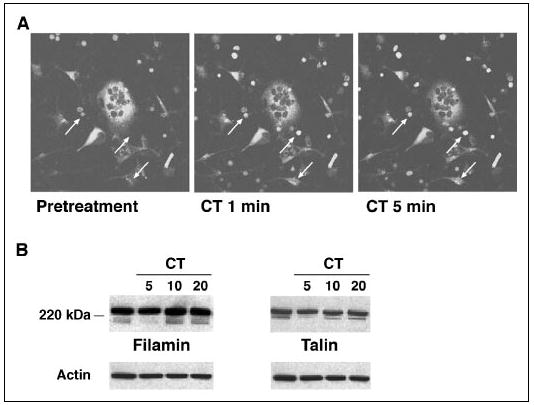
A, rabbit osteoclasts were preincubated with 30 μm Boc-LM-CMAC for 1 h and then treated with 10−8 m CT. Fluorescence images were taken before and after the addition of CT as indicated. The fluorescence intensity of the multinucleated osteoclasts decreased after the addition of CT. The progressive increase in the fluorescence in red blood cells and fibroblast-like cells (arrows) was not affected by the presence of CT. B, rabbit OCLs were treated with 10−8 m CT for the indicated times, and the lysates were blotted with antibodies against filamin and talin. CT transiently inhibited the constitutive cleavage of filamin and talin in OCLs.
Since calpain inhibitors blocked the constitutive fragmentation of filamin, talin, and Pyk2 in osteoclasts (Fig. 3), we asked whether CT also affected the proteolysis of talin and filamin in a similar manner. To address the question, rabbit OCLs were treated with 10−8 m CT for various times, and the lysates were blotted with antibodies against filamin and talin (Fig. 6B). As noted before, both the full-length proteins and high MW cleavage products were detected in untreated OCLs. CT treatment transiently reduced the amounts of the fragments, which were largely absent at 5 min after the addition of CT and present again by 10–20 min, providing further evidence that CT inhibits calpain activity in osteoclasts.
PKC Mediates the CT-induced Inhibition of Calpain
The CTR signals through several effectors that are known to inhibit calpain in other cell systems, including PKA and PKC (29, 34, 36, 37). In order to determine whether either PKA or PKC mediate the CT-induced decrease in calpain activity, we examined the effect of PKC and PKA inhibitors on the CT-induced inhibition of the constitutive cleavage of talin. Mouse OCLs were treated with 10−9 m CT for 5 min in the absence and in the presence of calphostin C (25, 50, or 100 nm), H89 (50 or 100 nm) or (Rp)-8-Br-cAMPS (1 or 5 μM) and then lysed and processed for Western blotting with anti-talin antibody. Three high molecular weight proteins were detected in lysates from untreated OCLs. CT treatment significantly reduced the lowest band but had little effect on the upper two bands. Calphostin C dose-dependently prevented the CT-induced disappearance of the talin cleavage product (Fig. 7A), whereas the PKA inhibitors had little effect (Fig. 7B), suggesting that CT acts via PKC but not PKA to inhibit calpain activity.
FIGURE 7. Calcitonin-induced PKC activity inhibits calpain.

OCLs were generated from murine bone marrow. Prior to treatment with CT and the inhibitors, OCLs were cultured for 18 h in 0.5% FBS. The supporting stromal cell layers were removed, and the OCLs were pretreated for 30 min with vehicle, calphostin C (CalC; 25, 50, and 100 nm), H-89 (50 and 100 nm), (Rp)-8-Br-cAMPS (Rp; 1 μM), or PD151746 (PD; 100 μm) followed by CT (10−9 m) for 5 min as indicated. Western blotting with anti-talin antibody was performed to observe the effect of PKA and PKC inhibitors on the CT-induced inhibition of talin cleavage. The membranes were stripped and reprobed for actin to show relative sample loading. A, three high molecular weight bands were observed in untreated cells. Treatment with PD151746 or CT largely eliminated the lowest band. The PKC inhibitor calphostin C dose-dependently blocked the CT-induced inhibition of talin cleavage. B, in contrast, neither PKA inhibitor (H-89 or (Rp)-8-Br-cAMPS) reversed the CT-induced inhibition of the talin cleavage. There was no effect of calphostin C, H-89, or (Rp)-8-Br-cAMPS alone on the presence of the talin cleavage product (not shown).
The Absence of μ-Calpain Causes Bone Loss in Mice
Based on the ability of calpain inhibitors to reduce osteoclast spreading, motility, and bone resorbing activity in vitro, the decreased motility and bone resorbing activity of μ-calpain−/− OCLs, and the CT-induced inhibition of calpain, we hypothesized that the μ-calpain−/− mice would have increased bone mass due to decreased bone resorption. Contrary to our prediction, however, Von Kossa staining of sections of tibias from 10 week-old μ-calpain−/− mice and genetically matched wild type mice revealed a marked decrease in the trabecular bone volume of the secondary spongiosa of the μ-calpain−/− bones (Fig. 8A). Histomorphometric analysis of 5- and 10-week-old μ-calpain−/− and wild type mice confirmed this observation. Trabecular bone volume (Fig. 8B) was markedly decreased at 10 weeks (but not at 5 weeks) in the μ-calpain−/− mice due to diminished trabecular number (Fig. 8, C and D). No difference was observed in the cortical bone thickness at either age (Fig. 8E). Both osteoclast numbers and the amount of bone surface covered by osteoclasts were strongly increased by 30–60% in μ-calpain−/− mice relative to wild type controls at both 5 and 10 weeks (Fig. 8, F and G), whereas no difference was observed in either osteoblast surface or number at either age (Fig. 8, H and I).
FIGURE 8. Bones of μ-calpain−/− mice are progressively osteopenic.
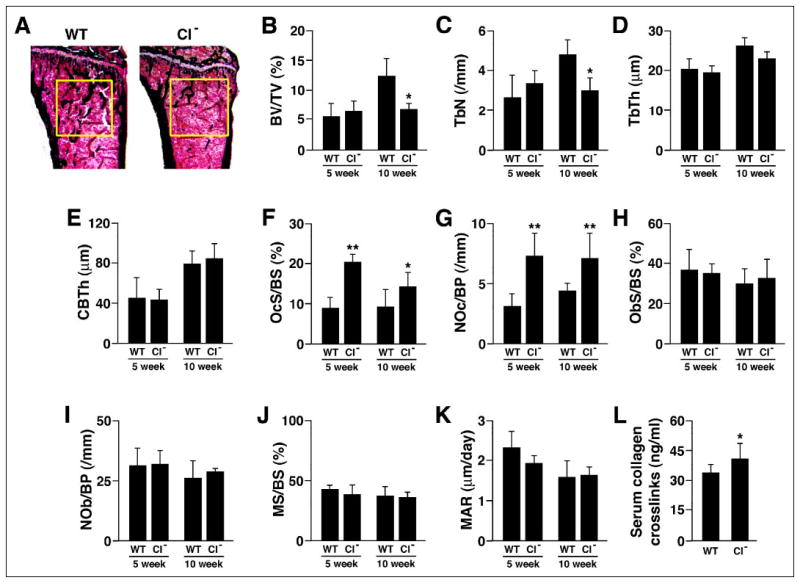
A, Von Kossa staining of tibial proximal metaphyses showing decreased trabeculation of the secondary spongiosa in bones from μ-calpain−/− (CI−) mice. B–E, structural histomorphometry of tibial proximal metaphyses. B, BV/TV, trabecular bone volume; C, TbN, trabecular number; D, TbTh, trabecular thickness; E, CBTh, cortical bone thickness. F–K, cellular and dynamic histomorphometric parameters in tibial proximal metaphyses. F, OcS/BS, osteoclast surface/ bone surface; G, NOc/BP, number of osteoclasts/bone perimeter; H, ObS/BS, osteoblast surface/bone surface; I, NOb/BP, number of osteoblasts/bone perimeter; J, MS/BS, mineralizing surface/bone surface; K, MAR, mineral apposition rate. *, p < 0.05 relative to age-matched WT; **, p < 0.01 relative to age-matched WT. L, serum levels of collagen-derived pyridinoline cross-links.
Bone formation was assessed in the same mice by examining fluorochrome labels incorporated during mineralization. There were no significant differences in either mineralizing surface or mineral apposition rate at either 5 or 10 weeks (Fig. 8, J and K). As a result, bone formation rate was also unchanged (data not shown).
To further assess whether bone resorption was increased in the absence of μ-calpain, we assayed the serum level of pyridinoline cross-links produced from collagen type I digestion, a well established serum marker of bone resorption, in 6-week-old μ-calpain−/− mice and found that this parameter was moderately increased (Fig. 8L). Given the marked elevation in the number of osteoclasts, the moderate increase in cross-links is consistent with the reduced bone resorbing activity of individual μ-calpain−/− OCLs that we observed in vitro. The results suggest that the reduced activity of the individual osteoclasts partly compensates for the greater number of osteoclasts, resulting in the slowly progressing osteopenia that was seen at 10 weeks but not at 5 weeks.
Inhibition or Disruption of Calpain Prolongs the Survival of OCLs
The increased number of osteoclasts in the spongiosa of μ-calpain−/−mice could be the consequence of either increased osteoclast differentiation or increased osteoclast survival. We therefore examined the effect of deleting μ-calpain on the formation of TRAP-positive multinucleated OCLs in the in vitro coculture system. Similar numbers of OCLs were formed in the cocultures of bone marrow cells from wild type and μ-calpain−/−mice (Fig. 9A), indicating that μ-calpain is not required for normal osteoclast differentiation and suggesting that the increased numbers of osteoclasts observed in the μ-calpain−/− mice was not due to increased recruitment. On the other hand, both the absence of μ-calpain and the pharmacological inhibition of calpain prolonged the survival of the OCLs following the removal of the supporting calvarial stromal cells (Fig. 9, B and C). The increased survival of the μ-calpain−/− OCLs may explain the increased number of osteoclasts observed in the μ-calpain−/− mice. CT prolonged the survival of the OCLs in the absence of supporting stromal cells to the same degree as the presence of calpain inhibitors. Interestingly, CT and the calpain inhibitors were more effective at prolonging the survival of the OCLs than the absence of the μ-calpain gene was, suggesting again that these agents are affecting more than μ-calpain in osteoclasts.
FIGURE 9. Reducing calpain activity enhances the survival of OCLs.
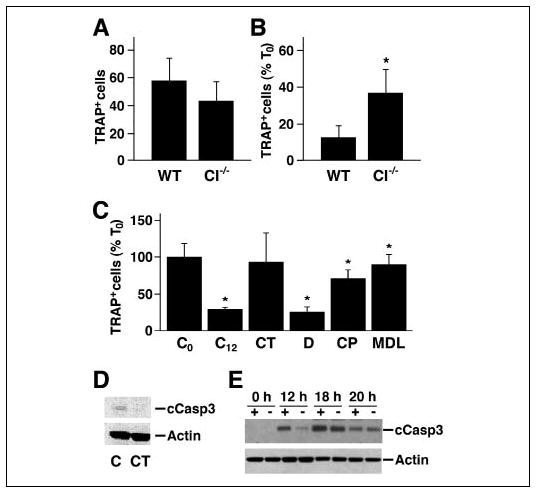
A, OCLs were generated from bone marrow obtained from μ-calpain−/− (CI−/−) and genetically matched wild type mice. After removal of the supporting stromal cell layer, the cells were fixed and stained for TRAP, and the TRAP-positive multinucleated cells were counted. The numbers in the WT and μ-calpain−/− cultures were similar at the time when the stromal cells layers were removed. B, OCLs were incubated in μ-MEM, 10% FBS for 12 h after removing the supporting stromal cell layer and then stained for TRAP and counted. The number of viable OCLs remaining in the WT and μ-calpain−/− cultures after 12 h is shown as a percentage of the number of cells when the stromal cell layer was removed (T0). *, p < 0.01 relative to the WT cells. C, wild type murine OCLs were generated in coculture. The supporting stromal cell layers were removed, and the OCLs were further incubated in the presence or absence of CT (10−9 M) or calpain inhibitors (40 μg/ml CP; 50 μm MDL) or dimethyl sulfoxide alone (D; 1 μl/ml) for 12 h and then TRAP-stained and counted. *, p < 0.05 relative to untreated controls. D, Western blotting of cleaved caspase-3 in WT OCLs treated with 10−9 m CT and untreated controls. E, wild type (+) and μ-calpain−/− (−) OCLs were cultured for the indicated times in the absence of the supporting stromal cell layer. The time-dependent change in cleaved caspase-3 was detected by Western blotting with anti-cleaved caspase-3 antibody. The membrane was stripped and reprobed for actin to show relative sample loading.
Calpains are reported to trigger apoptosis in various cell types by cleaving and activating caspase-3 (24–26), and our laboratory has shown that caspase-3 is cleaved during OCL apoptosis (74). We therefore examined the effect of CT on the presence of cleaved caspase-3 in wild type OCLs cultured in the absence of supporting stromal cells (Fig. 9D) and found that CT inhibited the production of activated caspase-3. We also examined the rate of appearance of cleaved caspase-3 in wild type and in μ-calpain−/−OCLs (Fig. 9E). Cleaved caspase-3 appeared in both the wild type and the μ-calpain−/− OCLs after the supporting stromal cell layer was removed, but the level of cleaved caspase-3 was higher in the wild type OCLs at 12 and 18 h, which would be predicted to promote the more rapid apoptosis of the wild type osteoclasts.
DISCUSSION
Calpains are a family of cytosolic cysteine proteases that catalyze the limited cleavage of specific proteins during regulatory signaling (9, 10). The most studied members of the calpain family are the ubiquitously expressed μ-calpain and m-calpain, which are heterodimeric proteins composed of large 80-kDa catalytic subunits that are unique for the two enzymes and a common 30-kDa regulatory subunit, calpain 4 (11). The proteolytic activities of both μ-calpain and m-calpain are influenced by Ca2−concentration. The two enzymes are also regulated by calpastatin, a highly specific endogenous peptide inhibitor of calpain, and by various signaling events, including PKA- and PKC-dependent mechanisms (43).
Calpains have previously been shown to influence spreading and motility of cells that form focal adhesions and to regulate focal complex assembly/disassembly (8). Calpains cleave several focal adhesion proteins and downstream effectors, although the identities of the specific proteins whose cleavage is most critical for calpain’s effects on the formation of focal adhesions and on cell detachment have not been firmly established (75). In addition, the effects of calpain on cell migration and cell spreading are greatly influenced by cellular context and can be quite different in different cell types (75). Finally, the regulatory mechanisms and the dynamics of assembly/disassembly of focal adhesion structures and podosomes are significantly different, and a role for calpain in podosome regulation has yet to be reported. Since high motility and the dynamic turnover of podosomes are required for efficient osteoclastic bone resorption (7, 76), we sought to test the hypothesis that calpains regulate osteoclast adhesion and motility and thereby possibly contribute to normal bone homeostasis.
Our results support this hypothesis. Pharmacological inhibition of calpain activity in authentic osteoclasts and OCLs using agents that inhibit calpains by two independent mechanisms (modifying a critical cysteine residue in the catalytic site or blocking the calcium-binding site) caused pronounced cell retraction and a sharp reduction in osteoclast motility and bone resorption. The inhibitors also reduced or prevented the constitutive cleavage of three known calpain substrates that are associated with podosomes, talin, filamin, and Pyk2. To test the physiological relevance of the results obtained with the calpain inhibitors, we examined OCLs derived from μ-calpain−/− mice. The μ-calpain−/− OCLs consistently displayed both reduced migration and reduced bone resorption compared with genetically matched wild type cells, providing further evidence that calpain activity is required for normal osteoclast function. Based on the results of these in vitro experiments, we hypothesized that the trabecular bone volume of the μ-calpain−/−mice would be increased. Contrary to our hypothesis, however, we found that bone volume was decreased in these mice at 10 weeks of age, and there was a large increase in the number of osteoclasts at both 5 and 10 weeks. The serum level of pyridinoline cross-links, a marker of bone resorption, was only moderately increased in the μ-calpain−/− mice, however, consistent with the reduced in vitro bone resorbing activity of the μ-calpain−/− osteoclasts. The opposing effects of the increased numbers of osteoclasts in the μ-calpain−/− bones and the reduced resorbing activity of the μ-calpain−/− osteoclasts may explain the relatively slow onset of detectable osteopenia. It should be noted that calpain activity is also required for the normal differentiation of osteoblast cell lines (77), and we cannot rule out the possibility that a change in osteoblast function contributes in some way to the μ-calpain−/− bone phenotype. However, the fact that the histomorphometric analysis revealed no difference in osteoblast number, osteoblast surface, mineralizing surface, or mineral apposition rate indicates that a contribution by the osteoblasts, if any, is extremely subtle. Conversely, the significantly increased values of osteoclast number, osteoclast surface, and pyridinoline cross-links provide strong evidence that bone resorption is increased in the μ-calpain−/− animals.
The increased number of osteoclasts in the bones of the μ-calpain−/− mice appears to be due to increased survival of the osteoclasts rather than to increased production of the cells (Fig. 9). Consistent with reports in the literature (43), the inhibition or absence of μ-calpain prolonged cell survival and reduced the production of caspase-3 when OCLs were cultured in the absence of supporting stromal cells. It is likely that the reduced production of caspase-3 is at least partly responsible for the increased survival, since the cleavage and activation of caspase-3 by calpain (24) is known to trigger apoptosis in other cell types (25, 26). Our finding that the bone marrow cells from μ-calpain−/− mice form osteoclasts as efficiently as those from wild type is contrary to a recently published report that μ-calpain is essential for RANK ligand-induced differentiation of RAW 264.7 cells (78). The opposite findings of the two studies regarding a role for μ-calpain in osteoclast differentiation may be due to unappreciated differences between authentic osteoclast precursors and the RAW 264.7 cell line.
We also found that CT, a potent inhibitor of bone resorption, inhibited osteoclast calpain activity, assessed both by monitoring intracellular calpain-catalyzed cleavage of a fluorogenic substrate and by immunoblotting of the calpain substrates talin and filamin. The effect of CT was rapid, occurring in less than 5 min. The rapid onset indicates that the CT-induced reduction in calpain activity is a consequence of signaling events downstream of the CTR, not changes in calpain gene transcription, a conclusion that is supported by our failure to detect a change in the amounts of μ-calpain and m-calpain in CT-treated OCLs (data not shown). CT and the calpain inhibitors induced similar cell retraction and reductions in osteoclast motility and bone resorption. In all three assays, CT had no further effect on wild type osteoclasts pre-treated with calpain inhibitors. However, CT did induce additional cell retraction and a further reduction of motility and bone resorbing activity in the μ-calpain−/−OCLs, in contrast to the lack of additive effects of CT and calpain inhibitors in wild type osteoclasts and OCLs. These results suggest that the calpain inhibitors and CT act on both μ-calpain and one or more other targets, probably including m-calpain, which was also identified in osteoclasts.
The absence of an additive effect of CT and calpain inhibitors on wild type osteoclast retraction, resorption, and motility suggests that the CT-induced inhibition of calpain contributes to the inhibitory effect of CT on osteoclast activity. This is not to suggest that the calpain inhibition is entirely responsible for the inhibitory effect of CT upon bone resorption. CT also modulates a number of important downstream signaling effectors in osteoclasts, including Erk1/Erk2, mitochondrial cyto-chrome C oxidase, and kinases and phosphatases that regulate tyrosine phosphorylation of a number of target proteins, including components of adhesion complexes (39, 74). The total inhibitory activity of CT depends on these activities as well. The transient effect of CT on the constitutive proteolysis of talin, filamin, and Pyk2 suggests that the inhibition by CT of calpain-catalyzed cleavage of cytoskeletal elements and components of attachment complexes may contribute particularly to the rapid onset of osteoclast retraction and decreased motility.
Our results confirm an earlier report that CT promotes osteoclast survival in vitro (79). The demonstration that CT inhibits both calpain activity and the generation of caspase-3 identifies a new component in the mechanism(s) by which CT affects osteoclast survival. Our laboratory has shown that CT induces the activation of Erk1/Erk2 in osteoclasts (39), which has been shown to promote osteoclast survival (45). Determining if and how CT-induced inhibition of calpain and activation of Erk combine to protect osteoclasts from apoptosis will be an important subject of future studies, particularly since studies in other cell types suggest that Erk activation promotes calpain activity (49, 80–82).
The calpain-dependent mechanisms that promote osteoclast attachment, motility, and bone resorption and the mechanisms by which CT inhibits calpain activity in osteoclasts remain to be characterized. We found evidence that at least three podosome-associated proteins, talin, filamin A, and Pyk2, are constitutively cleaved by calpain in osteoclasts, and it is likely that other podosome-associated calpain substrate proteins are also cleaved. Based on the reported roles of μ-calpain and m-calpain in regulating focal adhesion-based cell attachment and motility, we suggest that calpain-catalyzed cleavage of podosome-associated proteins is required for the rapid assembly and disassembly of podosomes, which in turn is required for normal cell attachment and the high motility that characterized osteoclasts. In fact, two of the cleaved proteins that we identified (talin and Pyk2) have been implicated in the regulation of podosome structure and function (6, 46, 64). Whereas the high molecular weight cleavage products that we observed represent only a small fraction of the amount of the parent proteins, particularly in the cases of talin and filamin A, this is not unexpected if the proteolytic products are generated during a single step in a dynamic and cyclic process, such as podosome assembly and disassembly. First, it is unlikely that more than a small fraction of the parent proteins will be undergoing cleavage at any given time, and second, it will be important to clear the resulting fragments as quickly as possible so that they do not accumulate and interfere with the incorporation of intact parent proteins in newly assembling complexes. In fact, at least in the cases of talin and filamin, the proteolytic fragments are rapidly cleared, as shown by their disappearance within 5 min from rabbit OCLs treated with CT (Fig. 6B). Whereas our results suggest an important role for calpain in osteoclasts, significant additional research will be required to define how calpain-catalyzed cleavage of one or more podosome-associated proteins affect podosome dynamics and osteoclast function.
Several signaling mechanisms that are activated downstream of the CTR have been reported to reduce calpain activity. Both PKA and PKC, which are activated downstream of the CT receptor (29, 34, 36, 37), phosphorylate the two calpains (42, 43, 83) and the endogenous calpain inhibitor calpastatin (84–86), whose specificity and inhibitory efficiency are modulated by both PKA and PKC (40, 41). In addition, calpastatin is activated by lower, more physiological cytosolic Ca2+ concentrations (87–89), suggesting that the increase in [Ca2+]i induced by CT (38) might contribute to calpain inhibition in osteoclasts. Although this study provides only preliminary evidence about the mechanisms responsible for CT-induced regulation of calpain activity, the experiments with the kinase inhibitors suggest that the mechanism involves PKC but not PKA. A role for PKC and not PKA is consistent with our earlier report that calcitonin-induced osteoclast retraction and inhibition of bone resorption is mediated by a PKC-dependent signaling pathway (30).
In conclusion, the present study provides evidence that calpain regulates osteoclast function in at least two somewhat antagonistic ways: first, by inducing cytoskeletal rearrangements and modulating cell attachment, thereby promoting osteoclast spreading, motility, and bone resorbing activity; and second, by limiting the life span of the osteoclast. Both of these activities contribute to determining the rate of overall bone resorption and thereby the balance point of skeletal homeostasis. Furthermore, the present study also suggests that calcitonin exerts its antiresorptive effect and promotes osteoclast survival in part by down-regulating calpain activity, possibly via the PKC signaling pathway.
Acknowledgments
We thank Proskelia (Romainville, France) for kindly providing 1,25-dihydroxyvitamin D3. We also thank Dr. Gianfranco Caselli and Rottapharm S.p.A. (Monza, Italy) for kindly providing materials and facilities and supporting this work.
Footnotes
This work was supported by NIAMS, National Institutes of Health (NIH), Public Health Service Grant AR-49879 (to W. C. H.), NIDCR, NIH, Public Health Service Grant DE-04724 (to R. B.), and NHLBI, NIH, Public Health Service Grant HL-51445 (to A. H. C.). Other support included NIH Grant AR-46032 (to the Yale Core Center for Musculoskeletal Diseases).
The abbreviations used are: CT, calcitonin; α-MEM, minimal essential medium, α modification; Boc-LM-CMAC, 7-amino-4-chloromethylcoumarin; CTR, calcitonin receptor; FBS, fetal bovine serum; OCL, osteoclast-like cell; PBS, phosphate-buffered saline; PKA, cyclic AMP-dependent protein kinase/protein kinase A; PKC, protein kinase C; TRAP, tartrate-resistant acid phosphatase; WT, wild type; (Rp)-8-Br-cAMP, (Rp)-8-bromo-adenosine 3′,5′-cyclic monophosphorothioate.
References
- 1.Marchisio PC, Cirillo D, Naldini L, Primavera MV, Teti A, Zambonin-Zallone A. J Cell Biol. 1984;99:1696–1705. doi: 10.1083/jcb.99.5.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tarone G, Cirillo D, Giancotti FG, Comoglio PM, Marchisio PC. Exp Cell Res. 1985;159:141–157. doi: 10.1016/s0014-4827(85)80044-6. [DOI] [PubMed] [Google Scholar]
- 3.Marchisio PC, Cirillo D, Teti A, Zambonin-Zallone A, Tarone G. Exp Cell Res. 1987;169:202–214. doi: 10.1016/0014-4827(87)90238-2. [DOI] [PubMed] [Google Scholar]
- 4.Nermut MV, Eason P, Hirst EMA, Kellie S. Exp Cell Res. 1991;193:382–397. doi: 10.1016/0014-4827(91)90111-7. [DOI] [PubMed] [Google Scholar]
- 5.Pfaff M, Jurdic P. J Cell Sci. 2001;114:2775–2786. doi: 10.1242/jcs.114.15.2775. [DOI] [PubMed] [Google Scholar]
- 6.Linder S, Aepfelbacher M. Trends Cell Biol. 2003;13:376–385. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 7.Destaing O, Saltel F, Geminard JC, Jurdic P, Bard F. Mol Biol Cell. 2003;14:407–416. doi: 10.1091/mbc.E02-07-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glading A, Lauffenburger DA, Wells A. Trends Cell Biol. 2002;12:46–54. doi: 10.1016/s0962-8924(01)02179-1. [DOI] [PubMed] [Google Scholar]
- 9.Wang KK, Yuen PW. Adv Pharmacol. 1997;37:117–152. doi: 10.1016/s1054-3589(08)60949-7. [DOI] [PubMed] [Google Scholar]
- 10.Pfaff M, Du X, Ginsberg MH. FEBS Lett. 1999;460:17–22. doi: 10.1016/s0014-5793(99)01250-8. [DOI] [PubMed] [Google Scholar]
- 11.Huang Y, Wang KKW. Trends Mol Med. 2001;7:355–362. doi: 10.1016/s1471-4914(01)02049-4. [DOI] [PubMed] [Google Scholar]
- 12.Shiraha H, Glading A, Gupta K, Wells A. J Cell Biol. 1999;146:243–253. doi: 10.1083/jcb.146.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A. Nat Cell Biol. 2004;6:977–983. doi: 10.1038/ncb1175. [DOI] [PubMed] [Google Scholar]
- 14.Tranqui L, Block MR. Exp Cell Res. 1995;217:149–156. doi: 10.1006/excr.1995.1074. [DOI] [PubMed] [Google Scholar]
- 15.Carragher NO, Levkau B, Ross R, Raines EW. J Cell Biol. 1999;147:619–629. doi: 10.1083/jcb.147.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi M, Suzuki H, Kawashima S, Saido TC, Inomata M. Arch Biochem Biophys. 1999;371:133–141. doi: 10.1006/abbi.1999.1427. [DOI] [PubMed] [Google Scholar]
- 17.Yan B, Calderwood DA, Yaspan B, Ginsberg MH. J Biol Chem. 2001;276:28164–28170. doi: 10.1074/jbc.M104161200. [DOI] [PubMed] [Google Scholar]
- 18.Dwyer-Nield LD, Miller AC, Neighbors BW, Dinsdale D, Malkinson AM. Am J Physiol. 1996;270:L526–L534. doi: 10.1152/ajplung.1996.270.4.L526. [DOI] [PubMed] [Google Scholar]
- 19.Bialkowska K, Kulkarni S, Du X, Goll DE, Saido TC, Fox JEB. J Cell Biol. 2000;151:685–696. doi: 10.1083/jcb.151.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoenwaelder SM, Yuan Y, Cooray P, Salem HH, Jackson SP. J Biol Chem. 1997;272:1694–1702. doi: 10.1074/jbc.272.3.1694. [DOI] [PubMed] [Google Scholar]
- 21.Raja S, Avraham S, Avraham H. J Biol Chem. 1997;272:10941–10947. doi: 10.1074/jbc.272.16.10941. [DOI] [PubMed] [Google Scholar]
- 22.Cooray P, Yuan Y, Schoenwaelder SM, Mitchell CA, Salem HH, Jackson SP. Biochem J. 1996;318:41–47. doi: 10.1042/bj3180041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carragher NO, Fincham VJ, Riley D, Frame MC. J Biol Chem. 2001;276:4270–4275. doi: 10.1074/jbc.M008972200. [DOI] [PubMed] [Google Scholar]
- 24.McGinnis KM, Gnegy ME, Park YH, Mukerjee N, Wang KKW. Biochem Biophys Res Commun. 1999;263:94–99. doi: 10.1006/bbrc.1999.1315. [DOI] [PubMed] [Google Scholar]
- 25.Chua BT, Guo K, Li P. J Biol Chem. 2000;275:5131–5135. doi: 10.1074/jbc.275.7.5131. [DOI] [PubMed] [Google Scholar]
- 26.Altznauer F, Conus S, Cavalli A, Folkers G, Simon HU. J Biol Chem. 2004;279:5947–5957. doi: 10.1074/jbc.M308576200. [DOI] [PubMed] [Google Scholar]
- 27.Zaidi M, Datta HK, Moonga BS, MacIntyre I. J Endocrinol. 1990;126:473–481. doi: 10.1677/joe.0.1260473. [DOI] [PubMed] [Google Scholar]
- 28.Chakraborty M, Chatterjee D, Kellokumpu S, Rasmussen H, Baron R. Science. 1991;251:1078–1082. doi: 10.1126/science.1847755. [DOI] [PubMed] [Google Scholar]
- 29.Chabre O, Conklin BR, Lin HY, Lodish HF, Wilson E, Ives HE, Catanzariti L, Hemmings BA, Bourne HR. Mol Endocrinol. 1992;6:551–556. doi: 10.1210/mend.6.4.1316547. [DOI] [PubMed] [Google Scholar]
- 30.Su Y, Chakraborty M, Nathanson MH, Baron R. Endocrinology. 1992;131:1497–1502. doi: 10.1210/endo.131.3.1324163. [DOI] [PubMed] [Google Scholar]
- 31.Chakraborty M, Chatterjee D, Gorelick FS, Baron R. Proc Natl Acad Sci U S A. 1994;91:2115–2119. doi: 10.1073/pnas.91.6.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Findlay DM, Houssami S, Lin HY, Myers DE, Brady CL, Darcy PK, Ikeda K, Martin TJ, Sexton PM. Mol Endocrinol. 1994;8:1691–1700. doi: 10.1210/mend.8.12.7708057. [DOI] [PubMed] [Google Scholar]
- 33.Moore EE, Kuestner RE, Stroop SD, Grant FJ, Matthewes SL, Brady CL, Sexton PM, Findlay DM. Mol Endocrinol. 1995;9:959–968. doi: 10.1210/mend.9.8.7476993. [DOI] [PubMed] [Google Scholar]
- 34.Shyu JF, Inoue D, Baron R, Horne WC. J Biol Chem. 1996;271:31127–31134. doi: 10.1074/jbc.271.49.31127. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, Shyu JF, Santhanagopal A, Inoue D, David JP, Dixon SJ, Horne WC, Baron R. J Biol Chem. 1998;273:19809–19816. doi: 10.1074/jbc.273.31.19809. [DOI] [PubMed] [Google Scholar]
- 36.Naro F, Perez M, Migliaccio S, Galson DL, Orcel P, Teti A, Goldring SR. Endocrinology. 1998;139:3241–3248. doi: 10.1210/endo.139.7.6112. [DOI] [PubMed] [Google Scholar]
- 37.Shyu JF, Zhang Z, Hernandez-Lagunas L, Camerino C, Chen Y, Inoue D, Baron R, Horne WC. Eur J Biochem. 1999;262:95–101. doi: 10.1046/j.1432-1327.1999.00346.x. [DOI] [PubMed] [Google Scholar]
- 38.Santhanagopal A, Chidiac P, Horne WC, Baron R, Dixon SJ. Endocrinology. 2001;142:4401–4413. doi: 10.1210/endo.142.10.8411. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Neff L, Bothwell ALM, Baron R, Horne WC. Bone. 2002;31:359–365. doi: 10.1016/s8756-3282(02)00834-7. [DOI] [PubMed] [Google Scholar]
- 40.Salamino F, De Tullio R, Mengotti P, Melloni E, Pontremoli S. Biochem Biophys Res Commun. 1994;202:1197–1203. doi: 10.1006/bbrc.1994.2057. [DOI] [PubMed] [Google Scholar]
- 41.Averna M, de Tullio R, Passalacqua M, Salamino F, Pontremoli S, Melloni E. Biochem J. 2001;354:25–30. doi: 10.1042/0264-6021:3540025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shiraha H, Glading A, Chou J, Jia Z, Wells A. Mol Cell Biol. 2002;22:2716–2727. doi: 10.1128/MCB.22.8.2716-2727.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goll DE, Thompson VF, Li H, Wei W, Cong J. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 44.David JP, Neff L, Chen Y, Rincon M, Horne WC, Baron R. J Bone Miner Res. 1998;13:1730–1738. doi: 10.1359/jbmr.1998.13.11.1730. [DOI] [PubMed] [Google Scholar]
- 45.Miyazaki T, Katagiri H, Kanegae Y, Takayanagi H, Sawada Y, Yamamoto A, Pando MP, Asano T, Verma IM, Oda H, Nakamura K, Tanaka S. J Cell Biol. 2000;148:333–342. doi: 10.1083/jcb.148.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanjay A, Houghton A, Neff L, Didomenico E, Bardelay C, Antoine E, Levy J, Gailit J, Bowtell D, Horne WC, Baron R. J Cell Biol. 2001;152:181–195. doi: 10.1083/jcb.152.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosser BG, Powers SP, Gores GJ. J Biol Chem. 1993;268:23593–23600. [PubMed] [Google Scholar]
- 48.Glading A, Chang P, Lauffenburger DA, Wells A. J Biol Chem. 2000;275:2390–2398. doi: 10.1074/jbc.275.4.2390. [DOI] [PubMed] [Google Scholar]
- 49.Glading A, Uberall F, Keyse SM, Lauffenburger DA, Wells A. J Biol Chem. 2001;276:23341–23348. doi: 10.1074/jbc.M008847200. [DOI] [PubMed] [Google Scholar]
- 50.Goldberg AF, Barka T. Nature. 1962;195:297. doi: 10.1038/195297a0. [DOI] [PubMed] [Google Scholar]
- 51.Janckila AJ, Li CY, Lam KW, Yam LT. Am J Clin Pathol. 1978;70:45–55. doi: 10.1093/ajcp/70.1.45. [DOI] [PubMed] [Google Scholar]
- 52.Chiusaroli R, Sanjay A, Henriksen K, Engsig MT, Horne WC, Gu H, Baron R. Dev Biol. 2003;261:537–547. doi: 10.1016/s0012-1606(03)00299-9. [DOI] [PubMed] [Google Scholar]
- 53.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 54.Potter DA, Tirnauer JS, Janssen R, Croall DE, Hughes CN, Fiacco KA, Mier JW, Maki M, Herman IM. J Cell Biol. 1998;141:647–662. doi: 10.1083/jcb.141.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carragher NO, Westhoff MA, Fincham VJ, Schaller MD, Frame MC. Curr Biol. 2003;13:1442–1450. doi: 10.1016/s0960-9822(03)00544-x. [DOI] [PubMed] [Google Scholar]
- 56.Sato K, Kawashima S. Biol Chem. 2001;382:743–751. doi: 10.1515/BC.2001.090. [DOI] [PubMed] [Google Scholar]
- 57.Pavalko FM, Otey CA, Burridge K. J Cell Sci. 1989;94:109–118. doi: 10.1242/jcs.94.1.109. [DOI] [PubMed] [Google Scholar]
- 58.Nikki M, Merilainen J, Lehto VP. J Biol Chem. 2002;277:11432–11440. doi: 10.1074/jbc.M111753200. [DOI] [PubMed] [Google Scholar]
- 59.Zaidel-Bar R, Ballestrem C, Kam Z, Geiger B. J Cell Sci. 2003;116:4605–4613. doi: 10.1242/jcs.00792. [DOI] [PubMed] [Google Scholar]
- 60.Marchisio PC, Bergui L, Corbascio GC, Cremona O, D’Urso N, Schena M, Tesio L, Caligaris-Cappio F. Blood. 1988;72:830–833. [PubMed] [Google Scholar]
- 61.Zambonin Zallone A, Teti A, Grano M, Rubinacci A, Abbadini M, Gaboli M, Marchisio PC. Exp Cell Res. 1989;182:645–652. doi: 10.1016/0014-4827(89)90266-8. [DOI] [PubMed] [Google Scholar]
- 62.Duong LT, Lakkakorpi PT, Nakamura I, Machwate M, Nagy RM, Rodan GA. J Clin Invest. 1998;102:881–892. doi: 10.1172/JCI3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duong LT, Nakamura I, Lakkakorpi PT, Lipfert L, Bett AJ, Rodan GA. J Biol Chem. 2001;276:7484–7492. doi: 10.1074/jbc.M008368200. [DOI] [PubMed] [Google Scholar]
- 64.Miyazaki T, Sanjay A, Neff L, Tanaka S, Horne WC, Baron R. J Biol Chem. 2004;279:17660–17666. doi: 10.1074/jbc.M311032200. [DOI] [PubMed] [Google Scholar]
- 65.Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH. J Cell Biol. 1990;111:1089–1105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mehdi S. Trends Biochem Sci. 1991;16:150–153. doi: 10.1016/0968-0004(91)90058-4. [DOI] [PubMed] [Google Scholar]
- 67.Squier MK, Sehnert AJ, Sellins KS, Malkinson AM, Takano E, Cohen JJ. J Cell Physiol. 1999;178:311–319. doi: 10.1002/(SICI)1097-4652(199903)178:3<311::AID-JCP5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 68.Burridge K, Chrzanowska-Wodnicka M. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- 69.Sheetz MP. Nat Rev Mol Cell Biol. 2001;2:392–396. doi: 10.1038/35073095. [DOI] [PubMed] [Google Scholar]
- 70.Robles E, Huttenlocher A, Gomez TM. Neuron. 2003;38:597–609. doi: 10.1016/s0896-6273(03)00260-5. [DOI] [PubMed] [Google Scholar]
- 71.Xi X, Bodnar RJ, Li Z, Lam SCT, Du X. J Cell Biol. 2003;162:329–339. doi: 10.1083/jcb.200303120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Azam M, Andrabi SS, Sahr KE, Kamath L, Kuliopulos A, Chishti AH. Mol Cell Biol. 2001;21:2213–2220. doi: 10.1128/MCB.21.6.2213-2220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baron R, Ravesloot J-H, Neff L, Chakraborty M, Chatterjee D, Lomri A, Horne W. In: Cellular and Molecular Biology of Bone. Noda M, editor. Academic Press, Inc.; San Diego, CA: 1993. pp. 445–495. [Google Scholar]
- 74.Miyazaki T, Neff L, Tanaka S, Horne WC, Baron R. J Cell Biol. 2003;160:709–718. doi: 10.1083/jcb.200209098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perrin BJ, Huttenlocher A. Int J Biochem Cell Biol. 2002;34:722–725. doi: 10.1016/s1357-2725(02)00009-2. [DOI] [PubMed] [Google Scholar]
- 76.Baron R, Horne WC. In: Bone Resorption. Bronner F, Farach-Carson MC, Rubin J, editors. Springer-Verlag; London: 2005. pp. 34–57. [Google Scholar]
- 77.Murray SS, Grisanti MS, Bentley GV, Kahn AJ, Urist MR, Murray EJ. Exp Cell Res. 1997;233:297–309. doi: 10.1006/excr.1997.3550. [DOI] [PubMed] [Google Scholar]
- 78.Lee FY, Kim DW, Karmin JA, Hong D, Chang SS, Fujisawa M, Takayanagi H, Bigliani LU, Blaine TA, Lee HJ. J Biol Chem. 2005;280:29929–29936. doi: 10.1074/jbc.M414600200. [DOI] [PubMed] [Google Scholar]
- 79.Selander KS, Harkonen PL, Valve E, Monkkonen J, Hannuniemi R, Vaananen HK. Mol Cell Endocrinol. 1996;122:119–129. doi: 10.1016/0303-7207(96)03870-1. [DOI] [PubMed] [Google Scholar]
- 80.Cuevas BD, Abell AN, Witowsky JA, Yujiri T, Johnson NL, Kesavan K, Ware M, Jones PL, Weed SA, DeBiasi RL, Oka Y, Tyler KL, Johnson GL. EMBO J. 2003;22:3346–3355. doi: 10.1093/emboj/cdg322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Delmas C, Aragou N, Poussard S, Cottin P, Darbon JM, Manenti S. J Biol Chem. 2003;278:12443–12451. doi: 10.1074/jbc.M209523200. [DOI] [PubMed] [Google Scholar]
- 82.Wong JK, Le HH, Zsarnovszky A, Belcher SM. J Neurosci. 2003;23:4984–4995. doi: 10.1523/JNEUROSCI.23-12-04984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith SD, Jia Z, Huynh KK, Wells A, Elce JS. FEBS Lett. 2003;542:115–118. doi: 10.1016/s0014-5793(03)00361-2. [DOI] [PubMed] [Google Scholar]
- 84.Melloni E, Salamino F, Sparatore B, Michetti M, Pontremoli S, Horecker BL. Arch Biochem Biophys. 1984;232:513–519. doi: 10.1016/0003-9861(84)90568-x. [DOI] [PubMed] [Google Scholar]
- 85.Cong J, Goll DE, Peterson AM, Kapprell HP. J Biol Chem. 1989;264:10096–10103. [PubMed] [Google Scholar]
- 86.Carafoli E, Molinari M. Biochem Biophys Res Commun. 1998;247:193–203. doi: 10.1006/bbrc.1998.8378. [DOI] [PubMed] [Google Scholar]
- 87.Cottin P, Vidalenc PL, Ducastaing A. FEBS Lett. 1981;136:221–224. doi: 10.1016/0014-5793(81)80622-9. [DOI] [PubMed] [Google Scholar]
- 88.Imajoh S, Suzuki K. FEBS Lett. 1985;87:47–50. doi: 10.1016/0014-5793(85)81211-4. [DOI] [PubMed] [Google Scholar]
- 89.Otsuka Y, Goll DE. J Biol Chem. 1987;262:5839–5851. [PubMed] [Google Scholar]