

Abstract
Defensins are endogenous antimicrobial peptides that protect the intestinal mucosa against bacterial invasion. It has been suggested that deficient defensin expression may underlie the chronic inflammation of Crohn disease (CD). The DNA copy number of the beta-defensin gene cluster on chromosome 8p23.1 is highly polymorphic within the healthy population, which suggests that the defective beta-defensin induction in colonic CD could be due to low beta-defensin–gene copy number. Here, we tested this hypothesis, using genomewide DNA copy number profiling by array-based comparative genomic hybridization and quantitative polymerase-chain-reaction analysis of the human beta-defensin 2 (HBD-2) gene. We showed that healthy individuals, as well as patients with ulcerative colitis, have a median of 4 (range 2–10) HBD-2 gene copies per genome. In a surgical cohort with ileal or colonic CD and in a second large cohort with inflammatory bowel diseases, those with ileal resections/disease exhibited a normal median HBD-2 copy number of 4, whereas those with colonic CD had a median of only 3 copies per genome (P=.008 for the surgical cohort; P=.032 for the second cohort). Overall, the copy number distribution in colonic CD was shifted to lower numbers compared with controls (P=.002 for both the surgical cohort and the cohort with inflammatory bowel diseases). Individuals with ⩽3 copies have a significantly higher risk of developing colonic CD than did individuals with ⩾4 copies (odds ratio 3.06; 95% confidence interval 1.46–6.45). An HBD-2 gene copy number of <4 was associated with diminished mucosal HBD-2 mRNA expression (P=.033). In conclusion, a lower HBD-2 gene copy number in the beta-defensin locus predisposes to colonic CD, most likely through diminished beta-defensin expression.
Crohn disease (CD [MIM 266600]) is a severe chronic inflammatory bowel disease characterized by intestinal ulceration that affects predominantly the ileum and colon.1,2 The cause of the disease is unknown. Recent findings have suggested that the mucosal immunological reaction is directed against the resident bacterial flora rather than against tissue antigens.3 This loss of tolerance to the normal flora may be due to a dysregulation of the gut mucosal immune response4 or, alternatively, a break in the antibacterial barrier where microbiota can trigger a deleterious immune response.5
CD appears to be a consequence of both genetic and environmental influences. The 50% concordance rate in MZ twins, who often exhibit the same phenotype,6 suggests a rather balanced impact of genetic and such environmental factors as smoking7,8 or childhood hygiene.9 During a genomewide search, several susceptibility loci and genes—including NOD2 (CARD15) (MIM 605956),10,11 DLG5 (MIM 604090),12 and OCTN (MIM 604190 and 603377)13—have been found to be associated with CD. The best replicated is NOD2, which is involved in intracellular sensing of bacterial muramyl dipeptide, prominently expressed in macrophages and in particular small intestinal Paneth cells.14,15 Despite these significant advances, the multiple susceptibility loci and other genetic factors hitherto identified16 do not satisfactorily explain the inheritance rates.
The clinical syndrome of CD is variable with respect to age at diagnosis, location (small and/or large intestine), and disease behavior (inflammatory, stricturing, or penetrating disease). Therefore, these behavior parameters were used in the Vienna classification,17 recently modified in Montreal.18 Location of disease involvement proved to be stable over time in individual patients, although the biological basis of small- versus large-intestinal involvement is unclear. NOD2 mutations have been shown to be predominantly associated with ileal disease and have been reported to be related to a relative lack of ileal Paneth-cell alpha-defensins HD-5 (MIM 600472) and HD-6 (MIM 600471).19 Defensins are endogenous antibiotic peptides that form a chemical barrier at the epithelial surface, and their relative deficiency may lead to bacterial adherence to the mucosa, slow invasion, and secondary mucosal inflammation.5,20
In contrast to ileal disease, colonic CD is characterized by an impaired induction of the epithelial beta-defensins HBD-2 (MIM 602215), HBD-3 (MIM 606611), and HBD-4.21–23 This relative deficiency of several beta-defensins is unlikely to be explained by multiple coincident mutations or other genetic alterations of all these genes. Detailed studies of the defensin locus on chromosome 8 have uncovered an extensive DNA copy number polymorphism (CNP) of a gene cluster, including, among others, the human beta-defensin genes HBD-2 (DEFB4), HBD-3 (DEFB103), and HBD-4 (DEFB104).24 It has been shown that the number of gene copies is positively correlated with the expression of HBD-2 in leukocytes.24 Since all these defensins were found to be coordinately underexpressed in colonic CD,21 we hypothesized that this particular phenotype may be associated with a low beta-defensin gene cluster copy number. We therefore measured DNA copy number in the beta-defensin cluster in two independent cohorts with inflammatory bowel diseases and related it to mucosal HBD-2 gene expression.
Material and Methods
Patients
The patients from three separate cohorts received diagnoses, with use of the same standard criteria, in Cleveland, Stuttgart, and Vienna and were treated in specialized tertiary-care out- and inpatient centers. Patients gave their informed consent, and local ethics committees approved the study protocols. A small cohort—of patients with colonic CD (n=10) and healthy control individuals (n=10)—was recruited in Stuttgart. Their blood-derived genomic DNA was subjected to the microarray analysis described below. The exploratory cohort was a surgical group of patients who underwent surgical resection at the Cleveland Clinic for treatment of ileal or colonic Crohn disease (table 1). The confirmatory cohort were all whites with CD or ulcerative colitis (UC [MIM 191930]) who were treated at the Robert-Bosch-Hospital (Stuttgart) or the University Hospital (Vienna) (table 1). Diagnostic and (Vienna) classification criteria were the same at both European centers.17 In this classification, L1 is defined as ileal disease only, L2 as colonic disease only, and L3 as ileal as well as colonic disease. Finally, the Stuttgart control group was combined with blood donors (n=103), individuals unaffected by inflammatory bowel disease who underwent surveillance colonoscopy (n=46), and 20 control individuals unaffected by intestinal disease from Cleveland. All colonoscopies that included biopsies were performed on patients from the Stuttgart cohort.
Table 1. .
Number of Patients in the Cohorts Examined for HBD-2 Gene Copy Number
| No. of Subjects |
|||
| Cohort | Cleveland | Stuttgart | Vienna |
| Control | 20 | 149 | |
| CD: | 85 | 54 | 111 |
| Ileal (L1) | 60 | 22 | 38 |
| Colonic (L2) | 25 | 10 | 36 |
| Ileal and colonic (L3) | 22 | 37 | |
| UC | 38 | 37 | |
Array CGH (Array-Based Comparative Genomic Hybridization) Analysis
DNA preparation, labeling, hybridization, and analysis procedures were performed as published elsewhere.25,26 In brief, genomic DNA from fresh-frozen blood obtained from 10 patients with colonic CD and from 10 healthy control individuals was isolated using the Blood and Cell Culture Kit (Qiagen) following the instructions of the supplier. Sample DNA and reference DNA (pooled DNA from six healthy individuals) were labeled differentially with use of the Bioprime Labelling Kit (Invitrogen) and were hybridized on a DNA microarray consisting of ∼8,000 genomic fragments covering the human genome at a resolution of ∼0.5 Mb.27 For the beta-defensin locus at 8p23.1, all additional genomic fragments that were available at the time were added to the microarray to enhance the resolution in the region of interest. The chromosomal mapping information was based on the Ensembl Genome Browser release 36.35i (December 2005). Arrays were scanned, and fluorescence intensities of all spots were filtered (intensity/local background >3; mean/median intensity <1.3; SD of genomic fragment log ratios <0.25) and were normalized blockwise. Chromosomal breakpoints delimiting regions of different copy number status were detected by GLAD (gain and loss analysis of DNA).28
Determination of HBD-2 Gene Copy Number
A TaqMan real-time PCR assay, specifically for amplification of genomic HBD-2, was established by using a specific set of amplification primers (forward 5′-CACCTGTGGTCTCCCTGGAA-3′; reverse 5′-AGCTTCTTGGCCTCCTCATG-3′) and a probe (6-FAM-ATGCTGCAAAAAG-MGB). Quantitative HBD-2 amplification data were normalized to ALB (albumin [MIM 103600]) as an internal reference gene, which was coamplified simultaneously in a single-tube biplex assay. The primers and probe for HBD-2 were designed using Primer Express software, version 1.5 (Applied Biosystems). For albumin, we used the primers and probe that were published elsewhere.29 Primers were purchased from MWG-Biotech, and probes were obtained from Applied Biosystems. Real-time PCR was performed using the ABI Prism 7700 sequence-detection system. Amplification reactions (25 μl each) were performed in triplicate with 20 ng of template DNA, 1× TaqMan Universal Master Mix buffer (Applied Biosystems), 300 nM of each primer, and 200 nM of each fluorogenic probe. Thermal cycling was initiated with a 2-min incubation at 50°C, followed by a first denaturation step of 10 min at 95°C and then by 40 cycles for 15 s at 95°C and for 1 min at 60°C. In each assay, a standard curve was recorded and a no-template control was included. To amplify HBD-2 and albumin in a one-tube biplex assay, limiting primer conditions were identified, to avoid competition of the two reactions. Quantification was performed by both the standard-curve method and the comparative CT (threshold cycle) method, as described elsewhere.29 The assay was validated with a selection of DNA samples, genotyped elsewhere (kindly provided by E. J. Hollox, Nottingham, United Kingdom), that contained 3, 4, 5, and 7 HBD-2 gene copies.24
Real-Time Quantitative PCR
Frozen biopsies were disrupted in 1 ml of Trizol (Gibco BRL) until complete fragmentation occurred. Total RNA was extracted according to the supplier’s protocol. RNA quality was determined by electrophoresis and was quantified by photometry. Subsequently, 2 μg of RNA was reverse transcribed with oligo-dT primers and 200 U Superscript (Gibco BRL), according to the routine procedure.
cDNA samples were subjected to real-time PCR as outlined elsewhere.21 In brief, an aliquot corresponding to 50 ng of RNA was set up in a 20-μl reaction mixture containing 4 mM MgCl2, 0.5 μM of each primer (for HBD-2, forward 5′-ATCAGCCATGAGGGTCTTGT-3′ and reverse 5′-GAGACCACAGGTGCCAATTT-3′ [annealing temperature 60°C; product 172 bp]; for HBD-3, forward 5′-TGAAGCCTAGCAGCTATGAGGATC-3′ and reverse 5′- CCGCCTCTGACTCTGCAATAA-3′ [annealing temperature 62°C; product 128 bp]; for interleukin 8 [IL-8 (MIM 146930)], forward 5′-ATGACTTCCAAGCTGGCCGTGGC-3′ and reverse 5′-TCTCAGCCCTCTTCAAAAACTTC-3′ [annealing temperature touch down protocol 66°C–60°C; product 292 bp] [Sigma]) and 1× LightCycler-FastStart DNA Master SYBR Green I Mix (Roche Molecular Biochemicals) and was loaded in capillary columns. PCR was performed for 45 cycles in a LightCycler (Roche Molecular Biochemicals). After each cycle, fluorescence emission readings reflecting the increase in PCR products were monitored and analyzed using LightCycler software (Roche Molecular Biochemicals).
DNA Sequencing
Exons 1 and 2 of the HBD2 gene and ∼50 bp of adjacent noncoding regions were PCR amplified from genomic DNA and were sequenced on an Applied Biosystems 3100 capillary sequencer with use of Big-Dye chemistry. The sequences of the amplification and nested sequencing primers are available on request.
Statistics
Statistical comparisons of copy numbers of (1) ileal and colonic subsets in the Cleveland cohort as well as (2) patients with L1, L2, and L3 UC and (3) control individuals from the European collective were performed with the (two-sided) Mann-Whitney test. Additionally, Kruskal-Wallis analysis of variance of ranks, including post hoc assessment by Dunn’s test, was performed to correct for multiple testing. Differences in copy number distribution among the clinical cohorts were assessed by the Pearson χ2 test, with continuity corrections. Again, the Mann-Whitney test was used to assess differences in HBD-2 mRNA expression with increasing HBD-2 gene copy numbers.
Results
Microarray Analysis
Using an array CGH with ∼8,000 genomic fragments covering the human genome, with an average resolution of ∼0.5 Mb, we screened 10 patients with colonic CD and 10 healthy control individuals for DNA copy number variations. No gross chromosomal aberrations were present in either group (data not shown), but we detected several known regions of CNP, such as the IGHG1 gene cluster at 14q32.33 (MIM 147100) (data not shown) or the amylase gene cluster at 1p21.1 (MIM 104700) (fig. 1A). Except for the beta-defensin gene cluster at 8p23.1 (fig. 1B), however, no region showed a bias toward copy number loss or gain in patients versus controls. In the Ensembl Genome Browser (release 36.35i), the beta-defensin cluster is shown to be organized as a pair of inverted repeats separated by a sequence gap (fig. 1C). This probably represents the minimal size of the locus in humans. In the patient and control samples that showed copy number variation compared with the control DNA, the size of the variable region was always the same, covering ∼900 kb (megabase 7.1–8.0), including BAC clones RP11-278P18, RP11-1005B13, and RP11-52B19 (Clone Registry). This region contains both copies of the beta-defensin cluster but not the alpha-defensin cluster, as is shown for patient X1604 in figure 1C. When comparing the copy number profiles of the patient and control groups, it became evident that patients with CD, on average, seemed to have fewer copies than did healthy individuals. Relative to a DNA pool from 6 healthy control individuals, 8 of 10 patients with CD displayed small copy number losses, and none displayed copy number gains (fig. 1D). In control individuals, no such bias was observed (fig. 1E). The neighboring alpha-defensin gene cluster (fig. 1C), which mapped to megabase 6.76–6.90, and other beta-defensin–like loci—identified on chromosomal bands 6p12, 20q11.1, and 20p13 by sequence similarity search30—did not show any copy number variation in patients or control individuals (data not shown).
Figure 1. .

A, Copy number ratios of ∼8,000 clones covering the whole genome, with a resolution of ∼0.5 Mb, are shown for patient X1604. The loci of the amylase and beta-defensin gene clusters are indicated. The red lines in panels A and B show the smoothed copy number ratio, as calculated by the GLAD algorithm. B, Copy number ratios of all clones on chromosome 8 of patient X1604. The beta-defensin locus on 8p23.1 is indicated. C, Detail of copy number ratios on 8p23.1 for patient X1604. The alpha-defensin and the two beta-defensin loci are shown by arrows, indicating the genomic orientation of the loci; “sequence gap” indicates a region where no human reference sequence is available. The genomic position, length, and name of genomic fragments covering the region of interest are displayed at the top. The red line indicates the average genomic position of the region that shows a CNP in our 20 hybridizations. D, Copy number ratios in the 8p23.1 region of 10 patients with colonic CD. E, Copy number ratios in the 8p23.1 region of 10 healthy control individuals.
HBD-2 Gene Copy Numbers in Inflammatory Bowel Diseases
Our array CGH analyses clearly indicated that low DNA copy number at the main beta-defensin locus might be connected to CD; however, the size of this initial sample was much too small for reliable correlation analysis. Furthermore, whereas array CGH is extremely useful for whole-genome scans, its quantitative performance is suboptimal at loci that are rich in low copy number DNA repeats (LCRs), such as the beta-defensin cluster. At such loci, the reported copy number ratios can be severely affected by cross-hybridization of the LCRs.25
Therefore, we decided, as an alternative, to use quantitative PCR analysis of the HBD-2 gene to estimate the DNA copy number of the beta-defensin cluster. This HBD2 gene–specific approach was applied to a control population and to two patient cohorts, one from the United States and one from Europe (table 1). In the control population (n=169) the copy numbers had a range of 2–10 per genome, with a median number of 4 copies (fig. 2A). The numbers of control individuals who carry the median (4), below-median (<4), or above-median (>4) number of copies were about equal. Details of the copy number frequencies of all cohorts and subgroups are given in table 2.
Figure 2. .
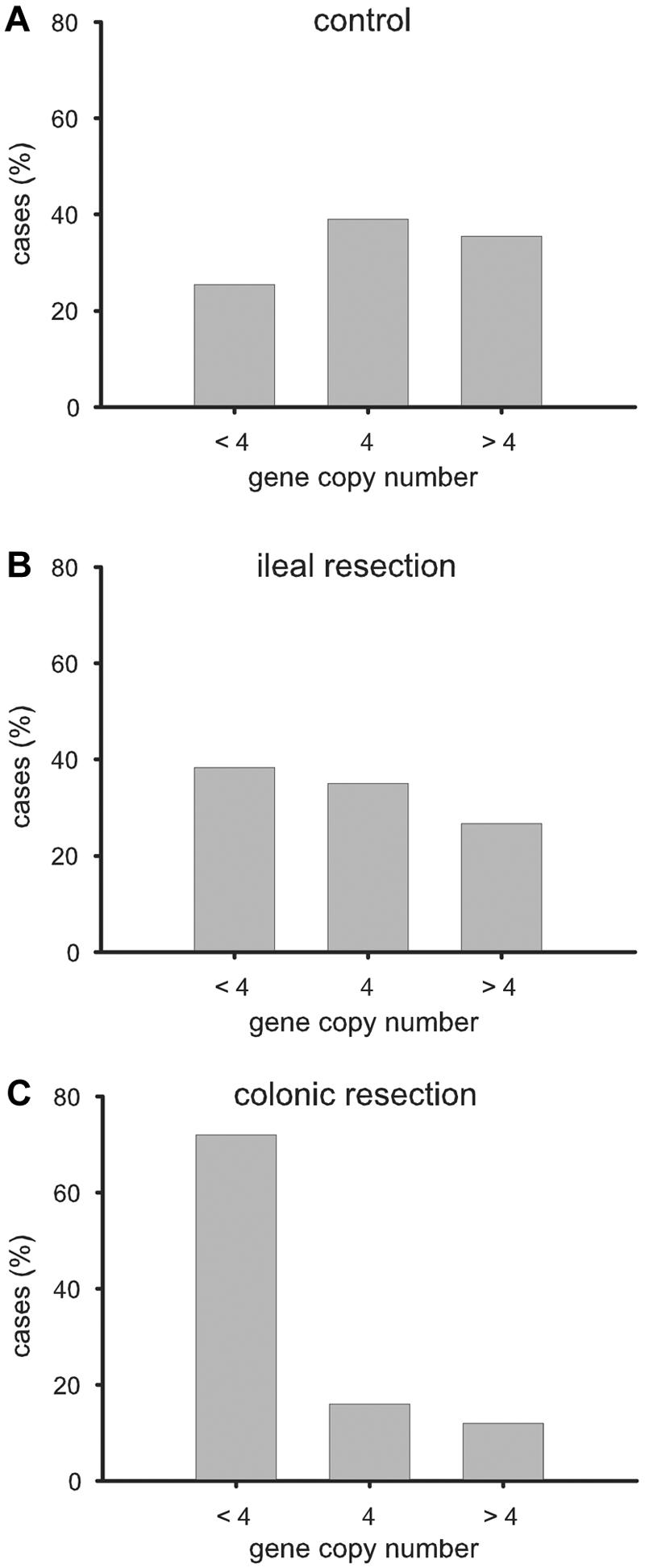
Distribution of HBD-2 gene copy numbers in the 169 controls from Stuttgart and Cleveland (A) and both surgical CD cohorts with ileal (B) and colonic resection (C) allocated to <4, 4, or >4 copies per genome. The difference in distribution between ileal and colonic resection was significant (P=.018 [χ2 test]).
Table 2. .
HBD-2 Gene Copy Number Frequencies[Note]
| Percentage of Cohort with HBD-2 Gene Copy Number of |
|||||||||
| Cohort | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Control: | |||||||||
| Cleveland | 5.0 | 15.0 | 45.0 | 25.0 | 5.0 | 5.0 | |||
| Europe | 2.7 | 23.5 | 38.3 | 23.5 | 10.1 | .7 | .7 | .7 | |
| Cleveland: | |||||||||
| Ileal | 1.7 | 36.7 | 35.0 | 20.0 | 6.7 | ||||
| Colonic | 12.0 | 60.0 | 16.0 | 8.0 | 4.0 | ||||
| European: | |||||||||
| L1 | 5.0 | 23.3 | 43.3 | 21.7 | 1.7 | 5.0 | |||
| L2 | 2.2 | 50.0 | 32.6 | 10.9 | 2.2 | 2.2 | |||
| L3 | 8.5 | 32.2 | 35.6 | 15.3 | 5.1 | 3.4 | |||
| UC | 5.3 | 33.3 | 33.3 | 14.7 | 9.3 | 2.7 | 1.3 | ||
Note.— For the number of patients in these subgroups, see table 1.
The U.S. patient cohort from the Cleveland Clinic consisted of 85 surgical patients with CD who had indications for ileal versus colonic resection. In patients with ileal resections (fig. 2B), the median number of copies was identical to that of the control group (4 copies); also, the frequency distribution of the three subgroups (with <4, with 4, and with >4 gene copy numbers) was not significantly different from that of the control group. In contrast, the majority (72%) of patients with colonic resections had a copy number <4, with a median of 3 copies (fig. 2C). The difference in copy numbers was highly significant (P=.008 [Mann-Whitney]) between the ileal and colonic subgroups with CD. This was maintained when tested by Kruskal-Wallis analysis of variance (P<.01 for both post hoc Dunn test ileal vs. colonic and colonic vs. control). Similarly, the proportion of the three copy number groups in the two subgroups with CD was significantly different (P=.018 [Pearson χ2]).
An independent second cohort of European patients with CD (n=165) from Stuttgart and Vienna was classified according to the Vienna classification of location into those with ileal disease only (L1), with colonic disease only (L2), or with ileal plus colonic disease (L3). Again, ileal CD (L1) exhibited a copy number distribution similar to controls (P>.05), with a majority in the group with 4 gene copies (fig. 3). The median was 4 copies in L1 and controls compared with 3 in L2 (P=.032 and P=.001, respectively [Mann-Whitney]). Analysis of variance and post hoc test identified a significant difference between L2 and controls only (P<.05). Colonic CD only (L2) was characterized by a shift to lower copy numbers, with the majority (52%) carrying <4 copies of HBD-2 (P=.002 vs. controls; P=.037 vs. L1 [Pearson χ2]). Individuals with ⩽3 copies have a significantly higher risk of developing colonic CD than do individuals with ⩾4 copies (odds ratio 3.06; 95% CI 1.46–6.45). Patients with L3 showed an intermediate distribution pattern between controls and L2, and, although the median was the normal 4 copies, the shift in distribution was significant (P=.034 [Mann-Whitney]). In UC, differences with controls were not significant. Detailed noncategorized copy number data are given in table 2.
Figure 3. .

Distribution of HBD-2 gene copy numbers (<4, 4, or >4 copies per genome) in the European cohort as categorized into ileal disease (L1) (A), colonic disease (L2) (B), ileal and colonic disease (L3) (C), and UC (D). L2 (colonic CD) differed significantly from L1 (ileal CD) and controls (P=.037 and P=.002, respectively [χ2 test]).
HBD-2 Expression Related to HBD-2 Gene Copy Numbers in Inflamed Mucosa
Mucosal biopsies were analyzed for HBD-2, HBD-3, and IL-8 expression in patients from the Stuttgart cohort. Since HBD-2 expression is negligible in normal mucosa but is enhanced during inflammation, the biopsy samples were taken from a subgroup of 44 patients with inflammation due to CD (n=17) or UC (n=27). HBD-2 and HBD-3 expression was highly correlated (r=0.84 in CD; r=0.86 in UC). When the mucosal mRNA expression of HBD-2 was related to the HBD-2 gene copy number in the same patients (fig. 4), HBD-2 mRNA expression was significantly diminished in the group with <4 compared with 4 copies (P=.023 [Mann-Whitney]) or ⩾4 (P=.033), whereas IL-8 expression in these copy number groups was not significantly different (data not shown).
Figure 4. .

HBD-2 mRNA expression, with respect to HBD-2 gene copy number, in mucosal specimens from patients with CD and UC with rectal inflammation.
HBD-2 Gene Sequencing
Finally, to investigate the possibility that HBD-2 gene expression might be affected by the presence of gene copies that are inactivated by point mutations, we sequenced exon 1 and exon 2 of the HBD-2 gene in eight patients with colonic CD and eight control individuals. We did not find any nonsense or missense mutations in the coding region. The only sequence variations present were one synonymous SNP in exon 2 (rs2740090) and four SNPs in noncoding sequences (rs2740086, rs2740091, rs2737912, and rs2737913). These SNPs occurred without predominance in either patients or control individuals (data not shown; SNPs were retrieved from dbSNP).
Discussion
CNPs and their effect on phenotypic variation and disease recently have become a major focus of attention.31–33 At present, >200 loci of large-scale CNPs have been detected using BAC arrays (Clone Registry)32,33 or oligonucleotide arrays.31 With use of SNP genotyping data, recent studies have found ∼1,000 fine-scale deletion variants in the human genome.34–36 The role of these genetic alterations and their impact on disease or disease susceptibility is not clear. Several examples show that copy number alterations can lead to altered gene expression and disease; for example, copy number increase at the alpha-synuclein locus (4q21 [MIM 163890]) causes Parkinson disease (MIM 168600),37 and duplication of PMP22 (17q11.2 [MIM 601097]) causes Charcot-Marie-Tooth disease (MIM 118220).38 Recently, a study showed that some cases of early-onset Alzheimer disease (MIM 104300) resulted from a duplication of the APP locus (MIM 104760).39 CNPs can also lead to disease susceptibility, which was shown, for example, for the possession of a low copy number of the CCL3L1 gene (MIM 601395) that is associated with markedly enhanced HIV/AIDS (MIM 609423) susceptibility.40 Gene CNPs, which are associated with variable drug response, have also been described for drug-metabolizing enzymes. For example, inherited amplification of an active gene in the cytochrome P450 CYP2D locus is a cause of ultrarapid metabolism of debrisoquine.41 We have recently reported the development of an RT-PCR–based assay to genotype CYP2D6 (MIM 124030) with respect to its gene copy number,29 similar to the technique used in the present study. A previous study of beta-defensin gene copy number in cystic fibrosis (MIM 219700) had negative results.42
Recently, the finished sequence and a gene catalogue of chromosome 8 were reported.43 A unique feature of the chromosome is a vast region of ∼15 Mb on distal 8p that appears to have a strikingly high mutation rate. Within this region, the CNPs in the defensin cluster on chromosome 8p23.1 are of special interest, since they may be linked to immune function and disease. The copy number variation in this region is complex, with two different clusters of defensins—alpha- and beta-defensins—apparently showing an independent variation of their copy numbers.44 In the alpha-defensin cluster, the genes DEFA1 (MIM 125220) and DEFA3 (MIM 604522) code for the neutrophil defensins HNP-1 and HNP-3, respectively. Their copy numbers, with a range of 5–14, were related to neutrophil HNP 1–3 levels.44 In contrast, copy numbers of DEFA5 (MIM 600472) and DEFA6 (MIM 600471)—coding for the alpha-defensins HD-5 and HD-6, respectively—appear to be stable, with two per diploid genome.44 This was also confirmed for the patients with CD, which suggests that the low HD-5 and HD-6 levels in ileal CD are not related to low copy numbers.45
Prompted by the diminished induction of beta-defensins in the colonic mucosa of patients with CD compared with patients with UC,21–23 we asked whether the copy numbers in the variable beta-defensin locus were low with respect to those in both healthy control individuals and subjects with inflammatory bowel disease (UC and ileal CD). The initial experiments, with use of high-resolution array CGH for genomic copy number profiling, suggested that, compared with controls. there indeed was a lower copy number of the beta-defensin locus in patients with colonic CD. All other regions of CNPs covered by the microarray showed equal copy number distributions between patients with colonic CD and control individuals. A sequencing approach of the HBD-2 gene in a limited number of patients with colonic CD and control individuals revealed several known SNPs but no nonsense or missense mutations. None of the SNPs was preferentially found in either patients or control individuals. Further haplotype analysis of the HBD-2 gene was not performed because of the immense difficulties of such analyses.46
It might seem surprising that the beta-defensin locus on chromosome 8 has not been identified in any of the previous linkage studies,10,11 in particular since deletions recently were shown to be in linkage disequilibrium with SNPs.35 The relevance of this finding for the present study is questionable, however, since the beta-defensin cluster consists of multiple gene copies and is expected to be much less stable than a simple deletion. Therefore, it is not clear whether gene CNPs are at all detectable by linkage analyses. Furthermore, previous studies might have overlooked a possible linkage, since they did not distinguish colonic and ileal CD.
Since DNA microarrays allow full coverage of the human genome but are less suitable for high-throughput studies in large patient cohorts, we developed a real-time PCR-based technique to quantitate HBD-2 copy number variations. Since HBD-2 and HBD-3 copy numbers covary in tandem, it may be assumed that the measured HBD-2 copy number reflects the whole beta-defensin gene cluster.24,44 This is also consistent with the close correlation of HBD-2 and HBD-3 expression demonstrated in the present and a previous study.21 A similar PCR-based technique was recently published by Chen et al.47 The normal median number of 4 copies found in the present study confirms the finding of Chen et al.47 and that of Hollox et al.42 but is slightly lower than the median of 5 observed by Linzmeier et al.44
Since the surgical patients with CD usually have a particularly severe disease course, we first studied patients with ileal versus colonic resections as typical phenotypes. A clear-cut difference by 1 gene copy was observed and was then confirmed in a second, larger cohort of patients classified clinically according to the Vienna classification.17 We focused on localization as a stable factor during the disease course and noted that the patient cohort with colonic disease experienced a shift to lower copy numbers. Although this shift was significant, there was clearly an overlap between the groups, which suggests that additional, still unknown factors of genetic or environmental origin impact CD. Interestingly, patients with both ileal and colonic CD (L3) had an intermediate copy number distribution.
Notably, this relatively small difference in HBD-2 copy numbers appeared to have an influence on mucosal HBD-2 expression by gene dosage effect,48 which was significantly higher in patients with 4 compared with those with fewer gene copies. The biological consequence will likely be an impaired antimicrobial defense in individuals with <4 copies. This is consistent with the previous finding of Hollox et al.24 that HBD-2 expression in lymphoblastoid cell lines was correlated linearly with the HBD-2 gene copy number in the range of 2–7 copies. In some contrast, we did not observe a linear increase in the higher range of copy numbers >4, which may be related to the present complex patient situation in which many other factors, including bacterial flora and inflammation, may affect defensin expression. Alternatively, the plateau in defensin gene expression we observe at copy numbers of ⩾4 might be due to some unknown mechanism limiting gene expression in the native colon tissue, which might not be active in lymphoblastoid cells. At any rate, despite these confounding factors, the gene dosage effect on HBD-2 expression prevailed for low-to-normal copy numbers.
In conclusion, the low beta-defensin gene copy number in CD was clearly associated with colonic disease localization—that is, a specific phenotype of CD. A normal beta-defensin copy number distribution was seen in CD of the ileum and also in UC. This study may provide the genetic basis for the diminished induction of beta-defensins in colonic CD compared with UC in which colonic beta-defensin induction is intact, as described elsewhere.21–23 We suggest that the antibacterial barrier in the colonic mucosa is weakened because of relative defensin deficiency in colonic CD. This finding complements our previous report of a diminished alpha-defensin synthesis in ileal CD, which was further pronounced with a concomitant NOD2 mutation.19,45 This differential compromise of the complex defensin system may explain the different phenotypes of colonic versus ileal CD20,49 as well as the observation that bacteria of the normal flora adhere and sometimes invade the epithelium in CD.50–52 Possibly, this barrier problem triggers the chronic inflammation known as CD.
Acknowledgments
This study was supported by the Robert Bosch Foundation, by National Institutes of Health grant AI32738 (to C.L.B.), by the Deutsche Forschungsgemeinschaft Graduiertenkolleg 886 (to D.E.S.), and by the Bundesministerium für Bildung und Forschung nationales Genomforschungsnetzwerk 01GS0460 (to P.L.) and 01GR0417 (to B.R.). We thank Falk Schubert (Division of Theoretical Bioinformatics, German Cancer Research Center, Heidelberg), for excellent support with the statistical analyses, and Daniel Mertens, for assistance with sequence analysis. We also thank Drs. Victor Fazio, Bo Shen, and others of the Inflammatory Bowel Disease Center (The Cleveland Clinic Foundation), and we thank the colleagues at the Robert-Bosch-Hospital (Stuttgart) for help with sample procurement. Last but not least, we are indebted to the excellent work of our technicians.
Web Resources
The URLs for data presented herein are as follows:
- Clone Registry, http://www.ncbi.nlm.nih.gov/genome/clone/
- Ensembl Genome Browser, http://dec2005.archive.ensembl.org/ (for release 36.35i [December 2005])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CD, NOD2 [CARD15], DLG5, OCTN, HD-5, HD-6, HBD-2, HBD-3, HBD-4, UC, albumin, IL-8, IGHG1, amylase, alpha-synuclein, Parkinson disease, PMP22, Charcot-Marie-Tooth disease, early-onset Alzheimer disease, APP, CCL3L1, HIV/AIDS, CYP2D6, cystic fibrosis, DEFA1, DEFA3, DEFA5, and DEFA6)
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/
References
- 1.Loftus EV Jr (2004) Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology 126:1504–1517 10.1053/j.gastro.2004.01.063 [DOI] [PubMed] [Google Scholar]
- 2.Shanahan F (2002) Crohn’s disease. Lancet 359:62–69 10.1016/S0140-6736(02)07284-7 [DOI] [PubMed] [Google Scholar]
- 3.Duchmann R, Kaiser I, Hermann E, Mayet W, Ewe K, Meyer zum Büschenfelde K-H (1995) Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease. Clin Exp Immunol 102:448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duchmann R, May E, Heike M, Knolle P, Neurath M, Meyer zum Büschenfelde K-H (1999) T cell specificity and cross reactivity towards enterobacteria, Bacteroides, Bifidobacterium, and antigens from resident intestinal flora in humans. Gut 44:812–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fellermann K, Wehkamp J, Herrlinger KR, Stange EF (2003) Crohn’s disease: a defensin deficiency syndrome? Eur J Gastroenterol Hepatol 15:627–634 10.1097/00042737-200306000-00008 [DOI] [PubMed] [Google Scholar]
- 6.Halfvarson J, Bodin L, Tysk C, Lindberg E, Jarnerot G (2003) Inflammatory bowel disease in a Swedish twin cohort: a long-term follow-up of concordance and clinical characteristics. Gastroenterology 124:1767–1773 10.1016/S0016-5085(03)00385-8 [DOI] [PubMed] [Google Scholar]
- 7.Tobin MV, Logan RF, Langman MJ, McConnell RB, Gilmore IT (1987) Cigarette smoking and inflammatory bowel disease. Gastroenterology 93:316–321 [DOI] [PubMed] [Google Scholar]
- 8.Bridger S, Lee JC, Bjarnason I, Jones JE, Macpherson AJ (2002) In siblings with similar genetic susceptibility for inflammatory bowel disease, smokers tend to develop Crohn’s disease and non-smokers develop ulcerative colitis. Gut 51:21–25 10.1136/gut.51.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gent AE, Hellier MD, Grace RH, Swarbrick ET, Coggon D (1994) Inflammatory bowel disease and domestic hygiene in infancy. Lancet 343:766–767 10.1016/S0140-6736(94)91841-4 [DOI] [PubMed] [Google Scholar]
- 10.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411:599–603 10.1038/35079107 [DOI] [PubMed] [Google Scholar]
- 11.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411:603–606 10.1038/35079114 [DOI] [PubMed] [Google Scholar]
- 12.Stoll M, Corneliussen B, Costello CM, Waetzig GH, Mellgard B, Koch WA, Rosenstiel P, Albrecht M, Croucher PJ, Seegert D, Nikolaus S, Hampe J, Lengauer T, Pierrou S, Foelsch UR, Mathew CG, Lagerstrom-Fermer M, Schreiber S (2004) Genetic variation in DLG5 is associated with inflammatory bowel disease. Nat Genet 36:476–480 10.1038/ng1345 [DOI] [PubMed] [Google Scholar]
- 13.Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G, Griffiths AM, George-Hyslop PH, Siminovitch KA (2004) Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet 36:471–475 10.1038/ng1339 [DOI] [PubMed] [Google Scholar]
- 14.Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, Ogunbiyi O, Nunez G, Keshav S (2003) Crohn’s disease and the NOD2 gene: a role for Paneth cells. Gastroenterology 125:47–57 10.1016/S0016-5085(03)00661-9 [DOI] [PubMed] [Google Scholar]
- 15.Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, Zimmermann E, Tretiakova M, Cho JH, Hart J, Greenson JK, Keshav S, Nunez G (2003) Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut 52:1591–1597 10.1136/gut.52.11.1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vermeire S, Rutgeerts P (2005) Current status of genetics research in inflammatory bowel disease. Genes Immun 6:637–645 [DOI] [PubMed] [Google Scholar]
- 17.Gasche C, Scholmerich J, Brynskov J, D’Haens G, Hanauer SB, Irvine EJ, Jewell DP, Rachmilewitz D, Sachar DB, Sandborn WJ, Sutherland LR (2000) A simple classification of Crohn’s disease: report of the Working Party for the World Congresses of Gastroenterology, Vienna 1998. Inflamm Bowel Dis 6:8–15 [DOI] [PubMed] [Google Scholar]
- 18.Silverberg MS, Satsangi J, Ahmad T, Arnott ID, Bernstein CN, Brant SR, Caprilli R, Colombel JF, Gasche C, Geboes K, Jewell DP, Karban A, Loftus EV Jr, Pena AS, Riddell RH, Sachar DB, Schreiber S, Steinhart AH, Targan SR, Vermeire S, Warren BF (2005) Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a working party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol Suppl A 19:5–36 [DOI] [PubMed] [Google Scholar]
- 19.Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M, Herrlinger KR, Stallmach A, Noack F, Fritz P, Schroder JM, Bevins CL, Fellermann K, Stange EF (2004) NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal α-defensin expression. Gut 53:1658–1664 10.1136/gut.2003.032805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wehkamp J, Schmid M, Fellermann K, Stange EF (2005) Defensin deficiency, intestinal microbes, and the clinical phenotypes of Crohn’s disease. J Leukoc Biol 77:460–465 10.1189/jlb.0904543 [DOI] [PubMed] [Google Scholar]
- 21.Wehkamp J, Harder J, Weichenthal M, Müller O, Herrlinger KR, Fellermann K, Schröder JM, Stange EF (2003) Inducible and constitutive b-defensins are differentially expressed in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 9:215–223 10.1097/00054725-200307000-00001 [DOI] [PubMed] [Google Scholar]
- 22.Fahlgren A, Hammarstrom S, Danielsson A, Hammarstrom ML (2003) Increased expression of antimicrobial peptides and lysozyme in colonic epithelial cells of patients with ulcerative colitis. Clin Exp Immunol 131:90–101 10.1046/j.1365-2249.2003.02035.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fahlgren A, Hammarstrom S, Danielsson A, Hammarstrom ML (2004) β-Defensin-3 and -4 in intestinal epithelial cells display increased mRNA expression in ulcerative colitis. Clin Exp Immunol 137:379–385 10.1111/j.1365-2249.2004.02543.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hollox EJ, Armour JAL, Barber JCK (2003) Extensive normal copy number variation of a β-defensin antimicrobial-gene cluster. Am J Hum Genet 73:591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendrzyk F, Korshunov A, Toedt G, Schwarz F, Korn B, Joos S, Hochhaus A, Schoch C, Lichter P, Radlwimmer B (2006) Isochromosome breakpoints on 17p in medulloblastoma are flanked by different classes of DNA sequence repeats. Genes Chromosomes Cancer 45:401–410 10.1002/gcc.20304 [DOI] [PubMed] [Google Scholar]
- 26.Stange DE, Radlwimmer B, Schubert F, Traub F, Pich A, Toedt G, Mendrzyk F, Lehmann U, Eils R, Kreipe H, Lichter P (2006) High-resolution genomic profiling reveals association of chromosomal aberrations on 1q and 16p with histologic and genetic subgroups of invasive breast cancer. Clin Cancer Res 12:345–352 10.1158/1078-0432.CCR-05-1633 [DOI] [PubMed] [Google Scholar]
- 27.Mendrzyk F, Radlwimmer B, Joos S, Kokocinski F, Benner A, Stange DE, Neben K, Fiegler H, Carter NP, Reifenberger G, Korshunov A, Lichter P (2005) Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J Clin Oncol 23:8853–8862 10.1200/JCO.2005.02.8589 [DOI] [PubMed] [Google Scholar]
- 28.Hupe P, Stransky N, Thiery JP, Radvanyi F, Barillot E (2004) Analysis of array CGH data: from signal ratio to gain and loss of DNA regions. Bioinformatics 20:3413–3422 10.1093/bioinformatics/bth418 [DOI] [PubMed] [Google Scholar]
- 29.Schaeffeler E, Schwab M, Eichelbaum M, Zanger UM (2003) CYP2D6 genotyping strategy based on gene copy number determination by TaqMan real-time PCR. Hum Mutat 22:476–485 10.1002/humu.10280 [DOI] [PubMed] [Google Scholar]
- 30.Schutte BC, Mitros JP, Bartlett JA, Walters JD, Jia HP, Welsh MJ, Casavant TL, McCray PB Jr (2002) Discovery of five conserved β-defensin gene clusters using a computational search strategy. Proc Natl Acad Sci USA 99:2129–2133 10.1073/pnas.042692699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M (2004) Large-scale copy number polymorphism in the human genome. Science 305:525–528 10.1126/science.1098918 [DOI] [PubMed] [Google Scholar]
- 32.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C (2004) Detection of large-scale variation in the human genome. Nat Genet 36:949–951 10.1038/ng1416 [DOI] [PubMed] [Google Scholar]
- 33.Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, Oseroff VV, Albertson DG, Pinkel D, Eichler EE (2005) Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77:78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCaroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, Dallaire S, Gabriel SB, Lee C, Daly MJ, Altshuler DM, International HapMap Consortium (2006) Common deletion polymorphisms in the human genome. Nat Genet 28:86–92 [DOI] [PubMed] [Google Scholar]
- 35.Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA (2006) Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet 38:82–85 [DOI] [PubMed] [Google Scholar]
- 36.Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK (2006) A high-resolution survey of deletion polymorphisms in the human genome. Nat Genet 38:75–81 [DOI] [PubMed] [Google Scholar]
- 37.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) α-Synuclein locus triplication causes Parkinson’s disease. Science 302:841 10.1126/science.1090278 [DOI] [PubMed] [Google Scholar]
- 38.Lupski JR, Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, Chakravarti A, Patel PI (1991) DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66:219–232 10.1016/0092-8674(91)90613-4 [DOI] [PubMed] [Google Scholar]
- 39.Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38:24–26 [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O’Connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK (2005) The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307:1434–1440 10.1126/science.1101160 [DOI] [PubMed] [Google Scholar]
- 41.Johansson I, Lundqvist E, Bertilsson L, Dahl ML, Sjoqvist F, Ingelman-Sundberg M (1993) Inherited amplification of an active gene in the cytochrome P450 CYP2D locus as a cause of ultrarapid metabolism of debrisoquine. Proc Natl Acad Sci USA 90:11825–11829 10.1073/pnas.90.24.11825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollox EJ, Davies J, Griesenbach U, Burgess J, Alton EW, Armour JA (2005) Beta-defensin genomic copy number is not a modifier locus for cystic fibrosis. J Negat Results Biomed 4:9 10.1186/1477-5751-4-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nusbaum C, Mikkelsen TS, Zody MC, Asakawa S, Taudien S, Garber M, Kodira CD, et al (2006) DNA sequence and analysis of human chromosome 8. Nature 439:331–335 10.1038/nature04406 [DOI] [PubMed] [Google Scholar]
- 44.Linzmeier RM, Ganz T (2005) Human defensin gene copy number polymorphisms: comprehensive analysis of independent variation in α- and β-defensin regions at 8p22-p23. Genomics 86:423–430 10.1016/j.ygeno.2005.06.003 [DOI] [PubMed] [Google Scholar]
- 45.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, Feathers RW, Chu H, Lima H Jr, Fellermann K, Ganz T, Stange EF, Bevins CL (2005) Reduced Paneth cell α-defensins in ileal Crohn’s disease. Proc Natl Acad Sci USA 102:18129–18134 10.1073/pnas.0505256102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taudien S, Galgoczy P, Huse K, Reichwald K, Schilhabel M, Szafranski K, Shimizu A, Asakawa S, Frankish A, Loncarevic IF, Shimizu N, Siddiqui R, Platzer M (2004) Polymorphic segmental duplications at 8p23.1 challenge the determination of individual defensin gene repertoires and the assembly of a contiguous human reference sequence. BMC Genomics 5:92 10.1186/1471-2164-5-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Q, Book M, Fang X, Hoeft A, Stuber F (2006) Screening of copy number polymorphisms in human β-defensin genes using modified real-time quantitative PCR. J Immunol Methods 308:231–240 10.1016/j.jim.2005.11.001 [DOI] [PubMed] [Google Scholar]
- 48.Lupski JR (1999) Charcot-Marie-Tooth polyneuropathy: duplication, gene dosage, and genetic heterogeneity. Pediatr Res 45:159–165 [DOI] [PubMed] [Google Scholar]
- 49.Wehkamp J, Fellermann K, Herrlinger KR, Bevins CL, Stange EF (2005) Mechanisms of disease: defensins in gastrointestinal diseases. Nat Clin Pract Gastroenterol Hepatol 2:406–415 10.1038/ncpgasthep0265 [DOI] [PubMed] [Google Scholar]
- 50.Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, Weber J, Hoffmann U, Schreiber S, Dietel M, Lochs H (2002) Mucosal flora in inflammatory bowel disease. Gastroenterology 122:44–54 10.1053/gast.2002.30294 [DOI] [PubMed] [Google Scholar]
- 51.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM (2004) Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 127:80–93 10.1053/j.gastro.2004.03.054 [DOI] [PubMed] [Google Scholar]
- 52.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF (2004) High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127:412–421 10.1053/j.gastro.2004.04.061 [DOI] [PubMed] [Google Scholar]