

Abstract
Alignments of homologous genomic sequences are widely used to identify functional genetic elements and study their evolution. Most studies tacitly equate homology of functional elements with sequence homology. This assumption is violated by the phenomenon of turnover, in which functionally equivalent elements reside at locations that are nonorthologous at the sequence level. Turnover has been demonstrated previously for transcriptionfactor-binding sites. Here, we show that transcription start sites of equivalent genes do not always reside at equivalent locations in the human and mouse genomes. We also identify two types of partial turnover, illustrating evolutionary pathways that could lead to complete turnover. These findings suggest that the signals encoding transcription start sites are highly flexible and evolvable, and have cautionary implications for the use of sequence-level conservation to detect gene regulatory elements.
Major goals of genomic biology include identifying the signals in genomes that encode specific biological functions, learning how they evolve, and understanding how and why they differ between species. A popular and promising approach to these questions is to analyze conservation and divergence in genome sequence alignments (Ureta-Vidal et al. 2003). It seems clear that functional genetic elements are supervenient on the genome sequence, but is there a straightforward relationship between the sequence and functional levels? In particular, can tracing the evolution of functional elements be reduced to tracing the evolution of the base pairs that lie within them? For sequences encoding globular protein domains, the answer appears to be yes: The residues in the cores of these domains are so intricately packed that it seems they can only evolve by in-place substitutions of amino acids and thus of the base pairs that encode them. On the other hand, there is evidence that transcription-factor-binding sites undergo turnover (Ludwig et al. 2000; Dermitzakis and Clark 2002). Since most transcription factors bind to short sites (∼4–12 bp) and tolerate considerable variation in target sequences (Wasserman and Sandelin 2004), novel binding sites can easily arise by random mutation (Stone and Wray 2001). Thus, compensatory evolution can occur, so that the loss or weakening of one binding site can be compensated by the gain or strengthening of another. Through this process, the biological function encoded by the sequence may remain the same, but the evolution of the base pairs does not trace out the evolution of the functional elements.
An appreciation of turnover not only reveals the limitations of sequence comparison, it also informs about the nature of the functional elements and how they can evolve. Globular protein domains do not undergo turnover because of their rigid encoding and the intricate interactions among their residues, whereas transcription-factor-binding sites are relatively unconstrained. It follows that we can obtain clues about the rigidity of the sequence requirements for encoding a type of functional element by observing whether turnover takes place. Thus far, turnover has only been described for two types of functional elements, to the best of our knowledge: transcription-factor-binding sites and, perhaps surprisingly, centromeres (Ventura et al. 2001).
In this study, we exploit the recently published, large-scale CAGE data sets (Carninci et al. 2005, 2006) that make it possible, for the first time, to assess turnover of transcription start sites (TSSs). The recent sequencing and analysis of 5,312,921 human and 7,151,511 mouse CAGE tags mapped to the respective genomes provides an unprecedented opportunity to study TSS evolution. Briefly, as described in Carninci et al. (2005, 2006), CAGE tags consist of 20- or 21-nt sequence tags that are derived from the 5′-edge of full-length cDNAs. cDNAs are constructed and specifically purified through selection of biotinylated caps as reviewed in Harbers and Carninci (2005). Protocols for CAGE are described in Kodzius et al. (2006) and Shiraki et al. (2003). A wide variety of cDNA libraries (209 from mouse, 43 from human) was used for CAGE sequencing (see Supplemental Tables S1 and S2): The content of the CAGE data repository has been described in detail elsewhere (Carninci et al. 2005, 2006; Kawaji et al. 2006). Most of the mouse data (84% of mapped tags) and all of the human data are from postnatal sources, or cell lines. Multiple lines of evidence indicate that even single CAGE tags are reliable indicators of TSS locations, including independent RACE verification, the high efficacy of cap selection, high interlibrary reproducibility rates, and comparisons with annotated promoters (Carninci et al. 2006) (see Supplemental material). As the selection of cDNA libraries used for CAGE production is not always comparable between species, sampling problems have to be considered. However, CAGE tags from liver, brain, and adipose tissue are available from both species.
Recent analyses of CAGE and other data have shown that most promoters have a wide distribution of closely located TSSs spanning a region wider than 50 bp, in contrast to the textbook-inflicted view that most genes have one distinct start site governed by a TATA-box (Carninci et al. 2006). The shape of the distribution of tags within such clusters is indicative of both promoter mechanisms and tissue specificity of the downstream gene. When such distributions are located in corresponding regions in human and mouse, the distribution shapes are generally very similar (Carninci et al. 2006). However, this study did not address the extent of cases in which TSSs are found in one genome but not at the corresponding location in the other: that is, the existence and extent of TSS turnover.
Here, we show that while most TSSs in mouse have a counterpart at the homologous location in the human genome sequence and vice versa, a significant number of cases of TSS turnover can be observed in which corresponding TSSs in human and mouse have been translocated by >100 bp. These range from extreme cases in which a TSS is observed in one species but not the other to intermediate instances in which a gene has two alternative start sites that are respectively dominant in each species. The existence of TSS turnover has implications for phylogenetic footprinting algorithms as well as promoter prediction tools that rely on cross-species comparison.
Results
Identification of TSS turnover events
In order to study TSS evolution, we analyzed alignments of human and mouse mRNAs and CAGE tags to their respective genomes. Homologous mRNAs were identified by the criterion that their protein-coding regions overlap by at least 90% in the human–mouse whole-genome BLASTZ NET alignment obtained from the UCSC Genome Browser Database (Karolchik et al. 2003; Schwartz et al. 2003). Thus, their TSSs may or may not reside at aligned positions. Since experimentally determined mRNA sequences may not extend all the way to the true 5′-ends, the mRNA start sites were verified by the presence of nearby CAGE tags (at least 10 tags within ±50 nt). Our analysis necessarily excludes TSSs with low expression levels, and hence low CAGE representation. Finally, we used the whole-genome alignments to find the aligned position in human of each mouse TSS and vice versa. After eliminating redundancy (see Methods), there were 7078 pairs of TSSs of homologous mRNAs satisfying these criteria. Supplemental Table S3 lists the pairs of homologous human and mouse mRNAs, their start sites, and the aligned positions in the other genome, and numbers of supporting CAGE tags.
Interestingly, there are a few cases in which the human TSS lies in a region that is unaligned to the mouse genome or vice versa. These might reflect gaps in the genome sequence, especially when the human TSS is unaligned, since the mouse genome is unfinished. Alternatively, the TSS may lie in an evolutionary hotspot that has diverged too much to be alignable: This might indicate positive selection for lineage-specific adaptations. The other possibilities are that the TSS resides in sequence that has been deleted in one lineage or inserted in the other. All options but the first represent drastic evolutionary changes that might indicate significant functional adaptations. In addition, there are cases in which the human TSS aligns to a different mouse chromosome or strand than the mouse TSS or vice versa. These could be alignment errors; a more interesting possibility is that they reflect recombinations that cause a TSS and its associated 5′-UTR to become linked to a different gene. However, most TSS pairs are consistently aligned: 6846 out of 7078 are mutually alignable, and 6823 align to the same chromosome and strand in the other species, this despite the fact that only ∼40% of each genome is alignable to the other (Waterston et al. 2002).
The TSSs are exactly aligned in only ∼2% of cases (Fig. 1). This is hardly surprising, since our CAGE validation step only determines TSS accuracy to within ±50 nt, and transcription often starts over a broad region rather than at one specific nucleotide (Carninci et al. 2006). In most cases (∼80%), the TSSs lie within 100 nt of one another (Fig. 1), most likely within the same broad promoter region. This typical situation is illustrated by the TSS of the PURA/Pura gene encoding purine-rich element binding protein A (Fig. 2A). Thus, most promoters corresponding to highly expressed transcripts do occur at homologous positions in human and mouse: a conclusion that would not be possible without large-scale TSS mapping.
Figure 1.

Histogram of distances between transcription start sites of homologous transcripts. The x-axis indicates the distance between the human TSS and the human position aligned to the mouse TSS.
Figure 2.

Examples of TSS usage and TSS turnover in promoters of homologous genes. Each histogram subplot is divided into two parts, displaying on the y-axis the CAGE tag distribution of the transcription start sites (TSSs) in the mouse genome (upper part) and of the homologous TSSs in the human genome (lower part). The number of CAGE tags originating from each analyzed tissue is indicated by the color legend (separate for mouse and human). The x-axis displays positions in the alignment between mouse and human; the line between the mouse and human TSS regions represents either aligned nucleotides (thick line) or insertions/deletions (thin line) in the BLASTZ promoter alignment. The homologous mRNAs used for defining the TSS pair (see text) are shown as black arrows with corresponding GenBank accession numbers, where the arrow indicates transcript direction. Full-size images with alignments are available in Supplemental Figure S1. The histograms illustrate different cases of TSS turnover. (A) No turnover (PURA/Pura gene). In most cases, homologous promoter regions have nearly identical TSS usage in the mouse and human genomes. (B) Complete TSS turnover (BCAP29/Bcap29 gene). The TSSs are separated by >100 nt and have zero CAGE tags proximal to the aligned regions in the other species. (C) Shift in alternate TSS usage (HIRIP3/C86302 gene). Similar to case B, but the aligned regions have retained a limited level of transcription initiation activity. (D) TSS sliding (WDR39/Wdr39 gene). The homologous TSS regions overlap, but the flanking regions have no TSSs at the aligned position in the other species.
However, ∼20% of homologous transcripts have TSSs separated by >100 nt, some of them much more (Fig. 1). These might reflect very broad TSS regions, alternative promoters used in both organisms, or TSS turnover. To identify cases of turnover, we counted human CAGE tags near (±50 nt) the aligned position in human of the mouse TSS, and vice versa. We found nine cases in which both aligned positions have zero CAGE tags. Since these TSSs are supported by at least 10 tags, which come from a variety of tissues, the lack of tags at the aligned positions is unlikely to result from chance undersampling. If each TSS is supported by 10 tags, the probability of observing zero tags at the aligned sites by chance undersampling is
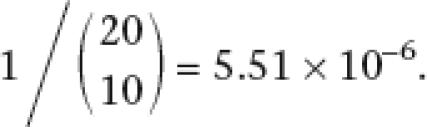 |
It is also unlikely that these TSS offsets are caused by misalignment, since these sequences have clear similarity (58% identity on average). We cannot rule out that the aligned positions initiate transcription at much lower levels. This situation of complete turnover is illustrated by the BCAP29/Bcap29 gene encoding B-cell receptor-associated protein 29 (Fig. 2B). Note that the large number of tags and the variety of tissues make chance undersampling an implausible explanation. A larger version of this figure, showing the aligned sequences, is provided as Supplemental Figure S1B.
Extent of TSS turnover
The criterion of zero tags at the aligned positions is very strict, given the depth of sampling in the CAGE libraries (in many cases >1 million tags per tissue), and that CAGE tags map at low levels to 3′-UTRs, exons, introns, and supposedly intergenic regions (Carninci et al. 2006). Several lines of evidence show that these low-abundance transcripts are unlikely to be due to experimental artifacts (Carninci et al. 2006). If we require that the human TSS has at least 10 times more CAGE tags than the human position aligned to the mouse TSS, and likewise in mouse mutatis mutandis, we find 44 cases of TSS turnover. If we require five times more, we find 87 cases. More generally, for each human and mouse TSS pair separated by >100 nt (in the human sequence), we calculated the probability of the observed skew in tag counts arising by chance undersampling (using the hypergeometric distribution). We found 1414 cases with probability ≤0.01, including 779 cases with probability ≤0.0001, out of 3841 TSS pairs with sufficient data to be considered (at least two tags near the mRNA-defined TSS in both species). Since not all the human and mouse data come from equivalent sources, some of these skews may reflect tissue differences in promoter usage rather than species differences. Nevertheless, it is clear that TSS turnover represents the extreme case of evolutionary shifts in usage of alternative promoters.
Gradual evolutionary shifts in TSS usage
The present CAGE data do not allow evolutionary shifts in promoter usage to be measured very precisely, because they generally do not originate from equivalent human and mouse sources, except in the case of liver, brain, and adipose tissues. (At a deeper level, there probably are no perfect equivalences: What is the human equivalent of 3-mo-old captive-raised, inbred mouse liver?) However, we can gain a rough picture for promoters that are broadly used in many tissues, so that changes in usage are less influenced by nonequivalent sources. To identify broadly used TSSs, we required that no more than a third of CAGE tags within ±50 nt come from any one tissue. Thus, there must be at least three tags from at least three tissues. We found 1263 TSS pairs of homologous transcripts that satisfy this criterion and are separated by >100 nt. To visualize shifts in promoter usage, we plotted the ratio of CAGE tags at the human TSS relative to the aligned position in human of the mouse TSS, against the equivalent ratio in mouse (Fig. 3). In many cases, the usage shifts by more than twofold or even by more than 10-fold. The same qualitative picture emerges when using a promoter separation of 50 nt and counting CAGE tags within ±25 nt, or a promoter separation of 150 nt and tags within ±75 nt, although as expected, the number of TSS pairs with larger separations is lower (Supplemental Fig. S2). These promoter usage differences are not caused by the 16% of mouse tags from pre- and perinatal developmental stages, since similar results are observed when these tags are excluded (Supplemental Fig. S3). This kind of shift in alternative promoter usage is illustrated by the HIRIP3/C86302 gene encoding HIRA interacting protein 3 (Fig. 2C). It is easy to imagine how the weaker promoters might degrade over evolutionary time to reach a situation of complete TSS turnover.
Figure 3.

Changes in usage of alternative promoters between human and mouse. We analyzed 1263 pairs of human and mouse TSSs of homologous transcripts. The x-axis indicates the number of CAGE tags at the human TSS region divided by the number of CAGE tags at the human position aligned to the mouse TSS region. The y-axis indicates the number of CAGE tags at the mouse TSS region divided by the number of CAGE tags at the mouse position aligned to the human TSS region. (Black line) No change in usage; (dashed line) twofold change in usage; (dotted line) 10-fold change in usage.
We also found a different intermediate form of TSS turnover, in which the two transcription start regions are partially shifted but still overlapping. This is illustrated by the WDR39/Wdr39 gene encoding a WD-repeat-containing protein (Fig. 2D). These cases suggest that transcription start regions can shift in the manner of a random walk, by losing transcription initiation activity at one side and gaining it at the other side. Again, it is easy to imagine how this process could lead to completely nonoverlapping TSS regions.
Tissue-specific TSS turnover
Since some genes possess alternative promoters that are differentially used in different tissues (e.g., human CALCR [Anusaksathien et al. 2001] and mouse Ache [Atanasova et al. 1999; large-scale analyses have been described in Kimura et al. [2006] and Carninci et al. [2006]), it would be instructive to dissect evolutionary shifts in promoter usage within specific cell types. To this end, we examined the usage in postnatal liver and brain of human and mouse TSS pairs separated by >100 nt (in the human sequence) (Supplemental Fig. S4). (The number of tags from mouse adipose tissue is somewhat low for this analysis [Supplemental Table S1]). Although many promoters have too few CAGE tags from these tissues (especially liver) to judge, there are clearly some species differences in promoter usage within each tissue. In brain, 246 TSS pairs have significantly skewed tag counts (probability ≤0.01), out of 1376 with sufficient data to be considered (at least two tags near the mRNA-defined TSS in both species). In liver, 45 pairs out of 406 have significant skews. Interestingly, genes with species differences in liver-specific TSS usage include ETFA (P = 3.6 × 10−7), mutations in which can cause lethal glutaric aciduria (Freneaux et al. 1992), and VKORC1 (P = 1.3 × 10−5), a blood clotting factor with human polymorphisms that affect its mRNA levels and sensitivity to the anticoagulant drug warfarin (Rieder et al. 2005). Thus, even essential genes can undergo TSS turnover, and the human VKORC1 promoter may still be evolving. The greater number of significant species differences in brain than in liver is intriguing, but it is simply due to there being more CAGE data from brain (Supplemental Fig. S5; Supplemental Tables S1, S2). There are several limitations to this analysis: (1) These organs contain a complex mixture of cell types. (2) It is unlikely that vital organ samples were obtained in comparable circumstances from humans and mice. (3) The human brain samples were obtained from the cerebrum, whereas many of the mouse samples come from more precisely defined regions such as the visual cortex, hippocampus, and so on (Supplemental Tables S1, S2).
TSSs with high turnover are less conserved
Intuitively, we would expect the genomic region around a TSS that has a significant usage shift to have a lower degree of conservation. In other words, we would expect that the number of evolutionary events since the mouse–human divergence would be higher in cases with complete or partial turnover. To verify this hypothesis, we assessed the conservation of the genomic regions surrounding TSS with a high degree of turnover (we required both tag ratios defined in Fig. 3 to be ≥3, and broad expression from multiple tissues as defined above), and compared to a reference set of TSS (see Methods). As above, we used the whole-genome alignments from the UCSC Genome Browser Database corresponding to ±50 nt from each analyzed TSS. We measured the number of (1) exact matches, (2) aligned but nonidentical bases, and (3) deletions in human, using the mouse sequence as a reference. Regardless of the statistic used, the degree of conservation was significantly higher in the reference set than in the set of TSSs with high turnover (see Fig. 4 for boxplots and Wilcoxon test P-values for all properties measured). In addition, there is a small but significant difference in the lengths of human deletions in the sets. Thus, the initial hypothesis is verified: As expected, the number of evolutionary events is in general higher in promoters where the TSS usage is different between species. (However, 82% of the regions analyzed in the turnover set have sequence identity ≥50%; thus, the turnover observations are unlikely to be artifacts of poor-quality alignment. Moreover, the fraction of alignments with sequence identity <25% in both the turnover and reference sets is below 5%.) Without an outgroup species in which CAGE tags are available, we cannot readily say whether the promoter changes primarily occurred in the mouse or human lineages, although it is recognized that the mouse genome on average has a greater number of evolutionary events since the human–mouse divergence (Waterston et al. 2002). An examination of SNP densities and derived allele frequencies across HapMap populations (see Supplemental material) does not suggest that turnover TSSs and the reference TSSs are subject to different evolutionary pressures in human populations.
Figure 4.

Sequence conservation is significantly lower in TSS regions with high levels of turnover. The degree of conservation in the genomic regions surrounding TSS (±50 bp) with high levels of turnover was investigated by using mouse/human BLASTZ NET alignments. Boxplots representing the (A) number of identical aligned nucleotides, (B) number of aligned nonidentical bases, and (C) number of deletions in the human genome using the mouse sequence as reference are shown for TSS regions exhibiting turnover and TSS regions from the reference set. (D) Distribution of lengths of deletions in the alignments. P-values resulting from a Wilcoxon two-tailed rank test between the sample and reference vectors are shown for each case.
Optimal initiation sites are subject to evolutionary change in promoters exhibiting turnover
We have previously shown that the preferred initiation sequence (the −1, +1 dinucleotide relative to the TSS) is pyrimidine–purine (PyPu) (this is equivalent to the Initiator element defined by Burke and Kadonaga [1997), while the least preferred is PuPy (Carninci et al. 2006). We analyzed the data set to see if certain initiation site changes were more prominent in promoters with a high degree of turnover. Specifically, we analyzed the nucleotide changes in each CAGE-defined TSS initiation site (the −1, +1 dinucleotide in relation to the CAGE-defined TSS; for clarity, each position defined as a TSS by CAGE was analyzed), using the species with the highest number of tag counts as the reference. This analysis was performed, as above, on both the turnover set (promoters with ≥3× turnover) and the reference set.
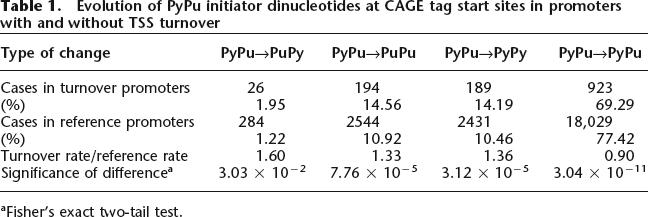
Since nucleotide changes in general are more common in the turnover set (as shown above), we would expect a higher number of changes among dinucleotides at CAGE initiation sites as well as dinucleotides not at CAGE initiation sites. In particular, changes from PyPu dinucleotides are highly overrepresented in CAGE-supported TSS in the turnover set (P = 2.308 × 10−10, χ2 test vs. the reference set). Double mutations from PyPu to PuPy are the most highly overrepresented change, occurring 60% more often in the turnover set (P = 0.01823, right tail Fisher exact test) (Table 1). We have previously shown that any change from a PyPu initiation site will on average decrease expression, and a change to PuPy will have the greatest impact (Carninci et al. 2006). Surprisingly, when we repeat the analysis on dinucleotides with no CAGE support, we observe similar rates of change (data not shown), which would imply that the dinucleotide change is not targeted toward the functional, CAGE-defined TSS. Nevertheless, it is clear that the changes result in a depletion of optimal initiation site dinucleotides (PyPu), which is entirely consistent with the lower number of initiation events measured by CAGE. The depletion of optimal start sites might be one of the functional explanations for TSS turnover.
Table 1.
Evolution of PyPu initiator dinucleotides at CAGE tag start sites in promoters with and without TSS turnover
aFisher’s exact two-tail test.
Sequence features and spatial constraint in promoters exhibiting turnover
While dinucleotide changes may in part explain the turnover phenomena, we also analyzed larger sequence features known to affect promoter function: CpG islands, TATA-boxes, and repetitive sequences, as well as the distance to the translation start site.
Approximately 50% of mammalian promoters are associated with one or more CpG islands (Gardiner-Garden and Frommer 1987; Antequera and Bird 1993). Generally, CpG islands are associated with highly expressed, ubiquitous genes (Schug et al. 2005; Carninci et al. 2006). To investigate if the frequency of CpG islands is altered in promoters with high degrees of TSS turnover, we examined the overlap of CpG islands around the TSS (±50 nt) with high turnover and the reference TSS in the mouse and human genomes (the test and reference sets used are the same as in the conservation analysis above).
For clarity, we refer to a schematic image for pairs of promoters in mouse and human (Fig. 5) in this and the following analyses. For all individual TSS regions (±50 nt), such as locations A, C in mouse (see Fig. 5) and locations B, D in human (see Fig. 5), we tested the distribution of the number of CpG-island-overlapping nucleotides in the turnover set versus the reference set. We found that in the mouse genome, the regions around the TSS with high turnover have a strongly significant decrease in CpG island coverage compared to the reference TSS (P = 0.000374, Wilcoxon two-tailed test). This phenomenon is also observed in human, where the difference is significant but less extreme (P = 0.0397, Wilcoxon two-tailed test). In addition, we retrieved the corresponding homologous dog TSS regions with and without turnover by aligning the mouse TSS to the respective dog genomic position (Methods), and performed the same test. The outcome is similar to that of human (P = 0.0348, Wilcoxon two-tailed test). This finding shows that TSSs with high turnover are less associated with CpG islands than expected, which is important to consider since promoter mapping approaches on a large scale often use CpG islands as a discriminator (Ioshikhes and Zhang 2000; Hannenhalli and Levy 2001).
Figure 5.

Schematic view of pairs of TSSs undergoing turnover. This generalized representation of the panels in Figure 2 provides a reference for method descriptions. Labels A–D correspond to the four different promoters in the two species. The vertical bars indicate supporting CAGE tags.
Since CpG islands are associated with enhanced promoter activity, we investigated whether TSS turnover correlates with turnover of CpG islands. We first measured the number of nucleotides covered by CpG islands in the mouse and human TSS surrounding region (±50 nt) separately for each homologous TSS pair (the pairs A, B and C, D, respectively, in Fig. 5). Subsequently, we calculated the absolute difference in CpG-island-covered nucleotides for each such pair. This analysis was performed on both the high turnover set and the reference set. In addition, we carried out the equivalent analysis for each homologous dog/human and dog/mouse TSS pair. Interestingly, the mouse/human and mouse/dog TSS pairs with high turnover have a greater difference in number of CpG-island-covered nucleotides than the corresponding reference TSS pairs (P = 0.00977 and P = 0.00853, respectively, two-tail Wilcoxon test). In contrast, the dog/human TSS pairs with high turnover show a comparable distribution to the corresponding reference set (P = 0.412, two-tail Wilcoxon test). Thus, the increased number of nucleotide changes in promoters with high turnover affects the CpG island coverage significantly in the mouse genome. As with the human SNP analysis, this observation suggests that the majority of turnover events have not occurred in the human lineage.
In contrast to CpG islands, the turnover and reference sets have no significant difference in occurrence of TATA-boxes in the proximal promoter (P = 0.30, Fisher’s exact test) (see Supplemental material).
As a next step, we investigated if interspersed repetitive elements occur more often at TSSs with high turnover. We identified the number of nucleotides covered by repeat sequences around (±50 bp) each TSS in the high turnover set and reference set, separately for each species. (For clarity, we considered each TSS region A, C in mouse and B, D in human separately for repeat coverage [Fig. 5]) On the contrary, we found significantly less repetitive sequence around the TSS with high turnover than around the reference TSS, in mouse (P = 0.00178, Wilcoxon test), and also in the corresponding regions of the dog genome (P = 0.00326, Wilcoxon test), whereas there is no significant difference in repeat element coverage in human (P = 0.271, Wilcoxon test).
Finally, we investigated if TSS turnover is correlated with the distance to the translation start site. There is a slight tendency for TSSs exhibiting turnover to be further from the translation start site than reference TSSs (P = 0.02, Wilcoxon two-sided test) (Supplemental Fig. S6). This may indicate that TSSs near translation starts are under more evolutionary constraint: Moving upstream or (especially) downstream is likely to interfere with the 5′-UTR and translation.
Discussion
Existence of transcription start site turnover
We have demonstrated that while human and mouse TSSs in most cases reside at equivalent locations, clear exceptions exist. Thus, at least a subset of TSSs is capable of undergoing evolutionary turnover. This suggests that straightforward analyses of genomic alignments, tracing the evolution of base pairs, is a reasonable approach for identifying and studying the evolution of many TSSs and proximal promoters, but it will not always be adequate in terms of function. Likewise, it will usually be correct to infer the location of a TSS in one organism from its location in another “model” organism, but not always.
This has cautionary implications for phylogenetic footprinting algorithms (Loots et al. 2002; Lenhard et al. 2003), where often a known TSS in one species is used to select upstream sequences and/or to anchor alignments. If TSS turnover is significant for a particular gene, this may result in suboptimal sequence selection; moreover, the identification of nontranscribed, conserved regions (a key step in interpreting results of phylogenetic footprinting results) (Wasserman and Sandelin 2004) will be harder since there will be regions that are transcribed in one species but not in the other. At present, we do not know if transcription-factor-binding site turnover is correlated with initiation site turnover. Such a correlation would severely decrease the utility of phylogenetic footprinting algorithms in promoters with significant TSS turnover.
Turnover reveals that at least some TSSs lack rigid sequence requirements; in other words, TSSs have a plasticity more similar to that of transcription-factor-binding sites than globular protein domains. This might be considered unexpected, since TSSs are the nexus where signals from multiple transcription regulatory elements converge, and the molecules that effect transcription initiation (transcription factors, coactivators, pre-initiation complex, etc.) are often viewed as forming a rigid, intricately packed complex resembling a larger version of a globular protein domain. However, we do not at present have a clear understanding of the interaction between bound transcription factors and the pre-initiation complex, and artificial insertions and deletions in proximal promoters are often tolerated. Moreover, the CAGE data show that the majority of promoters have a wide distribution of proximal start sites (Carninci et al. 2006), which implies that precise spacing to regulatory elements is generally not a large constraint (although obvious exceptions exist, most notably the TATA-box).
Analysis constraints due to sample collection
Given that the cDNA library collections in mouse and human are not fully equivalent, there is a risk of misinterpreting tissue and/or stage-specific alternative promoters that are only captured in one species because of the different sampling as evolutionary turnover events. To avoid this problem, we have focused on TSSs that are used in multiple tissues (no more than a third of tags from any one tissue). Since we also require that the pair of TSSs both have a high and opposite CAGE tag count discrepancy (see Methods), it is unlikely that a significant number of the turnover events reported are due to alternative promoter usage. In a more detailed study, we also established clear cases of evolutionary turnover by assessing directly comparable samples in terms of tissue and developmental stage. We have also established that the pre- and perinatal samples that are present in mouse but not human have no significant impact on the results in both these analyses. Nevertheless, the difference in library selection between the two species is a significant constraint with the current CAGE data for this type of study. To allow for a more fine-grained analysis of TSSs in human and mouse, a concerted effort to sample equivalent tissues in both species would be necessary.
Biological mechanisms underlying transcription start site turnover
Evolutionary turnover of TSSs is interesting from an evolutionary perspective but also from a molecular biology standpoint. What is the functional mechanism that accounts for a high level of transcription initiation in a given location for one species, but not the other? As our understanding of TSS selection in general is limited in any eukaryotic species, we can at present only infer certain clues. From a gene ontology (GO) term overrepresentation test (see Supplemental material), we conclude that there is no strong overrepresentation of particular types of genes in the turnover set compared to the reference set, with the possible exception of genes associated with receptor activity (P = 0.0426, two-tailed Fisher’s test, Bonferroni corrected P-value not significant). We have shown that the number of nucleotide changes in promoters with TSS turnover is significantly higher than in the reference set. A reasonable hypothesis is that the changes have an impact on the initiation sites in the promoter. Consistent with this, we have shown that the most optimal initiation site dinucleotide (PyPu) is often changed—in fact, the most overrepresented change is to the least optimal initiation site (PyPu→PuPy). In addition, there is a lower level of CpG island coverage in promoters with high turnover compared to the reference set, and there is a higher amount of change in CpG island coverage between species in aligned TSSs with high turnover. While these findings make it likely that part of the mechanical explanation of turnover lies in the properties of the proximal sequence, it will be important to consider other elements such as the impact of enhancers and chromatin states in future studies of the molecular mechanisms underlying the TSS turnover phenomena.
Gradual evolution of transcription start sites
The cases of partial turnover are interesting because they reveal how complete turnover can arise via gradual evolution. There appear to be two mechanisms: (1) gradual shifting of usage from an old promoter to an alternative promoter (which are separated to start with), and (2) sliding of the promoter along the sequence. The latter process may result from accretion of novel binding sites at one end of the proximal promoter (Ludwig et al. 2005). Our previous analyses of CAGE data have shown that 58% of protein-coding genes have two or more alternative promoters (Carninci et al. 2006), which is about three times more than estimated previously (Landry et al. 2003). The wealth of alternative start sites is consistent with the gradual shifting mechanism.
Turnover causes difficulties with the fundamental concepts of homology, orthology, and paralogy. Are the proximal promoters of human BCAP29 and mouse Bcap29 in Figure 2B homologous? Since homology means descent from a common ancestor (Fitch 2000), the base pairs within these promoters are not homologous. However, if the evolutionary path linking the promoters consists of gradual and compensatory changes, which allow the function to be “teleported” from one location to another, perhaps it is reasonable to consider the promoters regarded as entities to be homologous.
Future directions
To deepen our understanding of evolutionary turnover, it would be valuable to compare TSSs in a wider range of species, which would require CAGE or other comparable data sets from more species, and to examine turnover of other types of elements, such as splice sites and polyadenylation sites. Since it has been observed for transcription-factor-binding sites, centromeres, and now TSSs, turnover should no longer be viewed as peculiar and unexpected in genome evolution.
The flexibility of the signals encoding TSSs, demonstrated by their ability to undergo complete and partial turnover, makes them powerful substrates for evolution. If they can tolerate gradual, compensatory changes, they can equally tolerate gradual, adaptive changes. In this context, it is notable that, while promoters with high turnover are less conserved between human and mouse than other promoters, the SNP density in both sets in human is similar. The SNP data are far from complete, but suggest that the majority of changes we see either occurred in the mouse lineage or in early human and primate evolution: Thus, if the changes are adaptive, they are not recent in human (with possible exceptions such as VKORC1). Supporting this, we also observed a greater difference in CpG coverage in promoters undergoing turnover in mouse, compared to human or dog. However, without CAGE data for an outgroup species, we cannot resolve this question with certainty. It has been argued that purifying selection for regulatory elements is ineffective in hominids (Keightley et al. 2005); thus, TSS turnover may reflect weak selection on TSS locations. However, this study analyzed large (500 bp) upstream sequence blocks, whereas a later study (Bush and Lahn 2005), using smaller blocks (where the block sizes correspond to sizes of known functional regulatory elements), showed that purifying selection is at work in hominid non-coding sequences. Recent reports have revealed a large number of conserved non-coding elements in mammalian genomes (Dermitzakis et al. 2005). Some of these are remarkably conserved and cluster around developmental genes (Sandelin et al. 2004), but we can only confirm that such regions are enhancers in a handful of cases (Gomez-Skarmeta et al. 2006). Conversely, there also is evidence for compensatory changes and turnover of enhancers in eukaryotic promoters (Ludwig et al. 2000). As conservation between species often is used to locate enhancers, we do not at present know the general conservation level of enhancers. Thus, mammalian genomes contain a mixture of constrained and more freely evolving functional elements, and their relative contributions to different types of function (TSS, enhancer, etc.) remains unclear.
TSS and TFBS turnover in combination with the emerging importance of other regulatory systems, including the widespread occurrence of non-coding RNA (Carninci et al. 2005) and natural antisense transcripts (Katayama et al. 2005), hints at a regulatory machinery working at many levels with a high resilience for genetic changes (Pang et al. 2006). This fits with a picture in which globular protein domains are the most ancient and rigid elements encoded in genomes, whereas regulatory signals are more evolvable and likely to be responsible for most of the differences between species that shared a common ancestor relatively recently (Levine and Tjian 2003; Mattick 2004). Therefore, it is a clear possibility that the majority of functional genomic elements (e.g., non-protein-coding RNA, miRNA target sites) will undergo turnover to some degree.
Methods
Data sources
Alignments of human CAGE tags to version hg17 of the human genome and mouse CAGE tags to version mm5 of the mouse genome were obtained from the RIKEN CAGE Basic Database. Whole-genome alignments were obtained from http://hgdownload.cse.ucsc.edu/goldenPath/hg17/vsMm5/axtNet/ and http://hgdownload.cse.ucsc.edu/goldenPath/mm5/vsHg17/axtNet/. mRNA–genome alignments and CDS locations were obtained from the files all_mrna.txt, cds.txt and gbCdnaInfo.txt downloaded (on 9-16-2005) from http://hgdownload.cse.ucsc.edu/goldenPath/hg17/database/ and http://hgdownload.cse.ucsc.edu/goldenPath/mm5/database/. mRNAs aligned to multiple genome locations were discarded. Homologs were defined as mRNAs whose CDS regions overlap by at least 90% of the longer CDS, according to the mm5/vsHg17 alignment. A CAGE tag was considered to support an mRNA TSS if the CAGE tag start site lay within ≤50 nt of the mRNA start site on the same strand.
Availability of CAGE data
The CAGE basic viewer promoter sets (http://gerg01.gsc.riken.jp/cage/) contain all of the basic information on the CAGE libraries, mapped tags, and tissue information. Kawaji et al. (2006) describe the databases and connected interfaces.
TSS redundancy removal
There are often multiple mRNA sequences with identical or very nearby TSSs. This redundancy was removed in the following way. Every pair of mRNAs whose start sites map within 100 nt on the same strand of the same chromosome was compared, and the mRNA supported by fewer CAGE tags (±50 nt) was removed. In case of ties, the mRNA having the alphabetically later GenBank identifier was removed. For each analysis, redundancy removal was performed last (e.g., after finding cases with zero tags at the aligned positions in the other species).
Definition of turnover promoters and reference promoters for subsequent tests
For these analyses, we took pairs of broadly expressed promoters (no more than one-third of supporting CAGE tags from any one tissue) separated by >100 nt. The turnover set is those cases where both tag ratios (the number of CAGE tags at the human TSS divided by the number of CAGE tags at the human position aligned to the mouse TSS, and the number of CAGE tags at the mouse TSS divided by the number of CAGE tags at the mouse position aligned to the human TSS) are ≥3. The reference set consists of the remaining cases. Both reference and turnover sets consist of two pairs of aligned locations: one defined by the mouse cDNA and one by the human cDNA (Fig. 5).
Analysis of mouse–human conservation
We used the NET alignments from the UCSC Genome Browser Database (mouse assembly mm5 aligned to human assembly hg17) to evaluate the conservation properties of TSSs with high levels of turnover. For each pair of TSSs in mouse (as defined above) in the turnover and reference set, we extracted the alignment(s) in the ±50-bp region around the TSS. We analyzed several alignment characteristics using the mouse sequence as the reference in each alignment: (1) aligned nonidentical bases, defined as columns where mouse and human nucleotides are aligned but not identical; (2) exact nucleotide matches, defined as columns where mouse and human nucleotides are aligned and identical; (3) deletions in human, defined as columns where the human sequence has a gap and the mouse sequence has not. As the NET alignments are the product of a local alignment algorithm (BLASTZ) (Schwartz et al. 2003), there are regions that are not fully covered by the alignment(s). Mouse nucleotides within the regions that had no human counterpart in the alignment were counted as deletions in human.
CpG islands and repeats in promoters with transcription start site turnover
For each TSS pair (±50 nt) in mouse and human defined by cDNA (as above), we investigated the number of nucleotides covered by (1) CpG islands and (2) interspersed repeat elements. In both cases, we used the annotation from the UCSC Genome Browser Database (Karolchik et al. 2003) for the respective assemblies.
To define the homologous dog TSS position, we used the mouse TSS position as a reference and retrieved the corresponding dog genomic position using the mouse–dog BLASTZ NET alignments from the UCSC Genome Browser Database (assembly mm5 and canFam1). CpG islands were defined in the ±500-nt region relative to the dog TSS using the UCSC’s CpG program (http://www.cse.ucsc.edu/pipermail/genome/2004-July/005149.html). We then calculated the number of nucleotides in the dog TSS region (±50 nt) overlapped by CpG islands, as for the human and mouse sequences above. We used the annotation at the UCSC site for identifying overlapped interspersed repeat elements in the dog TSS region.
Initiation site evolution
For each TSS pair (mouse and human) with significant turnover (the “turnover” set used above), we used the TSS from the species with most CAGE tags (in this location) as the reference location. For analyzing changes of initiation sites, we defined a region of ±50 bp around the reference TSS location and within that region only considered the positions with CAGE tag support and defined these as [+1]. We then determined for each reference dinucleotide [−1, +1] the corresponding dinucleotide in the other species using the appropriate (i.e., mouse–human or human–mouse, depending on the reference species) BLASTZ NET alignments from the UCSC Genome Browser Database (assembly mm5 and hg17) (Karolchik et al. 2003). We discarded the alignments in those cases in which a reference dinucleotide did not map to an unambiguous, ungapped location in the other species. The analysis was repeated with the “reference set” defined above. In a separate analysis, we studied the remaining dinucleotides in the promoter with an analogous approach: All dinucleotides were considered unless they had a CAGE-supported TSS at position +1.
Acknowledgments
We thank Par G. Engström at the Center for Genomics and Bioinformatics at the Karolinska Institute and the Bergen Center for Computational Science at the University of Bergen for generously providing us with his scientific software libraries. J.P. gratefully acknowledges a joint research grant from the Studienstiftung des deutschen Volkes and the RIKEN Institute; a graduate scholarship by the Clarendon Fund Bursary, Balliol College (Domus Award), and the Studienstiftung des deutschen Volkes. M.C.F. is a University of Queensland Postdoctoral Fellow. This work is supported by (1) a research grant for the RIKEN Genome Exploration Research Project from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese Government to Y.H.; (2) a grant of the Genome Network Project from the Ministry of Education, Culture, Sports, Science and Technology, Japan to Y.H.; (3) a grant for the Strategic Programs for R&D of RIKEN to Y.H. We thank three anonymous referees for constructive criticism.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article is online at http://www.genome. org/cgi/doi/10.1101/gr.5031006
References
- Antequera F., Bird A., Bird A. Number of CpG islands and genes in human and mouse. Proc. Natl. Acad. Sci. 1993;90:11995–11999. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anusaksathien O., Laplace C., Li X., Ren Y., Peng L., Goldring S.R., Galson D.L., Laplace C., Li X., Ren Y., Peng L., Goldring S.R., Galson D.L., Li X., Ren Y., Peng L., Goldring S.R., Galson D.L., Ren Y., Peng L., Goldring S.R., Galson D.L., Peng L., Goldring S.R., Galson D.L., Goldring S.R., Galson D.L., Galson D.L. Tissue-specific and ubiquitous promoters direct the expression of alternatively spliced transcripts from the calcitonin receptor gene. J. Biol. Chem. 2001;276:22663–22674. doi: 10.1074/jbc.M007104200. [DOI] [PubMed] [Google Scholar]
- Atanasova E., Chiappa S., Wieben E., Brimijoin S., Chiappa S., Wieben E., Brimijoin S., Wieben E., Brimijoin S., Brimijoin S. Novel messenger RNA and alternative promoter for murine acetylcholinesterase. J. Biol. Chem. 1999;274:21078–21084. doi: 10.1074/jbc.274.30.21078. [DOI] [PubMed] [Google Scholar]
- Burke T.W., Kadonaga J.T., Kadonaga J.T. The downstream core promoter element, DPE, is conserved from Drosophila to humans and is recognized by TAFII60 of. Drosophila. Genes & Dev. 1997;11:3020–3031. doi: 10.1101/gad.11.22.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush E.C., Lahn B.T., Lahn B.T. Selective constraint on noncoding regions of hominid genomes. PLoS Comput. Biol. 2005;1:e73. doi: 10.1371/journal.pcbi.0010073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P.T., Kasukawa S., Katayama J., Gough M.C., Frith N., Maeda R., Oyama T., Ravasi B., Lenhard C., Wells R., Kasukawa S., Katayama J., Gough M.C., Frith N., Maeda R., Oyama T., Ravasi B., Lenhard C., Wells R., Katayama J., Gough M.C., Frith N., Maeda R., Oyama T., Ravasi B., Lenhard C., Wells R., Gough M.C., Frith N., Maeda R., Oyama T., Ravasi B., Lenhard C., Wells R., Frith N., Maeda R., Oyama T., Ravasi B., Lenhard C., Wells R., Maeda R., Oyama T., Ravasi B., Lenhard C., Wells R., Oyama T., Ravasi B., Lenhard C., Wells R., Ravasi B., Lenhard C., Wells R., Lenhard C., Wells R., Wells R., et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Carninci P., Sandelin A., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P., Frith M., Sandelin A., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P., Frith M., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P., Frith M., Katayama S., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P., Frith M., Shimokawa K., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P., Frith M., Ponjavic J., Semple C.A.M., Taylor M.S., Engstrom P., Frith M., Semple C.A.M., Taylor M.S., Engstrom P., Frith M., Taylor M.S., Engstrom P., Frith M., Engstrom P., Frith M., Frith M., et al. 2006Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. (in press). [DOI] [PubMed] [Google Scholar]
- Dermitzakis E.T., Clark A.G., Clark A.G. Evolution of transcription factor binding sites in mammalian gene regulatory regions: Conservation and turnover. Mol. Biol. Evol. 2002;19:1114–1121. doi: 10.1093/oxfordjournals.molbev.a004169. [DOI] [PubMed] [Google Scholar]
- Dermitzakis E.T., Reymond A., Antonarakis S.E., Reymond A., Antonarakis S.E., Antonarakis S.E. Conserved non-genic sequences—An unexpected feature of mammalian genomes. Nat. Rev. Genet. 2005;6:151–157. doi: 10.1038/nrg1527. [DOI] [PubMed] [Google Scholar]
- Fitch W.M. Homology: A personal view on some of the problems. Trends Genet. 2000;16:227–231. doi: 10.1016/s0168-9525(00)02005-9. [DOI] [PubMed] [Google Scholar]
- Freneaux E., Sheffield V.C., Molin L., Shires A., Rhead W.J., Sheffield V.C., Molin L., Shires A., Rhead W.J., Molin L., Shires A., Rhead W.J., Shires A., Rhead W.J., Rhead W.J. Glutaric acidemia type II. Heterogeneity in β-oxidation flux, polypeptide synthesis, and complementary DNA mutations in the α subunit of electron transfer flavoprotein in eight patients. J. Clin. Invest. 1992;90:1679–1686. doi: 10.1172/JCI116040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M., Frommer M., Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Gomez-Skarmeta J.L., Lenhard B., Becker T.S., Lenhard B., Becker T.S., Becker T.S. New technologies, new findings, and new concepts in the study of vertebrate cis-regulatory sequences. Dev. Dyn. 2006;235:870–885. doi: 10.1002/dvdy.20659. [DOI] [PubMed] [Google Scholar]
- Hannenhalli S., Levy S., Levy S.2001Promoter prediction in the human genome. Bioinformatics (Suppl 1) 17S90–S96. [DOI] [PubMed] [Google Scholar]
- Harbers M., Carninci P., Carninci P. Tag-based approaches for transcriptome research and genome annotation. Nat. Methods. 2005;2:495–502. doi: 10.1038/nmeth768. [DOI] [PubMed] [Google Scholar]
- Ioshikhes I.P., Zhang M.Q., Zhang M.Q. Large-scale human promoter mapping using CpG islands. Nat. Genet. 2000;26:61–63. doi: 10.1038/79189. [DOI] [PubMed] [Google Scholar]
- Karolchik D., Baertsch R., Diekhans M., Furey T.S., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., Baertsch R., Diekhans M., Furey T.S., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., Diekhans M., Furey T.S., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., Furey T.S., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., Schwartz M., Sugnet C.W., Thomas D.J., Sugnet C.W., Thomas D.J., Thomas D.J., et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama S., Tomaru Y., Kasukawa T., Waki K., Nakanishi M., Nakamura M., Nishida H., Yap C.C., Suzuki M., Kawai J., Tomaru Y., Kasukawa T., Waki K., Nakanishi M., Nakamura M., Nishida H., Yap C.C., Suzuki M., Kawai J., Kasukawa T., Waki K., Nakanishi M., Nakamura M., Nishida H., Yap C.C., Suzuki M., Kawai J., Waki K., Nakanishi M., Nakamura M., Nishida H., Yap C.C., Suzuki M., Kawai J., Nakanishi M., Nakamura M., Nishida H., Yap C.C., Suzuki M., Kawai J., Nakamura M., Nishida H., Yap C.C., Suzuki M., Kawai J., Nishida H., Yap C.C., Suzuki M., Kawai J., Yap C.C., Suzuki M., Kawai J., Suzuki M., Kawai J., Kawai J., et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- Kawaji H., Kasukawa T., Fukuda S., Katayama S., Kai C., Kawai J., Carninci P., Hayashizaki Y., Kasukawa T., Fukuda S., Katayama S., Kai C., Kawai J., Carninci P., Hayashizaki Y., Fukuda S., Katayama S., Kai C., Kawai J., Carninci P., Hayashizaki Y., Katayama S., Kai C., Kawai J., Carninci P., Hayashizaki Y., Kai C., Kawai J., Carninci P., Hayashizaki Y., Kawai J., Carninci P., Hayashizaki Y., Carninci P., Hayashizaki Y., Hayashizaki Y. CAGE basic/analysis databases: The CAGE resource for comprehensive promoter analysis. Nucleic Acids Res. 2006;34:D632–D636. doi: 10.1093/nar/gkj034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P.D., Lercher M.J., Eyre-Walker A., Lercher M.J., Eyre-Walker A., Eyre-Walker A. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol. 2005;3:e42. doi: 10.1371/journal.pbio.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K., Wakamatsu A., Suzuki Y., Ota T., Nishikawa T., Yamashita R., Yamamoto J., Sekine M., Tsuritani K., Wakaguri H., Wakamatsu A., Suzuki Y., Ota T., Nishikawa T., Yamashita R., Yamamoto J., Sekine M., Tsuritani K., Wakaguri H., Suzuki Y., Ota T., Nishikawa T., Yamashita R., Yamamoto J., Sekine M., Tsuritani K., Wakaguri H., Ota T., Nishikawa T., Yamashita R., Yamamoto J., Sekine M., Tsuritani K., Wakaguri H., Nishikawa T., Yamashita R., Yamamoto J., Sekine M., Tsuritani K., Wakaguri H., Yamashita R., Yamamoto J., Sekine M., Tsuritani K., Wakaguri H., Yamamoto J., Sekine M., Tsuritani K., Wakaguri H., Sekine M., Tsuritani K., Wakaguri H., Tsuritani K., Wakaguri H., Wakaguri H., et al. Diversification of transcriptional modulation: Large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2006;16:55–65. doi: 10.1101/gr.4039406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodzius R., Kojima M., Nishiyori H., Nakamura M., Fukuda S., Tagami M., Sasaki D., Imamura K., Kai C., Harbers M., Kojima M., Nishiyori H., Nakamura M., Fukuda S., Tagami M., Sasaki D., Imamura K., Kai C., Harbers M., Nishiyori H., Nakamura M., Fukuda S., Tagami M., Sasaki D., Imamura K., Kai C., Harbers M., Nakamura M., Fukuda S., Tagami M., Sasaki D., Imamura K., Kai C., Harbers M., Fukuda S., Tagami M., Sasaki D., Imamura K., Kai C., Harbers M., Tagami M., Sasaki D., Imamura K., Kai C., Harbers M., Sasaki D., Imamura K., Kai C., Harbers M., Imamura K., Kai C., Harbers M., Kai C., Harbers M., Harbers M., et al. CAGE: Cap analysis of gene expression. Nat. Methods. 2006;3:211–222. doi: 10.1038/nmeth0306-211. [DOI] [PubMed] [Google Scholar]
- Landry J.R., Mager D.L., Wilhelm B.T., Mager D.L., Wilhelm B.T., Wilhelm B.T. Complex controls: The role of alternative promoters in mammalian genomes. Trends Genet. 2003;19:640–648. doi: 10.1016/j.tig.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Lenhard B., Sandelin A., Mendoza L., Engstrom P., Jareborg N., Wasserman W.W., Sandelin A., Mendoza L., Engstrom P., Jareborg N., Wasserman W.W., Mendoza L., Engstrom P., Jareborg N., Wasserman W.W., Engstrom P., Jareborg N., Wasserman W.W., Jareborg N., Wasserman W.W., Wasserman W.W. Identification of conserved regulatory elements by comparative genome analysis. J. Biol. 2003;2:13. doi: 10.1186/1475-4924-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M., Tjian R., Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- Loots G.G., Ovcharenko I., Pachter L., Dubchak I., Rubin E.M., Ovcharenko I., Pachter L., Dubchak I., Rubin E.M., Pachter L., Dubchak I., Rubin E.M., Dubchak I., Rubin E.M., Rubin E.M. rVista for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res. 2002;12:832–839. doi: 10.1101/gr.225502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M.Z., Bergman C., Patel N.H., Kreitman M., Bergman C., Patel N.H., Kreitman M., Patel N.H., Kreitman M., Kreitman M. Evidence for stabilizing selection in a eukaryotic enhancer element. Nature. 2000;403:564–567. doi: 10.1038/35000615. [DOI] [PubMed] [Google Scholar]
- Ludwig M.Z., Palsson A., Alekseeva E., Bergman C.M., Nathan J., Kreitman M., Palsson A., Alekseeva E., Bergman C.M., Nathan J., Kreitman M., Alekseeva E., Bergman C.M., Nathan J., Kreitman M., Bergman C.M., Nathan J., Kreitman M., Nathan J., Kreitman M., Kreitman M. Functional evolution of a cis-regulatory module. PLoS Biol. 2005;3:e93. doi: 10.1371/journal.pbio.0030093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick J.S. RNA regulation: A new genetics? Nat. Rev. Genet. 2004;5:316–323. doi: 10.1038/nrg1321. [DOI] [PubMed] [Google Scholar]
- Pang K.C., Frith M.C., Mattick J.S., Frith M.C., Mattick J.S., Mattick J.S. Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet. 2006;22:1–5. doi: 10.1016/j.tig.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Rieder M.J., Reiner A.P., Gage B.F., Nickerson D.A., Eby C.S., McLeod H.L., Blough D.K., Thummel K.E., Veenstra D.L., Rettie A.E., Reiner A.P., Gage B.F., Nickerson D.A., Eby C.S., McLeod H.L., Blough D.K., Thummel K.E., Veenstra D.L., Rettie A.E., Gage B.F., Nickerson D.A., Eby C.S., McLeod H.L., Blough D.K., Thummel K.E., Veenstra D.L., Rettie A.E., Nickerson D.A., Eby C.S., McLeod H.L., Blough D.K., Thummel K.E., Veenstra D.L., Rettie A.E., Eby C.S., McLeod H.L., Blough D.K., Thummel K.E., Veenstra D.L., Rettie A.E., McLeod H.L., Blough D.K., Thummel K.E., Veenstra D.L., Rettie A.E., Blough D.K., Thummel K.E., Veenstra D.L., Rettie A.E., Thummel K.E., Veenstra D.L., Rettie A.E., Veenstra D.L., Rettie A.E., Rettie A.E. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005;352:2285–2293. doi: 10.1056/NEJMoa044503. [DOI] [PubMed] [Google Scholar]
- Sandelin A., Bailey P., Bruce S., Engstrom P.G., Klos J.M., Wasserman W.W., Ericson J., Lenhard B., Bailey P., Bruce S., Engstrom P.G., Klos J.M., Wasserman W.W., Ericson J., Lenhard B., Bruce S., Engstrom P.G., Klos J.M., Wasserman W.W., Ericson J., Lenhard B., Engstrom P.G., Klos J.M., Wasserman W.W., Ericson J., Lenhard B., Klos J.M., Wasserman W.W., Ericson J., Lenhard B., Wasserman W.W., Ericson J., Lenhard B., Ericson J., Lenhard B., Lenhard B. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics. 2004;5:99. doi: 10.1186/1471-2164-5-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug J., Schuller W.P., Kappen C., Salbaum J.M., Bucan M., Stoeckert C.J., Jr., Schuller W.P., Kappen C., Salbaum J.M., Bucan M., Stoeckert C.J., Jr., Kappen C., Salbaum J.M., Bucan M., Stoeckert C.J., Jr., Salbaum J.M., Bucan M., Stoeckert C.J., Jr., Bucan M., Stoeckert C.J., Jr., Stoeckert C.J., Jr. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 2005;6:R33. doi: 10.1186/gb-2005-6-4-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S., Kent W.J., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W., Kent W.J., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W., Baertsch R., Hardison R.C., Haussler D., Miller W., Hardison R.C., Haussler D., Miller W., Haussler D., Miller W., Miller W. Human–mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki T., Kondo S., Katayama S., Waki K., Kasukawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T., Kondo S., Katayama S., Waki K., Kasukawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T., Katayama S., Waki K., Kasukawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T., Waki K., Kasukawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T., Kasukawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T., Kodzius R., Watahiki A., Nakamura M., Arakawa T., Watahiki A., Nakamura M., Arakawa T., Nakamura M., Arakawa T., Arakawa T., et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. 2003;100:15776–15781. doi: 10.1073/pnas.2136655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J.R., Wray G.A., Wray G.A. Rapid evolution of cis-regulatory sequences via local point mutations. Mol. Biol. Evol. 2001;18:1764–1770. doi: 10.1093/oxfordjournals.molbev.a003964. [DOI] [PubMed] [Google Scholar]
- Ureta-Vidal A., Ettwiller L., Birney E., Ettwiller L., Birney E., Birney E. Comparative genomics: Genome-wide analysis in metazoan eukaryotes. Nat. Rev. Genet. 2003;4:251–262. doi: 10.1038/nrg1043. [DOI] [PubMed] [Google Scholar]
- Ventura M., Archidiacono N., Rocchi M., Archidiacono N., Rocchi M., Rocchi M. Centromere emergence in evolution. Genome Res. 2001;11:595–599. doi: 10.1101/gr.152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman W.W., Sandelin A., Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- Waterston R.H.K., Lindblad-Toh E., Birney J., Rogers J.F., Abril P., Agarwal R., Agarwala R., Ainscough M., Alexandersson P., An S.E., Lindblad-Toh E., Birney J., Rogers J.F., Abril P., Agarwal R., Agarwala R., Ainscough M., Alexandersson P., An S.E., Birney J., Rogers J.F., Abril P., Agarwal R., Agarwala R., Ainscough M., Alexandersson P., An S.E., Rogers J.F., Abril P., Agarwal R., Agarwala R., Ainscough M., Alexandersson P., An S.E., Abril P., Agarwal R., Agarwala R., Ainscough M., Alexandersson P., An S.E., Agarwal R., Agarwala R., Ainscough M., Alexandersson P., An S.E., Agarwala R., Ainscough M., Alexandersson P., An S.E., Ainscough M., Alexandersson P., An S.E., Alexandersson P., An S.E., An S.E., et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]