

Abstract
Cav2.1 channels, which mediate P/Q-type Ca2+ currents, undergo Ca2+/calmodulin (CaM)-dependent inactivation and facilitation that can significantly alter synaptic efficacy. Here we report that the neuronal Ca2+-binding protein 1 (CaBP1) modulates Cav2.1 channels in a manner that is markedly different from modulation by CaM. CaBP1 enhances inactivation, causes a depolarizing shift in the voltage dependence of activation, and does not support Ca2+-dependent facilitation of Cav2.1 channels. These inhibitory effects of CaBP1 do not require Ca2+, but depend on the CaM-binding domain in the α1 subunit of Cav2.1 channels (α12.1). CaBP1 binds to the CaM-binding domain, co-immunoprecipitates with α12.1 from transfected cells and brain extracts, and colocalizes with α12.1 in discrete microdomains of neurons in the hippocampus and cerebellum. Our results identify an interaction between Ca2+ channels and CaBP1 that may regulate Ca2+-dependent forms of synaptic plasticity by inhibiting Ca2+ influx into neurons.
Calcium entry into cells through voltage-gated Ca2+ channels initiates a wide range of cellular processes including protein phosphorylation, gene expression and neurotransmitter release1. Neuronal Ca2+ channels consist of a pore-forming α1 subunit and auxiliary β, α2δ and sometimes γ subunits2, and their function depends considerably on interactions with additional regulatory factors. For example, the activation of G-protein-coupled receptors by neurotransmitters inhibits Cav2.1 and Cav2.2 channels, which mediate P/Q-type and N-type Ca2+ currents, respectively, through the binding of G-protein βγ subunits to distinct sites on the Ca2+ channel α1 subunit3-5. These channels are also inhibited by direct interactions with synaptic SNARE (soluble NSF attachment protein receptor proteins)—a process that may optimize coupling between Ca2+ entry and synaptic vesicle fusion6-8. Characterizing the functional interactions between Ca2+ channels and other signaling molecules is therefore crucial to understanding how many Ca2+-dependent processes in neurons are regulated.
We have shown previously that the prominent Ca2+ sensor CaM binds to a CaM-binding site (CBD) in the carboxy-terminal domain of the α12.1 subunit and mediates the dual feedback regulation of Cav2.1 channels by Ca2+ ions9,10. A second site, located amino-terminal to the CBD, is analogous to the IQ domain that mediates Ca2+/CaM-dependent inactivation of Cav1 (L-type) channels11-13. The IQ domain of α12.1 interacts with CaM in vitro and also contributes to the regulation of Cav2.1 channels by CaM14,15. Ca2+/CaM mediates both facilitation and enhanced inactivation of Cav2.1 channels in transfected cells during trains of depolarizations10,15. Presynaptic Cav2.1 channels in the brain undergo similar forms of Ca2+-dependent modulation that can lead to both synaptic facilitation and depression16-18. Because Cav2.1 channels are essential to neurotransmitter release at most central synapses19,20, regulation by CaM may contribute widely to mechanisms of activity-dependent synaptic plasticity.
Calmodulin is the best characterized member of a superfamily of Ca2+-binding proteins that exhibit four EF-hand motifs, one or more of which may be nonfunctional in the coordination of Ca2+ (ref. 21). Included in this superfamily are the neuronal Ca2+-binding proteins (NCBPs) that, unlike CaM, are localized primarily in neurons22. Some NCBPs can substitute for CaM in vitro23,24, which suggests that NCBPs may regulate effectors that are typically thought to be modulated by CaM. Here we have studied the interaction of CaBP1, an NCBP located in the retina and brain25, with Cav2.1 channels. We show that CaBP1 binds to the CBD of the α12.1 subunit, but with properties and functional consequences that are different from those of CaM. Our findings expand the repertoire of modulatory interactions that take place between Ca2+ channels and Ca2+-binding proteins and indicate that NCBPs, in addition to CaM, may have a role in the activity-dependent regulation of neuronal Ca2+ influx.
RESULTS
CaBP1 interacts with the CBD of α12.1
Although CaBP1 is a neuron-specific Ca2+-binding protein that shares nearly 56% amino acid sequence identity with CaM25, it differs in having a consensus site for N-terminal myristoylation, an alternatively spliced region, inactivating amino acid substitutions in the second of the four EF-hand motifs, and an extra turn in the helical domain that links the N- and C-terminal lobes (Fig. 1a). To determine whether CaBP1 can substitute for CaM in interactions with Cav2.1 channels, we tested the ability of CaBP1 to interact with various intracellular domains of the α12.1 subunit. In yeast two-hybrid assays, CaBP1 activated transcription of HIS3 and lacZ reporter genes only in yeast that had been cotransformed with α12.1 constructs that included the CBD (Fig. 1b and c). CaBP1 did not interact with the IQ domain or with a control plasmid that lacked the CaBP1 coding region. These results indicated that CaBP1 may modulate Cav2.1 channel function through interactions with the CBD.
Fig. 1.
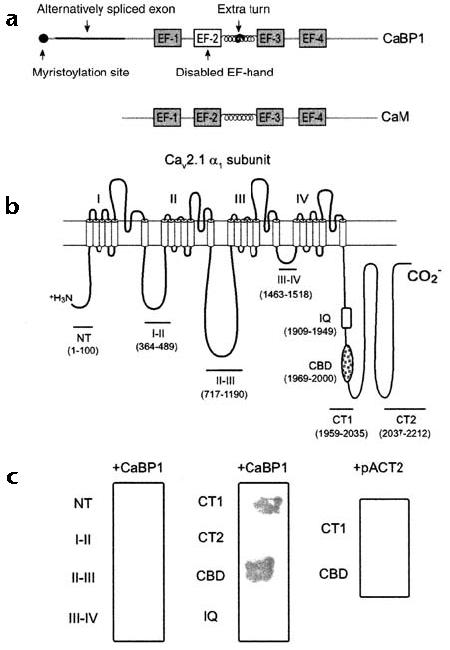
CaBP1 binds specifically to the CBD of α12.1. (a) Diagram of CaBP1 and CaM. The four Ca2+-binding EF-hand motifs are shown as boxes, and key structural differences between CaBP1 and CaM are indicated by arrows. (b) Diagram of the rat brain α12.1 subunit (rbA) showing the intracellular domains that were tested for interaction with CaBP1 in yeast two-hybrid assays. The amino acid boundaries of the indicated constructs are given in parentheses. (c) β-galactosidase assays of yeast cotransformed with the α12.1 constructs shown in (b) and either CaBP1 or control vector (pACT2).
To confirm that CaBP1 associated with the CBD in the intact channel, we tested whether CaBP1 co-immunoprecipitated with Cav2.1 channels from cotransfected tsA-201 cells. In this assay, CaM co-immunoprecipitates with α12.1 in a Ca2+-dependent manner only when the cells are exposed to Ca2+ ionophore9. Under these conditions CaBP1 also co-immunoprecipitated with α12.1. When Ca2+ was buffered with 10 mM EGTA, however, the association of CaBP1 with the channel was not affected (Fig. 2a). This co-immunoprecipitation of CaBP1 was specific because CaBP1 was not immunoprecipitated with control IgG or with α12.1-specific antibodies in cells transfected with CaBP1 alone, and CaBP1 did not co-immunoprecipitate with α12.1 subunits that lacked the CBD. Thus, despite its Ca2+ independence, the interaction between CaBP1 and Cav2.1 channels requires the same intracellular domain of Cav2.1 that binds CaM.
Fig. 2.
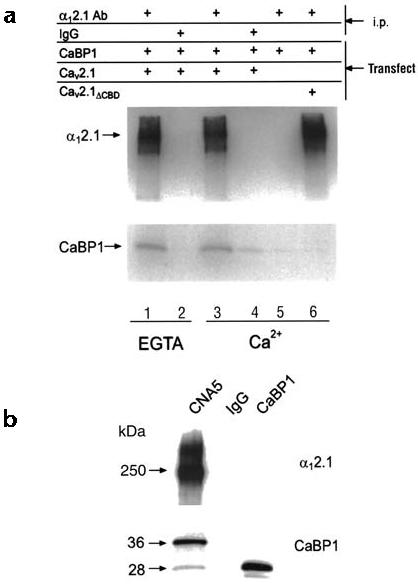
CaBP1 associates with the α12.1 subunit in tsA-201 cells and rat brain. (a) Lysates from cells transfected with Cav2.1 plus CaBP, CaBP1 alone or Cav2.1ΔCBD plus CaBP1 were subjected to immunoprecipitation (i.p.) with affinity-purified α12.1-specific antibodies or control IgG as indicated. Experiments were done with 10 mM EGTA (lanes 1 and 2) or 2 mM Ca2+ (lanes 3–6). Blots were probed with α12.1- (top) or CaBP1-specific antibodies (bottom). (b) Rat cerebellar proteins immunoprecipitated with α12.1-specific antibodies (CNA5) or control IgG were immunoblotted with α12.1- (top) or CaBP1-specific antibodies (bottom). Lysate from tsA-201 cells transfected with CaBP1 was used as a control.
CaBP1 associates with neuronal Cav2.1 channels
To determine whether CaBP1 associated with endogenous Cav2.1 Ca2+ channels, co-immunoprecipitation experiments were done with extracts from rat cerebellum, which contains high concentrations of α12.1 and CaBP1 mRNA25,26. Immunoblots of CaBP1 showed two proteins (28 and 36 kDa) that specifically co-immunoprecipitated with α12.1 (Fig. 2b, left) but not with control IgG (Fig. 2b, middle). The 36-kDa species might represent caldendrin, a larger isoform of CaBP1 that is produced from alternative splicing25,27. The 28-kDa species was consistent in size with the predicted molecular mass of the long CaBP1 isoform that we used in transfected cells (Fig. 2b, right), in support of a physiological interaction between neuronal Cav2.1 channels and CaBP1.
To identify the potential cellular sites of interaction between CaBP1 and α12.1, we immunostained rat brain sections with antibodies specific for both proteins. Compared with the immunostaining of α12.1, the immunostaining for CaBP1 was generally far more restricted within the brain and more commonly associated with somatodendritic regions than with nerve terminals. However, CaBP1 and α12.1 showed similar patterns of punctate staining in the CA1 region of the hippocampus and in the molecular layer of the cerebellum (Fig. 3a–f). As a large proportion of punctate labeling of α12.1 in the cerebellum colocalizes with that of syntaxin28, it is likely that CaBP1 and Cav2.1 channels coexist in at least some presynaptic nerve terminals. Immunostaining for CaBP1 and for α12.1 also overlapped in clusters along the dendrites of cerebellar Purkinje neurons and in structures that resembled dendritic spines (data not shown), however, which suggests that CaBP1 may associate with Cav2.1 channels in the post- as well as in the presynaptic membrane.
Fig. 3.
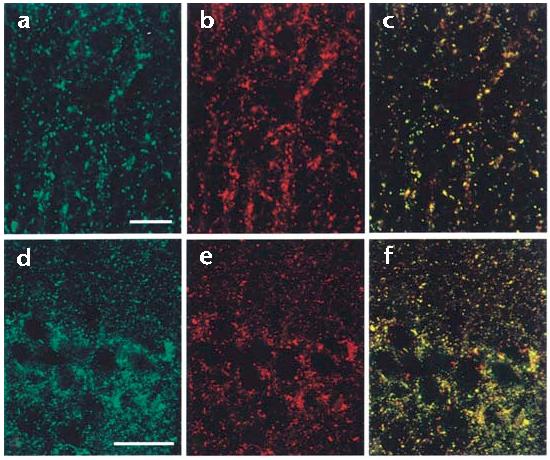
CaBP1 colocalizes with α12.1 in rat brain sections. Rat brain sections were double-labeled with antibodies specific for CaBP1 and α12.1. Labeling for CaBP1 is shown in green (a, d) and for α12.1 in red (b, e). In the merged images (c, f), double-labeled structures appear yellow. Representative examples are shown from the molecular layer of the cerebellum (a–c) and the CA1 region of the hippocampus (d–f). Scale bars, 5 μm (a–c) and 50 μm (d–f).
CaBP1 enhances inactivation of Cav2.1 channels
To elucidate the functional consequences of the interaction between CaBP1 and Cav2.1 channels, we determined the effect of CaBP1 on Ca2+ currents (ICa) in whole-cell patch-clamp recordings of transfected tsA-201 cells. We first compared the effects of transfected CaBP1 and endogenous CaM on inactivation of ICa. We have shown previously that Ca2+Ca/CaM enhances the inactivation of ICa during step depolarizations when intracellular recording solutions contain 0.5 mM EGTA9,10. Here, inactivation of ICa caused by Ca2+/CaM proceeded with a single exponential time course (τ = 852.3 ± 63.7 ms at +20 mV, n = 18) that was relatively insensitive to the test voltage (Fig. 4a and b). By contrast, CaBP1 caused ICa to decay significantly faster than in cells that were transfected with only Cav2.1.
Fig. 4.

CaBP1 enhances the inactivation of ICa in tsA-201 cells transfected with Cav2.1 channels. (a) Representative traces of ICa from cells transfected with Cav2.1 either alone (bottom) or with CaBP1 (top). Currents were evoked by 1-s pulses to the indicated voltages from a holding potential of −80 mV and were scaled for comparison. (b) Time constants for the inactivation of Cav2.1 channel currents in the absence of CaBP1. Test currents were evoked by pulses to the indicated voltages as described in (a) and fit with a single exponential function. Data were averaged from 6–18 cells. (c) Time constants for inactivation of ICa in cells cotransfected with CaBP1. Test currents were evoked by the same voltages as in (a) and (b), but current traces were fit with a double exponential function. Fast (τfast, filled bars) and slow time constants (τslow, open bars) were averaged from 7–20 cells.
In almost all of the cells that were cotransfected with CaBP1, the decay of ICa evoked by +20- and +30-mV pulses was best fit by a double exponential function, with a slow component similar to control and a fast component comprising 30–40% of the peak current (Fig. 4c). With a +10-mV test pulse, however, biphasic inactivation was detected in only 11 out of 20 cells that had been cotransfected with CaBP1. At this test voltage, which elicits the peak inward ICa, Ca2+/CaM-dependent inactivation is maximal10. Therefore, the absence of a fast phase of inactivation in some cells cotransfected with CaBP1 might have resulted from more effective competition by Ca2+/CaM.
Competition between CaM and CaBP1 for Cav2.1 channels was supported further by the observation of a marked reduction in Ca2+-dependent inactivation in cells cotransfected with CaBP1 (Fig. 5a and b). Enhanced inactivation caused by CaM results in a significant reduction in the residual current at the end of a 1-second depolarizing test pulse normalized to the peak current (Ires/Ipk) for ICa as compared with IBa (refs. 9, 10). By contrast, in cells cotransfected with CaBP1, Ires/Ipk was already reduced when Ba2+ was the charge carrier and was not significantly different for ICa and IBa (Fig. 5a and b). Ca2+-dependent inactivation was not affected in the same way by cotransfection with CaM instead of CaBP1, which indicated that the faster, Ca2+-independent inactivation was a specific consequence of the modulation of Cav2.1 channels by CaBP1.
Fig. 5.

Fast, Ca2+-independent inactivation of Cav2.1 channels by CaBP1 differs from the modulation of Cav2.1 channels by CaM. (a) Cav2.1 channel currents recorded with Ca2+ or Ba2+ as the permeant ion. Test pulses were applied from a holding voltage of −80 mV to +10 mV (Ca2+) or 0 mV (Ba2+) for Cav2.1 either alone or cotransfected with CaM, or to +20 mV (Ca2+) or +10 mV (Ba2+) for cells cotransfected with CaBP1, to account for the positive shift in voltage-dependent activation caused by CaBP1. The intracellular solution contained 0.5 mM EGTA. (b) The residual current amplitude at the end of a test pulse (Ires, indicated in a) was normalized to the peak current (Ipk) for cells transfected with Cav2.1 either alone or with CaBP1 or CaM. (c) Representative currents evoked by a test pulse to +30 mV (+20 mV for IBa) in cells transfected with wild-type or mutant Cav2.1 lacking the CBD (Cav2.1ΔCBD) either alone or cotransfected with CaBP1. Intracellular solutions contained 0.5 mM EGTA except where 10 mM BAPTA is indicated and extracellular solutions contained 10 mM Ca2+ except where Ba2+ is indicated. (d) Current amplitudes at 200 ms (I200, indicated in c) were normalized to the peak current (Ipk) and plotted for the different conditions. Recordings were from tsA-201 cells transfected with Cav2.1 or Cav2.1ΔCBD either alone (open bars) or with CaBP1 (filled bars). Results represent averages of 5–13 cells. Asterisks indicate statistically significant differences between the paired groups (p ≤ 0.05).
To clarify the effects of CaBP1 on fast inactivation of Cav2.1 channels, we measured the amplitude of ICa at the 200-ms time point during a 1-s test pulse and normalized this to the peak current (I200/Ipk, Fig. 5c and d). We used more positive test voltages to limit Ca2+ entry, thus minimizing the contribution of endogenous CaM in these experiments. With 0.5 mM EGTA, faster inactivation of ICa in cells with CaBP1 caused a significant decrease in I200/Ipk (0.45 ± 0.06 for CaBP1 versus 0.79 ± 0.03 for control, p < 0.01). CaBP1 significantly enhanced fast inactivation of IBa (I200/Ipk of 0.43 ± 0.09 for CaBP1 versus 0.74 ± 0.07 for control, p < 0.05) and also of ICa with 10 mM of the intracellular calcium chelator BAPTA (I200/Ipk of 0.56 ± 0.08 for CaBP1 versus 0.85 ± 0.03 for control, p < 0.02). The CBD was essential for these effects on inactivation, because CaBP1 had no effect on channels in which this domain had been deleted (Fig. 5c and d, Cav2.1ΔCBD; p > 0.3). Together with biochemical analyses, these results support a Ca2+-independent association of CaBP1 with the CBD, which mediates a strong acceleration of Cav2.1 channel inactivation that does not require Ca2+ influx or intracellular accumulation of Ca2+.
CaBP1 shifts voltage dependence of Cav2.1 activation
In cells cotransfected with CaBP1 and Cav2.1, the normalized tail current–voltage curve was shifted positively and was shallower than in cells transfected with Cav2.1 alone (Fig. 6a). CaBP1 caused significant increases in the half-activation voltage, V1/2 (12.8 ± 1.3 mV for CaBP1 versus 4.5 ± 0.9 mV for control, p < 0.01), and slope factor of the tail current–voltage curve (−8. 7 ± 0.5 mV for CaBP1 versus −5.2 ± 0.6 mV for control, p < 0.01). Similar to the other actions of CaBP1 on Cav2.1 channels, these effects on ICa activation were essentially reproduced with intracellular BAPTA and extracellular Ba2+ but were not observed with Cav2.1ΔCBD channels (Fig. 6b–d), which indicates that the Ca2+-independent association of CaBP1 with the CBD results in a newly identified, multifaceted regulation of Cav2.1 channels.
Fig. 6.
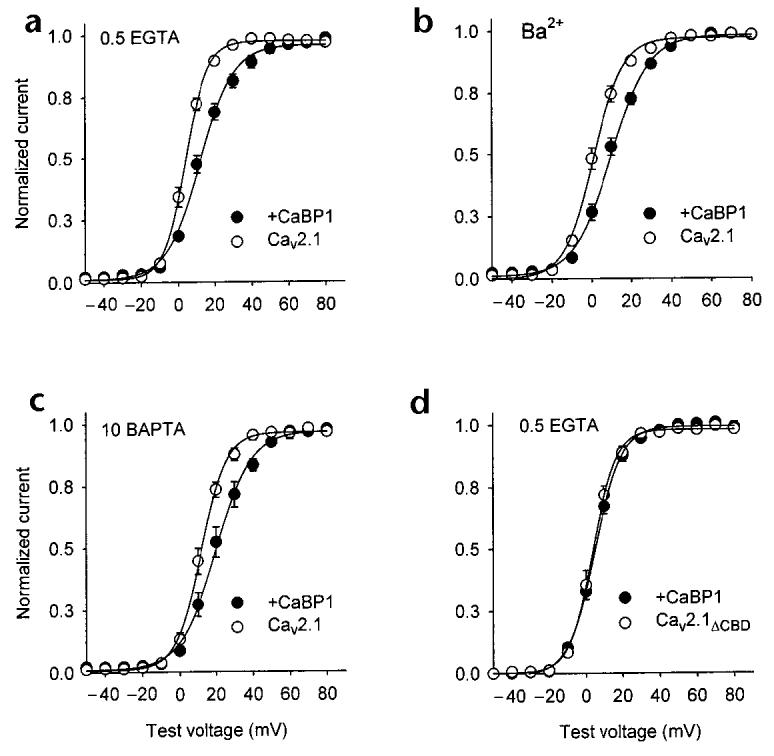
CaBP1 alters the voltage dependence of Cav2.1 activation. Tail current–voltage curves from tsA-201 cells transfected with Cav2.1 (a–c) or Cav2.1ΔCBD (d) either alone (open circles) or with CaBP1 (filled circles). Test pulses (10 ms) to the indicated voltages were applied from a holding voltage of −80 mV and peak tail currents were measured upon the repolarization of cells to −40 mV, normalized to the largest tail current in the series, and plotted against test voltage. Test pulses were held for 10 ms, as activation of currents was complete but inactivation was minimal during this time. Bath solutions contained 10 mM Ca2+ (a, c, d) or Ba2+ (b), and intracellular solutions contained 0.5 mM EGTA (a, b, d) or 10 mM BAPTA (c). Each point represents the mean of 7–20 cells.
Ca2+-dependent facilitation is not supported by CaBP1
Activity-dependent increases in intracellular Ca2+ cause an initial facilitation of ICa owing to the interaction of Ca2+/CaM with Cav2.1 channels (refs 10, 15). This Ca2+-dependent facilitation was evident with 0.5 mM intracellular EGTA in paired-pulse protocols, in which Ca2+ influx during a short prepulse induced a significant increase in the tail current elicited by a subsequent test pulse (Fig. 7a). With the same voltage protocol, no facilitation of ICa was observed in cells cotransfected with CaBP1 (Fig. 7b). Because of the strong voltage-dependent enhancement of ICa inactivation caused by CaBP1, it was possible that paired-pulse facilitation in cells cotransfected with CaBP1 might have been obscured by the onset of inactivation during the conditioning prepulse. Alternatively, CaBP1, unlike CaM, might not support Ca2+-dependent facilitation of ICa.
Fig. 7.

CaBP1 does not support Ca2+-dependent facilitation of Cav2.1 channels. (a, b) Voltage dependence of Cav2.1 Ca2+ currents evoked before (P1, filled circles) and after (P2, open circles) a depolarizing prepulse. Tail currents were measured by repolarizing cells to −40 mV for 5 ms after variable test voltages and normalized to the largest tail current evoked by P1. Inset, representative currents evoked by a test pulse to +10 mV before (filled circles) and after (open circles) the prepulse. Intracellular recording solution contained 0.5 mM EGTA. Results were obtained from cells transfected with Cav2.1 either alone (a, n = 7) or with CaBP1 (b, n = 10). (c, d) Cav2.1 channel currents elicited by repetitive depolarizations. Test pulses (+20 mV (c) or +10 mV (d) to account for voltage shifts cause by Ba2+ substitution) at a frequency of 100 Hz were applied to cells transfected with Cav2.1 either alone (open circles) or along with CaBP1 (filled circles). Peak current amplitudes were normalized to the first pulse in the series and plotted against time during the train. Every second data point is shown. Intracellular recording solutions contained 0.5 mM EGTA, and bath solutions contained 10 mM Ca2+ (c) or Ba2+ (d). In (c), n = 9 for open circles; n = 13 for closed circles. In (d), n = 5 for open circles; n = 11 for closed circles.
To distinguish between these possibilities, we analyzed the properties of ICa during trains of short (5-ms) repetitive depolarizations, which should initially minimize the impact of voltage-dependent inactivation and reveal facilitation of ICa early in the train. With 0.5 mM intracellular EGTA, Cav2.1 Ca2+ currents undergo a sustained facilitation and gradually inactivate below initial current amplitudes after 800 ms of repetitive pulses (Fig. 7c), an effect that depends on Ca2+/CaM10. In cells cotransfected with CaBP1, facilitation of ICa was reduced markedly, with current amplitudes rapidly inactivating below initial values only 200 ms into the train (Fig. 7c).
The maximum facilitated ICa amplitude at 50 ms in cells cotransfected with CaBP1 (1.04 ± 0.02, n = 13) was not significantly different from the Ca2+-independent facilitation of Ba2+ currents in cells transfected with Cav2.1 alone (1.04 ± 0.03, n =5, p = 0.80) or cotransfected with CaBP1 (1.02 ± 0.02, n = 11, p = 0.52; Fig. 7d), which indicated that CaBP1 does not support Ca2+-dependent facilitation of Cav2.1 channels. Together with the enhanced inactivation and positive shifts in activation caused by CaBP1, the absence of Ca2+-dependent facilitation would strongly limit voltage-dependent Ca2+ entry through Cav2.1 channels. These results highlight further the different modulation of these Ca2+ channels by CaBP1 and CaM.
Discussion
We have shown that the neuronal Ca2+-binding protein CaBP1 interacts with and modulates Cav2.1 channels in a manner that is markedly different from that of CaM. CaBP1 bound to the CBD of α12.1 but caused significantly faster inactivation of Cav2.1 channel currents than that caused by CaM. CaBP1 also positively shifted tail current-activation curves and did not support Ca2+-dependent facilitation of Cav2.1 currents. Neither the association of CaBP1 with the CBD nor the inhibitory modulation by CaBP1 required Ca2+, in contrast to the effects of CaM on Cav2.1 channels, which are strictly dependent on Ca2+. The observed association and colocalization of CaBP1 and Cav2.1 channels in neurons in the brain suggest that Ca2+ channel regulation by CaBP1 may be an important determinant of Ca2+ signaling pathways in neurons.
Ca2+-independent binding and modulation by CaBP1
The Ca2+ independence of the interaction between CaBP1 and Cav2.1 was unexpected given the previously observed Ca2+-dependent association of CaBP1 with other CaM targets25. It is possible that very local rises in Ca2+ might have escaped buffering by BAPTA in our experiments, which could have been sufficient for binding to CaBP1 and for causing Ca2+-dependent modulation of ICa. This possibility seems unlikely, however, because the modulation by CaBP1 did not change appreciably when Ba2+ was the permeant ion; Ba2+ ions bind to EF-hand motifs with relatively low affinity and so should not reproduce Ca2+-dependent regulation of target molecules29.
Calcium-free CaM can associate with and regulate several targets, including the ryanodine receptor RyR1, cyclic GMP kinase and a CaM-dependent adenylyl cyclase from Bordetella pertussis30-32. In addition, GCAPs—photoreceptor Ca2+-binding proteins—activate guanylyl cyclases in their Ca2+-free forms33,34. Thus, CaBP1 might have a similar flexibility and interact with and regulate some effectors without binding Ca2+. Although we cannot exclude the possibility that CaBP1 may modulate some aspects of Ca2+ channel function in a Ca2+-dependent manner, we found no evidence to support a requirement for Ca2+ in the effects of CaBP1 on the activation and inactivation of Cav2.1 channels. Thus, despite the Ca2+-sensing capability of CaBP1, we propose that CaBP1 itself does not mediate Ca2+-dependent regulation of Cav2.1 channels, but might indirectly influence feedback regulation by Ca2+ by competing with CaM. Thus, CaBP1 may act more like auxiliary Ca2+ channel β-subunits by altering the intrinsic properties of Cav2.1 channels to fine-tune voltage-gated Ca2+ entry in specific classes of neurons.
Distinct modulation of Cav2.1 by CaBP1 and CaM
We have shown that both CaM and CaBP1 interact with the CBD of the α12.1 subunit and that this site is essential for full channel regulation by both proteins. Conflicting evidence indicates that the IQ domain—a sequence that is N-terminal to the CBD—is involved in the modulation of Cav2.1 channels by Ca2+/CaM15. Our results do not support the importance of the IQ domain in modulation by CaBP1 because, first, CaBP1 interacted with the CBD but not the IQ domain in yeast two-hybrid assays (Fig. 1b and c); second, deleting the CBD prevented the co-immunoprecipitation of CaBP1 with α12.1 (Fig. 2a); and third, the fast inactivation and shifts in the voltage dependence of activation caused by CaBP1 were abolished in channels that lacked the CBD (Figs. 5 and 6). Notably, removing the CBD from α12.1 eliminated regulation by CaBP1 more completely than it eliminated regulation by CaM10. Together, our results indicate that the CBD may be the primary determinant for the functional effects of CaBP1 on Cav2.1 channels.
If both CaM and CaBP1 interact with the CBD, how is it that CaBP1 causes Ca2+-independent fast inactivation and positively shifted activation, whereas CaM causes Ca2+-dependent facilitation and inactivation of Cav2.1 channels? One possibility is that key structural features that distinguish CaBP1 from CaM, such as its extra-long central helical domain and N-terminal myristoylation (Fig. 1a), may permit Ca2+-independent binding of CaBP1 to the CBD, which might then lead to its unique inhibitory modulation of ICa. Future experiments that determine how such differences between CaBP1 and CaM contribute to specific forms of Cav2.1 regulation may reveal how ion channels and other signaling molecules are differentially modulated by CaM and related Ca2+-binding proteins.
Modulation of neuronal Cav2.1 channels by CaBP1
Our immunoprecipitation and immunofluorescence studies showed that CaBP1 and α12.1 associate physically in extracts of rat cerebellum and that their subcellular distributions overlap in this brain region, which indicates that CaBP1 may have a physiological role in the regulation of Cav2.1 channels. Because both CaM and CaBP1 interacted with the same site on the α12.1 subunit, an important issue is whether Cav2.1 would interact functionally with CaM and/or CaBP1 in neurons in which both Ca2+-binding proteins are expressed.
Although we do not know whether CaM and CaBP1 bind simultaneously to Cav2.1 channels, our electrophysiological studies suggested that CaM and CaBP1 might competitively regulate the channel. CaBP1 more strongly enhanced inactivation of ICa when the influence of Ca2+/CaM was suppressed either with extracellular Ba2+ or intracellular BAPTA, or with test voltages that elicited submaximal Ca2+ influx. These results imply that when intracellular Ca2+ concentrations are high Cav2.1 channels may be facilitated predominantly by CaM, and that the inactivating effects of CaBP1 become most prominent when cytoplasmic Ca2+ concentrations decline. In this way, CaM and CaBP1 may coordinately act as a molecular switch to intensify neuronal Ca2+ influx in response to activity-dependent alterations in intracellular concentrations of Ca2+.
NCBPs and synaptic transmission
Emerging evidence supports a role for NCBPs in the regulation of synaptic transmission. In particular, neuronal Ca2+ sensor-1 (NCS-1), which is more distantly related to CaM than is CaBP1, regulates neurotransmitter release35, synapse formation36 and neuronal circuits that control associative learning37. Notably, NCS-1 has been implicated in the negative regulation of Ca2+ channels in chromaffin cells38, which suggests that Cav2.1 channels may be modulated by NCBPs in addition to CaBP1.
Given the widespread distribution of Cav2.1 channels throughout the nervous system, the cell type–specific modulation of Cav2.1 by CaBP1, CaM or other NCBPs may fundamentally determine the nature of presynaptic and postsynaptic Ca2+ signals and the functional consequences of synaptic activity.
Methods
Yeast two-hybrid assays. We amplified cDNAs encoding the long isoform of human CaBP1 (ref. 25) and the cytoplasmic domains of α12.1 by polymerase chain reaction and subcloned them into the yeast two-hybrid vectors pACT2 and pAS2-1, respectively (Clontech, Palo Alto, California). To test for interactions between CaBP1 and specific domains of α12.1, the corresponding plasmids were cotransformed into yeast strain Y190. We assayed growth on medium lacking histidine and β-galactosidase to identify interacting proteins as described9.
Cell culture and transfection. We grew tsA-201 cells to ∼70% confluency and transfected them by the calcium phosphate method with an equimolar ratio of cDNAs encoding the rat brain Ca2+ channel subunits α12.1 (rbA), β2a and α2δ (ref. 26). The α12.1 construct that lacks amino acids 1969–2000 (α12.1ΔCBD) has been described10. The long isoform of human CaBP1 (ref. 25) was subcloned into the BamHI sites of pcDNA3.1+ (Invitrogen, Carlsbad, California) and transfected at a 5:1 molar excess with Ca2+ channel subunits. For electrophysiological experiments, we plated cells on 35-mm dishes and transfected them with 5 μg of total DNA, including 0.3 μg of a CD8 expression plasmid to allow the detection of transfected cells. For co-immunoprecipitation assays, we plated cells on 150-mm dishes and transfected them with 50 μg of total plasmid DNA.
Co-immunoprecipitation assays. At least 48 h after transfection, tsA-201 cells were homogenized in ice-cold lysis buffer (1% Nonidet P-40 in TBS (20 mM Tris-HCl, pH 7.3, 150 mM NaCl), 10 mM EGTA and protease inhibitors) and centrifuged at 1,000g for 5 min. To maintain Ca2+-dependent interactions, we pretreated some groups with 5 μM A23187 and 2 mM CaCl2 for 15 min but did not include EGTA in the lysis buffer. The postnuclear supernatant (300–400 μg of membrane protein) was incubated with 15 μg of α12.1-specific antibodies (raised against CNA5)39 for 2 h at 4°C. Immune complexes were separated on protein A–Sepharose, resolved by SDS–PAGE and transferred to nitrocellulose. For immunoblotting, we blocked nitrocellulose filters for 30 min in 5% milk/TBS and incubated them with the CaBP1 antiserum UW72 (ref. 25; 1:1,000 dilution) or with CNA5-specific antibodies (2.5 μg/ml) for 1 h. Blots were washed three times in TBS with 0.05% Tween 20 (TBST) and incubated with horse-radish peroxidase–linked protein A (Amersham, Piscataway, New Jersey; 1:2,000) for 40 min. We used ECL western blotting reagent (Amersham) for detection of chemiluminescence.
For co-immunoprecipitations from rat brain, we homogenized cerebellar tissue from two adult male rats in 0.3 M sucrose, 75 mM NaCl, 10 mM Tris-HCl, pH 7.4, and 10 mM EGTA. We included protease inhibitors in the homogenization buffer and in buffers used at all subsequent steps. Homogenates were centrifuged for 10 min at 1,000g, and membrane fractions were separated from the postnuclear supernatant at 100,000g for 30 min. Membrane proteins were solubilized with 4 ml of buffer A (1% Triton X-100, 10 mM Tris, pH 7.4, and 10 mM EGTA) and insoluble material was removed by further centrifugation (100,000g for 30 min). Ca2+ channels were immunoprecipitated with 15 μg of CNA5-specific antibodies per ml of solubilized membrane protein. We isolated immune complexes on protein A–Sepharose and detected the associated CaBP1 by immunoblotting as described above.
Immunocytochemistry. Anesthetized adult Sprague–Dawley rats were perfused intracardially with 4% paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.4. The brain was post-fixed and cryoprotected in 30% (w/v) sucrose, and tissue sections (35 μm) were cut on a sliding microtome in 0.1 M phosphate buffer. Tissue sections were rinsed with 0.1 M Tris-buffered saline (TBS) and blocked sequentially with 2% avidin and 2% biotin. For the double-labeling of CaBP1 and α12.1, we incubated tissue sections in UW72 antiserum (diluted 1:100) for 36 h at 4°C, biotinylated goat antibody against rabbit IgG (Vector Laboratories, Burlingame, California; 1:300) for 1 h at 37°C, and avidin D–fluorescein (Vector Laboratories; 1:300) for 1 h at 37°C, with rinsing between each step. The tissue was then blocked with 5% normal rabbit serum in TBS for 1 h and incubated with affinity-purified Fab fragments for 1 h at 37°C. After rinsing, tissue sections were incubated with antibodies specific for CNA5 (1:15) for 36 h at 4°C, biotinylated goat antibody against rabbit IgG (1:300) for 1 h at 37°C, and avidin D–Texas Red (1:300) for 1 h at 37°C. Tissue sections were mounted on gelatin-coated slides, protected with coverslips, and viewed with a Bio-Rad MRC 600 microscope in the W.M. Keck Imaging Facility at the University of Washington. All procedures conformed to protocols approved by the Animal Welfare Committee of the University of Washington.
Electrophysiology and data analysis. At least 48 h after transfection, tsA-201 cells were incubated with CD8-specific antibody–coated microspheres (Dynal, Oslo, Norway) to permit detection of transfected cells. We recorded whole-cell Ca2+ currents with a List EPC-7 patch-clamp amplifier and filtered them at 5 kHz. Leak and capacitive transients were subtracted using a P/–4 protocol. Extracellular recording solutions were composed of 150 mM Tris, 1 mM MgCl2 and 10 mM CaCl2 or BaCl2; intracellular solutions were composed of 120 mM N-methyl-d-glucamine, 60 mM HEPES, 1 mM MgCl2, 2 mM Mg-ATP and 0.5 mM EGTA or 10 mM BAPTA. The pH of all solutions was adjusted to 7.3 with methanesulfonic acid.
The time course of ICa decay was fit by either A[exp(−t/τ)] or Aslow[exp(−t/τslow)] + Afast[exp(t/τfast)], where t is time; Aslow and Afast are the amplitudes of the slow and fast exponentials, respectively, at t = 0; and τslow and τfast are the time constants of the decay of the two processes. Normalized tail current–voltage curves were fit with a single Boltzmann function: A/{1 + exp[(V − V1/2)/k] + b}, where V is test pulse voltage, V1/2 is the midpoint of the activation curve, k is a slope factor, A is the amplitude and b is the baseline. Curve fits and data analysis were done with Igor Pro software (Wavemetrics, Lake Oswego, Oregon). All averaged data are the mean ± s.e.m. We determined the statistical significance of differences between groups by Student's t-test (SigmaPlot, SPSS Science, Chicago, Illinois).
Acknowledgements
This work was supported by NIH Research Grant R01 NS22625 to W.A.C, a NSRA postdoctoral research fellowship from NIH (F32 NS10645) to A.L., NIH Research Grant R01 EY08061 to K.P. and research grants from Research to Prevent Blindness, Inc., the Alcon Research Institute and the E.K. Bishop Foundation to K.P.
References
- 1.Miller RJ. Multiple calcium channels and neuronal function. Science. 1987;235:46–52. doi: 10.1126/science.2432656. [DOI] [PubMed] [Google Scholar]
- 2.Catterall WA. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium. 1998;24:307–323. doi: 10.1016/s0143-4160(98)90055-0. [DOI] [PubMed] [Google Scholar]
- 3.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 4.Herlitze S, et al. Modulation of Ca2+ channels by G protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 5.De Waard M, et al. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 6.Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 7.Zhong H, Yokoyama CT, Scheuer T, Catterall WA. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat. Neurosci. 1999;2:939–941. doi: 10.1038/14721. [DOI] [PubMed] [Google Scholar]
- 8.Wiser O, Bennett MK, Atlas D. Functional interaction of syntaxin and SNAP-25 with voltage-sensitive L- and N-type Ca2+ channels. EMBO J. 1996;15:4100–4110. [PMC free article] [PubMed] [Google Scholar]
- 9.Lee A, et al. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;339:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 10.Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J. Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc. Natl. Acad. Sci. USA. 1999;96:2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 13.Zühlke RG, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–161. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 14.Pate P, et al. Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J. Biol. Chem. 2000;275:39786–39792. doi: 10.1074/jbc.M007158200. [DOI] [PubMed] [Google Scholar]
- 15.DeMaria CD, Soong T, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- 16.Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- 17.Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J. Physiol. (Lond.) 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borst JG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. J. Physiol. (Lond.) 1998;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 20.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 21.Polans A, Baehr W, Palczewski K. Turned on by Ca2+ǃ The physiology and pathology of Ca2+-binding proteins in the retina. Trends Neurosci. 1996;19:547–554. doi: 10.1016/s0166-2236(96)10059-x. [DOI] [PubMed] [Google Scholar]
- 22.Burgoyne RD, Weiss JL. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem. J. 2001;353:1–12. [PMC free article] [PubMed] [Google Scholar]
- 23.Sallese M, et al. The G-protein-coupled receptor kinase GRK4 mediates homologous desensitization of metabotropic glutamate receptor 1. FASEB J. 2000;14:2569–2580. doi: 10.1096/fj.00-0072com. [DOI] [PubMed] [Google Scholar]
- 24.Schaad NC, et al. Direct modulation of calmodulin targets by the neuronal calcium sensor NCS-1. Proc. Natl. Acad. Sci. USA. 1996;93:9253–9258. doi: 10.1073/pnas.93.17.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haeseleer F, et al. Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J. Biol. Chem. 2000;275:1247–1260. doi: 10.1074/jbc.275.2.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stea A, et al. The localization and functional properties of a rat brain α1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc. Natl. Acad. Sci. USA. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seidenbecher CI, et al. Caldendrin, a novel neuronal calcium-binding protein confined to the somato-dendritic compartment. J. Biol. Chem. 1998;273:21324–21331. doi: 10.1074/jbc.273.33.21324. [DOI] [PubMed] [Google Scholar]
- 28.Westenbroek RE, et al. Immunochemical identification and subcellular distribution of the α1A subunits of brain calcium channels. J. Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chao SH, Suzuki Y, Zysk JR, Cheung WY. Activation of calmodulin by various metal cations as a function of ionic radius. Mol. Pharmacol. 1984;26:75–82. [PubMed] [Google Scholar]
- 30.Rodney GG, Williams BY, Strasburg GM, Beckingham K, Hamilton SL. Regulation of RYR1 activity by Ca2+ and calmodulin. Biochemistry. 2000;39:7807–7812. doi: 10.1021/bi0005660. [DOI] [PubMed] [Google Scholar]
- 31.Yamaki T, Hidaka H. Ca2+-independent stimulation of cyclic GMP-dependent protein kinase by calmodulin. Biochem. Biophys. Res. Commun. 1980;94:727–733. doi: 10.1016/0006-291x(80)91293-0. [DOI] [PubMed] [Google Scholar]
- 32.Greenlee DV, Andreasen TJ, Storm DR. Calcium-independent stimulation of Bordetella pertussis adenylate cyclase. Biochemistry. 1982;21:2759–2764. doi: 10.1021/bi00540a028. [DOI] [PubMed] [Google Scholar]
- 33.Rudnicka-Nawrot M, et al. Changes in biological activity and folding of guanylate cyclase-activating protein 1 as a function of calcium. Biochemistry. 1998;37:248–257. doi: 10.1021/bi972306x. [DOI] [PubMed] [Google Scholar]
- 34.Haeseleer F, et al. Molecular characterization of a third member of the guanylyl cyclase-activating protein subfamily. J. Biol. Chem. 1999;274:6526–6535. doi: 10.1074/jbc.274.10.6526. [DOI] [PubMed] [Google Scholar]
- 35.McFerran BW, Graham ME, Burgoyne RD. Neuronal Ca2+ sensor 1, the mammalian homologue of frequenin, is expressed in chromaffin and PC12 cells and regulates neurosecretion from dense-core granules. J. Biol. Chem. 1998;273:22768–22772. doi: 10.1074/jbc.273.35.22768. [DOI] [PubMed] [Google Scholar]
- 36.Chen XL, et al. Overexpression of rat neuronal calcium sensor-1 in rodent NG108-15 cells enhances synapse formation and transmission. J. Physiol. (Lond.) 2001;532:649–659. doi: 10.1111/j.1469-7793.2001.0649e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomez M, et al. Ca2+ signaling via the neuronal calcium sensor-1 regulates associative learning and memory in C. elegans. Neuron. 2001;30:241–248. doi: 10.1016/s0896-6273(01)00276-8. [DOI] [PubMed] [Google Scholar]
- 38.Weiss JL, Archer DA, Burgoyne RD. Neuronal Ca2+ sensor-1/frequenin functions in an autocrine pathway regulating Ca2+ channels in bovine adrenal chromaffin cells. J. Biol. Chem. 2000;275:40082–40087. doi: 10.1074/jbc.M008603200. [DOI] [PubMed] [Google Scholar]
- 39.Sakurai T, Westenbroek RE, Rettig J, Hell J, Catterall WA. Biochemical properties and subcellular distribution of the BI and rbA isoforms of α1A subunits of brain calcium channels. J. Cell Biol. 1996;134:511–528. doi: 10.1083/jcb.134.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]