

Abstract
Parp-1 and Parp-2 are activated by DNA breaks and have been implicated in the repair of DNA single-strand breaks (SSB). Their involvement in double-strand break (DSB) repair mediated by homologous recombination (HR) or nonhomologous end joining (NHEJ) remains unclear. We addressed this question using chicken DT40 cells, which have the advantage of carrying only a PARP-1 gene but not a PARP-2 gene. We found that PARP-1−/− DT40 mutants show reduced levels of HR and are sensitive to various DSB-inducing genotoxic agents. Surprisingly, this phenotype was strictly dependent on the presence of Ku, a DSB-binding factor that mediates NHEJ. PARP-1/KU70 double mutants were proficient in the execution of HR and displayed elevated resistance to DSB-inducing drugs. Moreover, we found deletion of Ligase IV, another NHEJ gene, suppressed the camptothecin of PARP-1−/− cells. Our results suggest a new critical function for Parp in minimizing the suppressive effects of Ku and the NHEJ pathway on HR.
Keywords: base excision repair, camptothecin, homologous recombination, ionizing radiation, MMS
Introduction
Poly (ADP-ribosylation), the covalent attachment of ADP-ribose moieties derived from NAD to target proteins, is one of the earliest cellular responses to DNA-strand breaks. This post-translational modification of proteins sequesters negative charge at the site of DNA damage and thereby modulates the properties of the chromatin structure and directs the access and/or function of repair proteins (Huber et al, 2004; Rouleau et al, 2004). Among 17 potential Poly (ADP-ribosylation) proteins (Parps) in the mammalian genome, only Parp-1 and Parp-2 are known to act in the DNA damage response (Ame et al, 2004). Mice deficient in either of these enzymes are sensitive to ionizing radiation (IR) and alkylating agents (Wang et al, 1995; de Murcia et al, 1997; Masutani et al, 1999; Schreiber et al, 2002) and a simultaneous deletion of both impairs murine embryonic development (Menissier de Murcia et al, 2003). Parp-1 and Parp-2 interact with each other and are found in a larger protein complex including XRCC1 and DNA polymerase-β and DNA ligase III (Masson et al, 1998; Schreiber et al, 2002). Taken together, biochemical and genetic evidence firmly supports a role for Parp-1 and Parp-2 in the base excision repair (BER) repair pathway, in which it functions in damage detection, signaling, and recruitment of Xrcc1 to the lesion.
Besides its binding to BER proteins, biochemical studies revealed a physical interaction between Parp-1 and the Ku/DNA-PK complex that is involved in the nonhomologous end-joining (NHEJ) pathway of double-strand break (DSB) repair (Ariumi et al, 1999; Galande and Kohwi-Shigematsu, 1999; Li et al, 2004). These studies also established Ku and DNA-PK as a substrate of Parp-1. The in vivo relevance of this interaction is not understood, but Li et al's study suggests that Parp-1 could decrease the affinity of Ku to DSBs. It is, thus, conceivable that Parp-1 and/or Parp-2 play a role in the repair of DSBs by homologous recombination (HR) and NHEJ. HR repairs DSBs by using the information of a homologous sequence, preferentially the sister-chromatid, as a blueprint, while NHEJ directly ligates the broken ends, often resulting in the loss of genetic information (Paques and Haber, 1999). The initial step after DSB formation decides which pathway will be chosen. Access of an exonuclease creates a 3′ single strand tail that will be covered with Rad51 and initiate the subsequent HR reaction (Baumann and West, 1998). NHEJ, on the other hand, is initiated by the binding of the Ku heterodimer (consisting of Ku70 and Ku80) and DNA-PK to the double-strand break (DSB) (Downs and Jackson, 2004). Several studies have proposed a direct competition between NHEJ and HR (Takata et al, 1998; Fukushima et al, 2001; Pierce et al, 2001; Frank-Vaillant and Marcand, 2002), and a recent cytological study by Kim et al (2005) shows that even in S-phase cells NHEJ factors are recruited to sites of DSBs. The precise regulation of the balance between these two pathways is poorly understood.
Apart from its physical interaction with Ku, the following evidence links Parp with DSB repair: Parp has been proposed to carry out an antirecombinogenic function (Lindahl et al, 1995), because deletion or inhibition of Parp-1 results in an increase in sister-chromatid exchange (Oikawa et al, 1980; Wang et al, 1995; de Murcia et al, 1997; Masutani et al, 1999). Furthermore, cells carrying truncations of BRCA2, a protein that interacts with Rad51 and plays an important role in HR, are highly sensitive to inhibition or knock down of Parp-1 (Bryant et al, 2005; Farmer et al, 2005). Moreover, double deletions of PARP-1 and DSB repair and checkpoint genes such as KU80 and ATM are synthetically lethal in mice (Menisser-de Murcia et al, 2001; Henrie et al, 2003), while PARP-1 deletion partially suppresses the V(D)J recombination defect observed in DNA-PK-deficient SCID mice (Morrison et al, 1997). Taken together, these data suggest a complex relationship between poly(ADP-ribosylation) and DSB repair. However, several recent studies have come to the conclusion that Parp-1 is not directly involved in DSB repair (Noel et al, 2003; Schultz et al, 2003; Yang et al, 2004).
A PARP-1/2 double knock out cell line, where the repair-associated Parp activity is completely suppressed, might be necessary to obtain conclusive evidence about the DSB repair function of Parp. Here we use the chicken B-lymphocyte cell line DT40 (Sonoda et al, 2001) to reassess the question of Parp and DSB repair by reverse genetics. Chickens appear to lack a Parp-2 homolog, and the phenotype of PARP-1−/− DT40 cells could thus reflect the complete absence of repair-associated poly (ADP-ribosylating) activity, equivalent to a mammalian cell lacking Parp-1 and -2. We found that DT40 cells lacking Parp-1 were sensitive to IR, camptothecin (CPT), and DNA-methylating agents. Judged by impaired induction of sister chromatid exchange (SCE) in response to CPT, as well as by directly comparing levels of gene-conversion induced by the endonuclease I-Sce1, these cells appeared to have reduced levels of HR. Deletion of Ku70 in these PARP-1−/− cells re-established their ability to perform HR and conferred resistance to IR and CPT. We conclude that Parp-1 protects the HR pathway from toxic interference by Ku70, and suggest a new function for Parp in controlling DSB repair.
Results
Generation and characterization of PARP-1−/− DT40 cells
In order to delete Parp-1 in DT40 cells, we designed a gene-targeting construct to replace exons 3 and 4 of the chicken PARP-1 gene with selection marker cassettes (Supplementary Figure 1). These constructs were used to target the PARP-1 gene in wild-type (WT) cells and in KU70−/− cells (Takata et al, 1998). Confirmation of successful gene targeting by Southern blotting is shown in Supplementary Figure 1. Gene disruption resulted in a suppression of Parp-1 protein expression as judged by immunoblotting in both WT and KU70−/− cells (Figure 1A). Thus, we conclude that we successfully created PARP-1−/− DT40 cells.
Figure 1.
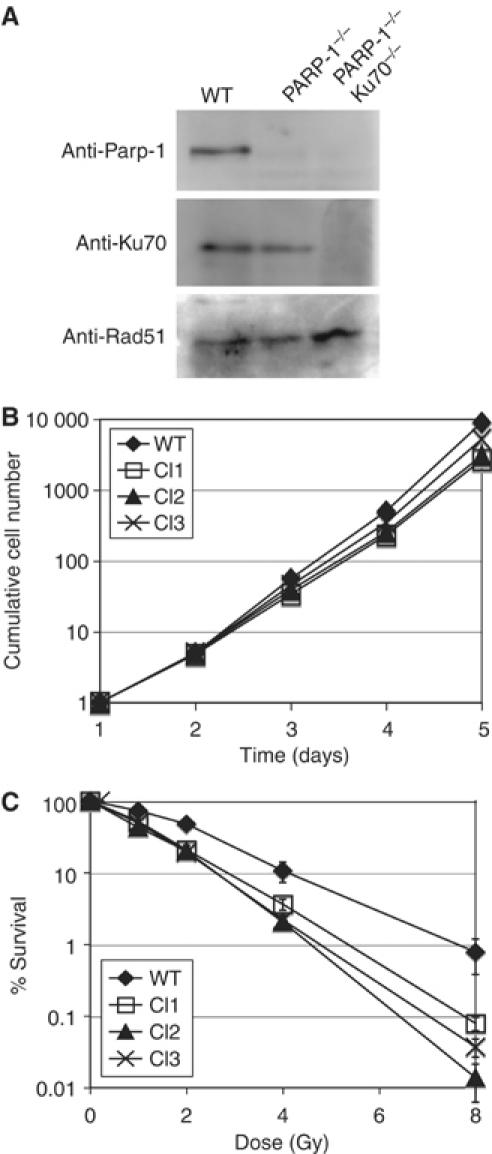
Characterization of PARP-1−/− cells in DT40. (A) Immunoblot analysis of DT40 WT and PARP-1−/− and PARP-1/KU70−/− cells using anti-Parp-1, Ku70 and Rad51 antibodies. (B) Growth curves of WT cells and three different PARP-1−/− clones. (C) Sensitivity of WT and PARP-1−/− clones to indicated doses of ionizing radiation, monitored by colony formation assay in methylcellulose. Values of the Y-axis indicate the percent ratio of the number of colonies after IR exposure versus the number of unexposed colonies. The means of three independent experiments, and standard deviation are shown.
PARP-1−/− clones were viable, but proliferated slightly more slowly than WT cells (Figure 1B) and were hypersensitive to IR (Figure 1C). We obtained three independent PARP-1−/− clones that showed a very similar phenotype with little clonal variation. All further experiments presented here were performed with Clone Nr.3. We next attempted to identify a chicken homolog of Parp-2 in order to generate Parp-1 and 2 double knock out DT40 cells. However, we could not find PARP-2 in the chicken genome or in EST databases, while other members of the Parp family such as Parp-3 were identified by homology searches. Parp-2 was originally discovered because high levels of cytotoxic treatment still triggered detectable levels of ADP-ribose polymer synthesis in PARP-1 −/− mouse cell lines (Ame et al, 1999). We reasoned that if Parp-2 was indeed absent in chicken, PARP-1−/− DT40 cells should lack any residual poly(ADP-ribosylating) activity in response to DNA damage. To confirm this, we probed WT and PARP-1−/− cells before and after H2O2 exposure by immunofluorescence using an anti-poly(ADP-ribose) (PAR) antibody. In WT cells, 1 mM H2O2 induced strong PAR synthesis (Figure 2A) that was absent in untreated cells and cells cotreated with 1 mM H2O2 and the Parp inhibitor 3-amino benzamide (3-AB) (data not shown). Unlike mouse embryonic fibroblast cell lines lacking Parp-1, our DT40 PARP-1−/− cells did not show a similar response to 1 mM H2O2 and appeared to be devoid of nuclear PAR (Figure 2A). We could, however, detect a single or double spot of immunofluorescence in the cytoplasm of anti-PAR-antibody-stained PARP-1−/− cells. This remaining cytoplasmic PAR could be a product of Parp-3 activity at the centrosomes (Augustin et al, 2003), but we did not further investigate this point. Next, we performed a biochemical assay to measure polymer formation in mutant and WT cells. We added radiolabeled [32P]NAD to permeabilized WT and PARP-1−/− cells that were pretreated with H2O2, and analyzed the resulting 32P-labeled PAR carrying proteins by SDS–PAGE and autoradiography. WT cells exhibited a major band of about 100 kDa that could be automodified Parp-1 itself and a smear of radiolabeled bands at lower molecular weights (Figure 2B). There was a slight induction of Parp activity after addition of H2O2, but Parp appeared to be already highly active in the untreated cells (Figure 2B WT lanes a and b). This activation may have been a result of DSBs introduced during the cell permeabilization. Parp activity dropped below detection limit in the presence of 3-AB (Figure 2B WT lane c). Confirming the results of our immunofluorescence experiment, PARP-1−/− cells did not contain any detectable poly(ADP-ribosylated) proteins in this assay (Figure 2B PARP-1−/− lanes a, b, and c).
Figure 2.
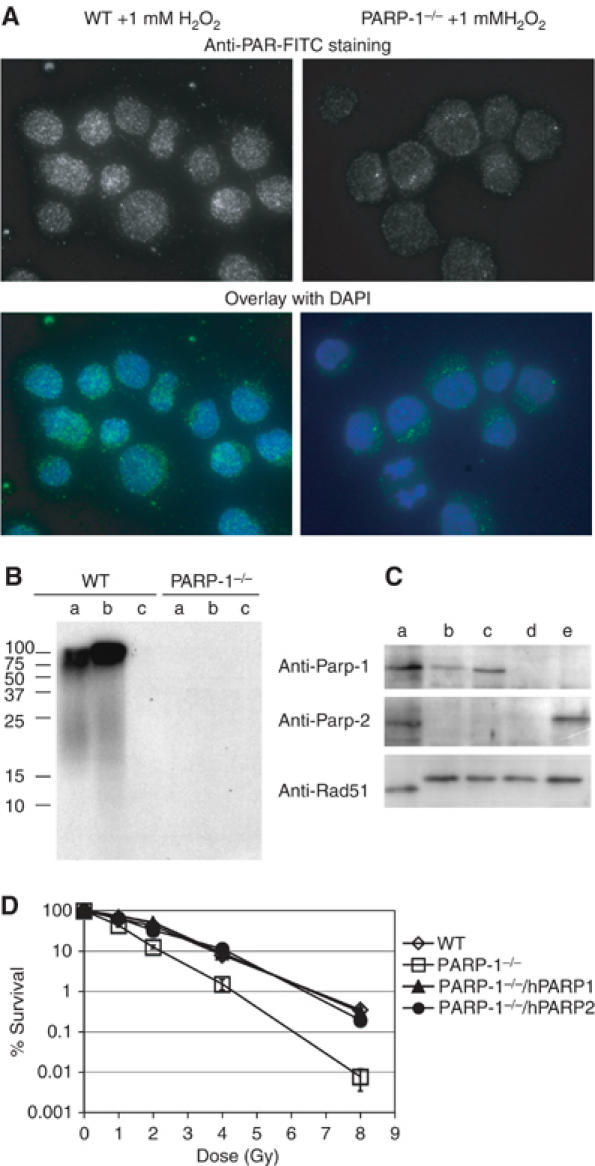
Apparent absence of a PARP-2 gene in PARP-1−/− cells. (A) Imaging of 1 mM H2O2 treated WT and PARP-1−/− cells by immunofluorescence using the 10H monoclonal anti-PAR antibody. Images of PAR staining with 10H primary and FITC-labeled secondary images, and merged FITC and DAPI images are shown. (B) Measuring polymer formation in DT40 cells. WT or PARP-1−/− cells were either untreated (a), exposed to 1 mM H2O2 (b), or 1 mM H2O2 and 3AB (c). Cells (106) of each sample were permeabilized and exposed to [32P]NAD. The cells were lysed in sample buffer and analyzed by SDS–PAGE and autoradiography; protein levels in each lane were compared by Coomassie staining (not shown). (C) Immunoblot analysis of human HeLa cells (a), DT40 WT cells (b), PARP-1−/− cells that stably expressed human Parp-1 (c), PARP-1−/− cells (d), or PARP-1−/− cells that stably expressed human Parp-2 (lane e) using the indicated antibodies. (Note that human Rad51 runs with higher velocity than chicken Rad51on the SDS–PAGE). (D) Sensitivity of the indicated cell lines to increasing doses of IR assayed by colony formation, as described in Figure 1 and Materials and methods; shown are the means of three independent experiments and standard deviation.
These results support our initial hypothesis that chickens might lack Parp-2. DT40 PARP-1−/− cells could thus be equivalents to a PARP-1/2 double mutant in a mammalian cell line. If these two Parp subtypes indeed share an overlapping function, overexpression of either of these proteins should revert the chicken PARP-1−/− phenotype. We tested this idea by establishing PARP-1−/− DT40 cells expressing human Parp-1 or Parp-2. Expression levels of the transgenes were confirmed by immunoblotting (Figure 2C). Expression of either hParp-1 or hParp-2 completely suppressed the IR sensitivity of PARP-1−/− DT40 cells (Figure 2D). We conclude that DT40 PARP-1−/− cells lack DNA damage-inducible Parp activity and thus appear to be the genetic equivalent of a mammalian PARP-1/2 double deletion.
Genetic interaction of chicken PARP-1 and KU70
Our primary aim in this study was to investigate the genetic interactions of Parp with players of the NHEJ pathways of DSB repair. We first focused on Ku, because this protein complex binds to Parp-1 and is poly(ADP-ribosylated) in vitro (Ariumi et al, 1999; Galande and Kohwi-Shigematsu, 1999; Li et al, 2004). We compared PARP-1−/− cells with PARP-1/KU70−/− cells (see above) with regard to their sensitivity to various genotoxic agents. To control for unexpected genetic alterations, we re-established Ku70 expression in this clone by stable integration of a vector carrying the Ku70 cDNA under the control of the CMV promoter. This cell line was thus expected to show the same phenotype as the PARP-1−/− single mutant.
We first tested the IR sensitivity of these various cell lines. Figure 3A shows that KU70−/− mutants are more than twice as sensitive as WT cells based on lethal dose (LD)10%. This is probably due to the high mortality of the G1 fraction of cells after low doses of irradiation (Takata et al, 1998). The remaining S- and G2-phase cells display elevated IR resistance, resulting in a decline of the slope of the sensitivity curve. This difference in resistance between WT and KU70−/− cells is likely due to the competition between Ku70 and the HR pathway (Takata et al, 1998; Fukushima et al, 2001; Pierce et al, 2001; Frank-Vaillant and Marcand, 2002). Surprisingly, KU70−/− had a suppressor effect on PARP-1−/− IR sensitivity at higher doses because the double mutants showed the same biphasic resistance pattern as the KU70−/− single mutant. Expression of the KU70 cDNA reversed this suppression, confirming that the IR sensitivity of PARP-1−/− indeed depends on the presence of Ku70. We next examined the interaction of Parp-1 and Ku in clonogenic survival after CPT and methyl-methanesulfonate (MMS) treatment. PARP-1−/− DT40 cells proved to be extremely sensitive to continuous CPT exposure, while KU70−/− cells were more resistant than WT cells as Adachi et al (2004) previously observed. Deletion of KU70 in PARP-1−/− cells restored CPT resistance to WT levels, but these mutants were still more sensitive than KU70−/− cells (Figure 3B). In the case of MMS, the Ku70 deletion also improved the clonogenic survival of PARP-1−/− cells, but the double mutants were still considerably more sensitive than WT DT40 cells (Figure 3C). We observed a similar partial reversal of sensitivity to other base-damaging drugs such as MNNG and H2O2 (data not shown).
Figure 3.
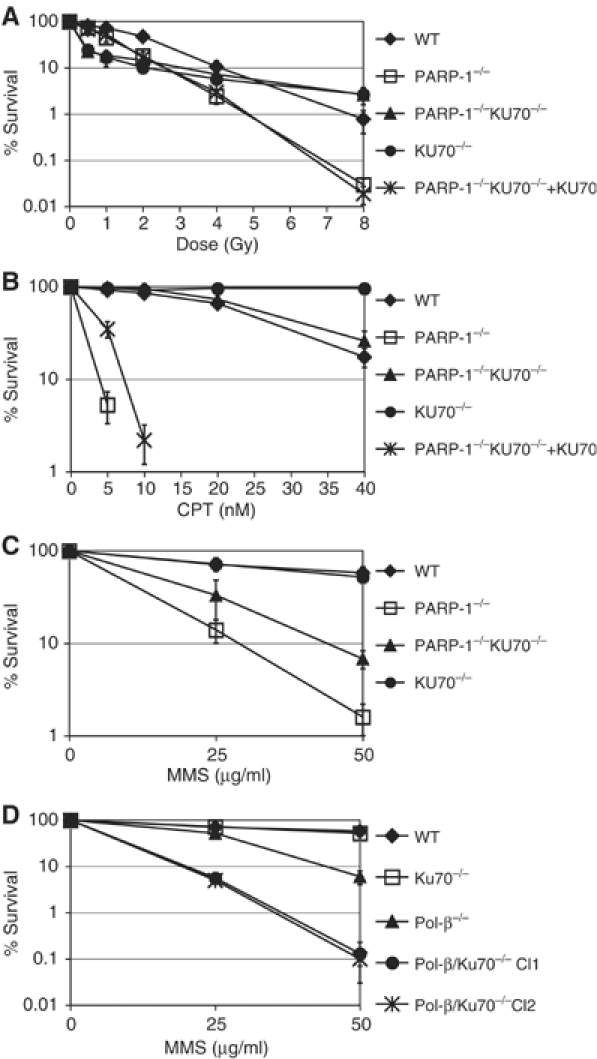
Differential interaction of KU70 with PARP-1 and POL-β (A) IR sensitivity of PARP-1−/−, PARP-1/KU70−/−, and other indicated cell lines. (B) Sensitivity of the same cell lines to continuous exposure of the indicated concentrations (nM) of CPT. (C) Sensitivity of the indicated cell lines to 1 h pulse exposure to 25 and 50 μg/μl MMS. (D) MMS sensitivity as in (C) of POL-β−/− and POL-β/KU70−/− cell lines. The means of three independent experiments and standard deviation are shown.
Deletion of KU70 does not suppress the polymerase-β phenotype
We first hypothesized that the suppression of the Parp-1 mutant phenotype by deletion of Ku could be an indirect result of more efficient HR in KU70−/− cells. A BER defect in PARP-1−/− cells is likely to lead to an accumulation of single strand breaks (SSBs) that are converted into DSBs during DNA replication. Deletion of Ku70 could allow more efficient repair of these DSBs by HR. If this were true, mutants in other BER repair genes such as polymerase-β (POL-β) should show a similar sensitivity reversal in the absence of Ku70. To test this idea, we established POL-β/KU70−/− cells by targeting the POL-β gene in KU70−/− cells (see Materials and methods) and probed the MMS sensitivity of the single and double mutants. Figure 3D shows that in contrast to PARP-1/KU70−/− cells, the MMS sensitivity of POL-β/KU70−/− was increased when compared to POL-β single mutants. Thus, the observed interaction of Parp-1 and Ku does not generally take place in BER repair mutants. We also tested the IR and CPT-sensitivity of POL-β single and POL-β/KU70 double mutants. POL-β−/− cells however, do not show a significant sensitivity to these agents and POL-β/KU70 double mutants show the same resistance as KU70 single mutant DT40 cells (data not shown).
Chicken Parp-1 protects HR from interference by Ku70
The suppression of the PARP-1−/− phenotype by inactivation of Ku70 could be explained by an inhibitory effect of Ku70 on HR that is counteracted by Parp-1. In order to test the HR efficacy in WT and mutant DT40 cells, we measured gene conversion induced by the rare-cutting endonuclease I-Sce1 using the SCneo construct (Johnson and Jasin, 2000). In this construct, two mutant neomycin allels that are complementary to each other are localized in tandem. One neo-coding region (S2neo) is disrupted by the I-SceI cleavage site, while the other serves as a donor in the HR reaction that restores a functional neomycin gene. We integrated this construct in the ovalbumin locus of DT40 WT, PARP-1−/− cells, and PARP-1−/− cells that expressed human Parp-1 (see Figure 2). Thus, each mutant had a single copy of the SCneo construct integrated at the same locus in the chicken genome. After transiently expressing the I-Sce1 endonuclease in these cells, we performed clonogenic survival assays in G418containing methylcellulose. Figure 4A shows that while 1.8% of the WT cells successfully underwent gene conversion and reconstituted G418 resistance, the same reaction occurred only in 0.5% of the PARP-1−/− cells. Overexpression of hParp-1 elevated the rate of I-Sce1-induced gene conversion to levels comparable to WT cells (1.6%). Hence, PARP-1−/− DT40 mutants are compromised in their ability to perform HR.
Figure 4.
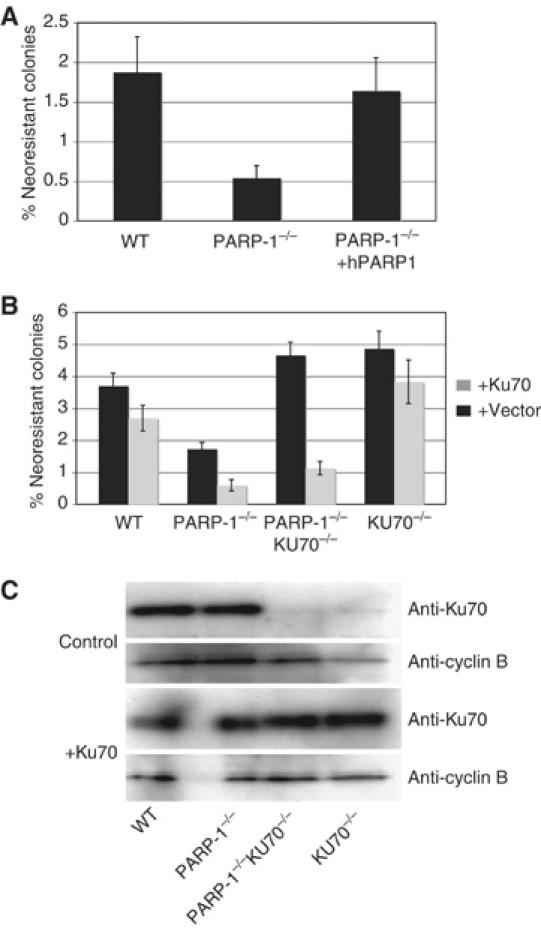
SCneo gene conversion assay. (A) Indicated cell lines carrying the scneo construct integrated in the OVA locus were transfected with I-Sce1 endonuclease and plated in methylcellulose with or without G418. Each bar indicates the percent ratio of the number of colonies growing with and without 6418. (B) Scneo gene conversion assay, as in (A), in indicated cell lines with (black bars) or without (gray bars) coexpression of KU70 transgene. Transfection of the I-Sce1 endonuclease and Ku70 in this experiment was performed using nucleofection (AMAXA) resulting in higher transfection efficacies than in (A) (see Materials and methods). In both (A) and (B), mean values of three independent experiments and standard deviation are shown. (C) Confirmation of Ku70 overexpression by analyzing extracts of cell lines with the indicated genotypes after transfection of control or Ku70 expression vector by immunoblotting with the anti-Ku70 antibody, and an anti-cyclin B2 antibody as a loading control.
Next, we investigated whether the reduction of HR in PARP-1−/− cells is a consequence of the presence of Ku70. We targeted the same SCneo construct as described above to the Ovalbumin locus of PARP-1/KU70−/− cells as well as KU70 single mutants and compared the I-Sce1-induced gene conversion rate of these mutants to PARP-1−/− and WT cells as described above. We also tested the effect of transient expression of Ku70 in each cell line, and Ku expression levels were confirmed by immunoblotting as shown in Figure 4C. In this experiment, the PARP-1−/− cells scored less than half of the HR events of WT cells (Figure 4B). Conversely, PARP-1/KU70−/− cells showed a higher gene conversion efficacy than WT DT40 cells, similar to KU70 single knock out cells. Transient expression of chicken Ku70 had a pronounced effect only on PARP-1−/− and PARP-1/KU70−/− cell lines, resulting in a three- and four-fold decrease in recombination efficacy, respectively. Thus, the presence or absence of Parp-1 drastically changes the inhibitory effect of Ku on HR in DT40 cells.
CPT induced SCE is decreased in PARP-1−/− but not PARP-1/Ku70−/− cells
Figure 3B shows that PARP-1−/− cells are highly sensitive to CPT. In fact, among all DT40 repair mutants tested in our laboratory, PARP-1-null mutants appear to display the lowest tolerance to CPT exposure (Takeda lab., unpublished results). We decided to investigate whether the reversal of CPT sensitivity in PARP-1/KU70−/− cells, shown in Figure 3B, could be the consequence of a direct involvement of Parp-1 in HR-mediated repair. CPT blocks Topoisomerase-I in a state where it is covalently linked to nicked DNA. The resulting protein/DNA crosslinks are obstacles to both DNA replication and transcription and appear to be repaired by the SSBR pathway (Pommier et al, 2003). Replication forks stalling at these lesions result in the formation of DSBs that are repaired by HR, using the intact sister chromatid as a template. Consequently, yeast mutants in the RAD52 epistasis group are also sensitive to CPT (Pommier et al, 2003), as are several DT40 cell lines defective in HR genes (Takeda lab., unpublished results). In order to examine the state of HR in PARP-1−/− and PARP-1/KU70−/− cells, we examined spontaneous and CPT-induced SCE. As shown in Figure 5, PARP-1−/− cells displayed an increase in spontaneous exchange events as compared to WT cells and SCE was further increased in PARP-1/KU70 double mutants. KU70−/− single mutants showed levels of SCE similar to WT cells. Exposure to 2.5 nM CPT induced, in average, 8 SCE events in WT cells. PARP-1−/− cells did not appear to respond to the same dose of CPT with elevated rates of SCE, while PARP-1/KU70 double-, as well as KU70 single mutants, had higher levels of induced SCE than WT cells, on average, 11.6 and 18.4 events per cell, respectively (the numbers are calculated from differences between the mean values of spontaneous and induced SCE in 50 nuclei as shown in Figure 5).
Figure 5.
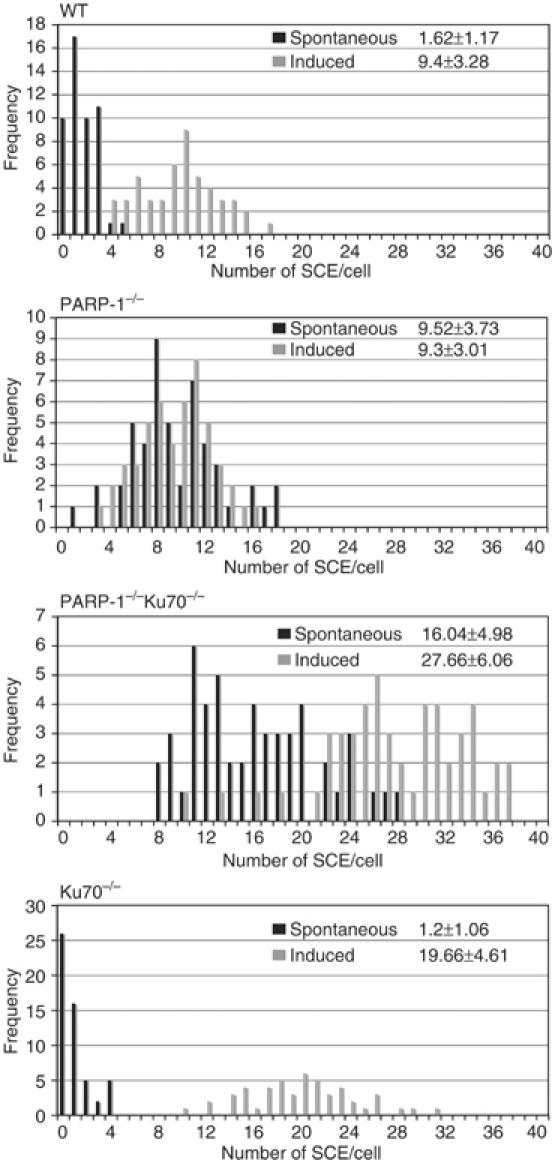
Spontaneous and CPT-induced SCE exchange. Cells were labeled with 1 mM BrdU during two cell cycle periods with (induced) or without (spontaneous) CPT treatment (2.5 nM) for the last 5.5 h. SCE in macrochromosomes of 50 nuclei were counted. The data are presented in histograms showing the frequency of nuclei with the indicated number of SCE. The mean value and standard deviation is shown in the upper right corner of each histogram.
Deletion of ligase IV (LIG4) is dominant over the PARP-1−/− phenotype
We next extended our genetic analysis by establishing PARP-1/LIG4−/− DT40 cells and testing their IR- and CPT sensitivity. We targeted both PARP-1 alleles in the previously described LIG4−/− cells (Adachi et al, 2001) and confirmed the successful deletion of Parp-1 by immunoblotting (Figure 6A). LIG4−/− cells are IR sensitive as shown in Figure 6B. Deletion of PARP-1 did not further sensitize these mutants to IR. In the case of CPT, LIG4−/− cells, similar to KU70−/− cells (see Figure 3), displayed an elevated resistance to CPT as previously observed by Adachi et al (2004). Similar to PARP-1/KU70−/− cells, PARP-1/LIG4 double mutants were more resistant to CPT than PARP-1 single mutants and showed a similar survival as WT cells. Thus, the phenotype of LIG4 is dominant over PARP-1 and suppresses the CPT sensitivity of cells lacking Parp-1.
Figure 6.
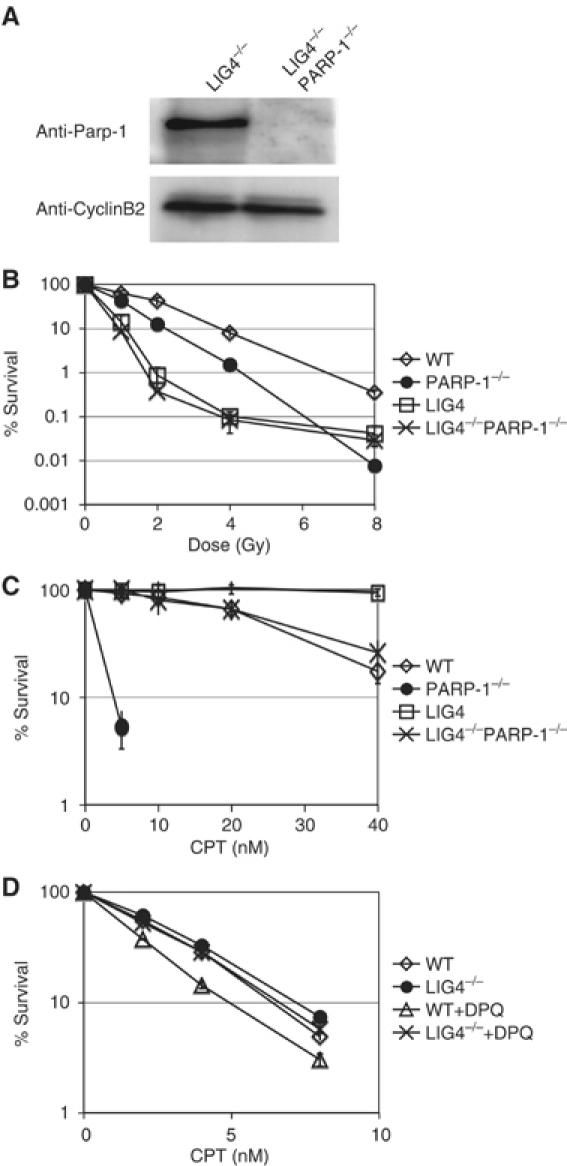
IR and CPT sensitivity in PARP/LIG4−/− cells. (A) Confirmation of the Parp-1 deletion in LIG4−/− cells by immunoblotting using anti-Parp-1 antibodies, and anti-cyclin B2 antibodies as a loading control. (B) Colony formation of indicated mutant DT40 cells after exposure to various doses of IR. (C) Colony formation assay of DT40 mutants in methylcellulose containing indicated concentrations of CPT. (D) Cell survival of NALM6 WT and LIG4−/− cells after 48 h growth in liquid medium containing indicated concentrations of CPT in the presence or absence of 10 μM DPQ. In experiments B, C and D, the means of three independent experiments and standard deviation are shown. In (D), standard deviation was very low and error bars are placed within the icons representing the various cell lines.
Inhibition of Parp in human NALM6 lymphocytes sensitizes WT but not LIG4−/− mutants to CPT
The results from our experiments with DT40 mutants, described in the sections above, suggest a novel, unexpected interaction between Parp and the NHEJ pathway in DSB repair. Experiments on DSB repair in DT40 cells, generally correlate well with findings in mammals, but the high recombination levels in this cell line could also result in phenotypes, which are irrelevant to mammalian cells, especially when the balance between HR and NHEJ is the subject of the investigation. In order to investigate the role of Parp in NHEJ mutants in a human cell line, we analyzed the effects of a chemical Parp inhibitor DPQ ([3,4-dihydro-5-[4-(1-piperidinyl)butoxyl-1(2H)-isoquinolin) (Eliasson et al, 1997) on CPT sensitivity in human NALM6 lymphocytes that were either WT or deficient in LIG4 (Grawunder et al, 1998; So et al, 2004). Figure 6D shows that DPQ treatment leads to a 50% drop in survival of WT but not LIG4 cells in the presence of CPT. It is, thus, likely that the observed interaction between Parp and NHEJ genes is a general feature of vertebrate cells. Inhibition of Parp in this human cell line, however, leads to a much less significant sensitization to CPT, then deletion of Parp-1 in DT40 cells. This difference may reflect the less potent HR-mediated repair of CPT-induced damage in WT NALM6 cells.
Discussion
Chicken DT40 cells lack a PARP-2 homolog
In this study, we analyzed the effect of Parp-1 deletion on chicken DT40 cells. Similar to the various Parp-1-deficient mouse models (Shall and de Murcia, 2000), PARP-1−/− DT40 cells were sensitive to IR, CPT, and DNA-methylating agents. In contrast to the mouse PARP-1−/− cells, we could not detect any residual DNA-strand-break-induced PAR-polymers in the DT40 PARP-1−/− mutants. Furthermore, we were not able to identify a chicken PARP-2 homolog in both EST libraries and the chicken genome. Other than in mammals, we identified a Parp-2 homolog in Xenopus EST libraries and in the Zebrafish genome. We do not know at this point what determines the evolutionary advantage of a Parp-2 gene in some species, but it will be of interest to study the presence or absence of this gene in various organisms as more and more genomic information becomes available. Clearly, mouse Parp-1 and Parp-2 share partially overlapping roles (Menissier de Murcia et al, 2003) and we could rescue the DT40 PARP-1−/− radiosensitivity by overexpression of either hParp-1or Parp-2. The lack of a cellular system in which both Parp-1 and Parp-2 are absent has been an obstacle to our understanding of the DNA-repair-associated role of Parp. The chicken DT40 PARP-1−/− cell line, which appears to lack Parp-2, is thus a valuable tool to further our understanding of the biology of PARP in DNA repair.
DT40 PARP-1−/− cells are defective in DSB repair mediated by HR
Several previous studies came to the conclusion that Parp-1 is not directly involved in the repair of DSBs induced by IR and I-Sce1 (Noel et al, 2003; Schultz et al, 2003; Yang et al, 2004). Conversely, we found that PARP-1−/− cells were less efficient in undergoing I-Sce1-induced gene conversion, and failed to induce SCE in response to CPT. Moreover, IR, CPT, and MMS sensitivity were at least partly suppressed by deletion of the DSB repair factor Ku. Taken together, these data suggest that DT40 cells lacking Parp-1 are compromised in their capacity to perform HR. The lack of Parp-2 might be a possible explanation for this difference between mouse and chicken PARP-1−/− cells. Alternatively, Parp-1 might have a separate function in HR in chicken. The high level of HR in DT40 cells might also contribute to differences in efficacy that would otherwise be below the detection limit. In general, however, DT40 cells have proved to be very useful to identify genes involved in HR and the data obtained from this cell line tend to correlate very well with results from mouse genetic studies. This issue will only be resolved once a mammalian PARP-1/Parp-2−/− cell line becomes available.
Ku is toxic in PARP-1−/− DT40 cells
Our analysis of PARP-1 single- and PARP-1/KU70 double mutants suggests that Parp-1 function is genetically intertwined with NHEJ and HR. We found that IR, CPT, and MMS sensitivity in PARP-1−/− cells were at least partially suppressed by the deletion of Ku, and that PARP-1/KU70 double mutants were in fact more IR and CPT resistant than WT cells. Our data suggest that the HR defect of PARP-1−/− DT40 cells depends entirely on the presence of Ku. Several previous studies suggested that Ku competes with HR by binding to DSBs and channeling the repair events toward the NHEJ pathway (Takata et al, 1998; Fukushima et al, 2001; Pierce, 2001; Frank-Vaillant and Marcand, 2002), so that deletion of Ku could confer elevated levels of resistance to CPT. In PARP-1−/− DT40 cells, the presence of Ku results in reduced HR efficacy, and, surprisingly, in dramatically increased DNA-damage sensitivity. Thus, Ku appears to exert a strong dominant negative effect on HR in DT40 cells lacking Parp-1. In other words, Parp-1 seems to be required to minimize the competitive effect of Ku on HR. A role for Parp-1 in protecting DNA breaks from unwanted Ku binding correlates well with biochemical results that suggest a direct interaction between these two proteins. According to Li et al's (2004) findings, the Ku complex is poly(ADP-ribosyl)ated by Parp-1 and this modification impairs its binding to DNA. These observations and our genetic data point to a scenario whereby Parp-1 inhibits Ku from binding to DSB and thereby allows access of the HR machinery.
Suppression of CPT sensitivity in PARP-1/LIG4 double mutants
Our analysis of the genetic interaction of PARP-1 and LIG4 in DT40 cells shows that the absence of NHEJ per se appears to suppress the sensitivity of Parp-1 mutants to CPT. In this case, it seems to be the execution of the NHEJ pathway, rather then the mere binding of Ku, that constitutes an unwanted effect as discussed by Adachi et al (2004). One could imagine that particular substrates that arise during replication fork collapse and subsequent repair by HR have to be protected from unwanted ligation, in order to allow the replication fork to proceed. However, other structures such as the hypothetical chicken foot could also be unwanted targets of NHEJ in the absence of Parp. The strong hypersensitivity of PARP-1−/− cells to CPT, and the striking suppression of this phenotype by both KU70 and LIG4 deletion, suggest that the protection from these aberrant ligation events might be a crucial function of Parp.
Interaction of Parp and NHEJ in mammals
In mice, a PARP-1/KU80 double deletion results in early embryonic lethality (Henrie et al, 2003). This may reflect the broad spectrum of functions of these two proteins in the maintenance of chromosome stability as well as other areas of biology such as cell contacts, telomere maintenance, and transcriptional regulation (Ame et al, 1999; Downs and Jackson, 2004). It is possible that Parp-1/2 play a similar role in minimizing the toxic effects of Ku, but synergies in other branches of function might nevertheless be detrimental to a developing embryo lacking both Parp and Ku. The genetic interactions that we observe in DT40 cells, on the other hand, have to be viewed in consideration of the hyper-recombination phenotype of this particular cell line. However, we could observe a similar interaction between Parp and NHEJ in human cells. It seems, thus, that in contrast to yeast, where NHEJ is only a minor pathway, vertebrates have evolved a mechanism to prevent the more dominant NHEJ pathway from interfering with essential HR reactions. This study points to a possible role of Parp in this mechanism.
Multiple DNA repair functions of Parp
While this study proposes a new role for Parp-1 in DSB repair, our findings also suggest multiple functions of this protein in other repair pathways, such as BER. Firstly, the observed high levels of SCE point to genetic instability in PARP-1−/− cells deriving from problems in repair pathways other than HR. High levels of SCE are likely to be the result of defects in repair pathways that cause an increase of HR substrates. A BER defect in PARP-1−/− cells, for example, would result in SSBs that are converted into DSBs during DNA replication. Cells lacking Parp could depend on HR as a back-up mechanism to deal with increased SSB. This dependence has been firmly established by two recent studies on the detrimental effect of Parp inhibitors on cells carrying BRCA2 mutations (Bryant et al, 2005; Farmer et al, 2005). The presence of Ku70, however, appears to suppress a subset of these HR reactions in PARP-1−/− DT40 cells, because the PARP-1/KU70−/− double mutants display a further increase in SCE levels. Secondly, the deletion of Ku only partially suppresses MMS sensitivity in PARP-1−/− cells. This remaining Ku-independent MMS sensitivity could reflect the BER repair function of Parp-1. Thus, PARP-1−/− cells might be compromised in multiple repair pathways, one of which results in an increase in spontaneous SCE, whereas at the same time HR efficacy is decreased. Figure 7 provides a model of Parp functions in SSB and DSB repair. This model might be further complicated by the recent finding of Levy et al (2006) that Xrcc1 is also closely linked to Ku and NHEJ. Similar to MMS, the difference in CPT sensitivity between NHEJ mutants and double mutants in both NHEJ and PARP-1 genes could be due to a Ku-independent CPT-repair function of Parp-1 such as the previously observed reactivation of topoisomerase-I by Parp-1 (Malanga and Althaus, 2004).
Figure 7.
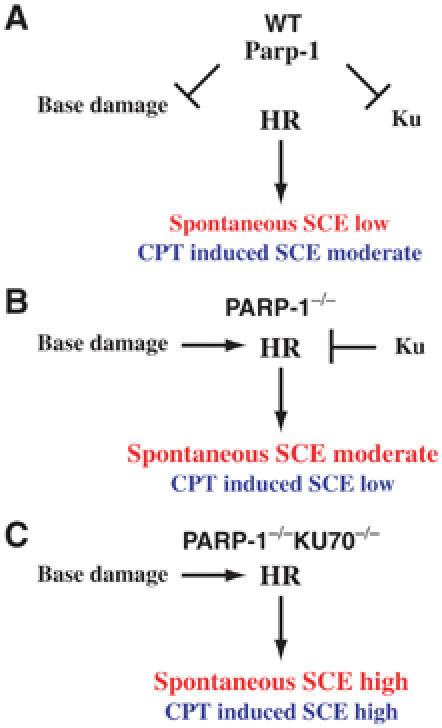
Model of Parp function in SSB and DSB repair in DT40 cells. One branch of Parp function is the stimulation of SSB repair. Remaining base damage will be converted into DSB during DNA replication and repaired by HR. The other branch concerns the role of Parp-1 in DSB repair in which Parp-1 minimizes the inhibitory effects of Ku on HR, as proposed in this study. Both functions affect the balance between HR and NHEJ differentially, as shown in (A) WT, (B) PARP-1−/− and (C) PARP-1/KU70−/− cells.
The results presented in this study point to a novel way of approaching Parp function. Parp inhibitors are promising therapeutic tools with a variety of applications such as cancer therapy and aging (Jagtap and Szabo, 2005). The finding that the deletion of Parp-1 in DT40 cells, or inhibition of Parp in NALM6 cells has different effects in different genetic backgrounds calls for caution, but also raises new possibilities. Cancer cells that are defective in HR genes might be highly sensitive to inhibition of Parp-1/2, whereas cells lacking Ku70 could display an increase in resistance. Clearly, Parp-1/2 are interconnected in different ways with a variety of repair pathways and represent ideal targets to modulate the effects of genotoxic treatment.
Materials and methods
Construction of gene targeting and expression vectors
To make the chicken PARP-1 gene-targeting construct, a 1 kb 5′ arm upstream of exons 2 and 3 was obtained by PCR (5′-AGGACTCGCTGCTGCGCCTG-3′; 5′-CGATGGAAAGTCCCTCA-3′) and cloned into the Not1 and BamH1 sites of pGEM-T Easy. The 4 kb 3′ arm was cut by Ssp1 and BamH1 from a longer genomic fragment that we obtained using 5′-AGCTACCAGCTACTAAGACTGAAG-3′ and 5′-ACGGGATGGCGTTTGGAGCTCTGCTTCCTT-3′ as primers and cloned into the pGEM vector containing the 5′arm. Various selection markers were cloned into the BamH1 site that separated the 5′ and 3′ genomic fragments of Parp-1 (see Supplementary Figure 1 for schematic representation of the PARP-1 locus and the targeting construct). The polymerase-β targeting construct was made by Eichiro Sonoda (Yoshimura et al; Tano et al, manuscripts in preparation). The Ovalbumin-targeting construct carrying Scneo was described by Fukushima et al (2001). hParp-1 expression vector was a gift from M Masutani; hParp-2 was cloned from a hParp-2 EGFP vector (Meder et al, 2005) into a pCR3 expression vector that contained an IRES-GFP element downstream of the multiple cloning site which was flanked by two loxP sites. To establish cell lines that stably express the chicken KU70 transgene, pAneo-GdKU70 (Fukushima et al, 2001) was used; for transient expression, the KU70 cDNA was cloned into the pCY4B vector (a gift from DrYasui Tohoku University).
Cell culture
DT40 cells were cultured and transfected as described previously (Sonoda et al, 1998). Gene targeting was confirmed by Southern blotting. Parp-1 deletion was confirmed by immunoblotting, and polymerase-β deletion by RT–PCR (data not shown). NALM6 cells were cultured as described by (So et al, 2004). To determine the CPT sensitivity, equal numbers of NALM6 cells were grown in medium containing various concentrations of CPT in the presence or absence of 10 μM DPQ (Alexis biochemicals). After 48 h, the number of live cells was determined by FACS analysis. The number of cells in medium without CPT was taken as a reference to calculate the percentage of survival.
Antibodies, immunoblotting, and immunofluorescence microscopy
Cells (105) were lysed in SDS sample buffer, separated by 12.5% SDS–PAGE, and transferred onto a membrane. Quality of transfer and amount of loaded protein was routinely checked by Ponceau staining. The following antibodies were used: anti-Parp-1 (dilution 1/2000 rabbit polyclonal, Monte (Schreiber et al, 2002)); anti-Parp-2 (1/2000 rabbit polyclonal, Yuc, Alexis Biochemicals); anti-Rad51 (1/1000 rabbit polyclonal AB-1 Oncogene); anti-chicken-Ku70 (1/500 rabbit polyclonal (Takata et al, 1998)), anti-cyclinB2 (1/1000 rabbit polyclonal (Gallant and Nigg, 1992)). For analysis of Rad51 foci, see Yamaguchi-Iwai et al (1999). To measure PAR by immunofluorecence, DT40 cells (105 cells/ml) were washed with PBS, either treated or not with 10 mM H2O2 for 5 min at room temperature and collected on a slide glass using a cytospin. Cells were fixed for 10 min with ice-cold methanol/acetone (1:1), and immunodetection of the PAR was performed as described by Dantzer et al (2004).
Biochemical detection of PAR in DT40 cells
Cells (106) were washed with PBS and incubated 10 min at 37°C in 56 mM Hepes pH 7.5, 28 mM KCl, 28 mM NaCl, 2 mM MgCl2, 0.01% digitonin, 0.125 uM NAD, 5 uCi [32P]NAD (3000 Ci/mmol), in the presence or absence of 1 mM H2O2 and 10 mM 3-AB. The reaction was stopped by the addition of 50 ul SDS-sample buffer. The samples were sonicated for 30 s and separated by 10% SDS–PAGE. The gel was dried and exposed O/N at −80°C.
Colony formation assay following genotoxic treatment
Colony formation assay was performed as described previously (Okada et al, 2002). Briefly, to assess IR sensitivity, serial diluted cells were plated in medium containing 1.5% (w/v) methylcellulose, incubated for 1 h at 39°C, and irradiated with a 137Cs source. For continuous CPT exposure, CPT was added at various concentrations to the medium containing methylcellulose and rotated at 4°C for at least 4 h. MMS pulse exposure was performed similar to CPT pulse exposure except that during incubation with MMS, serum was omitted.
Measurement of SCE levels
Measurements of SCE were performed as described previously (Sonoda et al, 1999). To detect CPT-induced SCE, cells were incubated in medium containing 10 μM BrdU and CPT was added at 2.5 nM for the last 5.5 h of the second cell cycle.
I-Sce1-induced gene conversion of SCneo
Modified SCneo was targeted into the OVALBUMIN locus as described previously (Fukushima et al, 2001). We used two different transfection systems: for the experiment shown in Figure 4A, cells were transfected using the Biorad electroporation system as described in Fukushima et al (2001). For the experiment in Figure 4B, 5 μg I-Sce1 endonuclease and Ku expression vectors were transfected into 5 × 106 cells using the Amaxa nucleofector kit (program B23). Electroporation was performed according to the manufacturer's instructions. In both cases, cells were incubated for 24 h, serially diluted, and plated into methylcellulose containing 2 mg/ml G418; a control set was plated into methylcellulose without G418. After 5–7 days, colonies appeared and were counted. We evaluated the gene conversion rate as the percent ratio of colonies growing in G418 to colonies growing in the control dished without G418.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank the members of Takeda, Masutani, and deMurcia labs for their help and support. Special thanks to Meiko Nakaoka for technical assistance, Jean-Christophe Amé for advice in Parp activity assays, and Jae Won Joh for critical reading of the MS. We thank Erich Nigg for sharing the chicken anti-cyclin B2 antibody. ST's lab is supported by COE and CREST grants. HH and CM were supported by a JSPS foreign postdoctoral fellowship, and HH by a COE grant. Work in CM's lab is supported by a Science Foundation Ireland Investigator award. VS and GdM were supported by Association pour la Recherche contre le Cancer, Ligue Contre le Cancer, Electricité de France and Commissariat a l'Energie Atomique.
References
- Adachi N, Ishino T, Ishii Y, Takeda S, Koyama H (2001) DNA ligase IV-deficient cells are more resistant to ionizing radiation in the absence of Ku70: implications for DNA double-strand break repair. Proc Natl Acad Sci USA 98: 12109–12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi N, So S, Koyama H (2004) Loss of nonhomologous end joining confers camptothecin resistance in DT40 cells. Implications for the repair of topoisomerase I-mediated DNA damage. J Biol Chem 279: 37343–37348 [DOI] [PubMed] [Google Scholar]
- Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Menissier-de Murcia J, de Murcia G (1999) PARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 274: 17860–17868 [DOI] [PubMed] [Google Scholar]
- Ame JC, Spenlehauer C, de Murcia G (2004) The PARP superfamily. BioEssays 26: 882–893 [DOI] [PubMed] [Google Scholar]
- Ariumi Y, Masutani M, Copeland TD, Mimori T, Sugimura T, Shimotohno K, Ueda K, Hatanaka M, Noda M (1999) Suppression of the poly(ADP-ribose) polymerase activity by DNA-dependent protein kinase in vitro. Oncogene 18: 4616–4625 [DOI] [PubMed] [Google Scholar]
- Augustin A, Spenlehauer C, Dumond H, Menissier-De Murcia J, Piel M, Schmit AC, Apiou F, Vonesch JL, Kock M, Bornens M, De Murcia G (2003) PARP-3 localizes preferentially to the daughter centriole and interferes with the G1/S cell cycle progression. J Cell Sci 116: 1551–1562 [DOI] [PubMed] [Google Scholar]
- Baumann P, West SC (1998) Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci 23: 247–251 [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917 [DOI] [PubMed] [Google Scholar]
- Dantzer F, Giraud-Panis MJ, Jaco I, Ame JC, Schultz I, Blasco M, Koering CE, Gilson E, Menissier-de Murcia J, de Murcia G, Schreiber V (2004) Functional interaction between poly(ADP-Ribose) polymerase 2 (PARP-2) and TRF2: PARP activity negatively regulates TRF2. Mol Cell Biol 24: 1595–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, Walztinger C, Chambon P, de Murcia G (1997) Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA 94: 7303–7307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JA, Jackson SP (2004) A means to a DNA end: the many roles of Ku. Nat Rev Mol Cell Biol 5: 367–378 [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL (1997) Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3: 1089–1095 [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921 [DOI] [PubMed] [Google Scholar]
- Frank-Vaillant M, Marcand S (2002) Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell 10: 1189–1199 [DOI] [PubMed] [Google Scholar]
- Fukushima T, Takata M, Morrison C, Araki R, Fujimori A, Abe M, Tatsumi K, Jasin M, Dhar PK, Sonoda E, Chiba T, Takeda S (2001) Genetic analysis of the DNA-dependent protein kinase reveals an inhibitory role of Ku in late S–G2 phase DNA double-strand break repair. J Biol Chem 276: 44413–44418 [DOI] [PubMed] [Google Scholar]
- Galande S, Kohwi-Shigematsu T (1999) Poly(ADP-ribose) polymerase and Ku autoantigen form a complex and synergistically bind to matrix attachment sequences. J Biol Chem 274: 20521–20528 [DOI] [PubMed] [Google Scholar]
- Gallant P, Nigg EA (1992) Cyclin B2 undergoes cell cycle-dependent nuclear translocation and, when expressed as a non-destructible mutant, causes mitotic arrest in HeLa cells. J Cell Biol 117: 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR (1998) DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell 2: 477–484 [DOI] [PubMed] [Google Scholar]
- Henrie MS, Kurimasa A, Burma S, Menissier-de Murcia J, de Murcia G, Li GC, Chen DJ (2003) Lethality in PARP-1/Ku80 double mutant mice reveals physiological synergy during early embryogenesis. DNA Repair (Amst) 2: 151–158 [DOI] [PubMed] [Google Scholar]
- Huber A, Bai P, de Murcia JM, de Murcia G (2004) PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair (Amst) 3: 1103–1108 [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabo C (2005) Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 4: 421–440 [DOI] [PubMed] [Google Scholar]
- Johnson RD, Jasin M (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 19: 3398–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Krasieva TB, Kurumizaka H, Chen DJ, Taylor AM, Yokomori K (2005) Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol 170: 341–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy N, Martz A, Bresson A, Spenlehauer C, de Murcia G, Menissier-de Murcia J (2006) XRCC1 is phosphorylated by DNA-dependent protein kinase in response to DNA damage. Nucleic Acids Res 34: 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Navarro S, Kasahara N, Comai L (2004) Identification and biochemical characterization of a Werner's syndrome protein complex with Ku70/80 and poly(ADP-ribose) polymerase-1. J Biol Chem 279: 13659–13667 [DOI] [PubMed] [Google Scholar]
- Lindahl T, Satoh MS, Poirier GG, Klungland A (1995) Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci 20: 405–411 [DOI] [PubMed] [Google Scholar]
- Malanga M, Althaus FR (2004) Poly(ADP-ribose) reactivates stalled DNA topoisomerase I and induces DNA strand break resealing. J Biol Chem 279: 5244–5248 [DOI] [PubMed] [Google Scholar]
- Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G (1998) XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol 18: 3563–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani M, Nozaki T, Nishiyama E, Shimokawa T, Tachi Y, Suzuki H, Nakagama H, Wakabayashi K, Sugimura T (1999) Function of poly(ADP-ribose) polymerase in response to DNA damage: gene-disruption study in mice. Mol Cell Biochem 193: 149–152 [PubMed] [Google Scholar]
- Meder VS, Boeglin M, de Murcia G, Schreiber V (2005) PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J Cell Sci 118: 211–222 [DOI] [PubMed] [Google Scholar]
- Menisser-de Murcia J, Mark M, Wendling O, Wynshaw-Boris A, de Murcia G (2001) Early embryonic lethality in PARP-1 Atm double-mutant mice suggests a functional synergy in cell proliferation during development. Mol Cell Biol 21: 1828–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G (2003) Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J 22: 2255–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison C, Smith GC, Stingl L, Jackson SP, Wagner EF, Wang ZQ (1997) Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nat Genet 17: 479–482 [DOI] [PubMed] [Google Scholar]
- Noel G, Giocanti N, Fernet M, Megnin-Chanet F, Favaudon V (2003) Poly(ADP-ribose) polymerase (PARP-1) is not involved in DNA double-strand break recovery. BMC Cell Biol 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa A, Tohda H, Kanai M, Miwa M, Sugimura T (1980) Inhibitors of poly(adenosine diphosphate ribose) polymerase induce sister chromatid exchanges. Biochem Biophys Res Commun 97: 1311–1316 [DOI] [PubMed] [Google Scholar]
- Okada T, Sonoda E, Yamashita YM, Koyoshi S, Tateishi S, Yamaizumi M, Takata M, Ogawa O, Takeda S (2002) Involvement of vertebrate polkappa in Rad18-independent postreplication repair of UV damage. J Biol Chem 277: 48690–48695 [DOI] [PubMed] [Google Scholar]
- Paques F, Haber JE (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63: 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Hu P, Han M, Ellis N, Jasin M (2001) Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev 15: 3237–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Redon C, Rao VA, Seiler JA, Sordet O, Takemura H, Antony S, Meng L, Liao Z, Kohlhagen G, Zhang H, Kohn KW (2003) Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat Res 532: 173–203 [DOI] [PubMed] [Google Scholar]
- Rouleau M, Aubin RA, Poirier GG (2004) Poly(ADP-ribosyl)ated chromatin domains: access granted. J Cell Sci 117: 815–825 [DOI] [PubMed] [Google Scholar]
- Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, Menissier-de Murcia J, de Murcia G (2002) Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem 277: 23028–23036 [DOI] [PubMed] [Google Scholar]
- Schultz N, Lopez E, Saleh-Gohari N, Helleday T (2003) Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res 31: 4959–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shall S, de Murcia G (2000) Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res 460: 1–15 [DOI] [PubMed] [Google Scholar]
- So S, Adachi N, Lieber MR, Koyama H (2004) Genetic interactions between BLM and DNA ligase IV in human cells. J Biol Chem 279: 55433–55442 [DOI] [PubMed] [Google Scholar]
- Sonoda E, Morrison C, Yamashita YM, Takata M, Takeda S (2001) Reverse genetic studies of homologous DNA recombination using the chicken B-lymphocyte line, DT40. Philos Trans R Soc Lond B Biol Sci 356: 111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S (1998) Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 17: 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Morrison C, Yamaguchi-Iwai Y, Takata M, Takeda S (1999) Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol Cell Biol 19: 5166–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17: 5497–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF (1995) Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev 9: 509–520 [DOI] [PubMed] [Google Scholar]
- Yamaguchi-Iwai Y, Sonoda E, Sasaki MS, Morrison C, Haraguchi T, Hiraoka Y, Yamashita YM, Yagi T, Takata M, Price C, Kakazu N, Takeda S (1999) Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J 18: 6619–6629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ (2004) Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 23: 3872–3882 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1