

Abstract
How sorting receptors recognize amino acid determinants on polypeptide ligands and respond to pH changes for ligand binding or release is unknown. The plant vacuolar sorting receptor BP-80 binds polypeptide ligands with a central Asn-Pro-Ile-Arg (NPIR) motif. tBP-80, a soluble form of the receptor lacking transmembrane and cytoplasmic sequences, binds the peptide SSSFADSNPIRPVTDRAASTYC as a monomer with a specificity indistinguishable from that of BP-80. tBP-80 contains an N-terminal region homologous to ReMembR-H2 (RMR) protein lumenal domains, a unique central region, and three C-terminal epidermal growth factor (EGF) repeats. By protease digestion of purified secreted tBP-80, and from ligand binding studies with a secreted protein lacking the EGF repeats, we defined three protease-resistant structural domains: an N-terminal/RMR homology domain connected to a central domain, which together determine the NPIR-specific ligand binding site, and a C-terminal EGF repeat domain that alters the conformation of the other two domains to enhance ligand binding. A fragment representing the central domain plus the C-terminal domain could bind ligand but was not specific for NPIR. These results indicate that two tBP-80 binding sites recognize two separate ligand determinants: a non-NPIR site defined by the central domain–EGF repeat domain structure and an NPIR-specific site contributed by the interaction of the N-terminal/RMR homology domain and the central domain.
INTRODUCTION
Animal, yeast, and plant cells have in common an organelle within their secretory pathways that maintains an acidic pH and functions as a terminal degradative compartment (Boller and Kende, 1979; Klionsky et al., 1990). Cells from the three types of organisms share a common mechanism for delivery of soluble proteins to the lysosome/lytic vacuole: an integral membrane receptor protein binds ligands at a relatively neutral pH in the Golgi/trans-Golgi network and delivers them to an endosomal/prevacuolar compartment, where the presence of an acidic pH causes their release from the receptor protein (Kornfeld, 1992; Kirsch et al., 1994; Cereghino et al., 1995; Hille-Rehfeld, 1995; Cooper and Stevens, 1996; Paris et al., 1997; Robinson and Hinz, 1997; Jiang and Rogers, 1998).
Interestingly, however, the three types of cells differ greatly in the structures and ligand specificities of their lysosomal/vacuolar sorting receptors (VSRs). The animal receptors recognize mannose 6-phosphate residues on Asn-linked oligosaccharides on proteins to be sorted to lysosomes (Kornfeld, 1992; Hille-Rehfeld, 1995). In contrast, the yeast VPS10p (Marcusson et al., 1994) and plant (Kirsch et al., 1994) receptors bind to amino acid sequence determinants within the polypeptide chains of the ligands, but the two families of proteins share no sequence homology (Paris et al., 1997). It is curious that two completely different receptor proteins would have evolved for relatively similar functions in the two types of organisms. We have speculated that plant cells, because they maintain two separate sorting pathways to two distinct types of vacuoles (Okita and Rogers, 1996; Neuhaus and Rogers, 1998), may have required a novel receptor that could distinguish proteins destined for the lytic vacuole from those to be sorted to a storage vacuole (Paris et al., 1997).
We identified the VSR family of proteins, of which BP-80 (VSRPs-1) is a prototype (Kirsch et al., 1994; Paris and Rogers, 1996; Paris et al., 1997), from strong but circumstantial evidence for that function. These are Type I integral membrane proteins (see Figure 1). BP-80 purified from clathrin-coated vesicle membranes of pea cotyledons was found to bind a synthetic peptide representing the vacuolar targeting determinant from the cysteine protease proaleurain with a Kd of 37 nM; binding was optimal at pH 6.0 to 6.5 and was abolished at pH 4.0 (Kirsch et al., 1994). Binding was specific for known vacuolar targeting determinants that contained a central, conserved central amino acid motif of Asn-Pro-Ile-Arg (NPIR), and mutations within that motif that abolished vacuolar targeting also abolished binding (Kirsch et al., 1994, 1996). BP-80 was abundant in highly purified clathrin-coated vesicles that lacked detectable storage proteins (Hohl et al., 1996), whereas highly purified dense vesicles that transported seed-type storage proteins to protein storage vacuoles in pea cotyledon cells contained little or no BP-80 (Hinz et al., 1999). BP-80 was localized to the dilated ends of Golgi cisternae and to “prevacuoles”—structures ∼250 nm in size that were adjacent to and appeared to be able to fuse with large lytic vacuoles—but it was not detected in the tonoplast membrane of vacuoles (Paris et al., 1997). Finally, a chimeric reporter protein was constructed that contained a mutated form of proaleurain (i.e., lacking vacuolar targeting determinants) connected through its C terminus to the transmembrane domain and cytoplasmic tail of BP-80; when this reporter was expressed in tobacco suspension culture cells, the BP-80 transmembrane domain/cytoplasmic tail sequences directed it through the Golgi to a prevacuolar compartment, where the proaleurain moiety was processed to mature form (Jiang and Rogers, 1998). Thus, BP-80 has all the structure, ligand binding characteristics, intracellular location, and pattern of traffic within the plant secretory pathway that would be expected for a VSR. Final proof of receptor function, as in the example of yeast VPS10p (Marcusson et al., 1994), will require demonstration that BP-80 can bind its proaleurain ligand within the secretory pathway, but we believe the evidence for that function is sufficiently strong to justify a detailed study of structural features that mediate its ligand binding functions.
Figure 1.

Secretion of Truncated Forms of BP-80 from Drosophila S2 Cells.
(A) Structure of truncations. Drawn to scale at the top is a model of BP-80; the open rectangle is the transmembrane domain, the oval represents the EGF-CB repeat (Davis, 1990), the black rectangles represent the EGF repeats, and the checked rectangle represents the domain found in RMR proteins. The circled C symbols identify the positions of Cys residues in the portion of the protein that is N-terminal to the EGF repeats. Shown below, numbered (1) to (4), are the four truncated forms of BP-80 expressed in Drosophila S2 cells. MAbs refers to monoclonal antibodies that recognize BP-80, and brackets above the four truncations indicate the regions of the proteins containing epitopes for the MAbs. At right, kD refers to the predicted molecular masses of the four truncations (minus their signal peptides) as calculated from their amino acid sequences.
(B) Binding to different truncated forms of BP-80 by the MAbs. Aliquots of Drosophila S2 cell media from cultures expressing tBP-80 (lanes 1), Δ1EGFR (lanes 2), Δ2EGFR (lanes 3), and Δ3EGFR (lanes 4) were denatured in the absence of disulfide-reducing agents, electrophoresed on a 4 to 20% SDS–polyacrylamide gel, and transferred to nitrocellulose, where replicate blots were incubated with each MAb as indicated. Volumes of culture media used were adjusted to yield roughly equal amounts of each truncated protein; the abundance of Δ1EGFR and Δ2EGFR was ∼0.10 times that of the other two proteins. A secondary antibody coupled to alkaline phosphatase was used to detect the antibody complexes. M indicates prestained blue molecular mass markers, with their size in kilodaltons indicated at left.
The VSR proteins are composed of a unique region ∼400 amino acids long at their N termini (which previously was thought to lack homologs in the animal and yeast databases), followed by three epidermal growth factor (EGF) repeats connected sequentially to a short Ser/Thr–rich sequence, a transmembrane domain, and a cytoplasmic tail (Paris et al., 1997). Two domains of the VSR proteins are of particular interest with respect to the studies that follow.
Within the unique region, ∼100 residues near the N terminus define a domain that is highly conserved in the lumenal sequences of what we have termed ReMembR-H2 (for receptor–transmembrane sequence–RING H2, abbreviated RMR) proteins (S.W. Rogers and J.C. Rogers, unpublished data). These receptor-like proteins are expressed in mammals, birds, plants, and Schizosaccharomyces pombe and appear to traffic to endosomal/prevacuolar compartments; their functions are unknown. We designate this N-terminal region within the VSR proteins as the RMR homology domain.
A second domain of interest is defined by the three EGF repeats. The EGF repeat motif is found in numerous proteins, including growth factors, transmembrane proteins that function as receptors (or as homeotic gene products), extracellular matrix proteins, and soluble secreted proteins that have highly regulated protease functions (Davis, 1990). To the best of our knowledge, however, the VSR proteins are the only examples of proteins carrying EGF repeats that are not directed to the cell exterior to perform their functions (Davis, 1990). VSR proteins contain two different types of EGF repeats, the first two having a B.1 consensus and the third a B.2 consensus (Herz et al., 1988). The functional difference in the two motifs is more easily appreciated by considering that B.2 contains conserved residues known to permit high-affinity calcium binding; accordingly, the two types should be designated EGF and EGF-CB (for calcium binding) motifs (Davis, 1990). In other studies of receptors or receptor-like proteins, EGF repeats have variously been shown to make up the ligand binding domain of thrombomodulin (Stearns et al., 1989; Zushi et al., 1989), to affect acid-dependent ligand dissociation from the low-density lipoprotein receptor (Davis et al., 1987), and to alter the conformation of a lectin domain in a peripheral lymph node homing receptor (Bowen et al., 1990). Thus, they participate in protein–protein interactions, whether cis or trans.
An understanding of how VSR proteins interact with their ligands, and of how pH affects these interactions, should provide insight into the role the RMR homology domain might have in protein–protein interactions and into the functions of the EGF repeats in this novel protein family. To investigate these questions, we have expressed four different soluble truncated forms of the protein in Drosophila S2 tissue culture cells: tBP-80 (lacking transmembrane domain and cytoplasmic tail), Δ1EGFR (tBP-80 with the additional deletion of one EGF repeat), Δ2EGFR (tBP-80 with the additional deletion of two EGF repeats), and Δ3EGFR (tBP-80 with all EGF repeats deleted). All four forms of the protein were secreted from the cells. We utilized ligand binding studies and studies of interactions of each form with four different monoclonal antibodies (MAbs) to probe the structural requirements for ligand binding and specificity. tBP-80 had a ligand binding specificity that was indistinguishable from that previously described for full-length BP-80 (Kirsch et al., 1994). In contrast, Δ3EGFR demonstrated very weak ligand binding. However, when either of two MAbs that specifically recognized the unique region of Δ3EGFR was included in the binding assay, the specific, high-affinity binding of the ligand was restored.
In a complementary approach, structural domains of tBP-80 were defined by digesting the protein with endoproteinase Asp-N, and protease-resistant fragments were mapped by their reactivity with different antibodies. tBP-80, which was protease sensitive, was rapidly degraded to two predominant size classes of products of similar abundance that were stable on continued exposure to the enzyme: one represented approximately two-thirds of the N-terminal region of the protein similar to Δ3EGFR, whereas the second represented approximately three-fifths of the C-terminal region of the protein and lacked the N-terminal RMR homology domain. Fragments in both classes retained the ability to bind proaleurain peptide ligand, although the latter appeared to have a lower affinity than the former and lacked NPIR sequence specificity.
These results indicate that tBP-80 consists of three domains: an N-terminal/RMR homology domain, a central domain, and a C-terminal EGF repeat domain. Protease-accessible sequences (termed “loops” for simplicity) connect each of the flanking domains to the central domain; their accessibility depends on the conformation of the protein, and the conformation in turn depends on which domains are present. The EGF repeats serve to keep the protein in an optimal conformation for ligand binding; in their absence, little binding was observed, but high-affinity binding was restored by interaction of the central domain with either of two different MAbs that appeared to mimic the effects of the EGF repeats. Ligand binding that is specific for the presence of the NPIR motif requires both the N-terminal/RMR homology domain and the central domain.
RESULTS
We expressed four different C-terminal truncations of BP-80 in Drosophila S2 cells. The structures of these proteins, truncated at their C termini, are shown in Figure 1A, in comparison with full-length BP-80. The positions of Cys residues within the unique region of the full-length protein are identified to emphasize the likelihood that this portion of the protein has a complex structure supported by seven intramolecular disulfide bonds. tBP-80 (Figure 1A, truncated protein 1), with 562 amino acids, is identical to the truncated protein described by Paris et al. (1997) and lacks the transmembrane domain/cytoplasmic tail sequences of the intact protein. Δ1EGFR (Figure 1A, protein 2), with 517 amino acids, additionally lacks the third EGF repeat; Δ2EGFR (Figure 1A, protein 3), with 468 amino acids, additionally lacks two EGF repeats; and Δ3EGFR (Figure 1A, protein 4), with 414 amino acids, additionally lacks all three EGF repeats. The predicted molecular masses for each of the truncated proteins after cleavage of their signal peptides (Paris et al., 1997) are also indicated.
All four proteins were secreted from the cells and accumulated in the culture medium. We detected the proteins on protein gel blots after SDS-PAGE with four different MAbs specific for different domains of the BP-80 protein (Figure 1B). Because the epitopes recognized by all four MAbs required preservation of intramolecular disulfide bonds for their integrity, the proteins were denatured by heating in SDS-PAGE sample buffer but were not treated with a disulfide-reducing agent before electrophoresis. All four MAbs recognized tBP-80 (Figure 1B, lanes 1). MAb 14G7, however, did not recognize any of the truncated forms (14G7, lanes 2 to 4); this result indicates that the epitope for 14G7 requires the presence of the third EGF repeat or the Ser/Thr–rich sequence (or both), as indicated by the bracket in Figure 1A. 14G7 therefore is a sensitive probe for the presence of the C terminus of the protein. MAb 19F2 did not recognize the shortest protein (the form lacking all EGF repeats) (Figure 1B, 19F2, lane 4). This result indicates that the epitope for MAb 19F2 is dependent on the presence of the first two EGF repeats, as indicated by the bracket in Figure 1A. The fact that 19F2 recognized Δ1EGFR poorly (19F2, lane 2) in comparison with Δ2EGFR (19F2, lane 3) may indicate that its epitope is predominantly dependent on the presence of the first EGF repeat and that the presence of the second EGF repeat partially obscures that epitope. In contrast, MAbs 17F9 and 18E7 recognized all four of the truncated proteins (Figure 1B), which indicates that their epitopes are contained entirely within the unique domain.
We wanted to use these truncated proteins to learn more about the structural requirements for ligand binding. Our previous ligand binding assay utilized a synthetic peptide with the sequence SSSFADSNPIRPVTDRAASTYC (Kirsch et al., 1994), of which the first 20 residues define the proaleurain vacuolar targeting determinant (note the central NPIR motif, which is underlined) and Y and C represent residues added to assist in coupling or labeling the peptide. When we again used this peptide, we coupled it through the Cys residue to the fluorescent tag, N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-yl)methyl) iodoacetamide (BODIPY-FL). In the previous study using full-length BP-80 in a detergent lysate of membrane proteins, the BP-80 protein with bound ligand could be separated from free peptide by precipitating the protein with polyethylene glycol (Kirsch et al., 1994). However, tBP-80 would not precipitate in this manner (data not shown). We therefore utilized chromatography with gel filtration through Superdex 200 (Amersham Pharmacia) to separate the BP-80 truncations and free peptide.
Ligand Binding Assay with Fluorescent Proaleurain Peptide
The Superdex 200 column was calibrated with markers of different molecular weights, and the elution positions of those proteins were compared with that of tBP-80 (Figure 2A). BSA, with a molecular weight of ∼66,000, eluted with a peak positioned between fractions 31 and 32. tBP-80, as detected by protein gel blot analysis (Figure 2A, bottom), eluted with a peak at fraction 31. This result, a size of ⩽66 kD for tBP-80, is consistent with that shown in Figure 1B and indicates that tBP-80 chromatographed as a monomer in the Superdex 200 system. If tBP-80 were a dimer under these conditions, it would have eluted at a position close to the peak obtained for alcohol dehydrogenase (150 kD) at fraction 27 (data not shown). The sensitivity of the assay system would have readily permitted detection of a shift of three or four fractions in its elution position.
Figure 2.

Superdex 200 Chromatography of tBP-80.
(A) tBP-80 chromatographs as a monomer. At top is shown a tracing of the A280 profile from calibration of the column by chromatography of four different size standards: a blue dextran peak defines the void volume (Vo), BSA provides a 66-kD peak, carbonic anhydrase provides a 29-kD peak, and cytochrome c provides a 12.4-kD peak. The elution position of 150-kD alcohol dehydrogenase from a separate run is also indicated. The vertical lines beneath the absorbance tracing indicate 0.5-mL fractions, with fraction numbers shown beneath. At bottom is shown protein gel blot detection, by enhanced chemiluminescence, of the elution position of tBP-80 when 0.2 mL of Drosophila S2 medium that had been concentrated fivefold was chromatographed on the column. tBP-80 eluted with a peak in fraction 31; in comparison, the position of elution of 66-kD BSA is indicated by the arrow.
(B) Fluorescent ligand binding assay. Medium containing tBP-80 was incubated with 10−7 M BODIPY-FL–labeled proaleurain peptide (*Peptide), as described in Methods, and then chromatographed on the Superdex 200 column. The vertical axis indicates fluorescence intensity for each fraction. Circles indicate *Peptide without competing peptide; X's indicate *Peptide plus a final concentration of 1 mM Spo peptide (sequence SRFNPIRLPT); squares indicate *Peptide plus 1 mM Spo-G peptide (sequence SRFNPgRLPT); Bound indicates the position of *Peptide bound to tBP-80; and Free indicates the elution position of *Peptide not bound to protein.
The BODIPY-FL–labeled peptide was then used to characterize ligand binding by tBP-80; the results are presented in Figure 2B. We used medium from Drosophila S2 cells expressing the protein, which had been concentrated approximately fivefold for the assays. When an aliquot was incubated with 10−7 M labeled peptide and then chromatographed on the Superdex 200 column, two fluorescent peaks were detected (open circles). The one at approximately fraction 50 corresponded to free peptide, whereas the second peak, at fraction 31, corresponded to the elution position of tBP-80 (Figure 2A, bottom). Similar results were obtained when the proaleurain peptide was labeled with iodine-125, and the presence of 104 M excess unlabeled peptide completely abolished the peak centered on fraction 31 (data not presented). Thus, the peak centered at fraction 31 was not dependent on any single peptide-labeling strategy. We hypothesized that it represented peptide bound by tBP-80, and used assays of sequence specificity requirements for binding and peak shifts induced by anti–BP-80 MAbs to confirm the hypothesis.
Sequence Specificity of Binding
To determine whether this binding depended on the presence of a functional vacuolar targeting motif in the peptide, we used different synthetic peptides to compete with the labeled peptide for binding (Kirsch et al., 1994). As shown in Figure 2B, when a competitor peptide, sequence SRFNPgRLPT (designated Spo-G), was included in the incubation at a concentration of 1 mM, no alteration of the fluorescent peaks was observed (Figure 2B, squares). In contrast, when a competitor peptide, sequence SRFNPIRLPT (designated Spo), was included, the peak representing bound fluorescent peptide (Figure 2B, X symbols, Bound) was diminished, whereas the peak representing free fluorescent peptide (X symbols, Free) was proportionately increased. The Spo-G peptide represents an Ile-to-Gly mutation of the central NPIR motif of the prosporamin vacuolar targeting determinant, a mutation that abolished targeting in vivo (Nakamura et al., 1993), whereas the Spo peptide represents the intact targeting determinant. The mutated motif did not compete with labeled proaleurain peptide for binding to full-length BP-80, whereas the intact motif did, albeit incompletely, in a manner similar to that observed here for tBP-80 (Kirsch et al., 1994). We conclude that sequence specificity of binding to tBP-80 is similar to that determined for full-length BP-80.
Effects of MAbs on Binding of Labeled Peptide to tBP-80
We used the anti–BP-80 MAbs to confirm that binding of the fluorescent peptide was attributable to tBP-80 and not to some other protein in the Drosophila cell culture medium. In each instance, 10 μg of purified MAb was incubated in 200 μL of concentrated medium containing tBP-80 plus 10−7 M labeled peptide. Results are shown in Figure 3, in which elution profiles obtained in the presence of different MAbs are compared with the superimposed profile of fluorescent peptide binding obtained in the absence of antibodies (open circles). MAb 14G7 chromatographed in a peak within fractions 19 to 24 and caused a shift of ∼40% of tBP-80 to the same position as detected by protein gel blot (Figure 3A, bottom); the remaining ∼60% of tBP-80 eluted at its normal position (Figure 3A, bottom, peak centered on fraction 31). At the same positions were peaks of fluorescence (Figure 3A) distributed in similar proportions, corresponding to a complex of labeled peptide plus tBP-80 plus 14G7 in fractions 20 to 24 and the labeled peptide plus tBP-80 in a peak centered on fraction 31 (Figure 3A, Bound + MAb and Bound, respectively). This result indicated that MAb14G7 bound to tBP-80 with a relatively weak affinity such that only ∼40% of the tBP-80 remained complexed to the antibody during chromatography. In contrast, MAbs 17F9 and 18E7 shifted essentially all of the fluorescent peptide binding activity to a peak centered on fraction 21 (Figure 3B), a position that corresponded to tBP-80 detected by protein gel blot analysis (Figure 3B, bottom, shown for 17F9; similar results obtained for 18E7 are not shown). Thus, 17F9 and 18E7 appeared to have higher affinities for tBP-80 such that most tBP-80 remained complexed with the antibodies during chromatography. The patterns shown in Figure 3 were consistent regardless of whether the antibodies were preincubated with tBP-80 before ligand was added or were added after the tBP-80–ligand interaction had been allowed to occur. Thus, interaction with the antibodies did not detectably affect ligand binding by tBP-80. MAb 19F2 appeared to have the least affinity for tBP-80 in a native conformation, causing neither a detectable shift in the elution position of tBP-80 nor a detectable effect on ligand binding by tBP-80 (data not shown).
Figure 3.

Effects of MAbs on Fluorescent Peptide Binding by tBP-80.
(A) Effect of 14G7. At the top, the elution profile of fluorescent peptide incubated with tBP-80 (*Peptide, open circles) is compared with the profile obtained when 10 μg of purified MAb 14G7 was added to the incubation mixture (*Peptide + 14G7, closed circles). Below is shown enhanced chemiluminescence detection on the same protein gel blot of MAb 14G7 (upper panel) and tBP-80 (lower panel). Because samples were not reduced before SDS-PAGE, the antibodies electrophoresed as a large disulfide-linked heavy chain–light chain complex separate from the position of ∼66-kD tBP-80. Bound + MAb indicates the fluorescent peak resulting from *Peptide plus tBP-80 plus antibody; Bound and Free are as given in Figure 2.
(B) Effects of 17F9 and 18E7. The elution profile of *Peptide plus tBP-80 (open circles) is compared with profiles obtained when 10 μg of 17F9 (*Peptide + 17F9, closed circles) or of 18E7 (*Peptide + 18E7, open squares) was added to the incubation mixtures. Below is shown protein gel blot detection of 17F9, and tBP-80 in the indicated fractions from the *Peptide + 17F9 chromatography run.
Ligand Binding by Δ3EGFR Is Enhanced by Antibody Interactions
The preceding experiments served as controls against which the results obtained by utilizing Δ3EGFR could be compared. In similar experiments, we used dialyzed culture medium containing Δ3EGFR that had been concentrated approximately fourfold. The concentration of Δ3EGFR in this medium was approximately half that of tBP-80 in medium used in the preceding experiments, as determined by comparing serial dilutions of the two media on protein gel blots (data not shown).
We first determined the ability of Δ3EGFR to bind the fluorescent ligand in the presence of MAb 14G7 as a control, given that 14G7 does not interact with that protein (Figure 1B). The fluorescence profile from that assay is presented in Figure 4, which indicates the presence of a very small peak of bound peptide in fractions 31 to 35 (indicated by Δ3EGFR above the bracket); these fractions corresponded to the position of Δ3EGFR protein detected by protein gel blot (Figure 4, bottom). This result was the best we obtained; in other experiments, a peak for fluorescence from the bound peptide could not be reliably detected (not shown). These results indicated that Δ3EGFR could bind the fluorescent peptide but at a much lower affinity than that demonstrated by tBP-80. In striking contrast, however, was the ability of Δ3EGFR to bind ligand in the presence of either 17F9 or 18E7 MAbs (Figure 4); both antibodies caused a substantial increase in the amount of ligand bound by Δ3EGFR, as demonstrated by the fluorescent peaks centering on fraction 21. Consistent with previous results, these fluorescent peaks corresponded to the presence of antibody plus Δ3EGFR in those fractions (Figure 4, bottom, shown for 17F9; similar results for 18E7 are not shown) and represented a complex of antibody plus Δ3EGFR plus bound ligand. Given that approximately half as much Δ3EGFR as tBP-80 was used in these assays and that peak area for bound peptide relative to free peptide for Δ3EGFR + 18E7 or 17F9 was approximately one-third of that obtained with tBP-80, we concluded that the ligand binding affinity of Δ3EGFR was similar to or only slightly less than that of tBP-80.
Figure 4.

Binding Assays for Δ3EGFR.
(Top) Medium containing Δ3EGFR protein was incubated with fluorescent proaleurain peptide plus 14G7 MAb (open circles), the peptide plus 18E7 MAb (yellow squares), the peptide plus 17F9 MAb (blue circles), the peptide plus 17F9 MAb plus 1 mM Spo-G peptide (green squares), or proaleurain peptide plus 17F9 MAb plus 1 mM Spo peptide (red triangles), and then chromatographed as in Figure 3. Because 14G7 MAb does not interact with Δ3EGFR, the open circle elution profile shows the ability of Δ3EGFR not complexed with antibodies to bind only a very small amount of labeled peptide (peak indicated by the bracket). (+) shows the elution profile of labeled peptide incubated with 17F9 MAb in the absence of culture medium.
(Bottom) Protein gel blot detection of 17F9 MAb (top) and of Δ3EGFR (middle) from the chromatography fractions indicated by the closed circles and of Δ3EGFR (bottom) from the chromatography run shown by the open circles. Bound and Free are as given in Figure 2.
The specificity of peptide binding by Δ3EGFR was tested with competition assays utilizing Spo-G (Figure 4) and Spo peptides. The presence of 1 mM Spo-G peptide had no effect on ligand binding by Δ3EGFR in the presence of 17F9 MAb, but the same concentration of Spo completely abolished the fluorescent peak centered on fraction 21. This result is substantially different from the partial competition obtained with tBP-80 (above) or with full-length BP-80 (Kirsch et al., 1994); its implications are discussed below. When 17F9 was incubated with labeled peptide in the absence of Δ3EGFR, all of the fluorescence eluted in a peak corresponding to unbound peptide (Figure 4), demonstrating that the MAb itself did not have any ligand binding activity.
Structural Domains of tBP-80 Defined by Protease Digestion
We isolated tBP-80 from Drosophila S2 cell medium, as described in Methods. An extensive purification protocol was necessary because even though tBP-80 in medium bound the proaleurain peptide ligand with high affinity (as judged from the fluorescent peptide binding assays), it interacted poorly with the proaleurain peptide–agarose affinity column used to purify intact BP-80 (Kirsch et al., 1994; see Figure 6). This indicates that the loss of attachment of BP-80 to the membrane altered the access of the ligand binding site to the peptide on a rigid matrix but not to the peptide in solution. The final tBP-80 preparation was estimated to be ∼95% pure (Figure 5A, lane 1), and its N-terminal amino acid sequence was identical to that of BP-80 (Paris et al., 1997; data not shown).
Figure 6.

Affinity Column Assay for Ligand Binding by Protease-Resistant Fragments.
Fifteen micrograms of tBP-80 was digested in 50 μL of 10 mM Tris-HCl, pH 7.5, with endoproteinase Asp-N at 22°C for 1 hr. Protease activity was stopped with EDTA, and the mixture was diluted to 1 mL with column buffer containing 100 μg/mL gelatin. To each of three 250-μL aliquots was added 50 μL of proaleurain peptide–agarose (3 mg of peptide per mL of gel; Kirsch et al., 1994). For the aliquot providing the samples analyzed in lanes 1 and 2, no competing peptide was added; for the aliquot analyzed in lanes 3 and 4, 0.5 mM Spo peptide was added; and for the aliquot analyzed in lanes 5 and 6, 0.5 mM proaleurain peptide was added. The mixtures were incubated on a rotating shaker at 4°C overnight, after which the agarose for each was collected by centrifugation and the supernates containing unbound proteins (U) were saved. After washing three times with column buffer at 4°C, bound proteins (B) were eluted from the agarose by heating it in 150 μL of SDS-PAGE sample buffer at 100°C for 10 min. Samples representing 10% of the volume of each fraction were denatured in the absence of βME and electrophoresed as in Figure 5. After transfer to nitrocellulose, protease-resistant fragments were detected with MAb 14G7 to allow mapping from the C terminus of tBP-80. Open arrow, position of fragments consistent with full-length tBP-80; solid arrow, position of ∼43-kD fragments; open arrowhead, position of ∼38-kD fragments. M, prestained molecular mass markers with sizes in kilodaltons indicated at left.
Figure 5.

Mapping of Structural Domains in tBP-80.
(A) Digestion of tBP-80 with endoproteinase Asp-N. Ten-microgram samples of purified tBP-80 were incubated with endoproteinase Asp-N in a total reaction volume of 50 μL for the times and at the pH values indicated at the top of the figure. For lanes 1, 2, and 4, the incubation temperature was 22°C. For lanes 3 and 5, the samples were incubated for 1 hr at 22°C and then for a second hour at 37°C. Protease activity was stopped by the addition of EDTA, and the samples were denatured (in the absence of β-mercaptoethanol [βME] for lanes 1 and 2 or with βME for lanes 3 to 5), electrophoresed on 4 to 20% SDS–polyacrylamide gels, and stained with Coomassie Brilliant Blue R 250. M, prestained molecular mass markers with sizes in kilodaltons indicated at left; *, intact tBP-80; arrows, ∼43-kD fragments; arrowheads, ∼38-kD fragments.
(B) tBP-80 drawn to scale: open rectangle, sequence recognized by RA3 antibodies; solid circles, Cys residues; open circles, Asp residues (those inside EGF repeats not shown); triangle, potential Asn-glycosylation site; small numbers, positions of residues indicated by open circles with longer stems; large numbers, approximate fragment sizes for fragments indicated by designated symbols in (C); other symbols are as given in Figure 1. (Note that Figure 1 of Paris et al. [1997] contains a typographical error; the residue at position 37 of NP471 [BP-80] is Glu, not Asp.)
(C) Antibody mapping of protease-resistant fragments. Twelve micrograms of tBP-80 in 50 μL of 0.01 M Tris-HCl, pH 7.5, was digested with 0.012 μg of Asp-N for 1 hr at 22°C plus 1 hr at 37°C. The reactions were stopped with EDTA; the solutions were brought to final concentrations of 1% SDS and 0.1 M Tris-HCl, pH 7.5, and heated at 65°C for 10 min; then 200 μL of Tris-buffered saline containing 1.25% Nonidet P-40 was added, followed by 5 μg of affinity-purified RA3 antibodies, and the mixture was incubated at 4°C overnight. Antibodies were removed in two sequential treatments with protein A–Sepharose, and the beads were washed four times with the solution of Tris-buffered saline and Nonidet P-40. Proteins in the antibody bound and unbound fractions were then prepared for SDS-PAGE by heating at 100°C for 10 min in the absence of disulfide-reducing agents. Equal aliquots of bound (B) or unbound (U) fractions were loaded on 4 to 20% gradient gels in pairs separated by a lane of molecular mass markers (M) prestained blue (masses are given in kilodaltons at left).
(D) Effects of βME on ∼43-kD N-terminal protease-resistant fragments. Equal aliquots of RA3 unbound (U) fractions from 1- and 2-hr (h) incubations were used. For the gels at left, samples were not treated with βME before electrophoresis (indicated by NO at bottom); for the gels on the right, samples were reduced in the presence of βME before electrophoresis (indicated by YES at bottom). The N-terminal protease-resistant fragments were detected either with polyclonal RA3 antibodies or with MAb 2D9, as indicated above each set of gels. Solid arrows indicate the broad ∼43-kD bands detected by the antibodies.
For both (C) and (D), after transfer to nitrocellulose, blots were cut down the middle of the marker lanes to separate them for treatment with the different antibodies, as indicated above each set of gels, and antibody complexes were detected with chemiluminescence. The positions of the labeled bands on x-ray film could be precisely aligned with the blots because the peroxidase reaction generated a yellow-brown color on the membrane on the most heavily labeled bands. The hollow appearance of labeled bands in (D) is an artifact of the presence of very large amounts of antigen on the blots at those positions. Symbols are as given in (A); other symbols are described in the text.
Digestion with endoproteinase Asp-N, which cuts to the N-terminal side of Asp residues, was utilized to probe tBP-80 structure; resistant fragments were identified on Coomassie blue–stained SDS–polyacrylamide gels and by probing protein gel blot transfers from those gels on membranes with different antibodies. We initially tested different conditions for digestion of the protein, as shown in Figure 5A. When a ratio of protease to tBP-80 of 1:1000 was used and the mixture was incubated at 22°C for 15 min, ∼75% was digested to yield two major products of ∼43 kD (Figure 5A, lane 2, arrow) and ∼38 kD (solid triangle), as assessed by electrophoresis in the absence of disulfide-reducing agents. The bands at ∼43 and ∼38 kD could represent collections of smaller fragments held together by multiple intramolecular disulfide bonds, or they could represent intact fragments resistant to protease.
To determine the size of protease-resistant polypeptides more stringently, we then performed SDS-PAGE on digests after disulfide reduction. Results are presented in Figure 5A, lanes 3 to 5. When the protein was digested at pH 7.5 for 1 hr at 22°C followed by 1 hr at 37°C (lane 3), resistant fragments presented bands at ∼43 and ∼38 kD that stained with approximately similar intensity. In contrast, when the protein was digested at pH 5.0 for 1 hr at 22°C (Figure 5A, lane 4) or for 1 hr at 22°C followed by 1 hr at 37°C (lane 5), the staining intensity of the ∼43-kD bands relative to that of the ∼38-kD bands appeared to be diminished at each time point. The intensity of staining for the 38-kD band in the 2-hr incubation samples for each pH was similar (Figure 5A, cf. lanes 3 and 5), whereas the intensity of staining of the ∼43-kD band in the pH 5.0 sample appeared to be less than that in the pH 7.5 sample (cf. lanes 3 and 5).
These results led to two conclusions. First, SDS-PAGE gave similar results whether or not the proteins were first treated with a disulfide-reducing agent. Thus, most protease-resistant fragments detected by electrophoresis of unreduced samples, as was necessary for mapping with the MAbs (see below), could be interpreted to represent intact polypeptide chains. Second, the decrease in pH may have affected the conformation of fragments in the ∼43-kD band such that they became more susceptible to protease digestion; however, this possibility requires confirmation by alternative experimental strategies before it can be accepted.
We then mapped the protease-resistant fragments by means of structural data presented in the diagram of tBP-80 in Figure 5B. The open rectangle at the left indicates the epitope for RA3 rabbit polyclonal antibodies raised against a 15–amino acid synthetic peptide representing the N terminus of BP-80 after cleavage of the signal peptide (Paris et al., 1997). Fragments recognized by RA3 therefore map from the N terminus of the protein. The oval to the right indicates the EGF-CB motif necessary for binding MAb 14G7; fragments recognized by 14G7 map from the C terminus of the protein. Lines terminated with open circles indicate the positions of all Asp residues outside of the EGF repeats; closed circles indicate the positions of Cys residues.
Fifteen micrograms of tBP-80 was digested at pH 7.5, protease activity was destroyed by addition of EDTA, the fragments were fractionated by incubation with 5 μg of affinity-purified RA3 antibodies, and the immune complexes were removed by treatment with protein A–Sepharose. This strategy was designed to produce two fractions, one with predominantly N-terminal fragments (selected by RA3) and one of fragments that predominantly lacked the N terminus. Proteins in the RA3 bound (Figures 5C and 5B) and unbound fractions were separated by SDS-PAGE after denaturation at 100°C in the absence of disulfide-reducing agents and then transferred to membranes; the resulting protein gel blots were probed with different antibodies. Digestion with the protease for 1 hr at 22°C, or for 1 hr at 22°C and then 1 hr at 37°C, gave results that were essentially indistinguishable for all antibodies (data not shown); therefore, only the samples digested for 2 hr are shown. Two patterns for N-terminal fragments were obtained. One constituted a species of ∼43 kD that remained unbound by RA3 in solution but displayed RA3 epitopes when the protein was in denatured form on the blots (Figure 5C, RA3, black arrow at right). This is similar in size to that of Δ3EGFR (Figure 1) and must represent Asp-N cleavage near residue 410 (Figure 5B). The fact that this fragment remained unbound by RA3 in solution, coupled with the fact that we were unable to demonstrate any binding of RA3 by tBP-80 in the fluorescent ligand assay (data not shown), argues that the N terminus of tBP-80 is ordinarily inaccessible. In contrast, a band of slightly faster mobility was in the bound fraction selected by RA3 (Figure 5C, open arrow at left); we hypothesize that the slight increase in mobility resulted from another proteolytic event within the molecule that caused a change in conformation and allowed access by the antibodies to the N terminus. The third most abundant fragment, of ∼27 kD (Figure 5C, open triangle), most likely represents a cleavage near residue 260 (Figure 5B).
In contrast, all four MAbs predominantly recognized a different fragment of ∼38 kD in the RA3 unbound fraction. Because a fragment this size did not react with RA3 (Figure 5C, lane U, right) and because a band this size was strongly labeled by 14G7 (Figure 5C, lane U, marked with filled arrowhead), it must represent the C-terminal portion of the protein with its N terminus near residue 210 (Figure 5B). We hypothesize that the identically sized (∼38 kD) fragments recognized by the other three MAbs also represent C-terminal fragments; however, the validity of this hypothesis is not required for interpretation of any of the subsequent experiments. These results indicate that the epitopes for both 17F9 and 18E7 reside within the C-terminal half of the unique region but differ because 18E7, but not 17F9, reproducibly labeled a fragment of ∼27 kD in the bound fraction (Figure 5C). Additionally, faint bands of ∼43 kD were recognized by 14G7, 19F2, and 18E7. The significance of the 14G7 band is investigated, as shown in Figure 6; bands recognized by the other two MAbs were not studied further.
We wanted to determine whether the polypeptide fragments making up the ∼43-kD band (Figure 5C, RA3) in the unbound fraction were intact, as the results shown in Figure 5A indicate. Protein gel blots carrying proteins from the RA3 unbound fraction from both 1- and 2-hr digestions were therefore prepared from proteins either not treated (Figure 5D, gels at left; RA3 and 2D9) or treated with β-mercaptoethanol (βME) (Figure 5D, gels at right) and probed with two antibodies that recognized N-terminal epitopes: the RA3 polyclonals and MAb 2D9. Both antibodies recognized the 43-kD band in samples not treated with βME (Figure 5D, gels at left panel), and both antibodies recognized 43-kD bands that appeared unchanged after reduction in the presence of βME (Figure 5D, gels at right panel). Therefore, most 43-kD proteolytic fragments were fully protease resistant. We could not repeat the experiment with the other MAbs because their epitopes required the presence of intact disulfide bonds.
Localization of the Ligand Binding Site
We combined protease digestion with assays for ligand binding. Fifteen micrograms of tBP-80 was digested with endoproteinase Asp-N for 1 hr at 22°C at pH 7.5; EDTA was then added, and the digest was incubated in three separate reaction mixtures overnight with proaleurain peptide–agarose. (In the fluorescent peptide binding assay, the absence of divalent cations plus the presence of 5 mM EDTA did not interfere with ligand binding by tBP-80 [data not shown].) In one mixture, no competitor peptide was added; in the second mixture, 0.5 mM Spo peptide was added; and in the third mixture, 0.5 mM proaleurain peptide was added. For each mixture, the resin was collected by centrifugation, and the buffer containing unbound proteins was removed and saved; the resin remaining was then washed, and the bound proteins were eluted at 100°C in SDS-PAGE sample buffer without βME. Bound and unbound proteins were then fractionated without βME treatment on replicate SDS–polyacrylamide gels, transferred to membranes, and probed with MAb 14G7 to map fragments from the C terminus of tBP-80. Results are presented in Figure 6.
Fragments of three different size classes were recognized by 14G7. One was slightly smaller than the 68-kD marker and represented full-length, or nearly full-length, tBP-80 that had not been digested by the protease (Figure 6). A second, less abundant fragment was ∼43 kD (solid arrow); the third was ∼38 kD (open triangle). Most of the full-length and ∼38-kD molecules were recovered in the unbound fractions in each set (Figure 6, lanes 2, 4, and 6); in the absence of competing peptide, however, ∼10% of each was retained on the affinity resin (lane 1). Retention of the ∼38-kD fragment on the resin was the result of interactions that did not depend on the covalently attached proaleurain peptide because the fragment's abundance in the bound fraction was not appreciably diminished by the presence of a vast excess of either Spo (Figure 6, lane 3) or proaleurain (lane 5) peptides. In contrast, retention of the full-length molecules was prevented by both Spo and proaleurain peptides (Figure 6, cf. lane 1 with lanes 3 and 5). Because NPIR is the only sequence motif shared by the two peptides, high-affinity binding by the full-length protein may be determined by that sequence. Interestingly, the ∼43-kD fragment showed a different pattern. Its binding was not prevented by excess Spo peptide (Figure 6, lane 3) but was prevented by excess proaleurain peptide (lane 5). Thus, the binding specificity of a fragment lacking the N-terminal ∼150 amino acids of tBP-80 (the ∼43-kD fragment recognized by 14G7) depended on sequences in the proaleurain peptide that differed from NPIR. Sequences other than NPIR within the proaleurain vacuolar targeting determinant (represented by the proaleurain peptide) that contribute to vacuolar sorting are known to exist (Holwerda et al., 1992; see Discussion).
DISCUSSION
The protease digestion experiments provide important information about the organization of the three domains in tBP-80. Full-length tBP-80 is not protease resistant and is rapidly cleaved to yield two major groups of protease resistant-products: one group constitutes the polypeptides of ∼43 kD that derive from the N terminus of the protein; the other, the ∼38-kD fragments from the C terminus. These are best described as groups because there is some heterogeneity of size among them, even though the fragments form relatively discrete bands on gels. Therefore, tBP-80 has two regions in which protease-accessible sequences (loops) connect three distinct structural domains. The domains are defined broadly as an N-terminal/RMR homology domain, a central domain, and a domain consisting of the EGF repeats. A model for this organization is presented in Figure 7.
Figure 7.

Structural Model.
The N-terminal/RMR homology domain is indicated in red, the central domain in yellow, the EFG repeat domain in blue, and the NPIR-specific ligand binding site in white; the shapes of the objects are solely for the purposes of illustration and probably have no relationship to reality. Scissors indicate regions digested by endoproteinase Asp-N to give products by way of reactions 1 and 2, and the protease-accessible sequences are depicted by black loops. In reaction 3, a MAb (green) interacts with the central domain of Δ3EGFR.
With protease digestion, a loop in the vicinity of residues 184 to 210, between the RMR homology region (Figure 5B) and the C-terminal two-thirds of the protein, was cleaved to give the 38-kD fragment recognized by MAbs 14G7, 19F2, 18E7, and 17F9 (Figure 7, pathway 1). Alternatively, a loop connecting the EGF repeats and the unique region was cleaved near residue 410 to give the fragment of ∼43 kD recognized by RA3 and 2D9 (Figure 7, pathway 2). These two alternatives appear to be mutually exclusive in that once formed, the ∼43- and ∼38-kD fragments are relatively stable. The protease-resistant fragments have in common the central domain connected to one of the flanking domains. Thus, the central third of tBP-80 makes up a protease-resistant domain, and the accessibility of the loops depends on which of the other domains is attached. The N-terminal fragments of ∼43 kD detected by RA3 and 2D9 were protease resistant, demonstrating that loss of the EGF repeats prevented protease accessibility to the loop between the RMR homology and central domains. Thus, the N-terminal/RMR homology domain interacts tightly with the central domain in a conformation-dependent manner that is facilitated by loss of the EGF repeats (Figure 7, pathway 2). The C-terminal 38-kD fragment was also protease resistant, consistent with the hypothesis that loss of the N-terminal/RMR homology domain prevented protease accessibility to the loop between the central domain and the EGF repeats (Figure 7, pathway 1) and indicating that interactions with the N-terminal/RMR homology domain profoundly affected how the central domain, in turn, interacted with the C-terminal region of the protein.
Because the intact protein functions as a receptor, the ligand binding activity of which is modulated by pH, we searched for indications that pH might affect its conformation. At pH 5.0, where ligand binding is ∼50% of that obtained at optimal pH (Kirsch et al., 1994), the fragments of ∼43 kD appeared to be more susceptible to proteolysis than at pH 7.5, whereas the fragments of ∼38 kD showed no change in stability. This observation may indicate that pH affects the interaction of the N-terminal/RMR homology domain with the central domain, perhaps by leading to changes in protein conformation, and would support the hypothesis that the NPIR-specific ligand binding site might be formed by interaction of the two domains as depicted in Figure 7. Further studies are necessary to confirm this preliminary observation.
We must keep in mind that the NPIR-specific ligand binding site is contained entirely within Δ3EGFR (as shown in Figure 4), which is similar in size to the N-terminal ∼43-kD fragments, but Δ3EGFR binds this ligand poorly. Clearly, the presence of the EGF repeat domain in intact tBP-80 both allows ligand binding and changes the conformation of the protein to make the loop between the N-terminal/RMR homology domain and the central domain protease accessible. Thus, protease accessibility and ligand binding appear to be linked in some way, a concept that would support the hypothesis that NPIR-specific ligand binding is determined by interactions between the N-terminal/RMR homology domain and the central domain. Under this model, one would expect that in tBP-80 with bound ligand, the loop between the N-terminal/RMR homology domain and the central domain would be fully accessible and would predominantly yield fragments of ∼38 kD with endoproteinase Asp-N digestion. We did not test this prediction experimentally because the proaleurain peptide ligand used in these experiments itself contains two Asp residues.
This concept would be consistent with the observed effects of MAbs 17F9 and 18E7 on ligand binding by Δ3EGFR. We hypothesize that the MAbs, by binding to the central domain, mimic the effects of the EGF repeats and induce a conformational change that diminishes interactions with the N-terminal/RMR homology domain and makes the ligand binding site accessible (Figure 7, pathway 3). The term conformational change would include steric effects by which a MAb might help push the N-terminal/RMR homology domain away. An alternative explanation for why interaction with the MAbs facilitated ligand binding by Δ3EGFR would be that they displaced an inhibitor that occluded the binding site. Although we cannot formally exclude this latter possibility, the following points argue against it. First, Δ3EGFR eluted from the Superdex 200 column at a position consistent with its predicted molecular mass; therefore, any inhibitor bound to it would have to be small. Second, such an inhibitor would have to interact preferentially with Δ3EGFR and not with tBP-80, because ligand binding by tBP-80 was not affected by the MAbs, even though they shifted the position at which it eluted from the column. Third, ligand binding by Δ3EGFR was enhanced by two separate MAbs that have different epitopes; for the inhibitor displacement theory to apply, these epitopes would have to be positioned such that both antibodies could displace the inhibitor. The results also would not formally exclude the possibility that the MAbs functioned to bring together two Δ3EGFR molecules that then could interact in some way to bind a ligand molecule.
However, in apparent contradiction to the model that N-terminal/RMR homology domain and central domain interactions determine the ligand binding site, ∼43-kD C-terminal fragments lacking the N-terminal/RMR homology domain were retained on the proaleurain peptide affinity resin. These fragments were retained because they interacted with the proaleurain peptide, as demonstrated by the ability of excess free proaleurain peptide to prevent their retention. This contradiction could be explained if there were two separate ligand binding sites, each recognizing a separate motif on the proaleurain peptide. In this model, NPIR-specific binding would depend on the presence of the N-terminal/RMR homology domain, and the ∼43-kD C-terminal fragments would contain a non-NPIR binding region. This model would be supported by the inability of the Spo peptide, containing an NPIR motif, to compete for binding of the ∼43-kD C-terminal fragments to the proaleurain peptide affinity column.
Support for the concept of multiple ligand binding sites came from our previous functional studies of the vacuolar targeting determinant on proaleurain, which indicated that three different portions could each mediate vacuolar targeting, albeit at reduced efficiency. These portions were defined by testing mutated targeting determinants in both positive and negative assays (Holwerda et al., 1992). These assays, for example, indicated that the intact determinant SSSFADSNPIRPVTDRAAST gave 100% efficiency for targeting, whereas SSSFADSNPIRPmedkd (mutated residues in lower case) gave ∼60% targeting efficiency, SSSFADScsaipVTDRAAST gave ∼20%, SSSFADScsaipmedkd gave ∼10%, and avelcsaipmedkd had no function as a vacuolar targeting determinant. Thus, SSSFADS, NPIRP, and VTDRAAST all contributed information that directed proper targeting; however, only when the three motifs were together was targeting efficiency optimal. This finding raised the interesting possibility that one receptor might recognize three different determinants and that each determinant would then contribute to high-affinity binding.
There are two general models of how this might happen. In the first, each motif would be recognized by the same binding site; three motifs on one peptide would then simply increase the possibility that one of them would encounter the receptor binding site. This seems unlikely, both because of the conceptual difficulties in reconciling the high-affinity binding of each of such apparently different sequences and because the Spo peptide at 10,000-fold molar excess was able to compete only partially with proaleurain peptide for binding to tBP-80 (Figure 2) and to intact BP-80 (Kirsch et al., 1994). Under the second model, each domain would be recognized by its own unique binding site; three domains interacting at the same time with one receptor would mean a high affinity for binding. (Of course, there also could be two different binding sites, with one motif interacting with one and either of two motifs interacting with the other.) If this model pertains, the two or three binding sites would have to be close together such that the Spo peptide in the NPIR site could sterically interfere with access by the other two motifs of the proaleurain peptide to their sites. This model also implies that the concentration of a peptide with a single motif required to displace the proaleurain peptide would be greater than the Kd of binding for the single motif peptide alone, as is likely true for the Spo peptide, for which millimolar concentrations could only partially displace the proaleurain peptide. The Kd for Spo peptide binding to BP-80 has not been measured, but it functioned as efficiently as proaleurain peptide when used as an affinity column ligand for purification of BP-80 (Kirsch et al., 1996).
This model is consistent with results obtained from analyses of single amino acid mutations of the prosporamin vacuolar targeting determinant (Matsuoka and Nakamura, 1999). Those studies demonstrated that the Ile and Leu residues in the sequence NPIRL were crucial for proper function. The Ile was essential (only substitution with Leu maintained function), whereas a broader range of substitutions for the Leu residue were tolerated—although hydrophobic residues with bulky side chains were preferred. Interestingly, substitution of Ser for Leu, to give NPIRS, resulted in essentially complete mistargeting. The sequence NPIRS, corresponding to NPIRP in proaleurain, is present in the rice aleurain homolog oryzain γ (Watanabe et al., 1991). Those authors hypothesized that the BP-80 binding site for the sporamin propeptide overlapped that for proaleurain, but the sites were not identical. They also hypothesized that another portion of the proaleurain vacuolar targeting domain could substitute for the function provided by Leu in NPIRL (Matsuoka and Nakamura, 1999). Binding of BP-80 or homologs from other plants to protein sequences lacking an NPIR motif has been demonstrated in vitro (Kirsch et al., 1996; Shimada et al., 1997) and in vivo (Miller et al., 1999). Further characterization of the sequence or sequences recognized in vivo, and analysis of the functional importance of sequences studied in vitro, should better define the VSR protein ligand binding sites (Matsuoka and Neuhaus, 1999).
We therefore hypothesize that the C-terminal fragments of ∼38 to 43 kD carry a binding site defined by the combination of the central and EGF repeat domains that recognizes a portion of the proaleurain peptide sequence other than NPIR; in the absence of the EGF repeat domains, as in Δ3EGFR, the site does not exist. That would explain why the Spo peptide could completely compete with proaleurain peptide for binding to Δ3EGFR (Figure 4); Δ3EGFR would have only the NPIR-specific site. In Δ3EGFR, the combination of the N-terminal/RMR homology domain and the central domain is necessary to define the NPIR-specific site. Whether the N-terminal/RMR homology domain provides part of the NPIR-specific site or, alternatively, causes a conformational change in the central domain to generate the site is not clear. In the future, when the crystal structure of tBP-80 is solved, this question should then be resolved.
METHODS
Expression of Truncated Proteins
Truncation of the BP-80 coding sequence to yield a construct expressing a 562–amino acid protein minus transmembrane domain and cytoplasmic tail sequences has been described (construct NP473; Paris et al., 1997). NP473 was removed from its plasmid by XbaI and SacI digestion and was ligated into pBlueBac4 (Invitrogen, Carlsbad, CA). This intermediate was excised as an EcoRI-XhoI fragment and inserted between those restriction sites in pMT/V5–His A (Invitrogen) to yield CX580; this construct encodes tBP-80 protein. Using a polymerase chain reaction (PCR) 5′ primer positioned just upstream from the BamHI site (Paris et al., 1997), we generated further truncations of the coding sequence with the following primers, which placed a translation stop codon after residues 414, 468, and 517, respectively: CX622, 5′-CCTCGAGCTCACTCATTTGTTTCCACATCATTG-3′; CX623, 5′-CCCCTCGAGCTCAACGCCCATGTCCACTAACTT-3′; and CX624, 5′-CCTCGAGCTCATTCATCAATGT-CTTCACAATTTT-3′. These PCR products were inserted into the BamHI-SacI interval of CX580 to generate constructs encoding Δ3EGFR, Δ2EGFR, and Δ1EGFR, respectively. The fidelity of the constructs was confirmed by sequencing the PCR-generated regions.
Plasmids were transfected into Drosophila melanogaster S2 cells by using protocols supplied with the Drosophila Expression System kit (Invitrogen). We found it important to use culture medium from JRH Biosciences (Lenexa, KS) or from Hyclone (Logan, UT) for optimal results. Stably transformed cell lines were selected with hygromycin and gradually transferred to serum-free medium over a month's time, according to kit protocols. When cells were at a density of 4 × 106 to 6 × 106 per mL, expression of the recombinant proteins was induced by treatment with 500 μM copper sulfate for 36 to 40 hr. The medium was harvested, and protease inhibitors (Sigma) were added to give the following final concentrations: 5 μg/mL leupeptin; 5 μg/mL pepstatin; 5 μg/mL antipain; 1 μg/mL trans-epoxysuccinyl-l-leucylamido-(4-guanidino)butane; and 0.2 mM 4-(2-aminoethyl)benzenesulfonyl fluoride. When necessary, the medium was concentrated under nitrogen gas pressure with an Amicon concentrator (Amicon Inc., Beverly, MA) using a membrane with a 10-kD limit. Glycerol was added to a final concentration of 10%, and the medium was stored at −80°C.
Protein Gel Blots
Methods for SDS-PAGE, transfer of proteins to nitrocellulose membranes, incubation with primary antibodies, and detection with alkaline phosphatase–linked secondary antibodies have been described elsewhere (Rogers et al., 1997). For detection of antibody complexes by enhanced chemiluminescence (Pierce Chemical Co., Rockford, IL), the secondary antibodies were conjugated to horseradish peroxidase, and the treated membranes were exposed to x-ray film according to the manufacturer's instructions.
Purification of tBP-80 from S2 Cell Medium
At each step, tBP-80 was detected on protein gel blots by using enhanced chemiluminescence. Ammonium sulfate was added to 3.6 liters of medium to a final concentration of 2.4 M, and the resulting precipitate was discarded. The supernate was applied with use of a peristaltic pump to a 1.2 × 8-cm column of phenyl–Sepharose (Amersham Pharmacia Biotechnology, Arlington Heights, IL) at 22°C, after which the column was washed sequentially with 2.4 M ammonium sulfate and 1.3 M ammonium sulfate, both in PB (0.05 M sodium phosphate, pH 7.0). All of the following steps were performed at 4°C, and a fast-performance liquid chromatography system (Amersham Pharmacia) was used. The protein retained was batch-eluted with 15 mL of PB and dialyzed extensively against 0.05 M sodium acetate solution, pH 4.5, containing 0.05 M NaCl. After centrifugation at 10,000g for 20 min, the supernate was applied to a Mono S HR 5/5 column (Amersham Pharmacia) equilibrated with the same buffer, and the protein retained was eluted with a 10-mL linear gradient from 0.05 M sodium acetate solution, pH 4.5, containing 0.05 M NaCl to 0.05 M sodium Hepes solution, pH 7.4, containing 0.05 M NaCl; tBP-80 eluted in a narrow peak as the gradient reached completion. The pooled 0.5-mL fractions were made to 3.3 M with ammonium sulfate and applied to a phenyl–Superose HR 5/5 column (Amersham Pharmacia); the column was washed with 1.3 M ammonium sulfate in PB, and the proteins retained were eluted with a 10-mL linear gradient from 1.3 M ammonium sulfate in PB to PB and collected in 0.5-mL fractions; tBP-80 eluted in the fractions between 0.9 and 0.7 M ammonium sulfate. Pooled fractions were dialyzed against 1 liter of 0.05 M Hepes-NaOH, pH 7.9, and applied to a 0.46 × 5-cm Poros 20 HQ column (Perseptive Biosystems, Cambridge, MA) equilibrated with the same buffer. Retained protein was eluted with a 10-mL linear gradient from 0 to 0.5 M NaCl in 0.05 M Hepes-NaOH, pH 7.9, and collected in 0.5-mL fractions; tBP-80 eluting in fractions 9 and 10 represented a total of 0.49 mg of protein, which was pooled and stored at 4°C. The concentrations of the purified protein of the proaleurain peptide were calculated from their A280 after dilution into a solution of 6 M guanidine HCl, 0.05 M phosphate buffer, pH 7.0, and 1 mM DTT (Gill and von Hippel, 1989).
Binding Assays
Proaleurain peptide, sequence SSSFADSNPIRPVTDRAASTYC, 200 nmol in 2 mL of 0.05 M Tris-HCl solution, pH 8.0, containing 5 mM EDTA, was mixed with 400 nmol of N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-yl)methyl) iodoacetamide (BODIPY-FL; Molecular Probes, Eugene, OR) in 0.1 mL of dimethylformamide and 0.04 mL of 0.5 M tris-(2-carboxyethyl)phosphine hydrochloride and incubated at 22°C in the dark for 90 min. Peptide coupled to BODIPY-FL through the Cys residue was separated from free BODIPY-FL by chromatography on a C18 reversed-phase HPLC column by using a gradient from 0.1% trifluoroacetic acid in water to 0.1% trifluoroacetic acid in acetonitrile. The fractions containing peptide coupled to BODIPY-FL were pooled and lyophilized; the peptide was resuspended in sterile water, and its concentration was determined by amino acid analysis.
For the fluorescent peptide binding assay, the labeled peptide was added to 0.3 mL of S2 cell culture medium (pH 6.5 to 6.7) to a final concentration of 10−7 M and incubated at 22°C for 1 hr; 0.2 mL was then applied to a 30 × 1-cm column of Superdex 200 (Amersham Pharmacia) and eluted with column buffer (0.05 M Hepes-NaOH, pH 7.0, 0.05 M NaCl, 1 mM CaCl2, and 1 mM MgCl2) at a flow rate of 1 mL/min. The fluorescence of 0.2-mL aliquots of each fraction was measured with a Luminescence Spectrometer LS50B (Perkin-Elmer Corp., Norwalk, CT), using 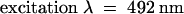 and
and 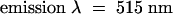 and expressed in units of fluorescence intensity.
and expressed in units of fluorescence intensity.
Anti–BP-80 Monoclonal Antibodies
Monoclonal antibody (MAb) 14G7 has been described previously (Paris et al., 1997); 2D9 was isolated in the same screen. MAbs 19F2, 18E7, and 17F9 were isolated as follows. Membranes from the less dense membrane fraction were prepared from developing pea cotyledons, as previously described (Kirsch et al., 1994). A suspension of the intact membranes was used to immunize mice, and hybridoma cells were prepared at the Cell and Immunobiology Core Facility (University of Missouri, Columbia), as previously described (Rogers et al., 1997). Positive clones were identified by using an ELISA, and the identity of the proteins within the less dense membranes that reacted with the antibodies in the hybridoma media samples was determined by SDS-PAGE and protein gel blot analysis.
Digestion of Purified tBP-80 with Endoproteinase Asp-N
Endoproteinase Asp-N, sequencing grade, was purchased from Roche Molecular Biochemicals (Indianapolis, IN); 2 μg was taken up in 50 μL of sterile water, according to the supplier's directions, and incubated with purified tBP-80 at a ratio of 1:1000 (w/w, protease:tBP-80). The incubation buffer was 10 mM Tris-HCl, pH 7.5, except for the experiment shown in Figure 5A, lanes 3 to 5, for which the buffers were 20 mM Tris-HCl, pH 7.5, or 20 mM sodium acetate, pH 5.0. Protease activity was terminated by bringing the reaction mixture to a final concentration of 10 mM EDTA.
Acknowledgments
This research was supported by Grant No. DE-FG 95ER 20165 to J.C.R. and Grant No. MCB-9304578 to L.B. from the U.S. Department of Energy. We thank Justin Scheer and Greg Pearce for their kind assistance with BODIPY-FL–peptide coupling and purification.
References
- Boller, T., and Kende, H. (1979). Hydrolytic enzymes in the central vacuole of plant cells. Plant Physiol. 63, 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen, B.R., Fennie, C., and Lasky, L.A. (1990). The Mel 14 antibody binds to the lectin domain of the murine peripheral lymph node homing receptor. J. Cell Biol. 110, 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereghino, J.L., Marcusson, E.G., and Emr, S.D. (1995). The cytoplasmic tail domain of the vacuolar protein sorting receptor Vps10p and a subset of VPS gene products regulate receptor stability, function, and localization. Mol. Biol. Cell 6, 1089–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, A.A., and Stevens, T.H. (1996). Vps10p cycles between the late-Golgi and prevacuolar compartments in its function as the sorting receptor for multiple yeast vacuolar hydrolases. J. Cell Biol. 133, 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, C.G. (1990). The many faces of epidermal growth factor repeats. New Biol. 2, 410–419. [PubMed] [Google Scholar]
- Davis, C.G., Goldstein, J.L., Südhof, T.C., Anderson, R.G.W., Russell, D.W., and Brown, M.S. (1987). Acid-dependent ligand dissociation and recycling of LDL receptor mediated by growth factor homology region. Nature 326, 760–765. [DOI] [PubMed] [Google Scholar]
- Gill, S.C., and von Hippel, P.H. (1989). Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326. [DOI] [PubMed] [Google Scholar]
- Herz, J., Hamann, U., Rogne, S., Myklebost, O., Gausepohl, H., and Stanley, K.K. (1988). Surface location and high affinity for calcium of a 500-kD liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. EMBO J. 7, 4119–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille-Rehfeld, A. (1995). Mannose 6-phosphate receptors in sorting and transport of lysosomal enzymes. Biochim. Biophys. Acta 1241, 177–194. [DOI] [PubMed] [Google Scholar]
- Hinz, G., Hillmer, S., Bäumer, M., and Hohl, I. (1999). Vacuolar storage proteins and the putative sorting receptor BP-80 exit the Golgi apparatus of developing pea cotyledons in different transport vesicles. Plant Cell 11, 1509–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl, I., Robinson, D.G., Chrispeels, M.C., and Hinz, G. (1996). Transport of storage proteins to the vacuole is mediated by vesicles without a clathrin coat. J. Cell Sci. 109, 2539–2550. [DOI] [PubMed] [Google Scholar]
- Holwerda, B.C., Padgett, H.S., and Rogers, J.C. (1992). Proaleurain vacuolar targeting is mediated by short contiguous peptide interactions. Plant Cell 4, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, L., and Rogers, J.C. (1998). Integral membrane protein sorting to vacuoles in plant cells: Evidence for two pathways. J. Cell Biol. 143, 1183–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch, T., Paris, N., Butler, J.M., Beevers, L., and Rogers, J.C. (1994). Purification and initial characterization of a potential plant vacuolar targeting receptor. Proc. Natl. Acad. Sci. USA 91, 3403–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch, T., Saalbach, G., Raikhel, N.V., and Beevers, L. (1996). Interaction of a potential vacuolar targeting receptor with amino- and carboxyl-terminal targeting determinants. Plant Physiol. 111, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J., Herman, P.K., and Emr, S.D. (1990). The fungal vacuole: Composition, function and biogenesis. Microbiol. Rev. 54, 266–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld, S. (1992). Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu. Rev. Biochem. 61, 307–330. [DOI] [PubMed] [Google Scholar]
- Marcusson, E.G., Horazdovsky, B.F., Cereghino, J.L., Gharakhanian, E., and Emr, S.D. (1994). The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by VPS10 gene. Cell 77, 579–586. [DOI] [PubMed] [Google Scholar]
- Matsuoka, K., and Nakamura, K. (1999). Large alkyl side chains of isoleucine and leucine in the NPIRL region constitute the core of the vacuolar sorting determinant of sporamin precursor. Plant Mol. Biol. 41, 825–835. [DOI] [PubMed] [Google Scholar]
- Matsuoka, K., and Neuhaus, J.-M. (1999). Cis-elements of targeting to the vacuole. J. Exp. Bot. 50, 165–174. [Google Scholar]
- Miller, E.A., Lee, M.C.S., and Anderson, M.A. (1999). Identification and characterization of a prevacuolar compartment in stigmas of Nicotiana alata. Plant Cell 11, 1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, K., Matsuoka, K., Mukumoto, F., and Watanabe, N. (1993). Processing and transport to the vacuole of a precursor to sweet potato sporamin in transformed tobacco cell line BY-2. J. Exp. Bot. 44 (suppl.), 331.–338. [Google Scholar]
- Neuhaus, J.M., and Rogers, J.C. (1998). Sorting of proteins to vacuoles in plant cells. Plant Mol. Biol. 38, 127–144. [PubMed] [Google Scholar]
- Okita, T.W., and Rogers, J.C. (1996). Compartmentation of proteins in the endomembrane system of plant cells. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 327–350. [DOI] [PubMed] [Google Scholar]
- Paris, N., and Rogers, J.C. (1996). The role of receptors in targeting soluble proteins from the secretory pathway to the vacuole. Plant Physiol. Biochem. 34, 223–227. [Google Scholar]
- Paris, N., Rogers, S.W., Jiang, L., Kirsch, T., Beevers, L., Phillips, T.E., and Rogers, J.C. (1997). Molecular cloning and further characterization of a probable plant vacuolar sorting receptor. Plant Physiol. 115, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, D.G., and Hinz, G. (1997). Vacuole biogenesis and protein transport to the plant vacuole: A comparison with the yeast vacuole and the mammalian lysosome. Protoplasma 197, 1–25. [Google Scholar]
- Rogers, S.W., Burks, M., and Rogers, J.C. (1997). Monoclonal antibodies to barley aleurain and homologues from other plants. Plant J. 11, 1359–1368. [DOI] [PubMed] [Google Scholar]
- Shimada, T., Kuroyanagi, M., Nishimura, M., and Hara-Nishimura, I. (1997). A pumpkin 72-kDa membrane protein of precursor-accumulating vesicles has characteristics of a vacuolar sorting receptor. Plant Cell Physiol. 38, 1414–1420. [DOI] [PubMed] [Google Scholar]
- Stearns, D.J., Kurosawa, S., and Esmon, C.T. (1989). Microthrombomodulin: Residues 310–486 from the epidermal growth factor precursor homology domain of thrombomodulin will accelerate protein C activation. J. Biol. Chem. 264, 3352–3356. [PubMed] [Google Scholar]
- Watanabe, H., Abe, K., Emori, Y., Hosoyama, H., and Arai, S. (1991). Molecular cloning and gibberellin-induced expression of multiple cysteine proteinases of rice seeds (Oryzains). J. Biol. Chem. 266, 16897–16902. [PubMed] [Google Scholar]
- Zushi, M., Gomi, K., Yamamoto, S., Maruyama, I., Hayashi, T., and Suzuki, K. (1989). The last three consecutive epidermal growth factor–like structures of human thrombomodulin comprise the minimum functional domain for protein C–activating cofactor activity and anticoagulant activity. J. Biol. Chem. 264, 10351–10353. [PubMed] [Google Scholar]