

Abstract
Human immunodeficiency virus type 1 (HIV-1) neutralizing antibodies are thought be distinguished from nonneutralizing antibodies by their ability to recognize functional gp120/gp41 envelope glycoprotein (Env) trimers. The antibody responses induced by natural HIV-1 infection or by vaccine candidates tested to date consist largely of nonneutralizing antibodies. One might have expected a more vigorous neutralizing response, particularly against virus particles that bear functional trimers. The recent surprising observation that nonneutralizing antibodies can specifically capture HIV-1 may provide a clue relating to this paradox. Specifically, it was suggested that forms of Env, to which nonneutralizing antibodies can bind, exist on virus surfaces. Here, we present evidence that HIV-1 particles bear nonfunctional gp120/gp41 monomers and gp120-depleted gp41 stumps. Using a native electrophoresis band shift assay, we show that antibody-trimer binding predicts neutralization and that the nonfunctional forms of Env may account for virus capture by nonneutralizing antibodies. We hypothesize that these nonfunctional forms of Env on particle surfaces serve to divert the antibody response, helping the virus to evade neutralization.
An effective human immunodeficiency virus type 1 (HIV-1) vaccine will likely need a component that is able to stimulate broadly neutralizing antibodies (45). However, progress in this area of vaccine research has been slow (26). A better understanding of the mechanisms by which the virus evades neutralization may provide key information necessary to accelerate progress in vaccine design.
Functional HIV-1 envelope surface glycoprotein (Env) spikes consist of compact trimers of noncovalently associated gp120 (surface subunit) and gp41 (transmembrane subunit) (29) (depicted schematically in Fig. 1A). If we assume that antibody binding to these trimers predicts neutralization, as has been proposed (28), then functional trimers would appear to be a logical basis for a vaccine. However, without exception, all vaccine approaches based on this premise, as well as natural HIV infection, result in antibody responses directed to Env that efficiently recognize nonfunctional forms of Env, for example, monomeric gp120, but which are largely nonneutralizing (7). If trimer binding is a prerequisite for neutralization, it appears then that nonneutralizing antibodies are somehow generated against a form of Env other than the trimer (53).
FIG. 1.

Potential forms of Env on the HIV-1 membrane. gp120 is shown in red with the outer neutralizing face in light shading and the inner nonneutralizing face in darker shading. Carbohydrate moieties are depicted as “tree”-like structures. gp41 is comprised of N-terminal (yellow) and C-terminal (green) transmembrane domains, separated by a disulfide-constrained loop. The membrane-proximal gp41 region exposed on the trimer is depicted in dark green. A) Functional Env trimer; B) uncleaved gp160 precursor (depicted here as a trimer; however, it may also exist as other oligomeric forms); C) gp120 shedding; D) alternative trimer isoform exposing the nonneutralizing face of gp120; E) gp120/gp41 monomers.
Possible alternative immunogenic forms of Env include soluble monomeric gp120 and gp160. However, it is also possible that nonfunctional forms of Env exist on the surfaces of virus particles. Relating to this possibility, nonneutralizing monoclonal antibodies (MAbs) have been shown to capture infectious virus in a highly specific manner (13, 17, 50, 54). Initially, it was proposed that nonneutralizing MAbs somehow capture the virus through functional trimers (17, 50). However, the behavior of two MAbs, b6 and b12, suggested otherwise. These MAbs are both directed to epitopes that overlap the CD4 binding site of gp120 but differ in that the former is nonneutralizing but the latter is potently neutralizing. Although b6 can inhibit virus capture by immobilized b12, it does not affect b12's neutralization activity (31, 54). Thus, an alternative explanation for virus capture by nonneutralizing antibodies is that it occurs via an as yet unidentified alternative form of Env that is recognized by both neutralizing and nonneutralizing MAbs.
Here, we further investigated the possibility of nonfunctional Env on HIV surfaces. Some potential candidates are depicted in Fig. 1B to E. To explain virus capture by nonneutralizing anti-gp120 MAbs, the nonneutralizing face of gp120 (71) would be expected to be exposed on the nonfunctional Env. One possibility is uncleaved gp160 (Fig. 1B), the Env precursor (9, 44, 52). In natural infection, uncleaved gp160 may be released from infected cells (53). However, whole inactivated HIV particles incorporate only fully processed Env (44). Another possibility is provided by gp120 shedding from the virus surface, leaving behind depleted gp41 stumps (Fig. 1C) (13, 26, 46). A further possibility is an alternative trimer isoform (Fig. 1D), a nonfunctional conformational variant of the trimer, for which there is a precedent in rhabdoviruses (40). Finally, as a complement to gp120 shedding, trimers might dissociate along the axis of trimerization, resulting in gp120/gp41 monomers (Fig. 1E).
MATERIALS AND METHODS
Plasmids and mutagenesis.
Plasmid pCAGGS (49) was used to express membrane-bound forms of Env from the primary R5 isolate JR-FL expressing wild-type, full-length gp160, denoted gp160WT, and a mutant referred to as gp160ΔCT, truncated after amino acid 708 (HXB2 numbering system), leaving 3 amino acids of the gp41 cytoplasmic tail, as reported previously (6). The SOS mutant involved the introduction of cysteines at specific sites, resulting in an intermolecular disulfide bond between gp120 and gp41, as described previously (6, 9). A mutation, termed gp160UNC, to change the wild-type SU/TM cleavage site from REKR to GEKR, eliminating gp160 precursor processing to gp120/gp41, has also been described (9). All amino acid substitutions were made by Quikchange site-directed mutagenesis (Stratagene, Inc.).
Using similar methods, we constructed pCAGGS plasmids expressing the simian immunodeficiency virus (SIV) SIVmac239 and SIVmac316 gp160ΔCT Env genes. We used the plasmid pNL4-3.Luc.R-E- (10, 22) to induce particle budding. This plasmid expresses an HIV-1 genome that is truncated to remove the env and nef genes, has a frameshift mutation in the vpr gene and carries the luciferase gene in place of nef. A plasmid expressing vesicular stomatitis virus G protein (pVSV-G) has been described previously (22, 42).
VLPs and inactivated virus preparations.
Virus-like particles (VLPs) were produced by transient transfection of 293T cells with pNL4-3.Luc.R-E- (22), a pCAGGS-based (49) Env-expressing plasmid and, for certain virus capture experiments, also with pVSV-G, by calcium phosphate precipitation. Two days later, supernatants were collected. These VLP-containing supernatants were used directly for virus capture and infectivity analyses, but concentrated VLPs were required for blue native polyacrylamide gel electrophoresis (BN-PAGE) and sodium dodecyl sulfate (SDS)-PAGE. To concentrate VLPs, the method of Willey was adapted (70), first preclearing cell debris by low-speed centrifugation, filtration through a 0.45-μM filter, and then pelleting particles at 50,000 × g for 1 h, followed by a second spin in microcentrifuge tubes at 25,000 × g to remove residual culture medium. VLPs were resuspended in phosphate-buffered saline (PBS) to 1,000 times the original concentration in the supernatant. Where appropriate, VLPs were inactivated using 1 mM aldrithiol, as described previously (38). VLPs were referred to as WT-VLPs (Env is unmodified, wild type), SOS-VLPs (Env is stabilized by the SOS disulfide bridge), and UNC-VLPs (Env in VLPs is uncleaved by use of a plasmid expressing an Env cleavage site mutation) (6).
Large volumes of virus supernatants prepared from primary mitogen-stimulated peripheral blood mononuclear cell cultures of primary viruses were provided by Meng Wang (The Scripps Research Institute, La Jolla, CA) and David Montefiori (Duke University Medical Center, Durham, NC). These were inactivated and concentrated as described for VLPs. Concentrated inactivated preparations of HIV-1 ADA, BaL, and MN, and SIVmac239 gp160ΔCT viruses were also obtained from Larry Arthur and Jeff Lifson, AIDS Vaccine Program, National Cancer Institute, Frederick, MD. The viruses were grown in SupT1 cells (MN) or SupT1 cells engineered to express CCR5 (ADA, BaL, and SIVmac239).
Monoclonal antibodies and HIVIG.
A panel of anti-HIV-1 gp120 MAbs was obtained. Their epitopes are described with reference to the concept that gp120 comprises a conserved core comprising of domains C1 through C5, separated by variable loops V1 to V5 (64). MAbs were b12 and b6 (directed to conformation-dependent epitopes overlapping the CD4 binding site of gp120) (15); 2G12 (directed to a unique glycan-dependent epitope) (16, 56, 59, 67); X5 (directed to a CD4-inducible epitope) (37); 447-52D and PA1 (directed to the V3 loop) (61, 62); c11 (directed to a conformational epitope involving the C1 and C5 domains of gp120); B12, against an epitope in the C2 domain of gp120 (1); P7 (directed to a conformational epitope involving the C1 domain) (24); and 133/290 (directed to a linear C1 epitope) (12).
MAbs against gp41 were 2F5 (48) and Fab Z13 (74) (directed to neutralizing epitopes in the C-terminal region of the gp41 ectodomain); 7B2 (9) and Fab T2 (7) (directed to the cluster I region); and 2.2B (9) and T3 (7) (directed to the cluster II region). We used a variant of Z13, termed Z13e1, a high-affinity mutant of the parent Z13, selected by phage display (M.B.Z., unpublished data). The anti-SIV MAbs included 5B11, 11F12, 2.6C, 17A, 8C7, 3E9, 7D3, 311H, and 3.10A (21, 25). Fab LS4 directed to Ebola virus GP was used as a control (43).
MAbs b12, 2G12, Z13, and 2F5 are broadly neutralizing (11, 66). MAb 447-52D neutralizes a subset of viruses with the GPGR sequence motif in the V3 loop (11, 65). Fab X5 can neutralize HIV isolates adapted to growth in T-cell lines but is weak against primary isolates (37). MAbs c11, P7, 7B2, 2.2B, Fab T2, Fab T3, B12, and b6 are all weakly or nonneutralizing against primary viruses.
For some experiments, monovalent Fab fragments were prepared by digesting the parent immunoglobulin G (IgG) with immobilized pepsin (followed by reduction of the Fab2 disulfide bond to make Fab′) or papain (Pierce), according to the manufacturer's instructions.
MAbs 2F5 and 2G12 were provided by Hermann Katinger (Polymun Scientific Inc., Vienna, Austria). MAbs 7B2 and 2.2B were provided by James Robinson (Tulane University). MAb B12 was provided by George Lewis (Institute of Human Virology, Baltimore, MD). Mouse MAb PA1 was provided by Progenics Pharmaceuticals Inc. Purified human immune globulin from an HIV-1-infected donor (HIVIG) was provided by John Mascola (Vaccine Research Center, National Institutes of Health, Washington, DC). MAb 447-52D was obtained from the National Institutes of Health AIDS Reference and Reagent Program. The anti-SIV MAbs were provided by James Robinson (Tulane University) and James Hoxie (University of Pennsylvania).
Purified intravenous IgG collected from HIV-negative donors (IVIG) was obtained from Armour Pharmaceuticals, Kankakee, IL.
Purified recombinant gp120 and CD4-based proteins and gp120 ELISA.
Four-domain soluble CD4 (sCD4), CD4-IgG2, and JR-FL gp120 have been described elsewhere (4) and were provided by Progenics Pharmaceuticals, Inc. Two-domain sCD4, consisting of domains one and two of the CD4 ectodomain, was obtained from the AIDS Reference and Reagent Repository. An enzyme-linked immunosorbent assay (ELISA) to measure MAb binding to gp120 directly coated on Immulon II ELISA wells at 5 μg/ml using the AMPAK substrate and amplifier system has been described previously (8).
Methods to analyze Env in VLPs. (i) SDS-PAGE.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed as previously described (9). Reduced and nonreduced samples were prepared by boiling for 5 min in Laemmli sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 25% glycerol, 0.01% bromophenol blue) in the presence or absence of 20 mM dithiothreitol (9). Western blots were probed with MAbs PA1 and B12 diluted to 1 μg/ml in PBS containing 4% nonfat milk, then detected with a goat anti-mouse alkaline phosphatase conjugate (Jackson) (at 1:3,000) and developed using 5-bromo-4-chloro-3-indolylphosphate (BCIP)/nitroblue tetrazolium (NBT) colorimetric reagents (Sigma-Aldrich).
(ii) BN-PAGE.
To analyze Env derived from VLPs under native conditions, we devised a modified BN-PAGE protocol (57, 60, 61). To liberate Env, VLPs were incubated in an equal volume of solubilization buffer (0.12% Triton X-100 in 1 mM EDTA/1.5 M aminocaproic acid) and one microliter of a protease inhibitor cocktail (Sigma). An equal volume of 2× sample buffer containing 100 mM morpholinepropanesulfonic acid (MOPS), 100 mM Tris-HCl, pH 7.7, 40% glycerol, and 0.1% Coomassie blue was then added. Samples were loaded onto a 4 to 12% Bis-Tris NuPAGE gel (Invitrogen). Ferritin (Amersham) was used as a size standard. Samples were electrophoresed at 4°C for 3 h at 100 V with 50 mM MOPS/50 mM Tris, pH 7.7, containing 0.002% Coomassie blue as cathode buffer and the same buffer without Coomassie blue as the anode buffer. The gel was then Western blotted onto polyvinylidene difluoride. Excess Coomassie blue dye was removed after blotting by washing with 30% methanol/10% acetic acid then 100% methanol.
The blot was then transferred to blocking buffer (4% nonfat milk in PBS) for 30 min and probed using 1 μg/ml each of MAbs 2G12, b12, and 447-52D (anti-gp120 cocktail), or 20 μg/ml each of 4E10 and 2F5 (anti-gp41 cocktail). Goat anti-human and/or mouse Fc and Fab′2 alkaline phosphatase conjugates were used, as appropriate, to detect the primary MAbs at 1:3,000 (Jackson). In some experiments, Env was cross-linked on VLP membrane surfaces, prior to lysis and BN-PAGE. For this purpose, the cross-linker bis(sulfosuccinimidyl) suberate (BS3) was used, as previously described (19).
In BN-PAGE band shift experiments, VLPs were mixed with either sCD4 or a monovalent Fab for 5 min. The concentration of sCD4 or Fab used in band shift experiments was recorded as that in the final sample at the time of loading. Env was detected as above, but omitting the anti-Fab conjugates in order to avoid cross-reactivity to any unbound Fab present in the samples. The density of native and liganded Env bands was determined using ImageJ software v. 1.33u (NIH freeware; http://rsb.info.nih.gov/ij/).
(iii) Virus capture.
To analyze the antigenic properties of Env on VLP surfaces, we adopted the MAb-virus capture assay described previously (17, 50, 54). Briefly, MAbs were coated on Immulon II ELISA wells overnight at 5 μg/ml in PBS. The wells were then washed with PBS and blocked with 3% bovine serum albumin in PBS for 30 min. VLP supernatant was then added to the plate and incubated for 3 h, after which the wells were washed three times with PBS and then overlaid with susceptible CF2.CD4Th.synCCR5 (33) cell targets (104 cells/well) to measure MAb-captured virus. For SOS-VLP capture, cells were overlaid for 30 min, after which 5 mM dithiothreitol was added for 10 min to break the SOS bond (6), and then the medium was changed.
To determine the ability of MAb in solution to inhibit capture by immobilized MAb, competitive capture assays were performed (54), with some modifications. Competitive MAb (10 μg/ml) was first premixed with virus, and then the mixture was added to ELISA wells that had been previously coated with 5 μg/ml of the same or a different MAb and then blocked with 3% bovine serum albumin/PBS. In experiments where the virus Env was noninfectious (e.g., when using the uncleaved Env to pseudotype or when the VLPs might be neutralized by premixing with neutralizing MAb b12), we used VLPs that bear VSV-G as well as HIV-1 Env. Since VSV-G is fusogenic with all cell types, it provides an independent readout of capture, regardless of the fusion competency of the HIV-1 Env.
(iv) Neutralization assays.
Neutralization assays have been described previously (6). Briefly, VLPs were incubated with an equal volume of MAb for 1 h at 37°C and then added to microtiter wells containing CF2.CD4Th.synCCR5 cells in an equal volume of culture medium for 2 hours. The nonneutralized infectious fraction was then assayed two days later by measuring the luciferase activity in target cell lysates. The recorded concentration of MAb or sCD4 in each experiment was expressed as the final concentration after the MAb-virus mixture was added to the cells.
(v) Electron microscopy.
Virus particle preparations were attached to carbon membranes, subsequently reacted with MAbs, and labeled with protein G-coated 10 nm gold. Particles were initially washed by suspending in 200 μl PBS and pelleted at 100,000 × g in an Airfuge centrifuge (Beckman Coulter) and resuspended in PBS. Thin carbon films were attached to 600-mesh copper grids (Polysciences) and floated on 5 μl of the washed virus for 15 min. The grids were then blocked by floating on 20 μl of 0.1% bovine serum albumin, 0.1% cold water fish gelatin (Amersham Biosciences) in PBS for 30 min. Grids were then transferred to 10 μl of the appropriate MAb at 10 μg/ml diluted in 0.1% acylated bovine serum albumin/PBS (Aurion) and incubated for 2 h. Grids were then washed five times for 2 min on 20 μl 0.1% acylated bovine serum albumin. Protein G-conjugated 10-nm gold (Ted Pella) was reacted by floating the grid on 10 μl of gold conjugate diluted 1:20 in 0.1% acylated bovine serum albumin /PBS, incubating for 1 h, and washing five times as above. Gold-labeled viruses were fixed by incubation on 10 μl of 2.5% glutaraldehyde/1% paraformaldehyde in 75 mM PBS for 15 min. Phosphate ions were removed by incubation for 5 min on 20 μl 20% BSB (20% buffered saline borate, 20 mM boric acid, 0.5 mM sodium borate, 15 mM NaCl). Negative staining was accomplished by 2 min of incubation on 5 μl of 0.5% fresh uranyl formate. Excess stain was removed by filter paper blotting and the grid was allowed to air dry. All steps were carried out at room temperature. Samples were recorded at a nominal magnification of 100,000× on a JEOL 1200EX electron microscope.
RESULTS
Production of VLPs bearing authentic trimers.
We used HIV-1 VLPs to probe Env on particle membranes. Recombinant methods to generate particles provide a way to control particle production and Env expression as well as an opportunity to examine the effect of mutations. A subtype B primary isolate Env of moderate neutralization resistance, JR-FL, was selected as a prototype (6, 11). VLPs were expressed bearing a truncated form of Env, termed gp160ΔCT, terminating after three residues of the gp41 cytoplasmic tail (6). gp160ΔCT WT- and SOS-VLPs were previously shown to be functional for infection (SOS requiring exposure to a low concentration of reducing agent after receptor binding, to break the disulfide bond) (3, 6, 52), suggesting that the particles bear fusion-competent trimers. In contrast, UNC-VLPs were found to be nonfunctional.
Initially, we compared VLP-derived Env proteins by SDS-PAGE and Western blot (Fig. 2). gp160ΔCT WT-VLPs bore fully cleaved Env proteins that resolved as gp120 (Fig. 2A and B, lane 3). This VLP-derived gp120 separated as a doublet, suggesting that it may exist in two forms. One mutant with a gp120-gp41 disulfide bond to eliminate gp120 shedding had been introduced (the SOS mutant) (2, 6, 9) and another in which the gp120-gp41 cleavage site had been eliminated (UNC) (6) led to dithiothreitol-reducible and dithiothreitol-resistant gp120/gp41 complexes, as expected (Fig. 2A and B, lanes 2 and 4). The SOS bond was broken under reducing conditions to reveal a gp120 doublet, similar to that observed with the wild type (Fig. 2B, lane 2). Env derived from full-length gp160 WT-VLPs was expressed relatively poorly and was less efficiently cleaved (data not shown), as has been observed previously (72). Furthermore, unlike gp160ΔCT, full-length gp160 was secreted even when the pNL4-3.Luc.R-E- plasmid (induces VLP budding) was omitted from transfections (data not shown), implying that it could be released from cell surfaces as debris, perhaps in the form of vesicles.
FIG. 2.
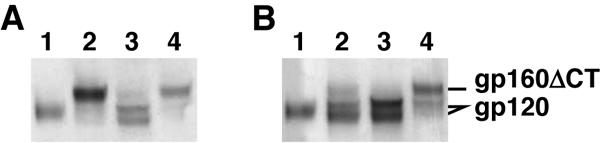
SDS-PAGE and Western blot analysis of Env derived from VLPs. Monomeric JR-FL gp120 and VLPs bearing JR-FLgp160ΔCT SOS, WT, and uncleaved Envs (lanes 1 to 4, respectively) were analyzed by SDS-PAGE and Western blot in A) nonreducing or B) reducing conditions. Blots were probed with anti-gp120 MAbs PA1 and B12.
Nonneutralizing MAbs specifically capture VLPs.
We compared the ability of various anti-Env MAbs to neutralize and to capture VLPs (Fig. 3). To assist clarity, the epitope of each MAb is depicted schematically in Fig. 4. MAbs b12, 2G12, and 2F5 effectively neutralized WT-VLPs and 447-52D had moderate neutralizing activity, while all of the other MAbs had no activity below 50 μg/ml (Fig. 3A). To measure virus capture, we used luciferase as a quantitative readout, as described previously (54). Initial reports of the virus capture technique, like traditional neutralization assays, used p24 as a measure of virus (50). However, because infectious virus constitutes only a fraction of the total particles in a virus preparation, p24 assay may not reflect the capture of fusion-competent virus. The use of luciferase as a readout therefore allows us to focus solely on the capture of infectious virus (54). Luciferase can also be used to measure infection in neutralization assays, ensuring consistency and allowing comparison of the effects of MAbs in these two assays.
FIG. 3.

Comparison of MAb-VLP capture and neutralization. A) Neutralization (50% inhibitory concentration [IC50] [μg/ml]) by a panel of MAbs, as indicated, against JR-FL WT-VLPs. B to E) Capture of various JR-FL VLPs by the same MAbs. B) WT-VLPs; C) SOS-VLPs; D) UNC-VLPs; E) UNC-VLPs which also bear VSV-G. RLU, relative light units.
FIG. 4.

MAb epitope availability on various forms of Env. The MAbs that are able to recognize each form of Env depicted are given below in parentheses. For some forms of Env, MAb recognition is inferred from virus capture and previous studies (7, 37, 58). A) gp120/gp41 trimers (sCD4, b12, 2G12, 2F5, and Z13); B) gp120/gp41 monomers (sCD4, b12, b6, 447-52D, P7, c11, X5, HIVIG, 2F5, and Z13); C) monomeric gp120 (sCD4, b12, b6, 447-52D, X5, P7); D) trimeric and monomeric gp41 stumps (7B2, 2.2B, T3, and Z13); E) uncleaved gp160 oligomers (sCD4, b12, b6, 2G12, 447-52D, P7, 2F5, Z13 and T3).
Previous analyses have been consistent in suggesting that nonneutralizing MAbs as well as neutralizing MAbs are able to capture virus. Here too, we found that the ability of each MAb to capture had no relationship with its ability to neutralize (compare Fig. 3A to Fig. 3B to E). Paradoxically, 2F5 did not capture efficiently, despite its neutralizing activity. Conversely, several non- or weakly neutralizing MAbs, including b6 and 447-52D, effectively captured both WT- and SOS-VLPs (Fig. 3B, C). As reported previously, the V3 loop MAb was highly effective (50, 54). Although MAbs b6, 7B2 and 2.2B capture WT-VLPs efficiently, they are completely nonneutralizing (Fig. 3) and would therefore not be expected to bind to the trimer. Virus capture was highly specific, as demonstrated by the lack of capture by SIV-specific MAb 17A.
One explanation for the discordance between neutralization and capture might be that capture does not require such high affinity trimer binding as neutralization. Multivalent low-affinity MAb-virus contacts available in capture assays might explain why virus mutants that have escaped b12 neutralization can still be captured by b12 (54). On the other hand, the observation that so many completely nonneutralizing MAbs (IC50, >50 μg/ml) that would not be expected to bind to trimers at all but nevertheless still captured virus efficiently (Fig. 3) is difficult to reconcile with a model of virus capture based solely on trimer binding.
It has been proposed that nonneutralizing MAbs capture virus through nonfunctional forms of Env on the virus surface (54). The patterns of capture provide some clues as to the nature of putative alternative form(s) of Env. The topology of various forms of Env is shown schematically in Fig. 4. The N and C termini of gp120 (represented by the epitope of MAb c11, Fig. 4C) and a large portion of the gp41 ectodomain (represented by the epitopes of MAbs 7B2 and 2.2B, Fig. 4D) are known to be important in stabilizing the gp120-gp41 association (9). We would expect these three epitopes to be occluded when gp120 and gp41 are properly associated (as in Fig. 4A and B). We found that c11 was unable to capture (Fig. 3), suggesting that no forms of Env exist on particles that resemble monomeric gp120 dissociated from gp41 (Fig. 4C). However, the capture of WT-VLPs by 7B2 and 2.2B suggests that gp120 shedding exposes gp41 stumps (Fig. 2B), perhaps explaining why antibodies with these specificities are so common in HIV-positive sera (13). The failure of 7B2 and 2.2B to capture SOS-VLPs (Fig. 3C) is fully consistent, because the SOS mutation eliminates gp120 shedding, so that these epitopes remain sequestered (Fig. 4B).
Effective readout of UNC-VLP virus capture was problematic because the uncleaved mutation rendered the particles noninfectious (Fig. 3D). As a solution, we generated UNC-VLPs that also bear VSV-G protein. VSV-G is a highly fusogenic amphotropic molecule that amplifies the readout of captured virus, irrespective of the functional status of the HIV spikes through which capture actually occurs. The pattern UNC+VSV-G-VLP capture was found to be similar to WT-VLPs, although 7B2 and 2.2B were less efficient (Fig. 3E). The similarity of WT- and UNC-VLPs in virus capture contrasts with studies suggesting that cleavage does affect epitope exposure (9, 52).
In keeping with these earlier analyses, we believe cleavage does in fact significantly affect epitope exposure, for reasons that will become clear in further experiments outlined below, using BN-PAGE. To determine whether the discordance between capture and neutralization extends to any other lentiviral Envs, we evaluated SIVmac239 gp160ΔCT WT-VLPs. Several anti-SIV MAbs, and the recombinant chimeric protein, CD4-IgG2, effectively captured these VLPs (not shown). However, only the CD4-IgG2 molecule was able to neutralize. Thus, virus capture by nonneutralizing MAbs may be a general phenomenon for lentiviruses.
Identification of possible nonfunctional forms of Env.
To attempt to identify alternative form(s) of Env that might account for our virus capture findings, we wished to analyze VLP-derived Env using the gentlest possible conditions to preserve protein-protein subunit associations. For this purpose we adapted a BN-PAGE protocol, described previously (60, 61). To obtain sufficient VLPs for BN-PAGE, preparations are routinely concentrated by centrifugation. It is possible that this procedure could adversely affect Env trimers. If this is the case, an effect might be observed on infectivity. However, comparison of centrifuged virus to uncentrifuged virus did not reveal a loss of infectivity (Fig. 5). We therefore conclude that centrifugation efficiently recovers undamaged particles. We cannot rule out that centrifugation may cause some gp120 shedding that may not manifest as a drop in infectivity, because virions retain sufficient trimers to meet a threshold necessary for infection. Indeed, as described above, a degree of gp120 shedding appeared to have taken place in WT-VLPs prior to centrifugation, since gp41 MAbs directed to clusters I and II can capture WT-VLPs but not SOS-VLPs (in which shedding is eliminated) (Fig. 3B).
FIG. 5.

Effect of centrifugal pelleting on VLP infectivity. WT-VLPs were concentrated by high-speed centrifugation and resuspended in the same starting volume of tissue culture medium. The infectivity of pelleted (circles) compared to unpelleted (triangles) VLPs was then assessed using CF2.CD4Th.synCCR5 cells.
To examine the Env proteins from concentrated VLPs we used BN-PAGE/Western blot. This entailed the use of Coomassie blue to bind hydrophobic domains of proteins, allowing their separation largely according to molecular weight (61). Initially, we screened several detergents to try to identify one that effectively liberates and resolves membrane-derived Env proteins without overtly destabilizing protein-protein associations (Fig. 6A). We selected Triton X-100 for further use. We titrated Triton X-100 to determine the minimum concentration required to isolate Env, and thereby limit the possibility of destabilizing protein-protein associations (Fig. 6B). Detergent titration did not greatly affect the intensity or relative proportion of each band. However, higher detergent concentrations appeared to retard the mobility of the putative gp120/gp41 monomer band. To minimize this effect, we used 0.12% Triton X-100 in subsequent experiments. As expected, when detergent was eliminated, no Env was resolved (Fig. 6B, lanes 6 and 11), confirming that the Env bands were liberated from VLP membranes.
FIG. 6.

Effect of detergent type and concentration on the pattern of Env separation by BN-PAGE. A) Various detergents were used to solubilize WT-VLPs, as follows: 0.5% Tween 80 (lane 1); 0.5% Tween 20 (lane 2); 10 mM β-octylglucoside (lane 3); 0.25% each of Igepal and Triton X-100 (lane 4); 0.25% Igepal (lane 5); 0.25% Triton X-100 (lane 6); 0.25% sodium deoxycholate (lane 7); 25 mM CHAPS (cholamidopropyldimethylammoniopropanesulfonate) (lane 8). Monomeric gp120 was included as a control (lane 9). B) Progressively lower concentrations of Triton X-100 were used to lyse WT-VLPs in each lane, as indicated. Monomeric gp120 was included as a control in lane 1. Western blots were probed with the anti-gp120 cocktail (b12, 447-52D, and 2G12) (lanes 1 to 6) or an anti-gp41 cocktail (2F5 and 4E10) (lanes 7 to 10).
Detection by anti-gp120 MAbs revealed that Env derived from WT-VLPs separated as two clear bands (Fig. 6B, lanes 2 to 5). The gp120 control contained a minor fraction of dimers as well as monomers (41). Anti-gp41 detection revealed 4 species in WT-VLPs (Fig. 6B, lanes 7 to 10), the top two of which corresponded to those also detected in the anti-gp120 blot. The largest VLP Env species ran close to the 439-kDa marker and is likely to be the gp120/gp41 trimer. The second largest species ran somewhat slower than the gp120 monomer control and appears to be gp120/gp41 monomer, judging by its detection by both anti-gp120 and gp41 MAbs.
The two smaller species detected by gp41 MAbs may be gp41 trimer and monomer stumps, produced by gp120 shedding from gp120/gp41 trimers and monomers (Fig. 6B, lanes 7 to 10). The presence of gp41 stumps implies that the cognate monomeric shed gp120 might also be present in the VLP sample. In the gp120 blot, a faint band beneath the gp120/gp41 band was distinguishable that could be monomeric gp120 (Fig. 6A, lanes 7 and 8; Fig. 6B, lanes 2 to 4). Detection via the gp41 MAb cocktail was generally less efficient than via the anti-gp120 cocktail, perhaps due to a lower affinity of the gp41 MAbs for their targets, or a decreased accessibility of their epitopes. In general, BN-PAGE blots gave far weaker bands than those typically observed in SDS-PAGE Western blots (Fig. 2), requiring the use of MAb cocktails at relatively high concentrations to optimize detection.
We next asked whether the species observed in Fig. 6 are truly present in intact particles, or if they arise from dissociation of the weakest protein-protein associations during sample preparation for BN-PAGE. To investigate this, we incubated WT- or SOS-VLPs with a cross-linker (Fig. 7). Effective cross-linking of WT-VLPs was confirmed by subjecting samples to denaturing and reducing conditions and probing with anti-gp120 MAbs, revealing three bands (Fig. 7A, compare lanes 5 to lane 6). These bands are likely to be trimers, dimers and monomers. The dimers probably reflect incompletely cross-linked trimer. The existence of a distinct monomer band even in the conditions of excess cross-linker is consistent with it being present on intact particles (Fig. 7A, lane 6). Without cross-linker, Env resolved as a gp120 doublet (Fig. 7A, lane 5), as observed previously (Fig. 2).
FIG. 7.

Effect of cross-linking on the proportion of functional and nonfunctional forms of Env. WT-VLPs (lanes 1, 2, 5, and 6 of part A; lanes 1 and 2 of part B) and SOS-VLPs (lanes 3, 4, 7, and 8 of part A; lanes 3 and 4 of part B) were incubated with (lanes 2, 4, 6, and 8) or without (lanes 1, 3, 5, and 7) 1 mM BS3 before BN-PAGE. Monomeric gp120 was included as a control (lane 9). Samples in lanes 5 to 8 were boiled in 1% SDS/50 mM dithiothreitol prior to loading. Western blots were probed with A) the anti-gp120 cocktail (b12, 447-52D, and 2G12) or B) the anti-gp41 cocktail (2F5 and 4E10).
In native conditions, cross-linking increased the proportion of trimer (Fig. 7A, compare lanes 1 to lane 2) but did not eliminate the monomer. Similarly, probing blots with gp41-specific antibodies revealed gp41 stumps in the wild type (visible as faint bands in Fig. 7B, lanes 1 and 2), but not SOS (Fig. 7B, lanes 3 and 4), regardless of cross-linking. Thus, although the conditions of BN-PAGE may cause some trimer dissociation, the gp120/gp41 monomer and gp41 stumps appear to exist on the surface of intact particles.
The SOS mutation was useful to simplify the identification of any putative defective form(s) of Env that did not arise from gp120 shedding. When probed with anti-gp120 MAbs, SOS Env also resolved as a mixture of gp120/gp41 trimers and monomers (Fig. 7A, lanes 3 and 4). Denaturing/reducing conditions, as for WT, resulted in a gp120 doublet (Fig. 7A, lane 7). The smallest two bands in the wild type probed with anti-gp41 MAbs (Fig. 7B, compare lanes 1 and 3) do not appear in SOS, consistent with their proposed identity as gp41 stumps.
To determine if the nonfunctional species is present on the surface of other virus isolates and, moreover, if it is relevant to live viruses, we examined concentrated, inactivated preparations of HIV-1ADA and HIV-1MN that had been grown in SupT1 cell lines in BN-PAGE. Like JR-FL VLPs, HIV-1ADA and HIV-1MN bore Env monomers and trimers (Fig. 8A and B). The monomers are likely to be gp120/gp41 and not solely monomeric (shed gp120), since they were detected by the anti-gp41 cocktail (Fig. 8B). The different migration properties of trimers and monomers may arise from variation in sequence and/or glycosylation. The MN trimer migrated relatively slowly in BN-PAGE compared to the other Envs (Fig. 8A and B, compare lane 3 to lanes 1 and 2), perhaps because MN trimers are less compact. To control for a possible producer cell effect, we also examined PBMC-grown JR-CSF and found that this also exhibited trimers and monomers (Fig. 8C). Finally, inactivated SIVmac239 particles also showed trimer and monomer bands, implying that the monomer is likely to be a broadly conserved species (not shown). Overall, these data indicate that nonfunctional forms of Env are relevant to infectious HIV.
FIG. 8.
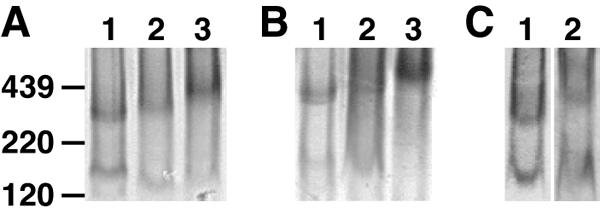
Behavior of inactivated HIV preparations in BN-PAGE. Env from VLPs and virus preparations was compared. A) JR-FL gp160ΔCT WT-VLPs (lane 1); HIV-1ADA (lane 2); and HIV-1MN (lane 3) Western blots were probed with an anti-gp120 cocktail, consisting of b12, 447-52D, and 2G12. B) The same samples in panel A were probed with the anti-gp41 cocktail of 2F5 and 4E10. C) JR-FL WT-VLPs (lane 1) and peripheral blood mononuclear cell-produced JR-CSF (lane 2) were probed with the anti-gp120 cocktail.
Band shift assays visualize sCD4 binding to native Env.
To investigate the possibility that the nontrimer Env species might account for the ability of nonneutralizing MAbs to capture virus, we wished to probe their topology using MAbs in BN-PAGE. Probing Western blot strips with various MAbs failed to show distinctions in MAb reactivities with gp120/gp41 trimers and monomers, probably because the Env is partially denatured during blotting (not shown). A native band shift BN-PAGE protocol was therefore developed to determine the true reactivity patterns of Env on intact particles. We initially tested the binding of two-domain and four-domain sCD4 to WT-VLPs (Fig. 9). The monomer and trimer bands were shifted as sCD4 bound and retarded their migration (Fig. 9). The magnitude of the shift was proportional to the size of the sCD4 ligand (molecular weight for two-domain sCD4 was for ∼25 kDa; four-domain sCD4, ∼50 kDa). Similar results using HIV-1ADA and HIV-1MN preparations (not shown) demonstrated that the technique was broadly applicable.
FIG. 9.
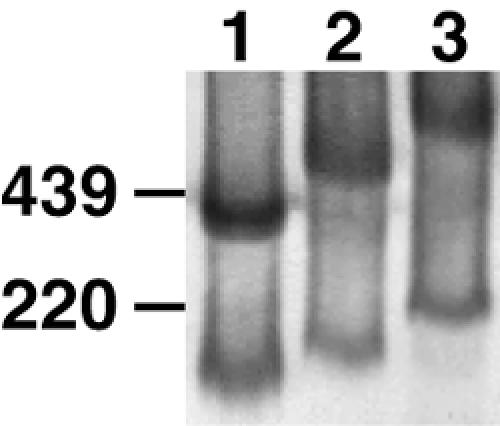
Analysis of sCD4 binding to native Env proteins in BN-PAGE band shifts. SOS-VLPs were incubated either alone (lane 1) or with two-domain (lane 2) or four-domain (lane 3) sCD4 at a final concentration of 20 μg/ml. Samples were then separated by BN-PAGE. Western blots were probed with an anti-gp120 MAb cocktail containing 2G12, b12, and 447-52D. Ferritin 12-mer (220 kDa) and 24-mer (439 kDa) served as molecular size markers.
Trimer binding correlates with neutralization whereas nonneutralizing MAbs recognize nonfunctional Env.
MAb binding assays using cells expressing surface Env have provided evidence for a correlation between neutralization and binding to trimers. However, the binding of some nonneutralizing MAbs revealed some apparent contradictions (28, 52). Nonneutralizing MAb binding might stem from heterogeneity of Env on cell surfaces, including uncleaved gp160 or other nonnative species. Examining MAb binding using BN-PAGE band shift assays might have the advantage of visually distinguishing the trimer from other forms of Env.
To develop an MAb-based band shift protocol, Env-MAb complexes should be detected via an Env component, so that liganded and unliganded Env fractions can be distinguished. To prevent interference by unbound MAb ligand in the sample, we used Fabs to form Env-Fab complexes and detected the Env component in blots using whole gG MAbs, followed by an anti-Fc conjugate. The use of monovalent Fabs also eliminated the possibility of multivalent Env-MAb complexes that might be difficult to identify.
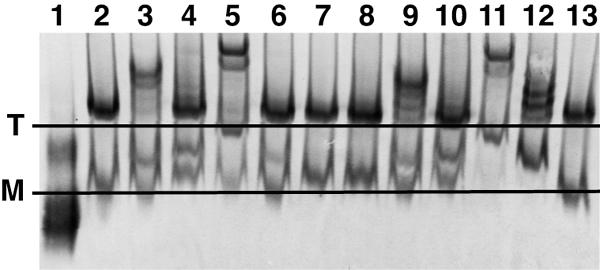
We analyzed the patterns of MAb band shifts with WT-, SOS-, and UNC-VLPs and HIV-1MN (Fig. 10). Western blots were probed either with anti-gp120 (Fig. 10A, B, E, and F) or anti-gp41 cocktails (Fig. 10C and D). The identity of the gp120/gp41 monomer band in WT-VLPs and HIV-1MN was confirmed by the observation that monomer shifts caused by gp41 MAbs Z13e1 and 2F5 could be detected by gp120 MAbs (Fig. 10A and E, lanes 9 and 10) and complementary data showing that monomer shifts caused by gp120 MAbs were detectable by the anti-gp41 cocktail (Fig. 10C, lanes 2 to 7).
FIG. 10.

MAb binding to native Env in BN-PAGE band shifts. WT-VLPs (A and C), SOS-VLPs (B and D), inactivated MN virus (E), and UNC-VLPs (F) were incubated with Fabs at a final concentration of 20 μg/ml and then processed for BN-PAGE. Western blots in panels A, B, E, and F were probed with the anti-gp120 cocktail containing 2G12, b12, and 447-52D; blots in panels C and D were probed with the anti-gp41 cocktail containing 2F5 and 4E10. Labels: T, gp120/gp41 trimer; M, gp120/gp41 monomer; O, uncleaved oligomers (most likely dimers, trimers, and tetramers). The Fabs used (neutralization IC50 titers against JR-FL gp160ΔCT WT in μg/ml) are as follows: lane 1, no Fab; lane 2, b12 (0.1 μg/ml); lane 3, b6 (>40 μg/ml); lane 4, 2G12 (0.5 μg/ml); lane 5, 447-52D (9 μg/ml); lane 6, P7 (>40 μg/ml); lane 7, X5 (>40 μg/ml); lane 8, HIVIG (>40 μg/ml); lane 9, 2F5 (8 μg/ml); lane 10, Z13e1 (19 μg/ml); lane 11, T2 (>40 μg/ml); lane 12, T3 (>40 μg/ml). In panels G and H four-domain sCD4 was titrated in band shifts (lanes 1 to 8 correspond to four-domain sCD4 concentrations of 0.003, 0.01, 0.03, 0.1, 0.3, 1, 3, and 10 μg/ml, respectively), and in a neutralization assay against WT-VLPs.
Bacterially produced Fabs (b12, b6, P7, X5, Z13e1, T2, and T3) gave cleaner shifts than Fabs prepared from IgG by papain digestion, probably because the former are inherently homogenous and free of any contaminating Fc. In general, blots from band shift experiments were slightly fainter than from regular BN-PAGE, because only anti-Fc conjugate was used (i.e., no anti-Fab conjugate) and also because, in some cases, Fab ligands obscure the epitopes of whole IgG probes used for detection. Overall, there was good consistency in the shift patterns of WT-VLPs and SOS-VLPs (Fig. 10A to D). The SOS monomer species was generally more distinct, probably because gp120 shedding is eliminated. SOS-VLPs are therefore particularly useful in determining the patterns of MAb binding to the gp120/gp41 monomer.
A comparison of the neutralizing and nonneutralizing prototype Fabs, b6 and b12, showed that both were able to shift monomers, but only the neutralizing Fab b12 shifted trimers (Fig. 10A to E, lanes 2 and 3). In contrast, b6 shifted the oligomeric uncleaved Env species (Fig. 10F, lane 3), consistent with exposure of the nonneutralizing face of gp120 (Fig. 10E) (9, 52). Analysis of other Fabs revealed that those that shifted native trimers (2G12, 2F5, and Z13e1) also had strong neutralizing activity (Fig. 10A to E, lanes 4, 9, and 10). Of particular interest, 2G12 induced “supershifts” compared to other Fabs, consistent with the notion that 2G12 exhibits a unique “domain exchange” conformation (16), in which the Fab arms are locked together in an unusually large 100-kDa Fab-Fab complex.
Fabs 447-52D and X5 appeared to partially shift SOS trimers (Fig. 10B and D, lanes 5 and 7). They did not, however, cause convincing shifts of WT-VLP trimers. This difference is reflected by a slightly increased neutralization sensitivity of SOS-VLPs compared to WT-VLPs to neutralization by certain MAbs (6). It is possible that MAb 447-52D neutralization occurs when it binds to trimers after CD4 engagement (39). However, band shift assays are limited to the native form of trimer and cannot fully recapitulate the patterns of epitope exposure that may occur during infection, when epitopes such as that of 447-52D might become exposed. The lack of X5 neutralization contrasts with a previous report using viruses bearing full-length JR-FL Env (37). This may be because gp160ΔCT Env fuses more rapidly than full-length gp160 (3), reducing the availability of fusion intermediates for X5 to neutralize. HIVIG is derived from pooled human HIV-positive sera and almost certainly contains a small fraction of neutralizing antibodies. However, these neutralizing antibodies appear to not be at a concentration high enough to shift the trimer in BN-PAGE, as evidenced by the fact that the IC50 of HIVIG exceeds 40 μg/ml.
Compared to the trimer, the monomeric gp120/gp41 species exhibited completely different binding patterns. Almost all Fabs were able to mediate shifts. The only exceptions were the gp41-specific Fabs T2 and T3 (Fig. 10A to D), whose epitopes appear to be obscured by proper gp120/gp41 association, as observed in SOS-VLP capture (Fig. 3C). However, MAb T3 recognized gp41 stumps from which gp120 had dissociated, as evidenced by a loss in native gp41 trimer stumps, accompanied by a gain in signal close to the gp120/gp41 monomer (Fig. 10C, lane 12). HIVIG and Z13e1 also appeared to shift the gp41 stumps, while 2F5 and T2 did not.
HIVMN shifts showed a similar pattern to JR-FL WT-VLPs (Fig. 10, compare parts A, C, and E). Because of the slower mobility of MN trimers (observed above in Fig. 8), electrophoresis was run longer to better resolve the bands. MAbs b12, 2G12, 2F5, and Z13e1 all clearly shifted MN trimers (Fig. 10E, lanes 2, 4, 9, and 10). The monomer bands were more difficult to resolve, as was discerning any shifts. However, close inspection of lanes containing ligands (Fig. 10E, lanes 2 to 12) compared to the unliganded control (Fig. 10E, lane 1) reveals that many of the Fabs (particularly lanes 4, 6, 8, 9, and 10) caused shifts similar to JR-FL WT-VLPs.
The oligomeric species of uncleaved protein exhibited a different pattern compared to native trimers and was recognized by all the Fabs except T2 (Fig. 10F, lane 11). This highlights the importance of gp120/gp41 cleavage in the proper folding of Env and indicates possible limitations in vaccine approaches that involve uncleaved Env proteins. However, the monomeric uncleaved species behaved very similarly to the monomers from WT-VLPs bearing cleaved Env and may possibly derive from a small amount of cleaved material in UNC-VLPs (Fig. 2). The similarities in overall behavior of uncleaved and cleaved gp120/gp41 monomers in WT-VLPs could explain the similarities noted earlier in the patterns of behavior between UNC- and WT-VLPs in virus capture (Fig. 3).
To investigate a possible quantitative relationship between neutralization and trimer binding, graded concentrations of four-domain sCD4 were added to WT-VLPs in parallel band shift and neutralization assays. The IC50 of trimer shift (0.5 μg/ml) and neutralization (IC50 = 0.8 μg/ml) closely corresponded (Fig. 10G and H). MAbs b12, 2G12, 2F5, and Z13e1 trimer shift IC50s similarly corresponded with neutralization IC50s (not shown). As documented above, monomer binding did not correlate with neutralization, because several Fabs recognized the monomer without neutralizing the virus. However, monomer binding did appear to correlate with virus capture. In particular, strong monomer binding by Fabs 447-52D, 2G12, and four-domain sCD4 reflected their efficient virus capture ability (Fig. 2 and 4 and data not shown).
Fabs T2 and T3 bound neither to trimers nor to monomers, but were nevertheless able to capture WT-VLPs with moderate efficiency (data not shown; capture by MAbs 7B2 and 2.2B that recognize similar epitopes is shown in Fig. 3). Capture by Fab T3 probably occurs through gp41 stumps (13), though it is unclear how T2 might capture. In contrast, Fabs 2F5 and Z13e1 bound monomer tightly (IC50 = 0.8 μg/ml each) but were unable to capture WT-VLPs efficiently. It is possible that the 2F5/Z13 epitopes on gp120/gp41 monomers (Fig. 10) are accessible to Fabs in solution, but when the Fabs are immobilized in capture wells, the gp120 moiety of gp120/gp41 monomers sterically limits access to the 2F5/Z13 epitopes on gp41.
Remaining questions concerning the existence of monomer on intact particles.
It could still be argued that the monomer observed in BN-PAGE is solely the product of treatment of VLPs with mild detergent and conditions of BN-PAGE. Undoubtedly, sample preparation does cause some trimer to fall apart into monomers (Fig. 7). To determine definitively if monomer is present on intact particles, it is important to eliminate the possibility of MAb binding to forms of Env that become available only after detergent treatment. This can be achieved by separating the MAb-virus incubation step from detergent treatment and sample preparation for BN-PAGE. Thus, we developed a new protocol. We incubated MAbs with SOS-VLPs, then pelleted virus and washed away any unbound MAb with two 1-ml washes of PBS, and then performed BN-PAGE. We prepared control samples in parallel, where VLPs were washed and spun in the same way, after which MAb was added, along with detergent, as per the original protocol. In the new format (Fig. 11, lanes 2 to 7), the neutralizing and nonneutralizing MAbs largely behaved as they did by the original protocol (Fig. 10B and Fig. 11, lanes 8 to 13). Fabs b12 and 2G12 shifted both trimers and monomers and the nonneutralizing Fab b6 shifted only the monomer. The fact that b6 recognized the monomer in this format suggests that the monomer is likely to be available on intact particles. Some unliganded monomer that resolved in this format may have been the product of trimer dissociation during sample preparation. Control Fab LS4 (Ebola virus GP-specific) did not cause any shift (Fig. 11, lanes 7 and 13).
FIG. 11.
Separation of the MAb-VLP binding step from detergent treatment and sample preparation indicates that monomer is present on intact particles. We further investigated whether gp120/gp41 monomers are a relevant species on intact particles by separating MAb binding from sample preparation for BN-PAGE. Various Fabs were incubated with SOS-VLPs either before (lanes 2 to 7) or after (lanes 8 to 13) two washes with 1 ml PBS, after which VLPs were detergent treated and run on BN-PAGE as usual (lane 1, JR-FL gp120; lanes 2 and 8, no Fab; lanes 3 and 9, Fab b12; lanes 4 and 10, Fab b6; lanes 5 and 11, Fab 2G12; lanes 6 and 12, Fab Z13 WT; lanes 7 and 13, Fab LS4 directed to Ebola virus GP) for 1 h before being pelleted, washed two times to completely remove any unbound Fab (verified in parallel controls), and then the washed VLP sample was resuspended in PBS and prepared for BN-PAGE as shown.
Interestingly, Fab Z13WT failed to cause convincing shifts in the new format (Fig. 11, lane 6), in contrast to the original format (Fig. 11, lane 12). This suggests that the Z13 epitope may be sequestered on native spikes and becomes fully available only after the Env trimers are liberated by detergent. Z13 may therefore exhibit greater neutralizing activity after Env has been activated, perhaps by receptor binding (6, 23). The fact that Z13 behaves differently in our two protocols also confirms that the washing procedure intended to remove unbound Fab was effective, providing further evidence that b6 binds to gp120/gp41 monomers on intact particles.
Immunoelectron microscopy reveals nonneutralizing MAb binding to VLPs.
We further investigated direct binding of nonneutralizing MAbs to intact particles by immunoelectron microscopy. We compared b6 and b12 as prototype monomer- and monomer/trimer-reactive MAbs. Both MAbs bind to monomeric JR-FL gp120 equally well (Fig. 12A), but only b12 neutralizes JR-FL WT-VLPs (Fig. 12B). In electron micrographs, we used MAbs b6 and b12 at 10 μg/ml each, a concentration at which both MAbs bind monomeric gp120 to saturation (Fig. 12A), but only b12 neutralizes (Fig. 12B). MAb binding was detected by subsequent incubation with 10- nm gold particles coupled with protein G (Fig. 12C and D).
FIG. 12.

Serologic and electron microscopic evidence that nonneutralizing MAb b6 binds to intact particles via a form of Env other than trimers. MAbs b12 (solid squares) and b6 (open squares) were titrated in ELISA against monomeric JR-FL gp120 (A) and in neutralization assays against JR-FL gp160ΔCT WT- (B). VLPs. Representative examples of electron micrographs of JR-FL gp160ΔCT WT-VLPs reacted with b12 (C; top panels) or b6 (D; bottom panels), at 10 μg/ml each, followed by protein G-conjugated 10-nm gold (one or two large black dots per virion were observed in these examples). Bar, 100 nm.
MAb b12 binding in fields encompassing 1,470 virions was scored. Of 182 gold particles observed, 131 were associated with 120 virions (i.e., 8.2% of virions were labeled) and 51 gold particles were not associated with virions. MAb b6 also bound to the particles, albeit to a lesser extent. Of 99 gold particles observed in fields encompassing 1,491 virions, 73 were associated with 71 virions (i.e., 4.9% of virions were labeled) and 26 gold particles were not associated with virions. The majority (∼75%) of the gold not bound to particles appeared to be associated with aggregated protein of cellular/viral debris and so may have been binding to legitimate target molecules. Representative examples of b12 and b6 immunogold-labeled particles are shown (Fig. 12C and D). Similar results were obtained with inactivated BaL virus particles (data not shown). A control in which virions were incubated with pooled normal intravenous IgG resulted in essentially no gold particle binding to JR-FL VLPs (of 1,500 virions, only four gold particles were virion associated and 17 gold particles were in the background, most of which were not associated with debris; not shown). This was also true for protein G-gold alone (not shown), confirming the specificity of binding. The fact that b6 bound to JR-FL VLPs suggests that its target, the gp120/gp41 monomer (Fig. 10), is present on intact particles. The observation that b6 binding was less pronounced compared to b12 is consistent with b12's having a greater range of targets, including both gp120/gp41 trimers and monomers.
Reciprocal competitive binding relationships of b6 and b12 in virus capture are consistent with the presence of gp120/gp41 monomers on intact particles.
Because b6 blocks b12 binding to monomeric Env but not trimeric Env, it might be expected that neutralizing MAb b12 can capture virus even in the presence of excess b6 (Fig. 10A and C, lanes 2 and 3). This would provide further evidence that the monomer exists on intact particles. To test this hypothesis, we analyzed MAb binding relationships in competitive virus capture assays, in which an inhibitory MAb was mixed with VLPs and then added to wells coated with the same or a different MAb. A previous study (54) demonstrated that capture by b12 could be inhibited by b6. However, that study did not investigate the ability of b12 to inhibit b6 capture, because b12 competitor neutralized the virus, and thus any virus captured by b6 was neutralized and therefore could not be detected using HIV infectivity as a readout.
We addressed this complication by using VLPs that bear VSV-G as well as JR-FL Env. As indicated above (Fig. 3E), VSV-G provided a means to detect captured virus, even when the HIV Env was neutralized or nonfunctional. Thus, for example, only a slight loss in detection (to 48.5 and 75.8% of controls) occurred, even when saturating concentrations of 2G12 or 2F5 neutralizing MAbs, respectively, were incubated with WT- and VSV-G-VLPs, followed by capture with a noncompeting MAb such as b6 (Fig. 13A). Similarly, 2G12 and 2F5 competitors only partially reduced the b12 capture signal (Fig. 13C) and b12 and 2F5 competitors only partially reduced the 2G12 capture signal (Fig. 13E). In contrast, self-competition of b6 capture gave a much more dramatic loss in capture signal (2.7% of controls, Fig. 13A). A similar effect was observed for b12 (Fig. 13C) and 2G12 (Fig. 13E).
FIG. 13.

Competitive virus capture. The effect of incubating VLPs with MAbs on their ability to be captured by the same or different MAbs was investigated. WT- and VSV-G-VLPs (A, C, and E) or UNC- and VSV-G-VLPs (B, D, and F) were incubated with 10 μg/ml of MAb competitor (or none), as indicated, and then capture by MAbs b6 (A and B), b12 (C and D), and 2G12 (E and F) was determined.
Examination of the patterns of CD4 binding site MAb competition revealed that b6 capture was inhibited by b12 (3.6% of control; Fig. 13A) but b6 was only partially effective at blocking b12 capture (15.3% of the control; Fig. 13C). This reflects b6's ability to block the b12 epitope on monomers, but not trimers. In control experiments using UNC- and VSV-G-VLPs (Fig. 6B, D, and F), b6 effectively inhibited b12 capture (Fig. 13C and D). This is consistent with exposure of the nonneutralizing face, allowing nonneutralizing MAbs to bind, as observed in BN-PAGE (Fig. 10F). In controls, 2G12 inhibited capture of both WT- and UNC-VLPs by immobilized 2G12, as expected (Fig. 13E and F).
DISCUSSION
Why have gp120/gp41 monomers not been described previously?
Collectively, our data support gp120/gp41 monomers as a primary nonfunctional form of Env on the surface of HIV-1. Less surprisingly, gp41 stumps from which gp120 has shed also decorate the particle surfaces (13). The reason why monomers have not been previously reported may be partly due to a lack of techniques available to investigate this possibility. Moreover, until recently, researchers had little reason to suspect that alternative forms of Env exist on the HIV-1 surface. Indeed, it is widely perceived that strong gp41-gp41 associations effectively stabilize trimers (20). However, in native trimers, gp41 is likely to be in a “prefusion” conformation that may be somewhat less stable than the fusion-active forms of gp41 previously studied. In fact, cryoelectron microscopic analysis of native virion-associated SIV Env spikes shows a pronounced gap separating the three membrane-proximal segments (i.e., the gp41 “stalk”) (Zhu et al., unpublished results). In fact, gp120-gp120 associations may be just as important in stabilizing native trimers (36, 41).
Electron microscopy studies have shed some light on alternative forms of Env on particle surfaces. Atomic force microscopy revealed what were interpreted as Env “monomers” (34). However, paradoxically, this method did not identify trimers. In contrast, electron tomographs of HIV and SIV particles revealed distinct Env trimers (73), but no other forms deemed to be Env were observed. This may be because (i) monomers are difficult to resolve because of their smaller size and less distinctive structure, (ii) they are simply too few in number, and/or (iii) they cannot be distinguished from other virion surface molecules. Indeed, electron micrographs of HIV-1 reveal some small, less regular structures that might be monomers (73 and unreported data).
In retrospect, the observation that soluble gp140SOS readily dissociates into gp120/gp41 monomers (9, 61) provided a paradigm for membrane-associated gp120/gp41 monomers observed here. There have been a few other hints in the literature regarding the possibility of gp120/gp41 monomers. For example, one group suggested that there might be a monomer-trimer equilibrium (5), based on the proposed inhibitory mechanism of a gp41 N-helix intercalating peptide that may bind to virus if trimers, at least temporarily, dissociate into monomers. In addition, a study of cross-linked Env derived from HIV preparations revealed a fraction of low-molecular-weight Env material that, considering the new information presented here, might well have been gp120/gp41 monomers (18).
How does the gp120/gp41 monomer originate?
Our analysis suggested the presence of gp120/gp41 monomers and gp41 stumps on virus surfaces, but no unbalanced gp120/gp41 oligomers (e.g., gp41 trimeric stumps from which gp120 has only partially shed). This indicates that the trimer dissociates in a concerted manner either at the gp120/gp41 interface (resulting in gp120 shedding) or at the vertices of oligomerization (to form gp120/gp41 monomers). While gp120 shedding probably occurs only after the virus has matured, it is possible that the gp120/gp41 monomer arises as a by-product of Env synthesis. Our detection of gp120 doublets in SDS-PAGE analysis of VLP Env (Fig. 2) provides a possible lead. gp120 doublets were observed previously in virus-derived Env (51) but, so far, an explanation for their existence has not been put forward. The present data suggest that gp120 doublet bands might originate from gp120/gp41 trimers and gp120/gp41 monomers. If we assume that monomers exist during the late stages of Env synthesis, we might expect that, due to the presumed increased accessibility of N-linked sites, they are glycosylated at a certain site(s) not exposed on trimers. Thus, the more heavily glycosylated upper band of the doublet might derive from monomer, and the lower band from trimer.
The possibility that the monomer arises during Env synthesis is rendered even more plausible when one considers that the uncleaved Env precursor exists as a heterogeneous mix of oligomers (Fig. 4F). It is possible that proteolytic cleavage of this mixed precursor leads to a similarly mixed final Env product of monomers and trimers. Other possibilities to account for the emergence of a subspecies of monomer during Env synthesis include incomplete signal peptide cleavage and partial cleavage of gp120/gp41 at a putative secondary cleavage site (44). On the other hand, much like gp120 shedding from mature virus, the gp120/gp41 monomer may progressively accumulate on the surface of mature virus. For example, premature “misfiring” of the metastable trimer complex or proteolytic cleavage of the V3 loop might induce dissociation of mature trimers.
What are the consequences of nonfunctional Env?
Since gp120/gp41 monomers and gp41 stumps are not thought to be involved in virus binding and fusion, nonneutralizing antibodies may bind to virus without directly affecting virus infection. However, antibody coating of nonfunctional Env on particle surfaces may have other consequences distinct from conventional neutralization (14). The recently described ability of nonneutralizing antibodies to potently inhibit HIV-1 infection of macrophages, most likely by Fc receptor-mediated phagocytosis (32), could be explained by the availability of nonfunctional targets on the virus, to which the nonneutralizing antibodies bind. Thus, even in the absence of conventional neutralization, antibody coating may impose certain restrictions on the cell targets the virus can infect. In addition, nonfunctional Env may well explain early reports of high concentrations of circulating immune complexes of infectious virus in HIV-positive subjects, despite the typically low concentration of neutralizing antibodies in sera (27, 47). Furthermore, nonneutralizing antibody coating may prolong the persistence of particles by facilitating their trapping by follicular dendritic cells (30, 63).
Nonfunctional Env may also have consequences for vaccine design. Since MAb binding to trimers is associated with neutralization, it has been difficult to reconcile why particles bearing trimers elicit mostly nonneutralizing antibodies. As the critical functional unit in viral infectivity, Env has been under intense pressure to evolve immune evasion tactics. The nonfunctional forms of Env we describe may prove to be one immune evasion tactic, serving to divert the attention of B cells from trimers.
One hypothetical problem with nonfunctional forms of Env acting as decoys is that we might expect B cells to respond equally to monomers, trimers and gp41 stumps on particle surfaces, in the same way that combination vaccines such as that for measles, mumps, and rubella induce equivalent antibody responses against each component as they do when each component is administered separately (68). However, the presentation of the various forms of Env together on the same particle creates an entirely different scenario. Virus particles can be considered as a single target. Affinity selection of germinal center B cells mandates the amplification of higher-affinity clones, at the expense of others. Therefore, the development of antibody responses toward each of the foreign antigens on particle surfaces might depend on their relative accessibility to antibody binding. Thus, the high accessibility of nonfunctional forms of Env may explain their apparent immunodominance. Antibodies that react with gp120/gp41 monomer or gp41 stumps do not tend to cross-react with the trimer, probably because their epitopes impinge on the nonneutralizing face (Fig. 4). Even B cells that recognize trimers tend to bind as well to monomers on the same particle and may lose their trimer-binding property during further maturation against the monomer.
Another question raised by these data is the significance of the fact that the monomer is a minor species. It is possible that the immunogenicity of monomers depends on expression above a certain threshold. However, our evidence that the monomer appears to play a dominant role in virus capture and is a major target of HIVIG prepared from HIV-positive donor plasma is consistent with the monomer eliciting strong antibody responses during natural infection. We therefore propose that accessibility, not quantity, is the major factor driving the relative immunogenicity of monomers and trimers. We suggest, then, that trimers are a secondary target for antibodies, consistent with the slow development of autologous neutralization in natural infection. In accordance with this idea, we have observed that immunizations of small animals with SOS-VLPs induced high titer anti-gp120 antibodies directed against epitopes that are highly accessible in virus capture and on monomers but ineffective at neutralizing the JR-FL parent virus (J. Binley, unpublished).
An awareness of HIV-1's full armory of evasion mechanisms may provide new solutions for vaccine research (35, 55, 69). The observation that simple trimer binding predicts neutralization (Fig. 10) reaffirms this as the primary target for antibody-based vaccine development. Devising ways to minimize or eliminate responses to nonfunctional Env and, at the same time, to focus responses on functional trimers is therefore worth investigating in future vaccine research.
Acknowledgments
This work was supported by NIH grants AI49566 and AI058763 and the AIDS and Infectious Disease Science Center (J.M.B.), AI055461 (K.H.R.), and AI33292 (D.R.B.). P.L.M. was supported through the Columbia University-Southern Africa Fogarty AIDS International Training and Research Program (grant number D.43 TW00231), funded by the Fogarty International Center. Additional support was provided by Progenics Pharmaceuticals, Inc.
We thank Rogier Sanders, Pascal Poignard, and Norbert Schulke for insightful discussions and advice; Welkin Johnson for providing a SIVmac316 molecular clone; Ned Landau for providing the pVSV-G plasmid; John Mascola for providing HIVIG; Jeff Robinson, George Lewis, James Hoxie, and Hermann Katinger for providing various antibodies; Jeff Lifson and Larry Arthur for providing inactivated HIV and SIV preparations; Meng Wang and David Montefiori for providing PBMC-produced viruses; the AIDS Reagent Repository for 447-52D and two-domain sCD4; and Michelle Nolasco for assistance with graphic artwork.
REFERENCES
- 1.Abacioglu, Y. H., T. R. Fouts, J. D. Laman, E. Claassen, S. H. Pincus, J. P. Moore, C. A. Roby, R. Kamin-Lewis, and G. K. Lewis. 1994. Epitope mapping and topology of baculovirus-expressed HIV-1 gp160 determined with a panel of murine monoclonal antibodies. AIDS Res. Hum. Retroviruses 10:371-381. [DOI] [PubMed] [Google Scholar]
- 2.Abrahamyan, L. G., R. M. Markosyan, J. P. Moore, F. S. Cohen, and G. B. Melikyan. 2003. Human immunodeficiency virus type 1 Env with an intersubunit disulfide bond engages coreceptors but requires bond reduction after engagement to induce fusion. J. Virol. 77:5829-5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abrahamyan, L. G., S. R. Mkrtchyan, J. Binley, M. Lu, G. B. Melikyan, and F. S. Cohen. 2005. The cytoplasmic tail slows the folding of human immunodeficiency virus type 1 Env from a late prebundle configuration into the six-helix bundle. J. Virol. 79:106-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allaway, G. P., K. L. Davis-Bruno, G. A. Beaudry, E. B. Garcia, E. L. Wong, A. M. Ryder, K. W. Hasel, M. C. Gauduin, R. A. Koup, J. S. McDougal, et al. 1995. Expression and characterization of CD4-IgG2, a novel heterotetramer that neutralizes primary HIV type 1 isolates. AIDS Res. Hum. Retrovir. 11:533-539. [DOI] [PubMed] [Google Scholar]
- 5.Bewley, C. A., J. M. Louis, R. Ghirlando, and G. M. Clore. 2002. Design of a novel peptide inhibitor of HIV fusion that disrupts the internal trimeric coiled-coil of gp41. J. Biol. Chem. 277:14238-14245. [DOI] [PubMed] [Google Scholar]
- 6.Binley, J. M., C. S. Cayanan, C. Wiley, N. Schulke, W. C. Olson, and D. R. Burton. 2003. Redox-triggered infection by disulfide-shackled human immunodeficiency virus type 1 pseudovirions. J. Virol. 77:5678-5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Binley, J. M., H. J. Ditzel, C. F. Barbas 3rd, N. Sullivan, J. Sodroski, P. W. Parren, and D. R. Burton. 1996. Human antibody responses to HIV type 1 glycoprotein 41 cloned in phage display libraries suggest three major epitopes are recognized and give evidence for conserved antibody motifs in antigen binding. AIDS Res. Hum. Retrovir. 12:911-924. [DOI] [PubMed] [Google Scholar]
- 8.Binley, J. M., P. J. Klasse, Y. Cao, I. Jones, M. Markowitz, D. D. Ho, and J. P. Moore. 1997. Differential regulation of the antibody responses to Gag and Env proteins of human immunodeficiency virus type 1. J. Virol. 71:2799-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binley, J. M., R. W. Sanders, B. Clas, N. Schuelke, A. Master, Y. Guo, F. Kajumo, D. J. Anselma, P. J. Maddon, W. C. Olson, and J. P. Moore. 2000. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J. Virol. 74:627-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Binley, J. M., R. W. Sanders, A. Master, C. S. Cayanan, C. L. Wiley, L. Schiffner, B. Travis, S. Kuhmann, D. R. Burton, S. L. Hu, W. C. Olson, and J. P. Moore. 2002. Enhancing the proteolytic maturation of human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 76:2606-2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Binley, J. M., T. Wrin, B. Korber, M. B. Zwick, M. Wang, C. Chappey, G. Stiegler, R. Kunert, S. Zolla-Pazner, H. Katinger, C. J. Petropoulos, and D. R. Burton. 2004. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 78:13232-13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binley, J. M., R. Wyatt, E. Desjardins, P. D. Kwong, W. Hendrickson, J. P. Moore, and J. Sodroski. 1998. Analysis of the interaction of antibodies with a conserved enzymatically deglycosylated core of the HIV type 1 envelope glycoprotein 120. AIDS Res. Hum. Retrovir. 14:191-198. [DOI] [PubMed] [Google Scholar]
- 13.Burrer, R., S. Haessig-Einius, A. M. Aubertin, and C. Moog. 2005. Neutralizing as well as non-neutralizing polyclonal immunoglobulin (Ig)G from infected patients capture HIV-1 via antibodies directed against the principal immunodominant domain of gp41. Virology 333:102-113. [DOI] [PubMed] [Google Scholar]
- 14.Burton, D. R. 2002. Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2:706-713. [DOI] [PubMed] [Google Scholar]
- 15.Burton, D. R., J. Pyati, R. Koduri, S. J. Sharp, G. B. Thornton, P. W. Parren, L. S. Sawyer, R. M. Hendry, N. Dunlop, P. L. Nara, et al. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024-1027. [DOI] [PubMed] [Google Scholar]
- 16.Calarese, D. A., C. N. Scanlan, M. B. Zwick, S. Deechongkit, Y. Mimura, R. Kunert, P. Zhu, M. R. Wormald, R. L. Stanfield, K. H. Roux, J. W. Kelly, P. M. Rudd, R. A. Dwek, H. Katinger, D. R. Burton, and I. A. Wilson. 2003. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science 300:2065-2071. [DOI] [PubMed] [Google Scholar]
- 17.Cavacini, L., and M. Posner. 2004. Native HIV type 1 virion surface structures: relationships between antibody binding and neutralization or lessons from the viral capture assay. AIDS Res. Hum. Retrovir. 20:435-441. [DOI] [PubMed] [Google Scholar]
- 18.Center, R. J., R. D. Leapman, J. Lebowitz, L. O. Arthur, P. L. Earl, and B. Moss. 2002. Oligomeric structure of the human immunodeficiency virus type 1 envelope protein on the virion surface. J. Virol. 76:7863-7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Center, R. J., P. Schuck, R. D. Leapman, L. O. Arthur, P. L. Earl, B. Moss, and J. Lebowitz. 2001. Oligomeric structure of virion-associated and soluble forms of the simian immunodeficiency virus envelope protein in the prefusion activated conformation. Proc. Natl. Acad. Sci. USA 98:14877-14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263-273. [DOI] [PubMed] [Google Scholar]
- 21.Cole, K. S., M. Alvarez, D. H. Elliott, H. Lam, E. Martin, T. Chau, K. Micken, J. L. Rowles, J. E. Clements, M. Murphey-Corb, R. C. Montelaro, and J. E. Robinson. 2001. Characterization of neutralization epitopes of simian immunodeficiency virus (SIV) recognized by rhesus monoclonal antibodies derived from monkeys infected with an attenuated SIV strain. Virology 290:59-73. [DOI] [PubMed] [Google Scholar]
- 22.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935-944. [DOI] [PubMed] [Google Scholar]
- 23.de Rosny, E., R. Vassell, S. Jiang, R. Kunert, and C. D. Weiss. 2004. Binding of the 2F5 monoclonal antibody to native and fusion-intermediate forms of human immunodeficiency virus type 1 gp41: implications for fusion-inducing conformational changes. J. Virol. 78:2627-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ditzel, H. J., P. W. Parren, J. M. Binley, J. Sodroski, J. P. Moore, C. F. Barbas 3rd, and D. R. Burton. 1997. Mapping the protein surface of human immunodeficiency virus type 1 gp120 using human monoclonal antibodies from phage display libraries. J. Mol. Biol. 267:684-695. [DOI] [PubMed] [Google Scholar]
- 25.Edinger, A. L., M. Ahuja, T. Sung, K. C. Baxter, B. Haggarty, R. W. Doms, and J. A. Hoxie. 2000. Characterization and epitope mapping of neutralizing monoclonal antibodies produced by immunization with oligomeric simian immunodeficiency virus envelope protein. J. Virol. 74:7922-7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emini, E. A., and W. C. Koff. 2004. AIDS/HIV. Developing an AIDS vaccine: need, uncertainty, hope. Science 304:1913-1914. [DOI] [PubMed] [Google Scholar]
- 27.Fiscus, S. A., E. B. Wallmark, J. D. Folds, J. Fryer, and C. M. van der Horst. 1991. Detection of infectious immune complexes in human immunodeficiency virus type 1 (HIV-1) infections: correlation with plasma viremia and CD4 cell counts. J. Infect. Dis. 164:765-769. [DOI] [PubMed] [Google Scholar]
- 28.Fouts, T. R., J. M. Binley, A. Trkola, J. E. Robinson, and J. P. Moore. 1997. Neutralization of the human immunodeficiency virus type 1 primary isolate JR-FL by human monoclonal antibodies correlates with antibody binding to the oligomeric form of the envelope glycoprotein complex. J. Virol. 71:2779-2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freed, E. O., and M. A. Martin. 2001. HIVs and their replication, p. 1971-2041. In D.M. Knipe et al. (ed.), Fields virology. Lippincott Williams and Wilkins, Philadelphia, Pa.
- 30.Heath, S. L., J. G. Tew, A. K. Szakal, and G. F. Burton. 1995. Follicular dendritic cells and human immunodeficiency virus infectivity. Nature 377:740-744. [DOI] [PubMed] [Google Scholar]
- 31.Herrera, C., C. Spenlehauer, M. S. Fung, D. R. Burton, S. Beddows, and J. P. Moore. 2003. Nonneutralizing antibodies to the CD4-binding site on the gp120 subunit of human immunodeficiency virus type 1 do not interfere with the activity of a neutralizing antibody against the same site. J. Virol. 77:1084-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holl, V., S. Hemmerter, R. Burrer, S. Schmidt, A. Bohbot, A. M. Aubertin, and C. Moog. 2004. Involvement of Fc gamma RI (CD64) in the mechanism of HIV-1 inhibition by polyclonal IgG purified from infected patients in cultured monocyte-derived macrophages. J. Immunol. 173:6274-6283. [DOI] [PubMed] [Google Scholar]
- 33.Kolchinsky, P., E. Kiprilov, and J. Sodroski. 2001. Increased neutralization sensitivity of CD4-independent human immunodeficiency virus variants. J. Virol. 75:2041-2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuznetsov, Y. G., J. G. Victoria, W. E. Robinson, Jr., and A. McPherson. 2003. Atomic force microscopy investigation of human immunodeficiency virus (HIV) and HIV-infected lymphocytes. J. Virol. 77:11896-11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwong, P. D., M. L. Doyle, D. J. Casper, C. Cicala, S. A. Leavitt, S. Majeed, T. D. Steenbeke, M. Venturi, I. Chaiken, M. Fung, H. Katinger, P. W. Parren, J. Robinson, D. Van Ryk, L. Wang, D. R. Burton, E. Freire, R. Wyatt, J. Sodroski, W. A. Hendrickson, and J. Arthos. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678-682. [DOI] [PubMed] [Google Scholar]
- 36.Kwong, P. D., R. Wyatt, Q. J. Sattentau, J. Sodroski, and W. A. Hendrickson. 2000. Oligomeric modeling and electrostatic analysis of the gp120 envelope glycoprotein of human immunodeficiency virus. J. Virol. 74:1961-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labrijn, A. F., P. Poignard, A. Raja, M. B. Zwick, K. Delgado, M. Franti, J. Binley, V. Vivona, C. Grundner, C. C. Huang, M. Venturi, C. J. Petropoulos, T. Wrin, D. S. Dimitrov, J. Robinson, P. D. Kwong, R. T. Wyatt, J. Sodroski, and D. R. Burton. 2003. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J. Virol. 77:10557-10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lifson, J. D., J. L. Rossio, M. Piatak, Jr., J. Bess, Jr., E. Chertova, D. K. Schneider, V. J. Coalter, B. Poore, R. F. Kiser, R. J. Imming, A. J. Scarzello, L. E. Henderson, W. G. Alvord, V. M. Hirsch, R. E. Benveniste, and L. O. Arthur. 2004. Evaluation of the safety, immunogenicity, and protective efficacy of whole inactivated simian immunodeficiency virus (SIV) vaccines with conformationally and functionally intact envelope glycoproteins. AIDS Res. Hum. Retrovir. 20:772-787. [DOI] [PubMed] [Google Scholar]
- 39.Lusso, P., P. L. Earl, F. Sironi, F. Santoro, C. Ripamonti, G. Scarlatti, R. Longhi, E. A. Berger, and S. E. Burastero. 2005. Cryptic nature of a conserved, CD4-inducible V3 loop neutralization epitope in the native envelope glycoprotein oligomer of CCR5-restricted, but not CXCR4-using, primary human immunodeficiency virus type 1 strains. J. Virol. 79:6957-6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maillard, A. P., and Y. Gaudin. 2002. Rabies virus glycoprotein can fold in two alternative, antigenically distinct conformations depending on membrane-anchor type. J. Gen. Virol. 83:1465-1476. [DOI] [PubMed] [Google Scholar]
- 41.Malvoisin, E., M. P. Kieny, and F. Wild. 1997. Self-association of truncated forms of HIV-1 gp120. Virus Res. 49:163-172. [DOI] [PubMed] [Google Scholar]
- 42.Mariani, R., G. Rutter, M. E. Harris, T. J. Hope, H. G. Krausslich, and N. R. Landau. 2000. A block to human immunodeficiency virus type 1 assembly in murine cells. J. Virol. 74:3859-3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maruyama, T., P. W. Parren, A. Sanchez, I. Rensink, L. L. Rodriguez, A. S. Khan, C. J. Peters, and D. R. Burton. 1999. Recombinant human monoclonal antibodies to Ebola virus. J. Infect. Dis. 179(Suppl. 1):S235-239. [DOI] [PubMed] [Google Scholar]
- 44.McCune, J. M., L. B. Rabin, M. B. Feinberg, M. Lieberman, J. C. Kosek, G. R. Reyes, and I. L. Weissman. 1988. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell 53:55-67. [DOI] [PubMed] [Google Scholar]
- 45.McMichael, A. J., and T. Hanke. 1999. Is an HIV vaccine possible? Nat. Med. 5:612-614. [DOI] [PubMed] [Google Scholar]
- 46.Moore, J., J. McKeating, R. Weiss, and Q. Sattentau. 1990. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 250:1139-1142. [DOI] [PubMed] [Google Scholar]
- 47.Morrow, W. J., M. Wharton, R. B. Stricker, and J. A. Levy. 1986. Circulating immune complexes in patients with acquired immune deficiency syndrome contain the AIDS-associated retrovirus. Clin. Immunol. Immunopathol. 40:515-524. [DOI] [PubMed] [Google Scholar]
- 48.Muster, T., F. Steindl, M. Purtscher, A. Trkola, A. Klima, G. Himmler, F. Ruker, and H. Katinger. 1993. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 67:6642-6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193-199. [DOI] [PubMed] [Google Scholar]
- 50.Nyambi, P. N., M. K. Gorny, L. Bastiani, G. van der Groen, C. Williams, and S. Zolla-Pazner. 1998. Mapping of epitopes exposed on intact human immunodeficiency virus type 1 (HIV-1) virions: a new strategy for studying the immunologic relatedness of HIV-1. J. Virol. 72:9384-9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Owens, R. J., and R. W. Compans. 1990. The human immunodeficiency virus type 1 envelope glycoprotein precursor acquires aberrant intermolecular disulfide bonds that may prevent normal proteolytic processing. Virology 179:827-833. [DOI] [PubMed] [Google Scholar]
- 52.Pancera, M., and R. Wyatt. 2005. Selective recognition of oligomeric HIV-1 primary isolate envelope glycoproteins by potently neutralizing ligands requires efficient precursor cleavage. Virology 332:145-156. [DOI] [PubMed] [Google Scholar]
- 53.Parren, P. W., D. R. Burton, and Q. J. Sattentau. 1997. HIV-1 antibody-debris or virion? Nat. Med. 3:366-367. [DOI] [PubMed] [Google Scholar]
- 54.Poignard, P., M. Moulard, E. Golez, V. Vivona, M. Franti, S. Venturini, M. Wang, P. W. Parren, and D. R. Burton. 2003. Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J. Virol. 77:353-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richman, D. D., T. Wrin, S. J. Little, and C. J. Petropoulos. 2003. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 100:4144-4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanders, R. W., M. Venturi, L. Schiffner, R. Kalyanaraman, H. Katinger, K. O. Lloyd, P. D. Kwong, and J. P. Moore. 2002. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J. Virol. 76:7293-7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanders, R. W., M. Vesanen, N. Schuelke, A. Master, L. Schiffner, R. Kalyanaraman, M. Paluch, B. Berkhout, P. J. Maddon, W. C. Olson, M. Lu, and J. P. Moore. 2002. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 76:8875-8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sattentau, Q. J., S. Zolla-Pazner, and P. Poignard. 1995. Epitope exposure on functional, oligomeric HIV-1 gp41 molecules. Virology 206:713-717. [DOI] [PubMed] [Google Scholar]
- 59.Scanlan, C. N., R. Pantophlet, M. R. Wormald, E. Ollmann Saphire, R. Stanfield, I. A. Wilson, H. Katinger, R. A. Dwek, P. M. Rudd, and D. R. Burton. 2002. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of α1->2 mannose residues on the outer face of gp120. J. Virol. 76:7306-7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schagger, H., W. A. Cramer, and G. von Jagow. 1994. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217:220-230. [DOI] [PubMed] [Google Scholar]
- 61.Schulke, N., M. S. Vesanen, R. W. Sanders, P. Zhu, M. Lu, D. J. Anselma, A. R. Villa, P. W. Parren, J. M. Binley, K. H. Roux, P. J. Maddon, J. P. Moore, and W. C. Olson. 2002. Oligomeric and conformational properties of a proteolytically mature, disulfide-stabilized human immunodeficiency virus type 1 gp140 envelope glycoprotein. J. Virol. 76:7760-7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharon, M., N. Kessler, R. Levy, S. Zolla-Pazner, M. Gorlach, and J. Anglister. 2003. Alternative conformations of HIV-1 V3 loops mimic beta hairpins in chemokines, suggesting a mechanism for coreceptor selectivity. Structure (Camb). 11:225-236. [DOI] [PubMed] [Google Scholar]
- 63.Smith-Franklin, B. A., B. F. Keele, J. G. Tew, S. Gartner, A. K. Szakal, J. D. Estes, T. C. Thacker, and G. F. Burton. 2002. Follicular dendritic cells and the persistence of HIV infectivity: the role of antibodies and Fc gamma receptors. J. Immunol. 168:2408-2414. [DOI] [PubMed] [Google Scholar]
- 64.Starcich, B. R., B. H. Hahn, G. M. Shaw, P. D. McNeely, S. Modrow, H. Wolf, E. S. Parks, W. P. Parks, S. F. Josephs, R. C. Gallo, et al. 1986. Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell 45:637-648. [DOI] [PubMed] [Google Scholar]
- 65.Trkola, A., T. Ketas, V. N. Kewalramani, F. Endorf, J. M. Binley, H. Katinger, J. Robinson, D. R. Littman, and J. P. Moore. 1998. Neutralization sensitivity of human immunodeficiency virus type 1 primary isolates to antibodies and CD4-based reagents is independent of coreceptor usage. J. Virol. 72:1876-1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trkola, A., A. B. Pomales, H. Yuan, B. Korber, P. J. Maddon, G. P. Allaway, H. Katinger, C. F. Barbas 3rd, D. R. Burton, D. D. Ho, et al. 1995. Cross-clade neutralization of primary isolates of human immunodeficiency virus type 1 by human monoclonal antibodies and tetrameric CD4-IgG. J. Virol. 69:6609-6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trkola, A., M. Purtscher, T. Muster, C. Ballaun, A. Buchacher, N. Sullivan, K. Srinivasan, J. Sodroski, J. P. Moore, and H. Katinger. 1996. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70:1100-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Usonis, V., S. Meriste, V. Bakasenas, I. Lutsar, F. Collard, M. Stoffel, and N. Tornieporth. 2005. Immunogenicity and safety of a combined hepatitis A and B vaccine administered concomitantly with either a measles-mumps-rubella or a diphtheria-tetanus-acellular pertussis-inactivated poliomyelitis vaccine mixed with a Haemophilus influenzae type b conjugate vaccine in infants aged 12-18 months. Vaccine 23:2602-2606. [DOI] [PubMed] [Google Scholar]
- 69.Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes, X. Wu, J. F. Salazar-Gonzalez, M. G. Salazar, J. M. Kilby, M. S. Saag, N. L. Komarova, M. A. Nowak, B. H. Hahn, P. D. Kwong, and G. M. Shaw. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307-312. [DOI] [PubMed] [Google Scholar]
- 70.Willey, R. L., M. A. Martin, and K. W. Peden. 1994. Increase in soluble CD4 binding to and CD4-induced dissociation of gp120 from virions correlates with infectivity of human immunodeficiency virus type 1. J. Virol. 68:1029-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wyatt, R., and J. Sodroski. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884-1888. [DOI] [PubMed] [Google Scholar]
- 72.Yamshchikov, G. V., G. D. Ritter, M. Vey, and R. W. Compans. 1995. Assembly of SIV virus-like particles containing envelope proteins using a baculovirus expression system. Virology 214:50-58. [DOI] [PubMed] [Google Scholar]
- 73.Zhu, P., E. Chertova, J. Bess, Jr., J. D. Lifson, L. O. Arthur, J. Liu, K. A. Taylor, and K. H. Roux. 2003. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 100:15812-15817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zwick, M. B., A. F. Labrijn, M. Wang, C. Spenlehauer, E. O. Saphire, J. M. Binley, J. P. Moore, G. Stiegler, H. Katinger, D. R. Burton, and P. W. Parren. 2001. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol. 75:10892-10905. [DOI] [PMC free article] [PubMed] [Google Scholar]