

Abstract
Mammals harbour multiple enzymes capable of excising uracil from DNA, although their distinct physiological roles remain uncertain. One of them (UNG) plays a critical role in antibody gene diversification, as UNG deficiency alone is sufficient to perturb the process. Here, we show this unique requirement for UNG does not reflect the fact that other glycosylases are unable to access the U:G lesion. SMUG1, if overexpressed, can partially substitute for UNG to assist antibody diversification as judged by its effect on somatic hypermutation patterns (in both DT40 B cells and mice) as well as a restoration of isotype switching in SMUG-transgenic msh2−/−ung−/− mice. However, SMUG1 plays little natural role in antibody diversification because (i) it is diminishingly expressed during B-cell activation and (ii) even if overexpressed, SMUG1 more appears to favour conventional repair of the uracil lesion than assist diversification. The distinction between UNG and overexpressed SMUG1 regarding the balance between antibody diversification and non-mutagenic repair of the U:G lesion could reflect the association of UNG (but not SMUG1) with sites of DNA replication.
Keywords: antibody diversification, DNA deamination, SMUG1, UNG, uracil-DNA glycosylase
Introduction
Although uracil is not a canonical component of DNA, it can arise in DNA either by spontaneous deamination of cytosine or by polymerase-catalysed misincorporation of dUTP (instead of dTTP) opposite a dA residue during replication (Lindahl, 2000; Krokan et al, 2002). Mammals harbour several enzymes with uracil-DNA glycosylase activity. Thus, UNG, SMUG1, TDG and MBD4 can all excise uracil from DNA as judged by in vitro assays (Krokan et al, 2002). Although the physiological roles of these glycosylases are not fully defined, current evidence suggests that UNG and SMUG1 are the major enzymes responsible for the repair of the U:G mispairs caused by spontaneous cytidine deamination, whereas uracil arising in DNA through dUTP misincorporation is mainly dealt with by UNG alone (Nilsen et al, 2000; Kavli et al, 2002, 2005). In contrast, MBD4 (and possibly TDG) appear to have a more specialised function in correcting T:G mismatches that arise from deamination of 5-methylC (especially in a CpG context) (Millar et al, 2002).
Uracil is also introduced into DNA as part of the physiological programme of antibody gene diversification where the B-cell-specific protein AID (activation-induced deaminase) acts to trigger somatic hypermutation or gene conversion of IgV genes as well as switch recombination at the IgC by targeted deamination of deoxycytidine residues to deoxyuridine within the immunoglobulin locus (reviewed in Neuberger et al, 2003). Here, antibody gene diversification depends on the resulting U:G lesion not being faithfully repaired: in fact, genetic evidence reveals that the UNG uracil-DNA glycosylase (which is conventionally associated with faithful repair) is actually co-opted to assist the diversification process. Thus, uracil removal by UNG is critical to antibody diversification as UNG deficiency is sufficient to perturb somatic hypermutation, gene conversion and switch recombination in a dramatic way, suggesting that none of the other enzymes with uracil excision activity provides a significant backup for UNG regarding its function in the antibody diversification process (Di Noia and Neuberger, 2002, 2004; Rada et al, 2002, 2004; Imai et al, 2003; Kavli et al, 2005).
These findings raise two questions. First, why do the other uracil-DNA glycosylases not provide backup activity for UNG in antibody gene diversification? This is particularly intriguing in the case of SMUG1, as it can apparently act as a backup for UNG in repair of the U:G lesions generated by spontaneous cytidine deamination (Nilsen et al, 2001; An et al, 2005). Furthermore, biochemical assays reveal that when UNG is depleted, SMUG1 contributes most of the residual uracil-excising activity in eukaryotic cell extracts with that due to TDG and MBD4 being scarcely detectable (Nilsen et al, 2001; Kavli et al, 2005). Second, how is it that UNG acts on the AID-generated U:G lesion to assist antibody gene diversification when the normal function of UNG is to repair U:G lesions by excising the U and allowing it to be replaced by a C using the G residue on the opposite strand as template (Lindahl, 2000)?
We have previously found that the enforced overexpression of SMUG1 does not restore detectable in vitro class switching in UNG-deficient mouse B lymphocytes (Rada et al, 2004). This could either indicate that SMUG1 cannot access the AID-generated U:G lesion in the Ig locus or that it can access it but does not do so in a way that potentiates effective switch recombination. Here, we provide evidence that although SMUG1 is normally downregulated as compared to UNG during B-cell activation, overexpressed SMUG1 can access the AID-generated U:G lesions in the Ig locus to potentiate antibody gene diversification. However, it does so less effectively than UNG and acts largely to diminish antibody gene diversification, presumably through enhancing repair. That handling by SMUG1 appears to favour repair whereas diversification is normally achieved through UNG could well reflect that UNG (unlike SMUG1) is associated with the replication fork (Otterlei et al, 1999; Akbari et al, 2004) and would therefore likely encounter the AID-generated uracil on the template strand, suggesting a model to explain the mutagenic outcome of UNG-mediated uracil excision at the immunoglobulin locus.
Results
Transfected SMUG1 affects IgV mutation spectrum but reduces mutation load in DT40 cells
SMUG1 is able to excise uracil from both single- and doubled-stranded DNA in vitro as judged by biochemical assays (Kavli et al, 2002). Furthermore, siRNA-mediated knockdown of SMUG1 expression in mouse embryo fibroblasts leads to increased C:G → T:A transition mutations, suggesting that endogenous SMUG1 can access and repair uracil in chromosomal DNA in mouse cells in vivo (An et al, 2005). We were therefore surprised by our recent finding (Rada et al, 2004) that enforced overexpression of SMUG1 does not restore efficient in vitro switching by UNG-deficient mouse splenic B cells. The observation suggested to us that either (i) SMUG1 is, for some unidentified reason, unable to gain access to the AID-generated U:G lesion in the immunoglobulin switch region, (ii) that is able to gain access but cannot do so in such a way as to potentiate switch recombination or (iii) that it can potentiate switch recombination but does so at an efficiency that scored below our sensitivity of detection.
To gain some insight into the possible explanation, we extended our studies to a different system and asked whether transfected SMUG1 could affect the pattern of IgV gene diversification in chicken DT40 cells. Parental DT40 cells diversify their IgV genes constitutively during in vitro culture through a gene conversion process that is triggered by AID-dependent cytidine deamination (Buerstedde et al, 1990; Arakawa et al, 2002; Harris et al, 2002). The DT40X2U derivative cell line carries targeted inactivation of the xrcc2 locus causing it instead to perform IgV gene somatic hypermutation at high frequency (Sale et al, 2001). DT40X2U cells also express a transfected Ugi cassette (which encodes a protein inhibitor of UNG; Friedberg et al, 1975; Wang and Mosbaugh, 1988) rendering them deficient in uracil excision and thereby causing the IgV gene mutations to be largely restricted to nucleotide transitions at C:G pairs, as the abasic site needed for the production of transversions is no longer generated (Di Noia and Neuberger, 2002).
Previous studies monitoring the ability of cell extracts to excise uracil from oligonucleotide substrates have revealed that, unlike the extracts from mouse and human cells, which have been examined where endogenous SMUG1 provides a backup for UNG activity (Nilsen et al, 2001; Kavli et al, 2005), DT40 cells exhibit little if any UNG-backup uracil excision activity (Di Noia and Neuberger, 2002, 2004). We have observed the same apparent lack of backup activity in other chicken cell lines and in chicken primary lymphoid tissue (not shown), suggesting that this lack of UNG-backup activity is not peculiar to DT40 but likely a characteristic of the species. Backup activity can, however, be provided by transfection of chicken DTX2U cells with a human SMUG1 expression cassette, with these transfectants exhibiting a ratio of SMUG1 to UNG activity similar to that observed in mammalian cell lines (Figure 1A).
Figure 1.
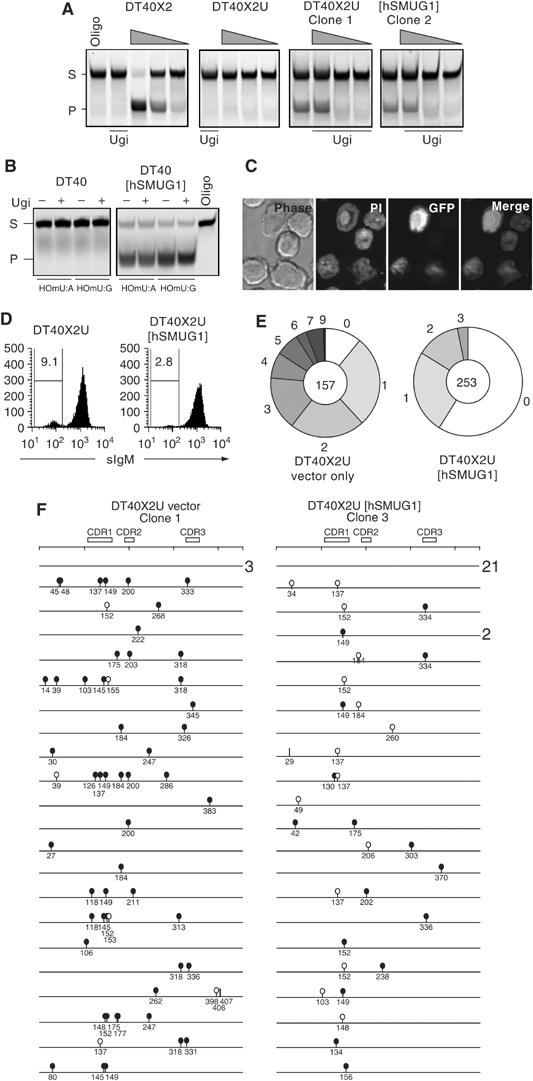
Expression of hSMUG1 in DT40 alters the mutation spectrum and reduces mutation accumulation. (A) Comparison of uracil excision activity in extracts of XRCC2-deficient derivatives of DT40 (DT40X2) that express Ugi (DT40X2U) or Ugi together with human SMUG1 (DT40X2U[hSMUG1]). Uracil excision activity is reflected by cleavage of the fluorescently labelled 42-mer U-containing oligonucleotide substrate (S) into a 26-nucleotide product (P). The gel was loaded with samples containing either oligonucleotide substrate alone (oligo) or following incubation with serial five-fold dilutions of extract (wedges). Extracts were preincubated on ice for 20 min with Ugi where indicated. (B) Assay for 5-hydroxymethyl uracil (HOmU) excision activity in extracts of DT40 transfectants expressing hSMUG1 as compared to controls. Activity was measured using double-stranded HOmU-containing oligonucleotide substrates in which the HOmU residue was paired with either A or G: extracts were preincubated with exogenous Ugi where indicated. (C) Confocal images of DT40 cells expressing an hSMUG1-GFP fusion protein: cellular DNA was stained with propidium iodide (PI). (D) Flow cytometric analysis of surface IgM expression in one representative DT40X2U[hSMUG1] and one vector-only-transfected DT40X2U control clone that had been expanded for 10 weeks following single cell cloning. The proportions of sIgM negatives (%) are indicated, with the results from multiple clones summarised in Table II. (E) Pie chart depicting the proportion of IgVλ sequences bearing the indicated number of mutations. The data from four DT40X2U[hSMUG1] and three DT40X2U control clone have been separately pooled. The total number of sequences in each pool is indicated at the centre of the pie. (F) Schematic depiction of the nature of the identified IgVλ mutations. All the 43 sequences obtained from one representative DT40X2U[hSMUG1] clone are shown (22 mutated; 21 unmutated) together with the first 24 sequences (21 mutated; three unmutated) from a vector-only control clone. Numbers at the right indicate when the same sequence was found more than once. Transition mutations at C:G are indicated by black lollipops, transversion mutations at C:G by white lollipops and mutations at A:T pairs by vertical lines.
SMUG1 from all species so far examined (but not UNG) is able to excise 5-hydroxy-me-U from DNA both in vitro and in vivo (Boorstein et al, 1992, 2001; Kavli et al, 2002; Ulbert et al, 2002). Consistent with their lack of SMUG1, DT40 cells lack 5-hydroxy-me-U-excising activity. However, introduction of the human SMUG1 expression vector confers 5-hydroxy-me-U excision activity, with the transfected SMUG1 being localised to the nucleus, as judged by use of an hSMUG1-GFP fusion gene (Figure 1B and C).
Although DT40X2U [hSMUG1] cells showed indistinguishable growth curves from the mock-transfected controls, we noticed that they consistently (in four independent transfectants) gave rise to a lower proportion of surface IgM-loss variants following clonal expansion (Figure 1D and Table I). Such loss variants are caused by mutation of the IgV genes, suggesting that hSMUG1 expression had reduced the mutation frequency. Sequencing the IgVλ genes amplified from sorted IgM-loss variants supported this interpretation. Thus, the hSMUG1 transfectants gave a higher proportion of unmutated sequences (despite comparable efficiencies of sorting for sIgM loss) and a lower mutation load than the DTX2U control clones (averaging 1.4 versus 2.6 mutations per mutated sequence; Figure 1E and F and Table I). It is also striking that the hSMUG1 transfectants exhibited a major shift in the mutation pattern. Although both hSMUG1 and control transfectants gave rise to mutations that were nearly exclusively at C:G pairs, 89% of these mutations in the controls were nucleotide transitions (consistent with previous work; Di Noia and Neuberger, 2002), but this dropped to only 45% in the DTX2U [hSMUG1] transfectants (Figure 1F and Table I).
Table 1.
IgVλ mutations in [hSMUG1] and control transfectants in DT40X2U
| Cells |
IgVλ sequencesa |
Mutation typeb |
Mutation load | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Expansion (week) | % sIgM− | Total (n) | Mutated (%) | Total (n) | Independent (n) | Transversions at C:G (n) | % Tvnsc | Mutations/mutated sequence | |
| hSMUG1 transfectant | |||||||||
| c1 | 8 | 0.5 | 87 | 26 | 32 | 19 | 18 | 72 | 1.4 |
| c2 | 8 | 2.5 | 43 | 51 | 32 | 23 | 22 | 46 | 1.5 |
| c3 | 10 | 6 | 57 | 28 | 20 | 15 | 15 | 53 | 1.3 |
| c4 | 10 | 2.8 | 67 | 78d | 73 | 20 | 18 | 50 | 1.4 |
| Pooled | 46 | 77 | 73 | 55 | 1.4 | ||||
| Vector-only transfectant | |||||||||
| c1 | 8 | 9.1 | 48 | 88 | 102 | 57 | 54 | 13 | 2.4 |
| c2 | 8 | 9 | 47 | 87 | 97 | 56 | 56 | 9 | 2.4 |
| c3 | 8 | 6 | 62 | 92 | 179 | 76 | 67 | 12 | 3.1 |
| Pooled | 89 | 189 | 177 | 11 | 2.6 | ||||
| aSequences obtained from subpopulations (2%) sorted as giving the dullest staining with anti-IgM. | |||||||||
| bAll the independent 266 mutational events observed in this study were single nucleotide substitutions except for six possible gene conversions and four deletions. | |||||||||
| cThe percentage of substitutions at C:G base pairs that are nucleotide transversions. | |||||||||
| dOne jackpot mutation in this clone was present in most of the sequences obtained. | |||||||||
The large increase in transversion mutations at C:G pairs provides good evidence that transfected hSMUG1 is able to excise the uracil produced by AID-mediated deamination at the IgVλ locus in DT40 cells. Nonetheless, the diminished mutation accumulation suggests that hSMUG1 not only acts to alter the pattern of IgV diversification but also to diminish the degree of mutation, possibly by stimulating repair.
SMUG1 overexpression partially restores isotype switching in msh2−/−ung−/− mice
The evidence from transfected DT40 cells that SMUG1 is able to access the AID-generated U:G lesion obviously led us to ask why UNG-deficient mouse B cells had exhibited a class-switching defect as judged by in vitro assays with neither endogenous mouse SMUG1 nor transgenically overexpressed SMUG1 apparently acting to compensate for the UNG deficiency (Rada et al, 2004).
A possible explanation relates to the sensitivity of the in vitro assay. We previously noted that, in UNG-deficient mice, there is an MSH2-dependent pathway of isotype switching, which is readily evident from analysis of serum immunoglobulin titres but is less apparent from in vitro switching assays (Rada et al, 2002, 2004). However, in order to use serum analysis as a test for the ability of overexpressed SMUG1 to potentiate switching, it was necessary to cross the SMUG1 transgene into an msh2−/−ung−/− double knockout background, as the high background of serum IgG in ung−/− single knockouts would otherwise render the assay insensitive.
The results were dramatic. Serum ELISA as well as SDS–PAGE analysis of total serum immunoglobulin that had been purified by use of Protein L revealed that the hSMUG1 transgene triggered the production of considerable quantities of IgG1 and IgG3 in msh2−/−ung−/− double knockout mice (Figure 2A and B). Consistent with previous findings (Rada et al, 2004), very little IgG is detectable in the sera of non-transgenic msh2−/−ung−/− double knockout controls (with the somewhat higher backgrounds detected in younger mice being likely largely attributable to maternal immunoglobulin). However, in the presence of the SMUG1 transgene, greatly elevated IgG1 and IgG3 levels are detected, although these levels are still several fold below those present in control animals.
Figure 2.
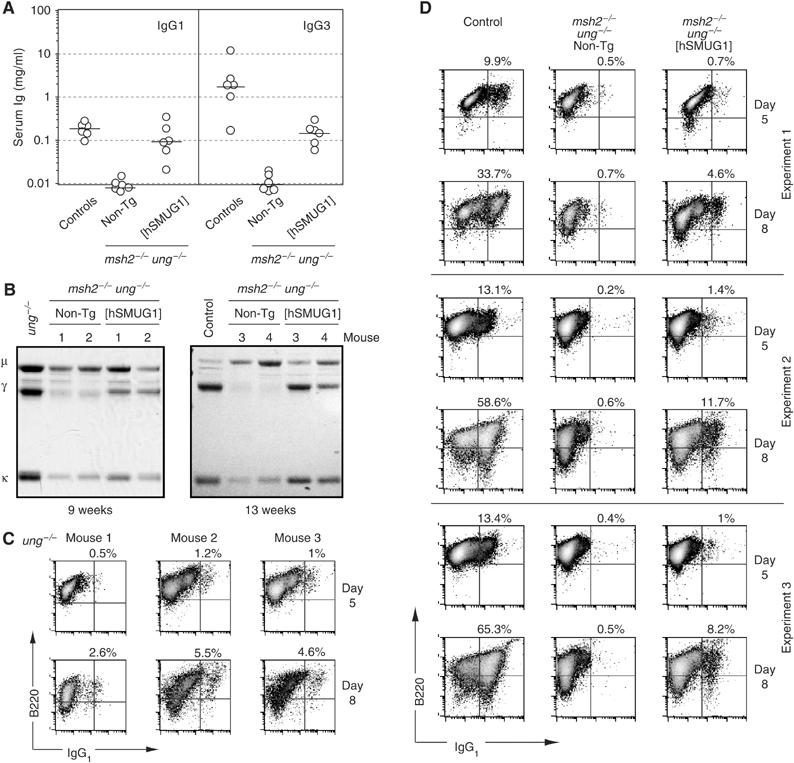
SMUG1 overexpression in msh2−/−ung−/− mice allows the accumulation of substantial levels of serum IgG. (A) Titres of IgG1 and IgG3 in sera of mice of different genotypes (aged 9–20 weeks) as determined by ELISA. The mice analysed were the products of crosses between msh2+/−ung−/− [hSMUG1] transgenic males and msh2+/−ung−/− non-transgenic females, with age-matched C57BL/6 mice providing controls. (B) Serum immunoglobulin profiles of four msh2−/−ung−/− control and four msh2−/−ung−/− [hSMUG1] transgenic mice analysed at the ages indicated. Representative examples of sera from age-matched normal and ung−/− mice are shown for comparison. Total serum immunoglobulin was precipitated using protein L-agarose, resolved by SDS–PAGE and stained with Coomassie blue. (C) Flow cytometric profiles of three independent experiments in which splenic B cells from UNG-deficient mice were cultured in the presence of LPS+IL4 for 5 or 8 days prior to staining for sIgG1 and CD45R(B220). Live cells were gated by staining with propidium iodide, and percentages of sIgG1+ B cells are indicated. (D) Flow cytometric profiles of three independent experiments in which splenic B cells from msh2−/−ung−/− [hSMUG1]-transgenic, msh2−/−ung−/− control or wild-type mice were cultured in the presence of LPS+IL4 for 5 or 8 days prior to staining for sIgG1 and CD45R(B220). Live cells were gated and percentages indicated as in panel C.
These findings led us to revisit the in vitro switching assay and ask whether it would be possible to detect UNG-independent switching by analysing the cultures after a longer period of culture with LPS+IL4. We found that by extending the period of in vitro culture from 5 to 8 days, significant switching could indeed be detected in the cultures from the ung−/− single knockout mice (Figure 2C) as well as in the culture from the hSMUG1-transgenic msh2−/−ung−/− double knockout mice, but not from the non-transgenic msh2−/−ung−/− double knockout controls (Figure 2D). These results indicate that the in vivo switching (as judged by serum IgG) that can be achieved in UNG-deficient mice through either the MSH2-dependent backup pathway or through enforced SMUG1 expression correlates with in vitro switching (as judged by the production of sIgG1+ cells during culture of spleen cells with LPS+IL4). It is, however, notable that a relatively weak restoration of in vitro switching correlates with a more substantial restoration of serum IgG titres. Analogous observations have been made with other mutant mouse lines and, as discussed previously (Rada et al, 2004), may well reflect in vivo selection for switched cells.
SMUG1 is downregulated during B-cell activation in normal mice
The observation that the hSMUG1 transgene restores significant amounts of serum IgG to msh2−/−ung−/− mice clearly reveals a distinction between endogenous and transgenic SMUG1. A likely explanation seemed that endogenous SMUG1 is expressed at too low a level to potentiate switching. Indeed, we have previously found that SMUG1 mRNA is poorly expressed in mouse lymphoid tissues as compared to heart, skeletal muscle, kidney and liver (Rada et al, 2004). However, activated B cells do display detectable Ugi-resistant uracil excision activity (Rada et al, 2004). This backup activity would appear to be largely due to SMUG1, as a combination of Ugi (to inhibit UNG) and the PSM1 anti-SMUG1 neutralising antibody (Kavli et al, 2002) eliminated nearly all detectable uracil excision activity in B-cell extracts (Figure 3A). Interestingly, though, while UNG activity increased following activation of resting B cells with LPS+IL4 (consistent with the association between UNG and DNA replication), SMUG1 activity actually diminished (Figure 3B). Thus, as judged by these types of assay, the ratio of UNG:SMUG1 activity increased from 4:1 to >50:1 following B-cell activation (Figure 3C). We have not discriminated nuclear from mitochondrial UNG in our assays. Given that nuclear UNG is practically absent from resting cells (Otterlei et al, 1999; Caradonna and Muller-Weeks, 2001; Imai et al, 2003; Fischer et al, 2004), the actual increase in the ratio of nuclear UNG:SMUG1 during B-cell activation is likely to be even more pronounced.
Figure 3.
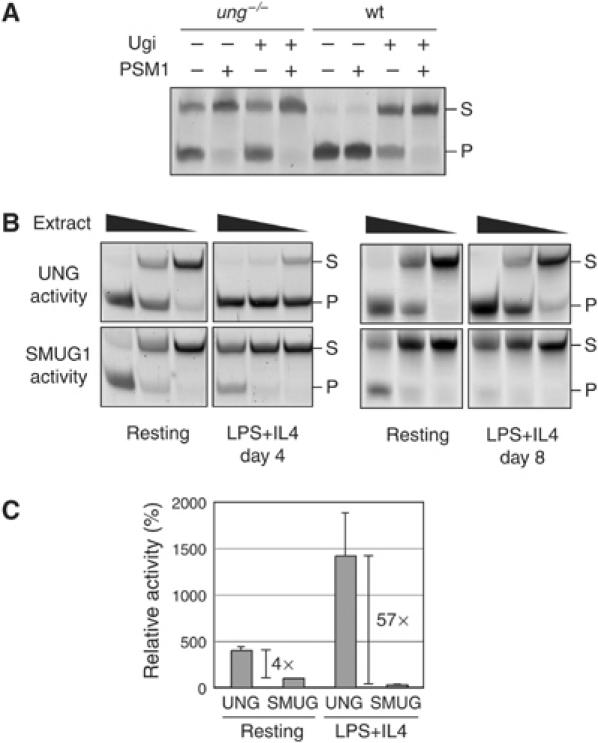
Comparison of SMUG1 and UNG uracil excision activity during in vitro B-cell activation. (A) Uracil excision activity in whole cell extracts of splenic B cells from either wild-type or ung−/− mice following activation for 4 days with LPS+IL4 was assayed using a 42-mer double-stranded oligonucleotide substrate. Where indicated, the extracts were incubated for 20 min on ice with the UNG inhibitor Ugi or with the PSM1 anti-SMUG1 antibody before adding the substrate. (B) Comparison of uracil excision activity in serial five-fold dilutions of whole cell extracts prepared from splenic B cells of a wild-type mouse after 0, 4 or 8 days of incubation with LPS+IL4. UNG activity was determined by performing the uracil excision assay in the presence of the PSM1 anti-SMUG1 antibody, whereas SMUG1 activity was determined by performing the assay in the presence of Ugi. (C) Quantitation of the relative levels of UNG and SMUG1 activity in splenic B cell extracts prepared after 0 or 4 days of incubation with LPS+IL4. The histogram is based on the results of four pairs of mice analysed as in panel B. UNG and SMUG1 activities were quantified by gel densitometry using the IQ software (Molecular Dynamics) and expressed as mean percentages (+s.d.) normalising the values to those of SMUG1 in resting B cells as 100%.
SMUG1 overexpression reduces the extent of switching and mutation in normal mice
The results indicate that SMUG1 is poorly expressed in activated mouse B cells but can, if overexpressed, access the AID-generated U:G lesion to potentiate class switching although it does not fully restore switching. However, our results with DT40 cells had raised the possibility that while SMUG1 can assist antibody diversification (as witnessed by the increase in transversion mutations in UNG-deficient DT40), it might often access the AID-generated U:G lesion to channel it into a repair pathway rather than potentiate antibody diversification.
We therefore wondered whether overexpressed SMUG1 might act similarly in mouse B cells—while able to assist diversification, acting more frequently to potentiate conventional repair. Thus, in an animal that is unable to switch (owing to deficiency in both UNG and MSH2), one would be well placed to detect the (albeit imperfect) switch-potentiating effect of SMUG1 overexpression. However, in a UNG-proficient wild-type background where switching and mutation occur normally, one might expect the repair activity of overexpressed SMUG1 to become apparent and lead to a consequent reduction in antibody gene diversification. This is indeed what is observed. Following in vitro incubation with LPS+IL4, splenic B cells from hSMUG1-transgenic UNG-proficient animals consistently exhibited an approximate three-fold reduction in switching to IgG1 as compared to B cells from non-transgenic littermate controls (Figure 4A). The B cells from the transgenic mice were indistinguishable from those of their non-transgenic littermates with respect to surface markers (not shown) as well as in their proliferative and blasting response to LPS (Figure 4B and Supplementary Figure 3A–C).
Figure 4.
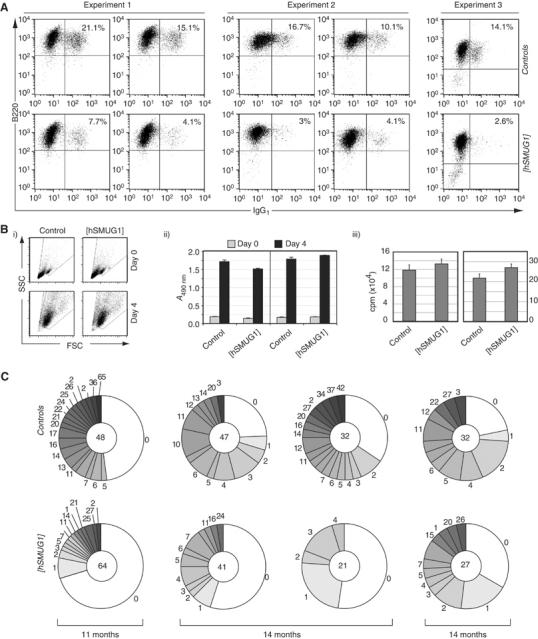
The hSMUG1 transgene inhibits in vitro switching and diminishes in vivo mutation accumulation in UNG-proficient mice. (A) Flow cytometric profiles of purified B cells from hSMUG1-transgenic and control littermates stained for surface IgG1 and CD45R(B220) after 5 days of culture in the presence of LPS+IL4. The percentages of sIgG1-positive cells are indicated. (B) Splenic B cells from [hSMUG1]-transgenic and control littermates give indistinguishable blasting and proliferative responses to LPS as judged by (i) scatter profiles of cell size and shape, (ii) metabolic activity as judged by reduction of MTS tetrazolium substrate and (iii) DNA synthesis as judged by [3H]thymidine incorporation monitored at day 4 of culture. (C) Mutation accumulation in the 3′-flanking region of VDJH rearrangements in Peyer's patch germinal centre B cells as analysed from four pairs of hSMUG1-transgenic ung+/+ versus control non-transgenic ung+/+ littermates of various ages. The pie charts indicate the proportion of sequences carrying the indicated number of mutations, with the total number of sequences analysed for each mouse indicated in the centre of the corresponding pie. Repeat counting of the same mutation was avoided by randomly eliminating from the analysis all but one of the sequences in each data set that shared the same VDJH rearrangement: this entailed removing 20 sequences from the database of 179 control sequences and 21 from the database of 184 sequences from transgenic animals. Insertions/deletions were found at similar frequencies in both groups (totalling 21 in the control and 20 in transgenic databases).
SMUG1 overexpression affects the IgV mutation spectrum in msh2−/−ung−/− mice
Sequence analysis of the IgVH 3′-intronic flank in germinal centre B cells from Peyer's patches also revealed that the presence of the SMUG1 transgene resulted in a substantial depression of mutation accumulation (Figure 4C). Thus, in all littermate pairs of UNG-proficient mice analysed, the hSMUG1-transgenic animal revealed a diminished mutation accumulation compared to its littermate control whether judged by the percentage of sequences that carried mutations (averaging 43% in SMUG1 transgenics versus 62% in controls) or by the mean number of mutations per mutated sequence (averaging 7.2 in hSMUG1 transgenics versus 12.7 in controls) (Table II). A similar effect of the hSMUG1 transgene on the depression of mutation accumulation was seen when analysed in both ung−/− and msh2−/−ung−/− knockout backgrounds (Table II and Supplementary Figure 1A). Consistent with the lack of impairment of B-cell proliferation (Figure 4B), the depression in mutation accumulation did not correlate with any alteration in the size or appearance of germinal centres (Supplementary Figure 3D). The presence of the hSMUG1 transgene also had no detectable effect on the distribution of mutations along the target sequence (Supplementary Figure 1B) and, despite the lower mutational load, the major hotspots were still conserved, with a major proportion of the mutations conforming to the hypermutation consensuses (75% of G:C mutations at WRC; 48% of A:T mutations at WA in hSMUG1-trasngenic msh2−/−ung−/− mice).
Table 2.
Pattern of VH 3′-flanking mutations in SMUG1-transgenic mice
| Mice | n | Age (months) | Sequences |
Mutationsa |
||||
|---|---|---|---|---|---|---|---|---|
| Total (n) | Mutated (%) | Total (n) | % at G:C | % Tvnsb | % at A:T | |||
| ung+/+ | ||||||||
| Control | 4 | 11–14 | 149 | 62 | 1169 | 42 | 47 | 57 |
| hSMUG1 | 4 | 11–14 | 153 | 43 | 468 | 40 | 46 | 60 |
| ung−/− | ||||||||
| Control | 3 | 9 | 214 | 66 | 1363 | 50 | 7 | 43 |
| hSMUG1 | 1 | 9 | 37 | 40 | 112 | 40 | 11 | 60 |
| hSMUG1 | 2 | 3.5 | 90 | 17 | 27 | 55 | 20 | 45 |
| msh2−/−/ung−/− | ||||||||
| Control | 2 | 5–6 | 148 | 65 | 520 | 98 | 0.2 | 2 |
| hSMUG1 | 3 | 5 | 232 | 61 | 100 | 75 | 28 | 25 |
| aThe stringent criteria described by Rada et al (2004) were used to avoid repeat counting of mutations in dynastically related sequences. | ||||||||
| bThe percentage of substitutions at C:G base pairs that are nucleotide transversions. | ||||||||
Although these results are consistent with the proposal that overexpression of SMUG1 in normal mice leads to a depression of mutation accumulation by increasing faithful repair of the initiating U:G lesion, one would expect in the light of our previous results that SMUG1 overexpression might restore some transversion mutations at C:G pairs in msh2−/−ung−/− mice. This is the case. Indeed, not only is there a restoration of transversions at C:G pairs in [hSMUG1] msh2−/−ung−/− transgenics, there is also some restoration of mutations at A:T pairs (Table II). Furthermore, while somatic mutation is msh2−/−ung−/− mice is not accompanied by the occasional deletions/insertions seen in normal mice (presumably reflecting the lack of induction of error-prone repair pathways), SMUG1 overexpression leads to a restoration of such aberrant events as witnessed by the identification of six insertion/deletion mutations.
Thus, overexpression of SMUG1 in msh2−/−ung−/− mice, while causing an overall depression of mutation (presumably through increased faithful repair), also leads to an alteration in the mutation pattern. This likely reflects that the SMUG1-generated abasic site can (like the UNG-generated abasic site; Rada et al, 2002, 2004) act as a template for the production of transversions at C:G pairs as well as a lesion that triggers (albeit somewhat poorly) mutation creation at A:T pairs. Interestingly, the presence of the SMUG1 transgene in ung−/− MSH2-proficient mice, while leading to the expected depression of mutation accumulation, gave only a mild increase in transversions at C:G pairs (Table II). We suspect that, in this background, MSH2 may compete with SMUG1 for the processing of the U:G lesion. In any case, the results provide clear evidence that overexpressed SMUG1 can access and excise the uracil from the immunoglobulin locus.
Discussion
Mice simultaneously deficient in both UNG and MSH2 exhibit a near-complete block of immunoglobulin class-switch recombination and a restriction of somatic hypermutation to transition substitutions at C:G pairs (Rada et al, 2004). It therefore appears that in the absence of UNG and MSH2, no other uracil excision enzyme or DNA repair protein accesses the AID-generated U:G lesion with significant efficacy in order to assist antibody gene diversification. Here, we have asked why the SMUG1 uracil-DNA glycosylase does not normally substitute for UNG and mask the phenotypic effects of UNG deficiency. We show that SMUG1 is indeed able to gain access to the AID-generated U:G lesion but is normally insufficiently expressed in activated B cells. Furthermore, in contrast to endogenous UNG, the overexpressed SMUG1 appears to act in large part to assist non-mutagenic repair of the U:G lesion. This gives possible insight into the mechanism by which UNG-mediated excision of the AID-generated uracil in the immunoglobulin locus normally leads to antibody diversification rather than non-mutagenic repair.
The results concerning SMUG1 expression are consistent with previous observations that UNG activity considerably exceeds that of SMUG1 in extracts of proliferating cells from different tissue types in man and mouse (Nilsen et al, 2000, 2001; Kavli et al, 2002, 2005; An et al, 2005). The difference in their relative activities does, however, appear to be particularly marked in lymphoid cells. Thus, we have previously noted that, at the RNA level, SMUG1 is particularly poorly expressed in spleen cells (Rada et al, 2004): it is clear from this work that SMUG1 actually diminishes in abundance relative to UNG during B-cell activation. We do not believe that this decrease in SMUG1:UNG ratio on cell blasting is peculiar to B cells, having made similar findings with concanavalin A-stimulated T lymphocytes (data not shown). The alteration in SMUG1:UNG ratio may in large part reflect a general difference between resting and dividing cell populations. Thus, UNG is known to increase in cycling cells and be associated with replication foci (Otterlei et al, 1999; Caradonna and Muller-Weeks, 2001; Imai et al, 2003; Fischer et al, 2004). In contrast, SMUG1 activity is not notably increased in proliferative tissues (Nilsen et al, 2001) and SMUG1 levels have been found to remain constant during the cell cycle in a human cell line (Kavli et al, 2002). The decrease in SMUG1:UNG ratio during B-cell activation may, however, be beneficial for antibody diversification.
The fact that msh2−/−ung−/− mice have such dramatic phenotypes regarding somatic hypermutation pattern and isotype switching despite exhibiting low but readily detectable endogenous SMUG1 activity strongly suggests that SMUG1 must normally play little if any role in antibody gene diversification. It could be argued that this is because SMUG1 is not able to see uracil in DNA in the context of the Ig locus. However, the results obtained here tell us that the enzyme is able to excise this uracil from chromosomal DNA in vivo, attacking the same lesion as UNG. This is not a trivial observation. Although biochemical assays have revealed that mammals harbour multiple enzymes that can excise uracil from oligonucleotide substrates in vitro, it does not follow that these enzymes actually access and excise uracil from chromosomal DNA in vivo. Indeed, the reason for the multiplicity of uracil excision enzymes is something of a mystery. With regard to SMUG1, genetic evidence certainly indicates that it acts in vivo to repair 5-hydroxymethyl uracil in DNA (Boorstein et al, 2001). The results here indicating that SMUG1 also works in vivo on uracil arising in DNA through endogenous dC deamination are consistent with assumptions based on the in vitro properties of SMUG1, the absence of a strong mutator phenotype in UNG-deficient mice and with the increased frequency of transition mutations at C:G observed following knockdown of SMUG1 expression in mouse embryo fibroblasts (Nilsen et al, 2001; An et al, 2005; Kavli et al, 2005).
The results reveal that the effects on antibody gene diversification of deficiency in one uracil excision enzyme (UNG) can be substantially overcome by enforced expression of another, essentially unrelated uracil excision enzyme (SMUG1). In an msh2−/−ung−/− background, SMUG1 overexpression can lead to a partial restoration of class switching as well as of transversions at C:G pairs, of substitutions at A:T pairs and somatic mutation-associated aberrant events. This provides further support to the contention that UNG functions in antibody diversification through its ability to recognise uracil rather than by virtue of some other distinctive feature of UNG such as, for example, a UNG-specific protein–protein interaction as suggested by Begum et al (2004).
The question nevertheless arises as to why enforced expression of SMUG1 does not entirely overcome the effects of deficiency in UNG. There could be multiple explanations. Thus, Kavli et al (2005) have shown that uracil can be efficiently excised from single-stranded DNA by UNG2 but only poorly by SMUG1, a distinction that could be critical if antibody gene diversification required the glycosylase to work on a single-stranded DNA substrate. Equally, UNG and SMUG1 could also differ in the efficiency with which they are recruited to the AID-generated uracil as well as in the efficacy with which they bias resolution of the U:G lesion towards antibody diversification rather than non-mutagenic repair.
Although SMUG1 overexpression in msh2−/−ung−/− mice gave a partial rescue of immunoglobulin class switching, enforced SMUG1 overexpression in normal mice led to diminished switching. Furthermore, SMUG1 overexpression led to a reduction in IgV gene mutation accumulation during somatic hypermutation (whether in a normal, ung−/− or msh2−/−ung−/− background) and a reduction in mutation accumulation was also seen in SMUG1-transfected DT40 cells. The mechanism by which SMUG1 acts to reduce antibody diversification in these different systems remains to be identified. However, given that SMUG1-overexpressing cells do not exhibit compromised viability or proliferation, we favour the interpretation that the overexpressed SMUG1 will preferentially act on the AID-generated U:G lesion to initiate conventional base excision repair.
Why then the difference between SMUG1 and UNG? As UNG (but not SMUG1) is regulated in a cell-cycle-dependent manner and is associated with replication foci (Otterlei et al, 1999; Caradonna and Muller-Weeks, 2001; Imai et al, 2003; Fischer et al, 2004), one can well imagine a model (Figure 5) in which UNG molecules at the replication fork encounter uracil residues located on the DNA template strand that have been generated (at some earlier time) by AID-mediated deoxycytidine deamination. Given its location on the template strand (in contrast to uracil arising through dUTP misincorporation, which would be on the newly synthesised strand), the abasic site generated by UNG-mediated excision might then preferentially act as a target for translesion synthesis rather than repair. In contrast, SMUG1 is not concentrated at replication foci and is likely to encounter the AID-generated U:G lesion in double-stranded DNA at another stage of the cell cycle: SMUG1 might therefore preferentially have conventional faithful (rather than mutation fixation) repair role. Conversely, consistent with the fact that UNG deficiency does not increase the IgV mutation load in chicken DT40 or mouse Peyer's patch B cells (Di Noia and Neuberger, 2002, 2004; Rada et al, 2002), UNG (by virtue of its cell cycle regulation) may be poorly available at this time and thereby play a lesser role in conventional repair of the U:G lesion. This proposal fits well with recent suggestions by Nilsen et al (2000, 2001) that SMUG1 has evolved in higher eukaryotes to excise the uracil generated by spontaneous dC deamination, whereas UNG has a major role at replication foci to repair uracil arising through dUTP incorporation.
Figure 5.
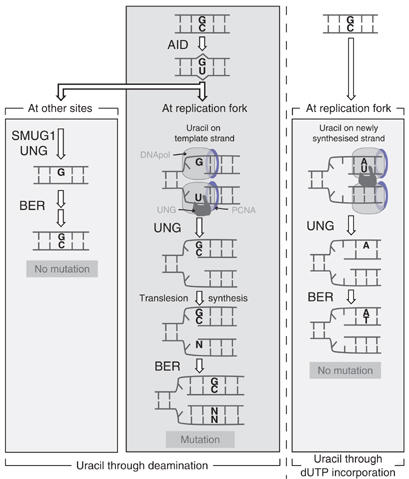
Model to suggest how the preferential association of UNG (but not SMUG1) with sites of DNA replication could bias resolution of the AID-generated uracil towards antibody diversification rather than non-mutagenic repair. Consistent with what is known about UNG localisation, it is envisaged that the major site at which UNG encounters the AID-generated uracil in the immunoglobulin locus is at the replication fork, where it will be located on the DNA template strand. If presented in a single-stranded form, the uracil will be a good substrate for UNG (although not, as noted by Kavli et al (2005) for SMUG1), whereas the resultant abasic site if also largely embedded in a single-stranded DNA structure would not constitute a suitable substrate for AP-endonuclease attack. This abasic site would presumably stall the replication fork and recruit proteins of the recombinational repair or translesion synthesis pathways, thereby triggering resolution by gene conversion or somatic mutation respectively, whereas polymerase stalling could trigger isotype switching through interlocus genomic rearrangement with a suitably presented downstream S region. This situation (central panel) is contrasted with what is likely to occur when uracil generated through deamination is encountered away from the replication fork (left-hand panel). Here, it is envisaged that SMUG1 or UNG would excise the uracil and trigger conventional base excision repair (BER). Similarly, if uracil arises through polymerase-catalysed dUTP incorporation (right-hand panel), the UNG at the replication fork will encounter the uracil on the newly synthesised DNA strand and trigger BER. Thus, the mutagenic outcome of uracil excision during antibody diversification is seen as being a consequence of uracil being encountered at the replication fork on the DNA template strand: the preferential association of UNG with replication forks is seen as being a main reason that UNG is the major glycosylase associated with the process. Endogenous or overexpressed SMUG1 associated with the fork might be able to trigger diversification but if SMUG1 or UNG encounters the uracil at other stages, their action would likely favour non-mutagenic repair.
In conclusion, both UNG and overexpressed SMUG1 can access the AID-generated U:G lesion in the immunoglobulin locus, but with UNG largely (but probably not exclusively) acting to potentiate antibody diversification and SMUG1 largely (but not exclusively) biases towards faithful repair. We propose that the different consequences of uracil excision by the two glycosylases reflect that UNG (but not SMUG1) is preferably associated with replication foci (Figure 5). Excision of uracil from the DNA template strand at the replication fork could also explain why such excision is mutagenic (rather than triggering faithful repair as one would expect in conventional base excision repair). It will be interesting to test whether UNG association with replication foci is essential for its contribution to antibody diversification.
Materials and methods
Mice and cell lines
Transgenic mice expressing human SMUG1 under the control of the mouse H2-Kk promoter and IgH intronic enhancer (which show a 25-fold increase over controls in SMUG1 enzymatic activity in splenocytes) as well as msh2−/−ung−/− double knockout mouse line have been previously described (Rada et al, 2004). The expression of hSMUG1 in lymphoid tissues produced no alterations in the abundance of B and T cells, as judged by flow cytometry with anti-CD45R(B220) and anti-CD3, nor in the relative proportions of CD4+ and CD8+ T cells (not shown).
The chicken B-cell line DT40CL18CL4 (an sIgM+ subclone of DT40CL18) is described by Sale et al (2001) as is its XRCC2-deficient derivative DT40X2. The Ugi-expressing transfectant of DT40X2 (designated DT40X2U here) is described by Di Noia and Neuberger (2002).
Expression of human SMUG1 in DT40
The open reading frame encoding human SMUG1 was PCR-amplified from IMAGE cDNA clone 726197 (MRC Geneservice, Cambridge, UK) using oligonucleotides TGGACATATGCCCCAGGCTTT and CCCCAAGGGCACTCATTTCAA, cloned into pCR-Blunt II TOPO (Invitrogen) and then subcloned as an EcoRI fragment into pIRES-Neo3 (BD Clontech). Chicken DT40 cells were transfected with the PvuI-linearised DNA of the resultant plasmid and G418-resistant clones were screened for Ugi-resistant uracil excision activity. Four clones showing such activity were selected and expanded.
For the subcellular localisation experiment, SMUG1 was PCR-amplified using the oligonucleotides GGAATTCTGGACATATGCCCCAGGCTT and CGGGATCCTTTCAACAGCAGTGGCAGCA, cloned into the EcoRI/BamHI sites of pEGFP-N3 (Clontech) and transfected into DT40. Cells from two GFP+ G418R clones (that exhibited SMUG1 activity) were incubated on slides coated with poly-L-lysine, fixed with 4% paraformaldehyde and analysed by confocal microscopy using propidium iodide to stain DNA.
Uracil excision assays
Uracil excision activity in whole cell extracts of DT40 and mouse B cells was measured essentially as described (Di Noia and Neuberger, 2002) except that the oligonucleotide CATAAAGTG(HOmU)AAAGCCTGG-FITC with a single 5′-hydroxymethyl deoxyuridine (HOmU) was annealed to CCAGGCTTTACACTTTATG or CCAGGCTTTGCACTTTATG and used as a substrate for the measurement of HOmU excision activity. Where indicated, extracts were preincubated on ice for 20 min with recombinant Ugi (NE Biolabs) and/or the PSM1 anti-SMUG1 antibody (a kind gift from Professor G Slupphaug, Institute of Cancer Research and Molecular Biology, Trondheim, Norway). UDG activity in resting versus activated mouse B cells was measured in extracts from normal mice splenocytes fractionated by CD43+ depletion using an AutoMacs device and anti-CD43 magnetic beads (Miltenyi Biotec). CD43-negative cells were >93% B cells (CD45R(B220)+) and >90% were arrested in G1 phase of the cell cycle as judged by propidium iodide staining. A fraction of these cells was stimulated to proliferate with LPS+IL4 for 4 days before processing for activity measurements, as described by Rada et al (2004).
Analysis of class switching
Serum immunoglobulin isotypes were quantified by ELISA (Rada et al, 2002) and immunoglobulin profiles were evaluated by Protein L-agarose precipitation followed by SDS–PAGE and staining with Coomassie blue (Rada et al, 2004). In vitro switch recombination to IgG1 was evaluated in samples of purified B cells that had been cultured for 4 days with LPS+IL4 as previously described (Rada et al, 2004). Cell blasting and proliferation following incubation with LPS was assayed by flow cytometric analysis of cell scatter and by monitoring incorporation of [3H]thymidine as previously described (Rada et al, 2002). The colorimetric assay for cell metabolic activity was carried out by measuring reduction of MTS tetrazolium CellTiter96 substrate (Promega) according to the manufacturer's instructions.
Analysis of mutation
The IgVλ region from DT40 cell populations was amplified by PCR and sequenced as described previously (Di Noia and Neuberger, 2002). Sequence determination of the JH4-proximal intronic region from sorted germinal centre B cells from Peyer's patches is described by Jolly et al (1997).
Supplementary Material
Supplementary Fig. 1
Supplementary Fig. 2
Supplementary Fig. 3
Acknowledgments
We are indebted to Sarah Davies and to Theresa Langford and her staff for assistance with animal care and Richard Grenfell for assistance with cell sorting. This work was in part supported by a grant from the Leukaemia Research Fund.
References
- Akbari M, Otterlei M, Pena-Diaz J, Aas PA, Kavli B, Liabakk NB, Hagen L, Imai K, Durandy A, Slupphaug G, Krokan HE (2004) Repair of U/G and U/A in DNA by UNG2-associated repair complexes takes place predominantly by short-patch repair both in proliferating and growth-arrested cells. Nucleic Acids Res 32: 5486–5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Q, Robins P, Lindahl T, Barnes DE (2005) C → T mutagenesis and gamma-radiation sensitivity due to deficiency in the Smug1 and Ung DNA glycosylases. EMBO J 24: 2205–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa H, Hauschild J, Buerstedde J M (2002) Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science 295: 1301–1306 [DOI] [PubMed] [Google Scholar]
- Begum NA, Kinoshita K, Kakazu N, Muramatsu M, Nagaoka H, Shinkura R, Biniszkiewicz D, Boyer LA, Jaenisch R, Honjo T (2004) Uracil DNA glycosylaser activity is dispensable for immunoglobulin class switch. Science 305: 1160–1163 [DOI] [PubMed] [Google Scholar]
- Boorstein RJ, Chiu LN, Teebor GW (1992) A mammalian cell line deficient in activity of the DNA repair enzyme 5-hydroxymethyluracil-DNA glycosylase is resistant to the toxic effects of the thymidine analog 5-hydroxymethyl-2′-deoxyuridine. Mol Cell Biol 12: 5536–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorstein RJ, Cummings A Jr, Marenstein DR, Chan MK, Ma Y, Neubert TA, Brown SM, Teebor GW (2001) Definitive identification of mammalian 5-hydroxymethyluracil DNA N-glycosylase activity as SMUG1. J Biol Chem 276: 41991–41997 [DOI] [PubMed] [Google Scholar]
- Buerstedde J M, Reynaud CA, Humphries EH, Olson W, Ewert DL, Weill JC (1990) Light chain gene conversion continues at high rate in an ALV-induced cell line. EMBO J 9: 921–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caradonna S, Muller-Weeks S (2001) The nature of enzymes involved in uracil-DNA repair: isoform characteristics of proteins responsible for nuclear and mitochondrial genomic integrity. Curr Protein Pept Sci 2: 335–347 [DOI] [PubMed] [Google Scholar]
- Di Noia J, Neuberger MS (2002) Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 419: 43–48 [DOI] [PubMed] [Google Scholar]
- Di Noia JM, Neuberger MS (2004) Immunoglobulin gene conversion in chicken DT40 cells largely proceeds through an abasic site intermediate generated by excision of the uracil produced by AID-mediated deoxycytidine deamination. Eur J Immunol 34: 504–508 [DOI] [PubMed] [Google Scholar]
- Fischer JA, Muller-Weeks S, Caradonna S (2004) Proteolytic degradation of the nuclear isoform of uracil-DNA glycosylase occurs during the S phase of the cell cycle. DNA Repair 3: 505–513 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Ganesan AK, Minton K (1975) N-glycosidase activity in extracts of Bacillus subtilis and its inhibition after infection with bacteriophage PBS2. J Virol 16: 315–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Sale JE, Petersen-Mahrt SK, Neuberger MS (2002) AID is essential for immunoglobulin V gene conversion in a cultured B cell line. Curr Biol 12: 435–438 [DOI] [PubMed] [Google Scholar]
- Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, Ochs HD, Fischer A, Durandy A (2003) Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol 4: 1023–1028 [DOI] [PubMed] [Google Scholar]
- Jolly C, Klix N, Neuberger MS (1997) Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic Acids Res 25: 1913–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Andersen S, Otterlei M, Liabakk NB, Imai K, Fischer A, Durandy A, Krokan HE, Slupphaug G (2005) B cells from hyper-IgM patients carrying UNG mutations lack ability to remove uracil from ssDNA and have elevated genomic uracil. J Exp Med 201: 2011–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G (2002) hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem 277: 39926–39936 [DOI] [PubMed] [Google Scholar]
- Krokan HE, Drablos F, Slupphaug G (2002) Uracil in DNA—occurrence, consequences and repair. Oncogene 21: 8935–8948 [DOI] [PubMed] [Google Scholar]
- Lindahl T (2000) Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat Res 462: 129–135 [DOI] [PubMed] [Google Scholar]
- Millar CB, Guy J, Samson OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A (2002) Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 297: 403–405 [DOI] [PubMed] [Google Scholar]
- Neuberger MS, Harris RS, Di Noia J, Petersen-Mahrt SK (2003) Immunity through DNA deamination. Trends Biochem Sci 28: 305–312 [DOI] [PubMed] [Google Scholar]
- Nilsen H, Haushalter KA, Robins P, Barnes DE, Verdine GL, Lindahl T (2001) Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J 20: 4278–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE (2000) Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell 5: 1059–1065 [DOI] [PubMed] [Google Scholar]
- Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakko O, Krokan HE (1999) Post-replicative base excision repair in replication foci. EMBO J 18: 3834–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Di Noia JM, Neuberger MS (2004) Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the a/t-focused phase of somatic mutation. Mol Cell 16: 163–171 [DOI] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS (2002) Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol 12: 1748–1755 [DOI] [PubMed] [Google Scholar]
- Sale JE, Calandrini DM, Takata M, Takeda S, Neuberger MS (2001) Ablation of XRCC2/3 transforms immunoglobulin V gene conversion into somatic hypermutation. Nature 412: 921–926 [DOI] [PubMed] [Google Scholar]
- Ulbert S, Cross M, Boorstein RJ, Teebor GW, Borst P (2002) Expression of the human DNA glycosylase hSMUG1 in Trypanosoma brucei causes DNA damage and interferes with J biosynthesis. Nucleic Acids Res 30: 3919–3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Mosbaugh DW (1988) Uracil-DNA glycosylase inhibitor of bacteriophage PBS2: cloning and effects of expression of the inhibitor gene in Escherichia coli. J Bacteriol 170: 1082–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1
Supplementary Fig. 2
Supplementary Fig. 3