

Abstract
The biophysical characterization of nonfunctional protein aggregates at physiologically relevant temperatures is much needed to gain deeper insights into the kinetic and thermodynamic relationships between protein folding and misfolding. Dynamic and static laser light scattering have been employed for the detection and detailed characterization of apomyoglobin (apoMb) soluble aggregates populated at room temperature upon dissolving the purified protein in buffer at pH 6.0, both in the presence and absence of high concentrations of urea. Unlike the β-sheet self-associated aggregates previously reported for this protein at high temperatures, the soluble aggregates detected here have either α-helical or random coil secondary structure, depending on solvent and solution conditions. Hydrodynamic diameters range from 80 to 130 nm, with semiflexible chain-like morphology. The combined use of low pH and high urea concentration leads to structural unfolding and complete elimination of the large aggregates. Even upon starting from this virtually monomeric unfolded state, however, protein refolding leads to the formation of severely self-associated species with native-like secondary structure. Under these conditions, kinetic apoMb refolding proceeds via two parallel routes: one leading to native monomer, and the other leading to a misfolded and heavily self-associated state bearing native-like secondary structure.
INTRODUCTION
Most biologically active proteins function as soluble monomers or low order self-associated species, e.g., dimers or tetramers. However, under special circumstances such as unusual solution conditions, chemical modification, effector binding or presence of a mutation, proteins and polypeptides misbehave in solution and give rise to the formation of higher order aggregates. In living organisms this phenomenon has severe consequences, leading to a number of deadly neurodegenerative disorders, including Alzheimer's (1), Huntington's (2,3) and Parkinson's disease (4). Protein aggregation is also a well-known problem in protein production and biotechnology. For instance, upon in vivo production of recombinant proteins, co- or posttranslational aggregation often leads to the formation of heavily insoluble inclusion bodies, which are notoriously hard to handle. Inclusion body solubilization may be difficult and require high concentrations of denaturing agent such as urea and guanidine hydrochloride (5). Upon removal of denaturing agent, refolding of proteins to their correct three-dimensional conformation competes with aggregation and precipitation, reducing the overall yields of properly folded products (6). Protein storage at high concentrations often favors aggregation, resulting in structural changes and loss of activity. This is an issue of concern in both basic research and in production of proteins for pharmaceutical applications (7). Despite the ongoing efforts in the area of protein misfolding and self-association, a coherent picture of the underlying causes and mechanisms for this process is still missing to date. This is due, in part, to the lack of accurate characterization of the aggregates in terms of size, morphology and degree of polydispersity. Therefore, it is extremely important to systematically analyze the types of aggregates formed under different conditions, for a given set of target model proteins. This will equip us with the necessary tools to improve our understanding of the underlying causes for the aggregation process.
Apomyoglobin (apoMb) is a well characterized α-helical globular protein that has been extensively employed as a model system for protein folding and stability studies (8–13). Hydrogen exchange pulse labeling and circular dichroism-detected stopped-flow refolding revealed the presence of a molten globular kinetic folding intermediate comprising the A, G, and H helices (10) and bearing some degree of conformational heterogeneity (14). Similarly, a thermodynamic intermediate was isolated under moderately acidic conditions (pH 4) and identified by tryptophan fluorescence, Fourier transform infrared and NMR spectroscopy (15). This molten globular intermediate appears almost as globular as the native state when probed by dynamic light scattering and x-ray scattering (16,17). The backbone dynamics of the acid- and denaturant-unfolded states have been studied by NMR (18,19). Fast temperature-induced unfolding of apoMb core formation was studied by laser-induced temperature jump, with detection by circular dichroism, Fourier transform infrared, and fluorescence spectroscopy (20). A phase diagram for the pH and ionic strength dependence of horse heart apoMb conformation has been constructed by Goto and co-workers (21).
In all the above studies, no appreciable amount of self-associated species has been reported, either in the unfolded, native, or partially folded states. This is likely because the techniques employed in most of these investigations were not able to specifically detect the presence of large soluble aggregates. In some cases, samples were depleted of some or all of the heavily aggregated species by filtration, Sephadex resin-based spin column treatment, and(or) traditional size exclusion chromatography, before data acquisition (13,17,20). Turbidity measurements, which detect the presence of macroscopic (1 μm or larger) aggregates, have shown that horse heart apoMb forms heavily self-associated species at moderate urea concentrations (22), reaching a minimum solubility at 2.4 M. However, the turbidity measurements carried out in this study do not allow detection of smaller soluble aggregates nor characterization of particle morphology. Horse heart apoMb is known to form large insoluble irreversible aggregates at temperatures above 50°C (23). Under more extreme conditions, i.e., at high pH and temperature, the protein irreversibly self-associates and forms amyloid fibrils, containing a significant amount of β-sheet secondary structure (24,25). Specific sperm whale apoMb mutations, such as the replacement of Trp-7 and Trp-14 by two Phe's, give rise to amyloid fibrils at pH 7 (26).
In this study, we report the detection of large soluble aggregates formed by wild-type apoMb under various solution conditions at room temperature. Protein concentration has been kept in the low micromolar range, which is typical for most spectroscopic studies in solution. Unlike prior reports on full-length apoMb self-association (24,25), the soluble self-associated species detected here do not require nonphysiologically relevant conditions (e.g., high temperature) to occur. We have investigated both mild conditions (i.e., pH 6.0, no urea, room temperature), and relatively harsher solution environments, yet of common usage within the protein science laboratory setting (i.e., high urea concentration, low pH, no extreme temperatures). The aggregates formed under these conditions have neither β-sheet nor amyloid-like character, but display semiflexible chain morphology. At high urea concentration they are devoid of any significant secondary structure, while under refolding conditions at pH 6.0 they have predominantly α-helical character. In addition, we show that kinetic refolding starting from a virtually monomeric unfolded state leads to the formation of self-associated species with native-like secondary structure. Under these conditions, the refolding time course proceeds via two parallel routes: one leading to native monomer, and the other leading to a misfolded, heavily aggregated, and soluble state bearing some native-like structure.
MATERIALS AND METHODS
Sample preparation
Urea and sodium chloride were purchased from EM Science (Gibbstown, NJ). Sodium acetate was purchased from Aldrich (Milwaukee, WI). Wild-type sperm whale apoMb was expressed and purified as described (13). All buffer and urea stock solutions were filtered through 0.22-μm membranes (Millipore, Billerica, MA). Lyophilized powder containing pure protein was dissolved in 10 mM sodium acetate in the presence of 0.0, 6.0, or 8.0 M urea at pH 6.0. Samples in 6.0 M urea and 10 mM sodium acetate at pH 2.3 were prepared similarly. Samples to be used for size exclusion (SE) chromatography were prepared by dissolving the lyophilized protein powder in 10 mM sodium acetate buffer (pH 6.0) followed by injection. Fractions corresponding to monomeric species, based on SE calibration profile, were collected and concentrated using a Centricon YM-10 device (Millipore). Protein concentrations were determined by ultraviolet (UV) absorbance at 280 nm as described (13). Sample concentrations were 38 μM (0.0 M urea, pH 6.0), 31 μM (6.0 M urea, pH 6.0), 21 μM (8.0 M urea, pH 6.0), 157 μM (6.0 M urea, pH 2.3), and 21 μM (0.0 M urea, pH 6.0, for the eluates collected from size exclusion column chromatography). The high sample concentration in 6.0 M urea at pH 2.3 was necessary to achieve a sufficient signal/noise ratio in the light scattering measurements.
ApoMb was refolded by dissolving protein powder into 6.0 M urea and 100 mM sodium phosphate buffer at pH 2.3, followed by a 10-fold dilution into 100 mM sodium phosphate buffer at pH 6.4. The final pH was 6.0, and the final protein and urea concentrations were 12 μM and 0.6 M, respectively.
Dynamic light scattering
Samples were filtered (0.45-μm pore size) directly into light scattering cuvettes and immediately placed in a temperature-controlled chamber (25°C) containing the refractive index-matching solvent decahydronapthalene. Data were acquired by a Malvern 4700 light scattering system equipped with a Coherent (Santa Clara, CA) argon ion laser operating at 488 nm. Scattered light intensity was detected at 90° from the incident light beam. Translational diffusion causes fluctuations in scattered light intensity. The frequency of the fluctuations depends on molecular size and is typically analyzed by plotting the raw data as autocorrelation functions. Autocorrelation functions were analyzed using the method of cumulants, to yield a z-average translational diffusion coefficient, 〈D〉z, defined as
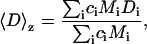 |
(1) |
where ci is the protein's mass concentration, Mi is the molecular weight, and Di is the translational diffusion coefficient, respectively, for species i. Upon substituting the experimentally measurable 〈D〉z into Stokes-Einstein's equation, one obtains the following simple correlation between the z-average translational diffusion coefficient and dsph
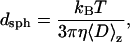 |
(2) |
where kB is the Boltzmann constant, T is the temperature (in Kelvin), η is the solvent viscosity, and dsph is the particle hydrodynamic diameter. Given the above assumptions, dsph represents the average hydrodynamic diameter of a hypothetical hard sphere with a translational diffusion coefficient identical to the measured value. The equation is formally correct only if the protein behaves as a point scatterer and if it undergoes no diffusional motions other than translational.
Dynamic light scattering data collection started ∼1.2 h after the solubilization of the lyophilized protein into buffer. This time interval accounts for solution mixing, pH adjustments, concentration determination and dynamic light scattering instrumental setup. The dead time for the refolding experiments was reduced to ∼10 min, since most of the sample characterization was performed on a previously prepared identical sample. The size distribution of the samples was further investigated by fitting the experimental autocorrelation profiles with the CONTIN algorithm (27).
Static light scattering
Static light scattering experiments were carried out by recording scattered light intensity at 24 angles from 20 to 135°. For each angle, five separate measurements were averaged. Scattered light intensities of reference buffer solutions and toluene were recorded at the same angles as the protein samples. The average scattering intensity of the buffer was subtracted from the sample scattering intensities. The resulting values were then normalized using the scattering intensity of toluene, serving as a reference solvent, to obtain the Rayleigh ratio Rs(q), which is defined as
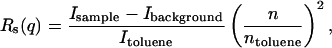 |
(3) |
where n is the refractive index of the buffer, ntoluene is the refractive index of toluene, Isample, Ibackground, and Itoluene are the scattered light intensities from the protein-containing solution, reference buffer and toluene, respectively. The Rayleigh ratio is a function of q, the scattering vector, defined as (4πn/λo)sin(θ/2), where λo is the laser excitation wavelength, and θ is the scattering angle. For dilute solutions, Rs(q) is related to the characteristic particle dimensions by
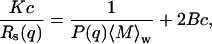 |
(4) |
where K is defined as 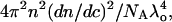 dn/dc is the refractive index increment with sample concentration c, NA is the Avogadro's number,
dn/dc is the refractive index increment with sample concentration c, NA is the Avogadro's number, 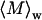 is the weight average molecular weight defined as
is the weight average molecular weight defined as 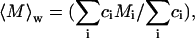 P(q) is the shape factor, and B is the thermodynamic second virial coefficient (28). A value of 0.181 cm3/g was used for dn/dc, which was assumed not to depend on solute aggregation state. For particles much smaller than λo (i.e., with a radius of gyration ≪ 488 nm, in our case), P(q) ∼ 1. For larger particles, constructive or destructive interference due to scattering from different parts of the target biomolecule takes place, and the scattered light intensity becomes a function of the scattering angle. For particles with
P(q) is the shape factor, and B is the thermodynamic second virial coefficient (28). A value of 0.181 cm3/g was used for dn/dc, which was assumed not to depend on solute aggregation state. For particles much smaller than λo (i.e., with a radius of gyration ≪ 488 nm, in our case), P(q) ∼ 1. For larger particles, constructive or destructive interference due to scattering from different parts of the target biomolecule takes place, and the scattered light intensity becomes a function of the scattering angle. For particles with 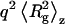 ∼ 1, the following relation applies
∼ 1, the following relation applies
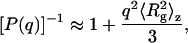 |
(5) |
where the z-average of the square radius of gyration 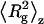 is defined as
is defined as
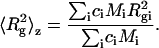 |
(6) |
Upon replacing Eq. 5 into Eq. 4, we get
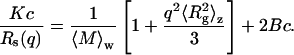 |
(7) |
This expression was employed for data analysis via Zimm plots, with Kc/Rs(q) plotted versus q2. Upon extrapolating q to zero, and assuming the “2Bc” term, which is mainly due to excluded-volume hard-core interaction effects, to be negligibly small, the y axis intercept and initial slope yield 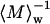 and
and 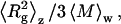 respectively. This enables determination of
respectively. This enables determination of 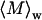 and average Rg, where Rg is the square root of
and average Rg, where Rg is the square root of 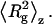 For species with
For species with 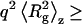 1.5, particle shape affects P(q). Therefore, both overall particle shapes and their associated characteristic dimensions can be determined. By plotting the data as q2Rs/Kc〈M〉w vs. q (Kratky plots), information on particle shape and characteristic dimension is derived (29,30). For results consistent with a semiflexible (worm-like) chain model, appropriate data fitting (31,32) enables determination of the Kuhn statistical segment length Lk, which provides a measure of chain stiffness, and the contour length Lc, which defines the total length of the extended chain.
1.5, particle shape affects P(q). Therefore, both overall particle shapes and their associated characteristic dimensions can be determined. By plotting the data as q2Rs/Kc〈M〉w vs. q (Kratky plots), information on particle shape and characteristic dimension is derived (29,30). For results consistent with a semiflexible (worm-like) chain model, appropriate data fitting (31,32) enables determination of the Kuhn statistical segment length Lk, which provides a measure of chain stiffness, and the contour length Lc, which defines the total length of the extended chain.
Size-exclusion chromatography
After completion of static light scattering data collection, a sample aliquot was injected into a Superdex 75 column (Amersham, Piscataway, NJ) on a LCC-500 FPLC system equipped with a LKB Uvicord SII detector operating at 226 nm. For each sample, an elution buffer of identical composition to the sample buffer was used, with additional 100 mM sodium chloride, which was added to minimize nonspecific interactions with the column. The size-exclusion column was calibrated with low molecular weight standards, i.e., ribonuclease A, 13.7 kDa; chymotrypsinogen A, 25 kDa; ovalbumin, 43 kDa; bovine serum albumin, 67 kDa (Amersham), and aprotinin, 6.5 kDa (USB, Cleveland, OH). Individual calibration curves were generated for each of the experimental conditions tested by size exclusion chromatography. The molecular weight of recombinant apoMb is 17,331 Da. Since large and misbehaved aggregates are prone to interacting with the column or being trapped by the filters, controls were performed by injecting all the samples into the identical FPLC setup in the absence of the column (i.e., system with only filters, tubing, etc.). This procedure enables assessing the extent of sample loss during size exclusion chromatography. The difference in the peak areas under both conditions yields the fraction of misbehaved protein originally present in the sample.
The samples refolded into pH 6.0 buffer from 6 M urea at pH 2.3 (1:10 dilution) were incubated at room temperature for 12 h before injection into the size-exclusion column.
Circular dichroism
Far-UV circular dichroism (CD) data were collected from aliquots of the samples used in the static light scattering experiments. CD measurements were performed on an MOS-450/AF-CD optical data collection system from Molecular Kinetics (original design by BioLogic, Claix, France); 0.1-cm pathlength quartz cuvettes were used. Data were collected as single scans (195–250 nm) with 1-nm step size and 20 s averaging time per point.
Electron microscopy
ApoMb-containing solutions were imaged using a JEOL 100CX transmission electron microscope (JEOL USA, Peabody, MA). Samples were stained with Nano-W (methylamine tungstate) negative stain (Nanoprobes, Yaphank, NY) and placed on a pioloform coating grid support film (Ted Pella, Redding, CA) before data analysis.
NMR spectroscopy
NMR data for refolded apoMb were collected on a 15N-labeled sample refolded from 6.0 M urea at pH 2.3 following identical procedures to those employed for dynamic light scattering. The final 15N-apoMb concentration was 50 μM. Sensitivity-enhanced 1H-15N heteronuclear single quantum correlation (HSQC) (33) data were collected on a Varian INOVA-600 MHz NMR spectrometer equipped with a Varian triple resonance 1H{13C, 15N} triple axis gradient probe. One-dimensional (1D) HSQC traces, collected by setting the 15N chemical shift evolution delay to zero, were acquired successively, starting from the approximate time of refolding initiation. Two-dimensional (2D) HSQC data acquisition was initiated 12 h after refolding; 296 transients were collected with 2000 complex points in the direct dimension. The relaxation delay was set to 1.8 s. The HSQC experiment included 96 complex t1 increments. An unshifted Gaussian function was applied to the time-domain data in both dimensions. Data were zero-filled twice. The NMRPipe (34) and NMRView (35) software packages were used for data processing.
RESULTS
Dynamic light scattering
The lyophilized protein powder was dissolved in 0.0, 6.0, and 8.0 M urea solutions at pH 6.0. As shown in Fig. 1, large aggregates are detected within the experimental dead time under all the above conditions, with dsph > 70 nm, compared to dsph of 4.2 and 8.6 nm for the apoMb native state and acid unfolded monomer, respectively (16). Over 14 h, dsph of the aggregates in 6.0 and 8.0 M urea did not change significantly whereas the dsph of species formed in 0.0 M urea increased slightly.
FIGURE 1.

ApoMb dynamic light scattering under different experimental conditions. (A) Time courses after protein solubilization in (▾) 10 mM sodium acetate buffer, 0.0 M urea, pH 6.0 (38 μM); (○) 10 mM sodium acetate buffer, 6.0 M urea, pH 6.0 (31 μM); (□) 10 mM sodium acetate buffer, 8.0 M urea, pH 6.0 (21 μM); (×) 10 mM sodium acetate buffer 0.0 M urea, pH 6.0, monomer peak collected during size-exclusion chromatography run (21 μM); (•)10 mM sodium acetate buffer, 6.0 M urea, pH 2.3 (157 μM). (B) ApoMb folding time course from 6.0 M urea and 100 mM sodium phosphate at pH 2.3. Refolding was initiated by 10-fold dilution into 100 mM sodium phosphate buffer at pH 6.4 (final pH 6.0). Final protein and urea concentrations were 12 μM and 0.6 M, respectively. Autocorrelation functions were analyzed by the cumulants method. Experimental dead times are not included in the plots.
To search for conditions where no aggregation is present, the lyophilized protein was dissolved in 10 mM sodium acetate (pH 6.0) and purified by size-exclusion chromatography. The collected peak for monomeric apoMb is characterized by a much smaller apparent hydrodynamic diameter, i.e., 18 nm (Table 1). This value is still high relative to the expected result for a globular native protein of the size of apoMb. We believe this is due to a small amount of aggregation during the protein concentration step following size-exclusion chromatography (see discussion of static light scattering data).
TABLE 1.
Static and dynamic light scattering analysis of apoMb aggregates
| Experimental Conditions* | Rg† (nm) | 〈M〉w† (kDa) | 〈M〉w‡ (kDa) | Lc‡ (nm) | Lk‡ (nm) | dsph§ (nm) | 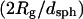 |
|---|---|---|---|---|---|---|---|
| 0 M urea,¶ pH 6.0 | ∼50 | ∼1500 | ∼1400 | ∼1200 | ∼100 | ∼130 | ∼0.8 |
| 6.0 M urea, pH 6.0 | 60 ± 10 | 2900 ± 700 | 2500 ± 400 | 1000 ± 100 | 150 ± 60 | 87 ± 5 | 1.3 ± 0.3 |
| 8.0 M urea, pH 6.0 | 80 ± 10 | 3300 ± 1100 | 2600 ± 750 | 1900 ± 1200 | 150 ± 70 | 78 ± 6 | 2.0 ± 0.5 |
| Peak collected from SE chromatography | – | 19 ± 3‖ | – | – | – | 18 ± 3 | – |
| 6.0 M urea, pH 2.3 | 14 ± 1‖ | 39 ± 1‖ | – | – | – | 14 ± 3 | – |
| Refolding from 6.0 M urea, pH 2.3 | – | Large | – | – | – | 500–2000 | – |
Errors refer to ± 1 SD from the mean for two or three independent experiments, unless otherwise stated.
Values obtained from initial slopes of Zimm plots (static light scattering data).
Molecular weight and characteristic dimensions; i.e., Kuhn statistical segment length Lk and contour length Lc, obtained from Kratky plot fits (static light scattering data) according to a semiflexible chain model.
Values obtained from dynamic light scattering data.
Data collected under these conditions were not reproducible after four independent runs; i.e., standard deviations from the mean are actually comparable to the mean itself. This is possibly due to heterogeneities within the lyophilized powder.
Errors from curve fitting.
Given the presence of large aggregates at high urea concentrations, i.e., under typical unfolding conditions, an even more strongly denaturing environment was explored. In 6 M urea and pH 2.3 the large aggregates are absent. Dynamic light scattering reveals an average apparent dsph of 14 nm (Table 1). CONTIN analysis shows a bimodal distribution with peaks characterized by dsph ∼8 and ∼20 nm. The smaller particle size is likely a monomer because this is similar to the reported value for apoMb's unfolded state at low pH (16). The slightly larger species is possibly a dimer or a small oligomer.
This virtually nonaggregated unfolded state was employed for apoMb refolding experiments. Folding was initiated by 10-fold dilution of apoMb in 6.0 M urea and pH 2.3 into pH 6.4 buffer. The final solution pH was 6.0. Refolding from this unfolded state leads to formation of extremely large aggregates that increase in size over time (Fig. 1 B). This supramolecular size is larger than that of any other species observed in this work. A significant overall dsph increase, from 14 (i.e., the unfolded state apparent dsph) to 400 nm (i.e., burst phase dsph value in Fig. 1 B), takes place in a fast process occurring within the dead time of the measurement, i.e., 10 min from refolding initiation. The slowest kinetic phase, nearly complete in 10 h, is adequately fit by a single exponential (Fig. 1 B), yielding a rate constant of 0.29 ± 0.03 h−1. This phase leads to a further increase in apparent dsph up to ∼2 μm.
Static light scattering
To further characterize the size and structure of the detected apoMb aggregates, static light scattering measurements were collected immediately after dynamic light scattering data collection. Data were analyzed by Zimm and Kratky plots for all samples, as shown in Fig. 2. Zimm plots were used to determine 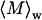 and Rg. The average molecular weights and radii of gyration determined from Zimm plots are shown in Table 1. For all samples at pH 6.0,
and Rg. The average molecular weights and radii of gyration determined from Zimm plots are shown in Table 1. For all samples at pH 6.0, 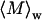 is greater than 1000 kDa, with Rg ranging between 50 and 80 nm, and a general trend of increasing average aggregate size with increasing urea concentration. Kratky plots provide a complementary representation of the data and can reveal morphological information. For instance, a straight line is characteristic of rigid rods while a curve with a plateau at intermediate q values is consistent with the presence of a linear semiflexible chain (29,30). The data for samples dissolved in pH 6.0 buffer with or without urea were most consistent with a semiflexible (worm-like) chain model, and corresponding Kratky plot fits were used to estimate the average molecular weights, contour lengths, and Kuhn statistical lengths (29). All values obtained from Zimm and Kratky plots are tabulated in Table 1. The estimated molecular weights from these two types of plots are in very good agreement. The fairly small Kuhn statistical length, Lk, values indicate that the aggregates have some flexibility. The ratio of Rg to
is greater than 1000 kDa, with Rg ranging between 50 and 80 nm, and a general trend of increasing average aggregate size with increasing urea concentration. Kratky plots provide a complementary representation of the data and can reveal morphological information. For instance, a straight line is characteristic of rigid rods while a curve with a plateau at intermediate q values is consistent with the presence of a linear semiflexible chain (29,30). The data for samples dissolved in pH 6.0 buffer with or without urea were most consistent with a semiflexible (worm-like) chain model, and corresponding Kratky plot fits were used to estimate the average molecular weights, contour lengths, and Kuhn statistical lengths (29). All values obtained from Zimm and Kratky plots are tabulated in Table 1. The estimated molecular weights from these two types of plots are in very good agreement. The fairly small Kuhn statistical length, Lk, values indicate that the aggregates have some flexibility. The ratio of Rg to 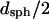 (i.e., the hydrodynamic radius) shows a decrease in overall particle compaction as urea concentration increases. At pH 2.3 and 6.0 M urea, the average molecular weight drastically decreases, and it roughly corresponds to an nonaggregated species. These results are consistent with the dynamic light scattering data where an apparent hydrodynamic diameter slightly larger than the expected values for a monodisperse monomer was observed (Fig. 1; Table 1). Interestingly, myoglobin from horse heart has also been reported to form dimers or higher order aggregates under partially denaturing conditions (36).
(i.e., the hydrodynamic radius) shows a decrease in overall particle compaction as urea concentration increases. At pH 2.3 and 6.0 M urea, the average molecular weight drastically decreases, and it roughly corresponds to an nonaggregated species. These results are consistent with the dynamic light scattering data where an apparent hydrodynamic diameter slightly larger than the expected values for a monodisperse monomer was observed (Fig. 1; Table 1). Interestingly, myoglobin from horse heart has also been reported to form dimers or higher order aggregates under partially denaturing conditions (36).
FIGURE 2.

(A) Zimm and (B) Kratky plots for apoMb-containing solution under different experimental conditions, denoted by symbols equivalent to those used in Fig. 1. Due to contributions from dust in the 0.0 M urea sample collected from SE chromatography, 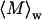 and Rg were determined from linear fits to the data at large q2. Since the other samples contained large aggregates, the limiting slope as q2 approaches zero was used to estimate
and Rg were determined from linear fits to the data at large q2. Since the other samples contained large aggregates, the limiting slope as q2 approaches zero was used to estimate 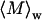 and Rg.
and Rg.
The static light scattering data for apoMb refolding from a low pH concentrated urea solution have larger error bars than the plots in Fig. 2, but clearly identifiable general features (data not shown). The Zimm plot has a small intercept from the initial slope, consistent with the presence of very large aggregates. The Kratky plot exhibits a large scatter in the data, but the general shape is more consistent with very large and macroscopically amorphous aggregates rather than the extended chain-like morphology of the pH 6.0 samples. This result was confirmed by transmission electron microscopy (EM). The corresponding electron micrograms for this sample, shown in Fig. 3, reveal the presence of amorphous aggregates close to micron size with no preferred three-dimensional shape and lacking any fibril-like character (Fig. 3). Additional EM data collected two weeks after sample preparation show no detectable changes in morphology (data not shown). This supports the idea that the amorphous self-associated species observed here are aggregation end points, and not transient intermediates evolving to fibril-type morphology.
FIGURE 3.

Transmission electron micrograms of apoMb aggregates formed upon refolding the protein from a 6.0 M urea solution at pH 2.5 into 100 mM sodium phosphate buffer at final pH 6.0 (12 μM apoMb concentration), at room temperature. Data were collected after 17 h from refolding initiation.
Size-exclusion chromatography
To determine the distribution of aggregate sizes, samples from light scattering experiments were subjected to size exclusion chromatography. Although elution volume changed with solvent, all proteins behaved as apparent monomers based on individual column calibrations, with little or no signal at the void volume (Fig. 4). However, some of the injected sample may not elute and therefore remain undetected. To gain additional insights into why the large aggregates observed by light scattering are not detected by size exclusion chromatography, samples were run through a size exclusion system without the column. Peak areas were compared to those for runs performed in the presence of a column. Consistent with previous work on both apomyoglobin (13) and amyloidogenic peptides (29,30), the observed losses suggest the presence of very large aggregates, which are presumably trapped by the column or its filter (Table 2). The losses may also be due to nonspecific adsorption of the protein to the column. At pH 6.0 there were significant sample losses at 0.0, 6.0, and 8.0 M urea concentrations. This result correlates well with the light scattering data. The pH 6.0 samples with no urea did not behave reproducibly. We hypothesize that this may be due to different amounts of self-associated species present in the lyophilized samples. Losses in the 6.0 M urea pH 2.3 sample were negligible. Consistent with the light scattering data, these results indicate the lack of detectable aggregates under these conditions.
FIGURE 4.

Size exclusion chromatography analysis of apoMb under different experimental conditions: (A) 10 mM sodium acetate buffer 0.0 M urea, pH 6.0 (38 μM); (B) 10 mM sodium acetate buffer, 6.0 M urea, pH 6.0 (31 μM); (C) 10 mM sodium acetate buffer, 8.0 M urea, pH 6.0 (21 μM); (D) 10 mM sodium acetate, 6.0 M urea, pH 2.3 (157 μM); (E) Refolded from 100 mM sodium phosphate buffer, 6.0 M urea, pH 2.3, to final pH 6.0 (12 μM). The predicted elution volumes for monomer (m) and the void volume (v) in each panel are based on calibrations with protein standards performed under each of the experimental conditions A–E.
TABLE 2.
Size-exclusion chromatography analysis of apoMb samples
| Experimental Conditions | Monomer (%) | Misbehaved species (%)† |
|---|---|---|
| 0 M urea, pH 6.0* | 40 ± 30 | 60 ± 30 |
| 6.0 M urea, pH 6.0 | 27 ± 3 | 73 ± 3 |
| 8.0 M urea, pH 6.0 | 58 ± 2 | 42 ± 2 |
| 6.0 M urea, pH 2.3 | ∼100 | ∼0 |
| Refolding from 6.0 M urea, pH 2.3 | 45 ± 5 | 55 ± 5 |
These data were not reproducible within four independent runs.
Determined from loss of material during size-exclusion chromatography runs in the presence and absence of column and precolumn filters. No significant void volume peaks were observed.
Upon dilution into pH 6.0 buffer to initiate refolding, a significant fraction of the material elutes as a monomer (Fig. 4 E). However, slightly >50% of the total protein is lost in the SE system (Table 2). This fraction of the protein is either self-associated or strongly interacting with the column.
Circular dichroism
ApoMb in 6.0 and 8.0 M urea displays little residual secondary structure (Fig. 5). On the other hand, apoMb dissolved in pH 6.0 buffer has a substantial degree of α-helical secondary structure (Fig. 5), in agreement with published data collected under similar conditions (11,16). The far-UV CD spectrum of monomeric native apoMb acquired by Damaschun and co-workers (16) is entirely similar to the data of Fig. 5. Since our sample contains a sizable fraction of aggregated protein (see light scattering and size-exclusion chromatography data), the concurrence of the two spectra suggests that the overall fraction of helical secondary structure present in the aggregates is likely similar to that of the folded protein.
FIGURE 5.

Far-UV circular dichroism spectra of apoMb samples under different experimental conditions: (▾) 10 mM sodium acetate buffer, 0.0 M urea, pH 6.0, 38 μM apoMb; (○) 10 mM sodium acetate buffer, 6.0 M urea, pH 6.0, 31 μM apoMb; (◊) 10 mM sodium acetate buffer, 8.0 M urea, pH 6.0, 21 μM apoMb; (•) 10 mM sodium acetate, 6.0 M urea, pH 2.3, 157 μM apoMb; (Δ) refolded from 100 mM sodium phosphate buffer, 6.0 M urea, pH 2.3, to final pH 6.0, 12 μM apoMb.
Further insights are provided by the refolded protein originating from a pH 2.3 unfolded state in 6.0 M urea. CD shows overall native-like secondary structure (Fig. 5). Dynamic light scattering indicates that refolding under these conditions gives rise to the formation of large soluble aggregates (Fig. 1 B). Therefore, similarly to the case discussed above, the CD spectrum of refolded apomyoglobin reflects the presence of both the soluble supramolecular aggregates detected by light scattering (Fig. 1) and the monomeric native protein. All species coexist in solution.
An overview of the secondary structure for the apoMb aggregates in relation to their morphology under different experimental conditions is provided in Table 3.
TABLE 3.
Structural properties of apoMb samples investigated in this study
| Experimental Conditions | Secondary structure (nm scale) | Particle morphology (μm scale) |
|---|---|---|
| 0 M urea, pH 6.0* | Mostly α-helix | Semiflexible chain |
| 6 M urea, pH 6.0 | Random coil | Semiflexible chain |
| 8 M urea, pH 6.0 | Random coil | Semiflexible chain |
| Peak collected from SE chromatography | Mostly α-helix | Monomer |
| 6 M urea, pH 2.3 | Random coil | Monomer/dimer |
| Refolding from 6.0 M urea, pH 2.3* | Mostly α-helix | Amorphous |
The samples also contain a fraction of correctly folded monomeric apoMb. This table reports the properties of the self-associated particles present in the sample.
NMR spectroscopy
Nuclear magnetic resonance was employed as a complementary tool to investigate the existence of correctly folded monomeric protein in a pool containing large soluble aggregates. ApoMb was examined upon refolding in pH 6.0 buffer, starting from a nonaggregated unfolded state in 6.0 M urea at pH 2.5. These experimental conditions are entirely similar to those employed for the data of Fig. 1 B except that the protein concentration had to be kept slightly higher, due to sensitivity requirements. A number of 1D HSQC traces were collected back-to-back, after refolding initiation; 2D NMR data were then acquired, 12 h after the start of refolding, at a time corresponding to the approximate endpoint of the kinetic data of Fig. 1 B. As previously stated, dynamic light scattering (Fig. 1 B) and gel filtration (Table 2) support the presence of large aggregates under these conditions. The representative 1D HSQC traces of Fig. 6 A indicate that a species bearing native-like chemical shift dispersion is present in solution ∼1 h after refolding initiation. The population of this species does not significantly vary over the following 10 h, despite the fact that large aggregates are generated and continue to grow during this time, as shown by dynamic light scattering (Fig. 1 B). The 2D 1H-15N HSQC spectrum is characterized by large chemical shift dispersion in the 1H dimension (Fig. 6 B). In addition, both 1H and 15N chemical shifts are similar to those of the published native apoMb HSQC spectrum (37). This confirms the presence of native protein in solution. Therefore, the refolded sample contains a substantial amount of correctly folded apoMb, along with the large aggregates detected by dynamic light scattering but largely undetectable by NMR spectroscopy.
FIGURE 6.

NMR analysis of apoMb refolding starting from an unfolded state in 6.0 M urea at pH 2.5. Refolding was triggered by a 1:10 dilution in buffer as described in the Materials and Methods section, under identical conditions to those of Fig. 1 B except that the final protein concentration was 50 μM. (A) Two representative 1H, 15N HSQC 1-dimensional (no 15N evolution) spectra taken during the time course of refolding. Data acquisition was initiated at the times indicated on the top left of the panels. (B) 2D 1H, 15N HSQC spectrum for apoMb refolding. Data acquisition was started 12 h after refolding initiation. This time is close to the end point for the aggregation time course of Fig. 1 B.
DISCUSSION
ApoMb structure and misfolding
Despite earlier claims that sperm whale apoMb may behave as a molten globule under physiologically relevant conditions (38), it is now well established that this protein fits all the characteristics of a typical globular protein and it has a well-defined secondary and tertiary structure at pH 6 (37). Hydrodynamic investigations have shown that the degree of compaction of folded apoMb is slightly smaller than the corresponding value for the parent holoprotein (39). The secondary structure of apomyoglobin, as assessed by circular dichroism and NMR spectroscopy, is very similar to that of the holoprotein, with the exception of the F helix, which is unstructured in the apoprotein (37).
Although the overall structural features of the native apoMb monomer are well defined and characterized, this protein also exhibits a tendency toward self-association. This trend has been addressed by a few recent studies, mostly focusing on either nonnative conditions (24,25) or native-like conditions applied to truncated apoMb fragments (13). However, despite its intrinsic importance for the delicate balance between folding and misfolding, this tendency toward aggregation has generally been largely neglected or underestimated so far. This work detects the presence and characterizes the size and secondary structure of large apoMb soluble aggregates. Three fundamentally different conditions often used in folding and refolding studies have been analyzed, namely buffered solution at pH 6, high urea concentration (6.0 and 8.0 M), and the protein refolding process starting from an unfolded state virtually devoid of aggregates.
Aggregate size, shape, and polydispersity
Significant amounts of soluble aggregates were identified in aqueous buffer and in concentrated urea solution under physiologically relevant pH conditions. The wide size range observed for the self-associated apoMb species detected here is a novel feature of the apoMb system. The average hydrodynamic diameter of the aggregates, generated upon dissolving the lyophilized powder at pH 6.0, is ∼130 nm. The previously reported apoMb self-associated species detected in urea solution (22) were considerably larger than those reported here (and possibly partially insoluble), because they were identified by turbidimetry, which is insensitive to the presence of nanometer-size particles. Upon refolding apoMb from a virtually nonaggregated unfolded state, we also observe very large self-associated species with a hydrodynamic diameter up to 2 μm.
Formation of large self-associated species was found to be more severe at pH 6.0 than at pH 2. Low pH and high urea concentrations lead to complete disappearance of large aggregates. Identification of these low pH/high urea concentration conditions, under which virtually no apomyoglobin aggregates are present, is a useful and reproducible starting point for all folding/misfolding studies on apomyoglobin. In addition, these “nonaggregated” initial conditions may prove effective for other proteins as well. At low pH, polypeptides acquire a net positive charge. This leads to an overall increase in intermolecular electrostatic repulsion, which is expected to reduce the tendency toward intermolecular complex formation. Therefore, the absence of large aggregates under low pH conditions is not entirely surprising.
The large aggregates observed at pH 6.0 in the absence of urea are either formed within the experimental dead time or may reflect the presence of heavily self-associated species in the lyophilized protein sample, persisting in solution after protein solubilization. The same argument applies to the large aggregates detected in 6.0 and 8.0 M urea at pH 6.0.
Interestingly, we detect severe self-association even upon apoMb refolding from a virtually nonaggregated unfolded state (i.e., at low pH and high urea concentration). This process takes place on a parallel route to the refolding to native monomeric structure, as further discussed below. Most importantly, given that there is no loss in the amount of monomeric structure over the course of large aggregate formation (Fig. 6 A) the aggregation observed by dynamic light scattering (Fig. 1 B) is interpreted here as due to the noncovalent rearrangement of preexisting large species to give rise to even larger entities (dsph up to 2 μm). This process is expected to be complete in ∼10–12 h, as the average aggregate size levels off in the dynamic light scattering results (Fig. 1 B). Electron micrograms taken 17 h, 41 h, and 2 weeks after refolding the sample confirm this finding, showing that the final aggregate size and morphology is reached at this time point.
Effect of urea on protein folding and self-association
High urea concentrations are known to dramatically affect protein conformation. Urea is typically regarded as a denaturing agent, and it is believed to disrupt protein structure by stabilizing the unfolded state. Despite the abundance of experimental data on the effect of urea on protein stability and folding, the physical bases for its action are still the subject of intense debate (40). Although structural analysis by NMR spectroscopy has shown that urea displays weak specific binding to protein aliphatic side chains (41,42), thermodynamic data support an alternative view envisioning urea as interacting with the polypeptide backbone by hydrogen bonding (43–46). In both cases, a high urea concentration is expected to structurally relax a polypeptide by preferentially binding to the solvent-exposed unfolded state. In principle, similar arguments should apply to self-association phenomena. Therefore, one would expect urea to exert a stronger stabilizing effect on the unfolded state than on the self-associated protein chains. According to this argument, it is hard to imagine how protein aggregates can be significantly populated in solution at high urea concentrations. Yet, the data presented here show evidence for large aggregates in 6.0 and 8.0 M urea aqueous solution. Although more research needs to be done to elucidate the origins of this unexpected effect, two possible explanations provide clues about its potential origin. In case the self-associated species is extremely thermodynamically stable (much more than a typical monomeric globular protein), relative to the urea-stabilized unfolded state, an overall larger population of aggregate may still persist in solution at equilibrium. Alternatively, kinetically trapped self-associated species present in the lyophilized powder may not be able to completely disassociate due to large free energies of activation involved in the solubilization process. In the case of a very compact self-associated species, urea may not be able to effectively penetrate into the aggregated structure. Careful experiments on free energies of transfer of model compounds from water to denaturant solution (47,48) indicate that urea stabilizes amino acids and polypeptides by increasing their solubility. This observation does not explain the increase in aggregate size at higher urea concentrations observed here (i.e., 6 vs. 8 M), arguing against the former thermodynamic argument. More studies are clearly needed to fully understand the origin of this phenomenon.
Self-association kinetics by dynamic light scattering and NMR
The large apoMb self-associated species generated from low pH and 6.0 M urea conditions are formed, at room temperature, within 10 min. Dynamic light scattering shows that the increase in size of the large species formed upon refolding is slow (Fig. 1 B), taking place over the course of a few hours. NMR has been instrumental in getting further insights into this process.
NMR spectroscopy is typically able to detect molecules of size up to 25–30 kDa. Due to their large rotational correlation times, the resonances associated with larger macromolecules or noncovalent aggregates experience severe line-broadening due to efficient transverse relaxation, and are typically filtered out by this technique. Although perdeuteration and TROSY-type data acquisition have the potential to partially overcome this problem (49,50), NMR is generally regarded as a spectroscopic technique insensitive to the presence of very large aggregates, as long as these are in slow exchange on the NMR chemical shift timescale. In the presence of fast or intermediate chemical exchange, large macromolecules are detectable as exchange-averaged spectral line-broadening (51). Therefore, NMR's ability to detect small-size species makes it a useful complementary tool to laser light scattering, which is specifically sensitive to the presence of large aggregates.
The 1D HSQC traces acquired during the first 12 h of refolding (Fig. 6 A) show that the total amount of folded protein does not vary as a function of time. On the other hand, dynamic light scattering shows that self-association continues during this time, resulting in an increase in the aggregate size. Therefore, the amount of correctly folded monomeric apoMb does not change while the aggregates get larger. This is possible only if the already misfolded and self-associated apoMb molecules come together to aggregate further and increase the observed average aggregate size, without recruiting any significant amounts of the correctly folded monomeric species. Interestingly, the presence of slow internal rearrangements among fast-forming aggregates has also been observed in amyloid fibril formation (29,52).
Secondary structure of ApoMb aggregates
Light scattering indicates that apoMb solutions in pH 6.0 buffer in the presence of 0, 6.0, or 8.0 M urea, and refolded from pH 2.3 in 6.0 M urea, contain large aggregates. As seen in the Results section, this population is likely to correspond, at least in part, to the population of misbehaved apoMb, which does not elute through the size exclusion chromatography column. The fraction of misbehaved species by SE chromatography can be tentatively assigned to the fractional population of large aggregates. On the other hand, the far-UV circular dichroism analysis is consistent with the presence of two widely different classes of secondary structure. The apoMb chains are either disordered (all samples in 6.0 and 8.0 M urea) or predominantly helical (samples in 0 and 0.6 M urea). Furthermore, the known CD spectrum of pure monomeric apoMb (corresponding to the eluted monomer peak from SE chromatography) (16) is very similar to the helical traces of Fig. 5, which contain a mixture of monomer and large soluble aggregates. The above observations can only be reconciled by recognizing that, under native conditions, both monomeric and aggregated apoMb must share common CD spectral features. An important implication of the above is that, under the conditions examined in this study, the soluble aggregates have a predominantly α-helical secondary structure.
In summary, we have identified two novel types of large apoMb aggregates with widely different secondary structure content (Table 3). At high urea concentrations and pH 6.0 the self-associated apoMb chains are conformationally disordered and largely devoid of a defined secondary structure. At low urea concentration, or in absence of urea, the aggregates are mostly helical, just as in the native state.
The structural plasticity of the apoMb polypeptide chain is further highlighted by the contrast with previously detected amyloid-like aggregates formed at high temperature and pH, which have a predominantly β-sheet conformation (24,25). Quite remarkably, apoMb, which is a predominantly α-helical protein in its native state, populates widely different types of secondary structure, i.e., α-helix, β-sheet, and random coil, in its self-associated states, depending on the specific environmental conditions.
Morphology of ApoMb aggregates
The Kratky plots of Fig. 2 show that the morphology of the aggregates formed at pH 6 can be described as an elongated semiflexible chain, independent of the urea concentration. The increase in both Lc and in the ratio 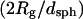 with urea concentration (Table 1) indicates that aggregate particles become more elongated with increasing urea concentration. The relatively high
with urea concentration (Table 1) indicates that aggregate particles become more elongated with increasing urea concentration. The relatively high 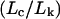 ratio is indicative that the aggregates have considerable flexibility rather than being rod-like. In contrast, the aggregates detected upon refolding of the monomer into pH 6 buffer are much larger and appear to be loosely globular with no distinct morphology (Fig. 3). This highlights an interesting distinction between particle morphology and secondary structure. In short, the self-associated species at pH 6 and in high urea concentration have a defined elongated shape but are devoid of secondary structure, whereas the aggregates detected upon refolding bear a significant degree of short-range secondary structure but have a poorly defined amorphous shape.
ratio is indicative that the aggregates have considerable flexibility rather than being rod-like. In contrast, the aggregates detected upon refolding of the monomer into pH 6 buffer are much larger and appear to be loosely globular with no distinct morphology (Fig. 3). This highlights an interesting distinction between particle morphology and secondary structure. In short, the self-associated species at pH 6 and in high urea concentration have a defined elongated shape but are devoid of secondary structure, whereas the aggregates detected upon refolding bear a significant degree of short-range secondary structure but have a poorly defined amorphous shape.
The gel filtration data show that the overall tertiary structure of monomeric apoMb also follows an interesting trend. Specifically, in the absence of urea, apoMb has a smaller apparent Stokes radius than in the presence of urea (Fig. 4). Small angle x-ray scattering experiments performed on native and unfolded apoMb are consistent with this view (16,53).
Parallel folding and misfolding pathways
Folding and misfolding are two phenomenologically distinct biophysical processes. Although the former gives rise to native protein conformation, the latter generates one or more nonnative, often self-associated, nonphysiologically relevant protein structures. From the physical viewpoint, folding and misfolding are nothing but related aspects of the same phenomenon, i.e., the conformational search leading to the kinetically accessible lowest free-energy structure(s). As such, the two events can, in principle, occur concurrently. Misfolding, however, often gives rise to irreversible aggregation, and this process ends up driving all the solution equilibria toward the irreversibly misfolded species. This often prevents the observation of intramolecular protein folding. In the present case, large aggregates form on a longer timescale relative to folding. Under these conditions, misfolding can be followed in the presence of folding, providing valuable insights on the kinetic and structural interplay between the two classes of events.
The combined use of low pH and high urea concentration leads to the nearly complete structural unfolding of apoMb, and to the complete suppression of the large aggregates detected at pH 6.0. Starting from this virtually monomeric unfolded state, refolding leads to the formation of severely self-associated species with native-like secondary structure. Under these conditions, kinetic refolding proceeds via two parallel routes: one leading to native monomer, and the other leading to a misfolded heavily self-associated state bearing native-like secondary structure. The concurrent fast formation of native apoMb, ensured by its direct detection via 2D 1H,15N-HSQC NMR, highlights the fact that the complex conformational energy landscape accounting for both folding and misfolding is heavily biased toward the rapid formation of native structure. However, even at the relatively low protein concentrations analyzed here, multimolecular encounters occur and give rise to the formation of very large aggregates. As seen above, these have secondary structure features similar to those of the native protein, suggesting that the aggregates may contain some aberrant global or local conformations, without substantial disruption of local short-range structural order.
The 1D NMR analysis shows no variation in population of folded protein over time, while large aggregate formation takes place. This implies that the kinetic discrimination between the populations committed to folding and those committed to misfolding must occur relatively early in time, within the folding/misfolding energy landscape. On the other hand, consolidation of misfolded self-associated intermediates into larger species is a much slower process, resulting in no significant perturbations in the overall protein secondary structure.
CONCLUSIONS
This study has focused on the detection and detailed characterization of the large apoMb soluble aggregates populated at room temperature in buffered solutions in the presence and absence of high urea concentrations. These species differ widely in secondary structure, being either α-helical or conformationally disordered. ApoMb refolding from a nonaggregated unfolded state gives rise to the parallel generation of native folded protein and self-associated intermediates. The intermediates slowly get larger until micron-size hydrodynamic diameter, and bear an overall native-like secondary structure. These results highlight the rich structural diversity of apoMb aggregates generated under different conditions, and advance our understanding of the correlations between morphology and secondary structure in large self-associated complexes. In addition, they provide an initial framework to investigate the symbiotic relationships between apoMb folding and misfolding. It is hoped that this report will stimulate additional future investigations aiming at an even higher resolution analysis of apoMb's self-association events at room temperature.
Acknowledgments
We are grateful to Lin Liu, Jin Ryoun Kim, Ye-Jin Eun, and Todd Gibson for technical assistance.
We thank the National Science Foundation (MCB-0215368 to S.C. and BES-0330537 to R.M.), the Milwaukee Foundation (Shaw Scientist Award to S.C.), and the American Heart Association (postdoctoral fellowship to N.K.) for supporting this research.
References
- 1.Jaochim, C. L., and D. J. Selkoe. 1992. The seminal role of β-amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 6:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Perutz, M. F. 1999. Glutamine repeats and neurodegenerative disease: molecular aspects. Trends Biochem. Sci. 24:56–63. [DOI] [PubMed] [Google Scholar]
- 3.Scherzinger, E., and E. E. Wanker. 1997. Huntingtin-encoded polyglutamine expansions form amyloid-like protein. Cell. 90:549–558. [DOI] [PubMed] [Google Scholar]
- 4.Lansbury, P. T. 1999. Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. USA. 96:3342–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hlodan, R., S. Craig, and R. H. Pain. 1991. Protein folding and its implications for the production of recombinant proteins. Biotechnol. Genet. Eng. Rev. 9:47–88. [PubMed] [Google Scholar]
- 6.Fink, A. L. 1998. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold. Des. 3:R9–R23. [DOI] [PubMed] [Google Scholar]
- 7.Jordon, G. M., S. Yoshioka, and T. Terao. 1994. The aggregation of bovine serum albumin in solution and in the solid state. J. Pharm. Pharmacol. 46:182–185. [DOI] [PubMed] [Google Scholar]
- 8.Griko, Y. V., P. L. Privalov, S. Y. Venyaminov, and V. P. Kutyshenko. 1988. Thermodynamic study of the apomyoglobin structure. J. Mol. Biol. 202:127–138. [DOI] [PubMed] [Google Scholar]
- 9.Hughson, F. M., P. E. Wright, and R. L. Baldwin. 1990. Structural characterization of a partly folded apomyoglobin intermediate. Science. 249:1544–1548. [DOI] [PubMed] [Google Scholar]
- 10.Jennings, P. A., and P. E. Wright. 1993. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science. 262:892–896. [DOI] [PubMed] [Google Scholar]
- 11.Cavagnero, S., H. J. Dyson, and P. E. Wright. 1999. Effect of H-helix destabilizing mutations on the kinetic and equilibrium folding of apomyoglobin. J. Mol. Biol. 285:269–282. [DOI] [PubMed] [Google Scholar]
- 12.Garcia, C., C. Nishimura, S. Cavagnero, H. J. Dyson, and P. E. Wright. 2000. Changes in the apomyoglobin folding pathway caused by mutation of the distal histidine residue. Biochemistry. 39:11227–11237. [DOI] [PubMed] [Google Scholar]
- 13.Chow, C. C., C. Chow, V. Raghunathan, T. J. Huppert, E. B. Kimball, and S. Cavagnero. 2003. The chain length dependence of apomyoglobin folding: structural evolution from misfolded sheets to native helices. Biochemistry. 42:7090–7099. [DOI] [PubMed] [Google Scholar]
- 14.Nishimura, C., H. J. Dyson, and P. E. Wright. 2003. The apomyoglobin folding pathway revisited: structural heterogeneity in the kinetic burst phase intermediate. J. Mol. Biol. 331:1171–1172. [DOI] [PubMed] [Google Scholar]
- 15.Gilmanshin, R., M. Gulotta, R. B. Dyer, and R. H. Callender. 2001. Structures of apomyoglobin's various acid-destabilized forms. Biochemistry. 40:5127–5136. [DOI] [PubMed] [Google Scholar]
- 16.Gast, K., H. Damaschun, R. Misselwitz, M. Muller-Frohne, D. Zirwer, and G. Damaschun. 1994. Compactness of protein molten globules: temperature-induced structural changes of the apomyoglobin folding intermediate. Eur. Biophys. J. 23:297–305. [DOI] [PubMed] [Google Scholar]
- 17.Kataoka, M., I. Nishii, T. Fujisawa, T. Ueki, F. Tokunaga, and Y. Goto. 1995. Structural characterization of the molten globule and native states of apomyoglobin by solution x-ray scattering. J. Mol. Biol. 249:215–228. [DOI] [PubMed] [Google Scholar]
- 18.Yao, J., J. Chung, D. Eliezer, P. Wright, and H. Dyson. 2001. NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into early events in protein folding. Biochemistry. 40:3561–3571. [DOI] [PubMed] [Google Scholar]
- 19.Schwarzinger, S., P. E. Wright, and H. J. Dyson. 2002. Molecular hinges in protein folding: the urea-denatured state of apomyoglobin. Biochemistry. 41:12681–12686. [DOI] [PubMed] [Google Scholar]
- 20.Gulotta, M., E. Rogatsky, R. H. Callender, and R. B. Dyer. 2003. Primary folding dynamics of sperm whale apomyoglobin: core formation. Biophys. J. 84:1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goto, Y., and A. L. Fink. 1990. Phase diagram for acidic conformational states of apomyoglobin. J. Mol. Biol. 214:803–805. [DOI] [PubMed] [Google Scholar]
- 22.De Young, L., K. A. Dill, and A. L. Fink. 1993. Aggregation and denaturation of apomyoglobin in aqueous urea solutions. Biochemistry. 32:3877–3886. [DOI] [PubMed] [Google Scholar]
- 23.Mohney, B. K., E. T. Petri, V. Uvarova, and G. C. Walker. 2000. Infrared absorption and ultraviolet-circular dichroism spectral studies of thermally induced unfolding of apomyoglobin. Appl. Spectrosc. 54:9–14. [Google Scholar]
- 24.Fandrich, M., M. A. Fletcher, and C. M. Dobson. 2001. Amyloid fibrils from muscle myoglobin. Nature. 410:165–166. [DOI] [PubMed] [Google Scholar]
- 25.Fandrich, M., V. Forge, K. Buder, M. Kittler, C. M. Dobson, and S. Diekmann. 2003. Myoglobin forms amyloid fibrils by association of unfolded polypeptide segments. Proc. Natl. Acad. Sci. USA. 100:15463–15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sirangelo, I., C. Malmo, M. Casillo, A. Mezzogiorno, M. Papa, and G. Irace. 2002. Tryptophanyl substitutions in apomyoglobin determine protein aggregation and amyloid-like fibril formation at physiological pH. J. Biol. Chem. 277:45887–45891. [DOI] [PubMed] [Google Scholar]
- 27.Provencher, S. W. 1982. A constrained regularization method for inverting data represented by linear algebraic or integral equations. Comput. Phys. Commun. 27:213–227. [Google Scholar]
- 28.Young, R. J., and P. A. Lovell. 1991. Introduction to Polymers. Chapman and Hall, London, UK; New York, NY.
- 29.Murphy, R. M., and M. M. Pallitto. 2000. Probing the kinetics of β-amyloid self-association. J. Struct. Biol. 130:109–122. [DOI] [PubMed] [Google Scholar]
- 30.Lowe, T. L., A. Strzelec, L. L. Kiessling, and R. M. Murphy. 2001. Structure-function relationships for inhibitors of β-amyloid toxicity containing the recognition sequence KLVFF. Biochemistry. 40:7882–7889. [DOI] [PubMed] [Google Scholar]
- 31.Koyama, R. 1973. Light scattering of stiff chain polymers. J. Phys. Soc. Jpn. 34:1029–1038. [Google Scholar]
- 32.Shen, C.-L., and R. M. Murphy. 1995. Solvent effects on self-assembly of β-amyloid peptide. Biophys. J. 69:640–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kay, L., P. Keifer, and T. Saarinen. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114:10663–10665. [Google Scholar]
- 34.Delaglio, F., S. Grzesiek, G. Vuister, G. Zhu, J. Pfeifer, and A. Bax. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6:277–293. [DOI] [PubMed] [Google Scholar]
- 35.Johnson, B., and R. Blevins. 1994. NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 4:603–614. [DOI] [PubMed] [Google Scholar]
- 36.Eliezer, D., K. Chiba, H. Tsuruta, S. Doniach, K. O. Hodgson, and H. Kihara. 1993. Evidence of an associative intermediate on the myoglobin refolding pathway. Biophys. J. 65:912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eliezer, D., and P. E. Wright. 1996. Is apomyoglobin a molten globule? Structural characterization by NMR. J. Mol. Biol. 263:531–538. [DOI] [PubMed] [Google Scholar]
- 38.Lin, L., R. J. Pinker, K. Forde, G. D. Rose, and N. R. Kallenbach. 1994. The molten globular characteristics of the native state of apomyoglobin. Nat. Struct. Biol. 1:447–452. [DOI] [PubMed] [Google Scholar]
- 39.Crumpton, M. J., and A. Polson. 1965. A comparison of the conformation of Sperm Whale metmyoglobin with that of apomyoglobin. J. Mol. Biol. 11:722–729. [DOI] [PubMed] [Google Scholar]
- 40.Schellman, J. A. 2002. Fifty years of solvent denaturation. Biophys. Chem. 96:91–101. [DOI] [PubMed] [Google Scholar]
- 41.Dotsch, V., G. Wider, G. Siegal, and K. Wuthrich. 1995. Interaction of urea with an unfolded protein: the DNA-binding domain of the 434-repressor. FEBS Lett. 366:6–10. [DOI] [PubMed] [Google Scholar]
- 42.Liepinsh, E., and G. Otting. 1994. Specificity of urea binding to proteins. J. Am. Chem. Soc. 116:9670–9674. [Google Scholar]
- 43.Schellman, J. A. 1955. The stability of hydrogen-bonded peptide structures in aqueous solutions. Comptes Rendus des Travaux du Laboratoire Carlsberg: Serie Chimique. 29:230–259. [PubMed] [Google Scholar]
- 44.Schellman, J. A. 1978. Solvent denaturation. Biopolymers. 17:1305–1322. [DOI] [PubMed] [Google Scholar]
- 45.Schellman, J. A. 1987. Selective binding and solvent denaturation. Biopolymers. 26:549–559. [DOI] [PubMed] [Google Scholar]
- 46.Courtenay, E. S., M. W. Capp, and M. T. Record, Jr. 2001. Thermodynamics of interactions of urea and guanidinium salts with protein surface: relationship between solute effects on protein processes and changes in water-accessible surface area. Protein Sci. 10:2485–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nozaki, Y., and C. Tanford. 1963. The solubility of amino acids and related compounds in aqueous urea solutions. J. Biol. Chem. 238:4074–4081. [PubMed] [Google Scholar]
- 48.Tanford, C. 1964. Isothermal unfolding of globular proteins in aqueous solutions. J. Am. Chem. Soc. 86:2050–2059. [Google Scholar]
- 49.Wütrich, K., G. Wider, K. Pervushin, H. Senn, and K. Wütrich. 1999. TROSY-type triple-resonance experiments for sequential NMR assignments of large proteins. J. Am. Chem. Soc. 121:844–848. [Google Scholar]
- 50.Pervushin, K., R. Riek, G. Wider, and K. Wüthrich. 1997. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA. 94:12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyer, B., and T. Peters. 2003. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem. Int. Ed. Engl. 42:864–890. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi, N., K. Hasegawa, I. Yamaguchi, H. Okada, T. Ueda, F. Gejyo, and H. Naiki. 2002. Establishment of a first-order kinetic model of light chain-associated amyloid fibril extension in Vitro. Biochim. Biophys. Acta. 1601:110–120. [DOI] [PubMed] [Google Scholar]
- 53.Eliezer, D., P. A. Jennings, P. E. Wright, S. Doniach, K. O. Hodgson, and H. Tsuruta. 1995. The radius of gyration of an apomyoglobin folding intermediate. Science. 270:487–488. [DOI] [PubMed] [Google Scholar]