

Abstract
The tissue inhibitors of metalloproteinases (TIMPs) regulate matrix metalloproteinase (MMP) activity required for cell migration/invasion associated with cancer progression and angiogenesis. TIMPs also modulate cell proliferation in vitro and angiogenesis in vivo independent of their MMP-inhibitory activity. Here, we show that TIMP-2 mediates G1 growth arrest in human endothelial cells through de novo synthesis of the cyclin-dependent kinase inhibitor p27Kip1. TIMP-2-mediated inhibition of Cdk4 and Cdk2 activity is associated with increased binding of p27Kip1 to these complexes in vivo. Protein tyrosine phosphatase inhibitors or expression of a dominant negative Shp-1 mutant ablates TIMP-2 induction of p27Kip1. Finally, angiogenic responses to FGF-2 and VEGF-A in ‘motheaten viable’ Shp-1 deficient mice are resistant to TIMP-2 inhibition, demonstrating that Shp-1 is an important negative regulator of angiogenesis in vivo.
Abbreviations: TIMP-2, tissue inhibitor of metalloproteinase-2; MMP, matrix metalloproteinase; Shp-1, SH2-containing protein tyrosine phosphatase-1; PTP, protein tyrosine phosphatase; hMVECs, human microvascular endothelial cells; Cdks, cyclin-dependent kinases; ECM, extracellular matrix; FGF-2, fibroblast growth factor; PDGF, platelet derived growth factor; EGF, epidermal growth factor; VEGF-A, vascular endothelial growth factor-A; INK4, inhibitors of Cdk4; PBS, phosphate-buffered saline; pRb, retinoblastoma protein
Angiogenesis, the formation of new blood vessels from pre-existing vessels, accompanies a variety of pathologic responses in the adult, such as wound healing, tumor growth, cancer progression, and many chronic inflammatory diseases (1). Angiogenesis requires the dissolution of existing extracellular matrix (ECM) and formation of new ECM in particular the subendothelial basement membrane (2). The matrix metalloproteinases (MMPs) have been demonstrated to play a pivotal role in angiogenesis through altering biological functions of ECM macromolecules by selectively degrading and/or releasing matrix- or membrane-anchored growth factors (3). Endogenous protease inhibitors, such as the tissue inhibitors of metalloproteinases (TIMPs), regulate the activities of these proteinases. TIMPs can suppress cell proliferation, invasion, and reduce metastasis formation through inhibition of MMP activity and prevention of ECM turnover (4). However, recent studies suggest that TIMPs also directly modulate cell growth and migration via MMP-independent mechanisms (5-11).
TIMPs 1-3 all inhibit angiogenesis, however, the mechanisms of these effects appear to be specific for each member of the TIMP family. TIMP-1 blocks tumor-associated angiogenesis via a mechanism involving inhibition of MMP-dependent endothelial cell migration (5,12). However, a recent report suggests that TIMP-1 may also inhibit endothelial cell migration by an MMP-independent mechanism as well (13). In contrast, TIMP-3 prevents vascular endothelial growth factor-A (VEGF-A) induced angiogenesis by direct antagonism of binding of this growth factor to its cognate receptor, VEGFR-2 (14). Previous studies have demonstrated that TIMP-2 inhibits the proliferation of endothelial cells, fibroblasts, and carcinoma cell lines in response to stimulation with mitogenic growth factors such as fibroblast growth factor-2 (FGF-2), platelet derived growth factor (PDGF), or epidermal growth factor (EGF) (6,9,10). TIMP-2 binds to the surface of human microvascular endothelial cells (hMVECs) through interaction with the integrin α3β1 and this interaction mediates suppression of FGF-2- or VEGF-A-induced endothelial cell proliferation in vitro and angiogenesis in vivo (6). These effects are entirely independent of TIMP-2 inhibitory activity against MMPs, as demonstrated by the Ala+TIMP-2 mutant that lacks MMP inhibitory function but retains anti-angiogenic activity in vitro and in vivo (6, 10). The mechanism of these effects involves integrin-mediated heterologous receptor inactivation, specifically, α3β1-mediated inhibition of receptor tyrosine kinase activation. In hMVECs, TIMP-2 induces protein tyrosine phosphatase (PTP) activity, independently of MMP inhibitory activity. The effects of TIMP-2 on endothelial cell growth and angiogenesis are sensitive to the non-specific PTP inhibitor orthovanadate (6). The relevant PTP activity was putatively identified as the SH2-containing PTP-1 (Shp-1), using a dominant-negative approach (6). TIMP-2 also inhibits migration of endothelial cells by directly enhancing cell surface expression of RECK (5), a novel, cell membrane-associated protease inhibitor. Enhanced expression of RECK reduces cell-associated MMP activity required for cell migration and also appears to involve an Shp-1-dependent mechanism.
These findings lead us to posit that Shp-1 is an important negative regulator of endothelial cell growth in vitro and angiogenesis in vivo. In addition, the observation that TIMP-2 can modulate gene expression (i.e. RECK) in an orthovanadate-sensitive manner through binding to α3β1 (5), suggests that TIMP-2 may also activate signal transduction pathways important in cell growth regulation. Here we investigate the cellular pathways and effectors responsible for TIMP-2-mediated suppression of hMVEC proliferation in response to mitogenic growth factor stimulation. TIMP-2 inhibits mitogenic stimulation of hMVECs resulting in G1 growth arrest. The TIMP-2-mediated arrest of hMVEC growth is Shp-1-dependent. Furthermore, TIMP-2 anti-angiogenic activity is completely ablated in Shp-1 deficient (motheaten viable) mice. These results lead us to suggest that Shp-1 is a critical negative regulator of endothelial cell growth in vitro and angiogenesis in vivo.
Materials and Methods
Cell Lines, Growth Factors, and Antibodies
Human microvascular endothelial cells (hMVECs) and growth factors were purchased from commercial sources and used as previously described (6). The following antibodies were also purchased from commercial sources; anti-PTP1C/Shp-1 and anti-p27Kip1 (BD transduction laboratories); anti-Cdk2, cyclin E, p16INK4a, Cdk4 and cyclin D (Upstate Biotechnology); anti-p27Kip1 (T187) and p27Kip1 (S10) (Zymed laboratories); anti-p21WAF1/CiP1, Shp-2, Skp2, α3, and lamin B, actin antibodies, mouse and rabbit IgG-horseradish peroxidase conjugate (Santa Cruz Biotechnology). Rb-C fusion protein and polyclonal antibodies against phospho-pRb (S780), phospho-pRb (S807/811) and pRb were from Cell Signaling. TIMP-2 and Ala+TIMP-2 were prepared and characterized as described previously (10).
Immunoprecipitation and Western Blot Analysis
Subconfluent cells in gelatin-coated 100 mm dishes were serum-starved for 24 h in endothelial cell basal medium and replaced with fresh media, at 37 °C. Cells were then pretreated with or without TIMP-2 or Ala+TIMP-2 (50 nM) for 15 min prior to growth factor (FGF-2 or VEGF-A, 50 ng/mL) stimulation for 24 h, unless otherwise noted. After treatment with growth factors and/or TIMP-2, cell lysates were prepared, clarified and the supernatants subjected to immunoprecipitation and Western blot as described previously (6).
Cell Cycle Assay by Flow Cytometry
For cell cycle analysis, hMVECs following FGF-2 stimulation in the presence or absence of TIMP-2 were harvested with trypsin-EDTA and washed in phosphate-buffered saline (PBS), and then fixed with ice-cold 70% ethanol for at least 1 h. After cells were treated with RNase, DNA was stained with propidium iodide. The profile of cells in the G0/G1, S, and G2/M phases of the cell cycle were analyzed with a FACScan flow cytometer.
Cell Growth Assays
The p27+/+ and p27−/− immortalized MEFs, plated on 96-well plates (4×103 cells/well) were serumstarved for 36 to 48 h prior to treatment with TIMP-2 or Ala+TIMP-2. Following subsequent growth factor stimulation and culture for 48 h, the cell numbers were quantified as previously described (6).
Northern Blot Analysis
Northern blots analysis was performed using standard techniques. Blots were sequentially hybridized with [32P]-labeled cDNA probes (p27Kip1 and GAPDH) for 14 h at 42°C in prehybridization/hybridization buffer (Quality Biologicals). The probes for p27Kip1 and GAPDH were prepared using PCR products by High Prime DNA labeling kit (Roche Diagnostics Corp.). After hybridization, the blots were washed in 2X SSC, 0.1% SDS at room temperature and in 0.2X SSC, 0.1% SDS at 42°C before autoradiography at −80°C.
In Vitro Cyclin-Dependent Kinase Assays
For pRb kinase assays, anti-Cdk4 and anti-Cdk2 immunoprecipitates were rinsed three times with lysis buffer, and once with kinase buffer (20 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 1 mM DTT, 0.1 mM EDTA, 10 μg/ml 4-(2-aminoethyl) benzene sulfonyl fluoride, 1 μg/ml aprotinin, 0.1 μg/ml pepstatin A, 0.05 μg/ml leupeptin, 8 mM β-glycerophosphate, 2.5 mM NaF and 0.1 mM sodium orthovanadate) and re-suspended in 40 μl of kinase buffer containing 20 μg/ml Rb-C fusion protein (Cell Signaling), 50 μM ATP, and 10 μCi of [γ-32P]ATP (ICN Biomedical Inc., 6000 Ci/mmol) and incubated for 20 min at 30 °C. The reaction was terminated with 10 μl of 5X SDS-sample loading buffer, boiled for 5 min, and resolved by SDS-PAGE. Phosphorylation of substrate was visualized by autoradiography and quantified by the use of National Institutes of Health (NIH) Image 1.6 software.
siRNA Preparation and Transfection
For design of siRNA inserts, a cDNA sequence of integrin α3 AGCAACACAGACTACCTGGAG was selected according to the InvivoGen’s siRNA Wizard program based on a BLAST search. As a control we used the following scrambled oligonucleotide sequence AGCATATGTGCGTACCTAGCT available pre-packaged in the psiRNA-hH1zeo vector (InvivoGen).
Both the siRNA-targeting integrin α3 gene and control sequences were cloned into psiRNA-hH1zeo vector (InvivoGen), and these vectors are designated psiRNA-hH1zeo-α3 and psiRNA-hH1zeo-scr, respectively. These vector constructs were transfected into A549 lung carcinoma cells using LyoVec (InvivoGen) according to the manufacturer’s protocol. After 48 h the cells were selected with zeocin in DMEM containing 10% FBS for 1-2 weeks until colony formation.
Dominant Negative Shp-1 Adenovirus
The role of Shp-1 on TIMP-2-mediated cell growth arrest was investigated using dominant negative Shp-1 adenovirus as previously described (6). Briefly, the pcDNA3.1 vector containing wild type human Shp-1 (PTPN6) cDNA was first constructed and the Shp-1 dominant-negative mutant was constructed by using the QuikChange site-directed mutagenesis kit (Stratagene) to convert the active site cysteine to a serine residue (C453S). Replication defective, human adenovirus (Ad5ΔE1ΔE3) with both wild type (wtShp-1) and the C453S mutation (dnShp-1) sequences of Shp-1 were generated using the AdEasy Vector System (Qbiogene). Expression of dn Shp-1 in adenovirus-infected cells was verified by Western blotting using anti-PTP1C/Shp-1 antibody (BD biosciences) and PTP assay (Upstate Biotechnology) of whole cell lysates.
Subcellular Fractionation
Following treatments as indicated, hMVECs were rinsed twice with ice-cold PBS and cytoplasmic and nuclear extracts were prepared using NE-PER nuclear and cytoplasmic extraction reagents (Pierce) following manufacturer’s instructions.
Immunofluorescence Analysis
For p27Kip1 immunofluorescence, the hMVECs were grown on glass cover slips in 6-well plates, following FGF-2 stimulation in the presence or absence of Ala+TIMP-2, were fixed with 4% paraformaldehyde for 20 min, washed with PBS, and incubated with PBS containing 10% normal goat serum (NGS) and 0.2% Triton X-100 for 30 min. Primary antibody diluted 1:500 in 1% NGS-PBS was incubated for overnight at 4°C followed by Alexa 488-conjugated anti-mouse antibody (Molecular Probes). The slides were sealed using Vectashield mounting medium with DAPI (Vectashield) for nuclei counterstaining. Images were obtained with a SPOT digital camera (Model 1.3.0, Diagnostic Instruments, Inc.) attached to Zeiss Axioskop microscope (Zeiss Plan-neofluor 100X/1, 30 oil objective lenses, Carl Zeiss Inc.).
Directed In Vivo Angiogenesis assays
Wild type and homozygous motheaten viable (mev/mev) mice were generated from the intercrosses of mev/+ mice and genotyped by PCR detection of wild type and mutant Shp-1 loci from the tail genomic DNA as previously reported (15). Quantification of the angiogenic responses in both wild type and motheaten viable mice were performed using the directed in vivo angiogenesis assay (DIVAA) as previously described (6,16). Briefly, angioreactors were filled with cold matrigel alone (negative controls), or matrigel containing 500 ng/mL of either FGF-2 or VEGF-A. To test for in vivo antiangiogenic activity, angioreactors were filled with matrigel containing FGF-2 or VEGF-A plus either TIMP-2 or Ala+TIMP-2 (200 nM). Filled angioreactors were implanted subcutaneously in the dorsal flank of wt or mev/mev mice. Angioreactors were recovered at day 11 following tail vein injection of 100 μL of 25 mg/mL FITC-dextran (MW 150 kDa). All data are obtained using a single angioreactor per mouse with experiments performed in quadruplicate. Negative controls show slight background fluorescence but contain no vessels or endothelial cells upon histological examination (16).
Results
TIMP-2 treatment results in pRb hypo-phosphorylation and inhibits G1 to S phase transition
In previous studies we demonstrate that TIMP-2 treatment of hMVECs prior to stimulation with angiogenic growth factors, such as FGF-2 or VEGF-A, inhibits both 3H-thymidine incorporation and cell growth (6,17). These studies suggest that TIMP-2 prevents entry of cells into the S phase of the cell cycle, which is usually associated with mitogenic growth factor stimulation. In the G1 phase of the cell cycle, retinoblastoma protein (pRb) is sequentially phosphorylated by cyclin-dependent kinase (Cdk) activities, and this hyper-phosphorylation of pRb controls the transition from G1 to S phase of the cell cycle (18). We examined the effect of TIMP-2 pretreatment on the pRb phosphorylation status in hMVECs. Specifically, we analyzed the phosphorylation of pRb on residues S780 and S807/811, sites specific for Cdk4 and Cdk2 phosphorylation, respectively (19). As anticipated, FGF-2 stimulation for 24 h significantly increases pRb phosphorylation at both sites when compared with quiescent hMVECs. However, pretreatment with TIMP-2 or the Ala+TIMP-2 mutant, lacking MMP inhibitory activity, completely blocks FGF-2-induced pRb phosphorylation to levels observed in untreated cells, Figure 1A. This finding is consistent with TIMP-2 preventing S phase entry, possibly through inhibition of Cdk activities.
Figure 1. TIMP-2 Induces G1 Arrest.
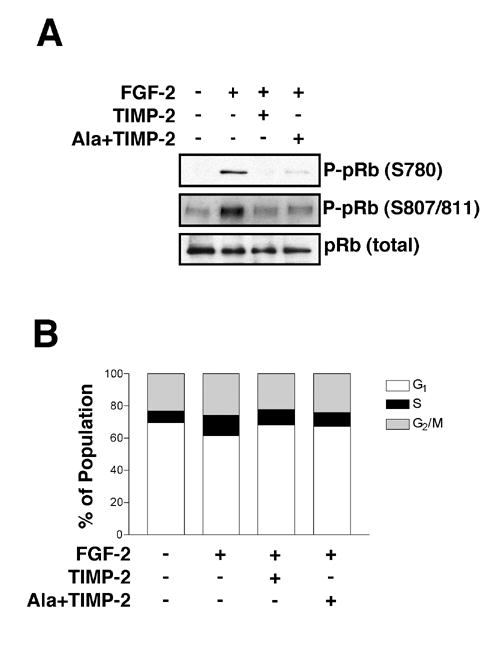
(A) TIMP-2 treatment inhibits the phosphorylation of pRb. Following FGF-2 (50 ng/ml) stimulation for 24 h with or without TIMP-2 and Ala+TIMP-2 (50 nM), cell lysates were separated by SDS-PAGE and Western blotted for phospho-pRb and pRb.
(B) Quiescent hMVECs were pretreated with TIMP-2 or Ala+TIMP-2 for 15 min followed by FGF-2 (50 ng/ml) stimulation for 24 h. DNA content was determined by flow cytometry. Results shown are representative of at least two independent experiments.
In addition, we studied the effects of TIMP-2 or Ala+TIMP-2 pretreatment on cell cycle by DNA content flow cytometry. Cell cycle analysis of hMVECs following FGF-2 stimulation for 24 h increases the percentage of cells in S phase approximately two-fold compared with untreated cells (6.6 % vs. 12.2%), along with a concomitant reduction of cells in G1 phase (70 % vs. 62 %), Figure 1B. This twofold increase in S phase is consistent with our previous cell growth experiments (6). However, pretreatment with TIMP-2 or Ala+TIMP-2 prior to FGF-2 stimulation prevents the anticipated increase in S phase associated with FGF-2 mitogenic stimulation and leads to a corresponding increase in cells in G1 phase (70%) compared to hMVECs treated with FGF-2 alone (62 % in G1). Pretreatment with either TIMP-2 or Ala+TIMP-2 alone did not alter the cell cycle pattern of hMVECs when compared with untreated cells (data not shown). These data are consistent with the pRb phosphorylation studies (Figure 1A), as well as previous reports demonstrating that TIMP-2 inhibits 3H-thymidine incorporation upon FGF-2 stimulation (17). Collectively, these data suggest that TIMP-2 pretreatment inhibits the G1 to S phase transition of the cell cycle, resulting in G1 arrest.
TIMP-2 enhances de novo synthesis of p27Kip1
It is currently appreciated that transition of cells from G1 to S phase specifically requires activation of Cdks, such as Cdk4 and Cdk2 through complex formation with their cognate regulatory subunits, cyclin D and cyclin E, respectively (18). The kinase activity of these complexes is regulated by Cdk inhibitors (CKIs), such as p21WAF1/Cip1 and p27Kip1, which inhibit a wide range of Cdks, as well as p15 and p16 members of the INK family that selectively inhibit cyclin D/Cdk4/6 complexes (20). Enhanced expression of these inhibitors attenuates proliferative responses in a variety of cells, whereas reduced levels of the CKIs are associated with S phase entry and increased cellular proliferation. Thus, we analyzed the expression of CKIs associated with the G1 to S phase transition. Following FGF-2 stimulation for 24 h the levels of p27Kip1 expression are markedly reduced to ~20 % of levels observed in untreated cells. In contrast the levels of p16INK4A and p21WAF1/Cip1 show little or no change following FGF-2 stimulation alone. This decrease in p27Kip1 following mitogenic stimulation is expected, as previous studies have demonstrated increased proteasomal degradation of this protein prior to S phase entry (21).
Ala+TIMP-2 prevented the FGF-2-mediated decrease in p27Kip1 protein levels, Figure 2A. Ala+TIMP-2 pretreatment also slightly reduced the levels of p21WAF1/Cip1, but no change in p21WAF1/Cip1 levels was observed following FGF-2 treatment alone. These findings suggest that TIMP-2 may suppress FGF-2-stimulated hMVEC proliferation by augmenting expression of the Cdk inhibitor p27Kip1. Increased cellular levels of p27Kip1 are indicative of G1 cell cycle arrest, as previously demonstrated (22), and are consistent with the G1 arrest of endothelial cell growth following TIMP-2 treatment.
Figure 2. TIMP-2 Enhances Expression of Cdk Inhibitor p27Kip1.
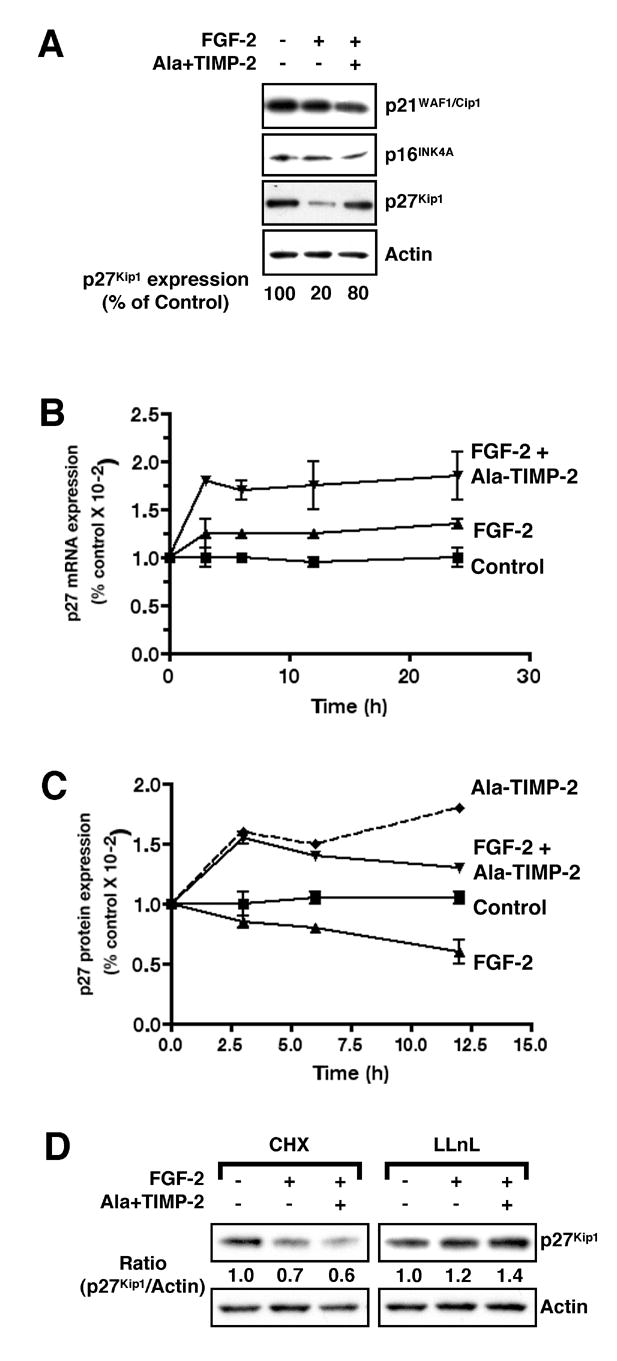
hMVECs stimulated with FGF-2 in the presence or absence of Ala+TIMP-2 for different time periods, as indicated, were subjected to Northern and Western blot analysis.
(A) Cells collected at 24 h time point were Western blotted with anti-Cdk inhibitors antibodies. Expression levels of p27Kip1 were normalized to actin loading control.
(B) TIMP-2 induces transcriptional activation of p27Kip1. Untreated hMVECs (control) were stimulated with FGF-2 in the absence (FGF-2) or presence (FGF-2 + Ala-TIMP-2) of Ala+TIMP-2 for the time indicated. Average p27 Kip1 mRNA ± SEM is expressed as the change relative to that observed in untreated hMVECs. Data shown are the results from three independent experiments.
(C) TIMP-2 increases p27Kip1 protein expression. Ala+TIMP-2 treatment results in increases of p27Kip1 protein level with (FGF-2 + Ala-TIMP-2) or without (Ala-TIMP-2) subsequent FGF-2 stimulation, compared to that in untreated (control) or FGF-2-stimulated (FGF-2) hMVECs. Values represent mean ± SEM from three independent experiments.
(D) TIMP-2 induction of p27Kip1 expression requires de novo protein synthesis. Cells were treated with Ala+TIMP-2 (50 nM), FGF-2 (50 ng/ml) alone or both Ala+TIMP-2 and FGF-2 in the presence of cycloheximide (CHX, 10 μM) or N-acetyl-Leu-Leu-norleucinal (LLnL, 20 μM) for 6 h. Results shown are representative of two independent experiments.
The regulation of cellular p27Kip1 levels is complex, involving both de novo protein synthesis and/or protein degradation via the proteasomal pathway (23). To examine possible mechanisms via which TIMP-2 increases cellular p27Kip1 levels we examined the time course of p27Kip1 expression during the growth inhibition experiments. FGF-2 stimulation of hMVECs results in a slight increase (< 30%) in p27Kip1 mRNA expression over 24 h, Figure 2B, whereas FGF-2 decreased p27Kip1 total protein levels below those observed in controlcells, Figure 2C. These findings are consistent with the patterns of p27Kip1 mRNA and total cellular protein levels observed following stimulation by a variety of mitogenic factors (21).
In contrast, pretreatment with Ala+TIMP-2 prior to FGF-2 stimulation results in statistically significant increases in both p27Kip1 transcripts and protein levels, Figure 2B and 2C. This increase is detectable as early as 3 h, with maximal p27Kip1 transcript levels increased ~2-fold over untreated cells. Treatment of hMVECs with Ala+TIMP-2 prior to FGF-2 stimulation results in a > 60 % increase in p27Kip1 protein levels as early as 3 h that gradually decreased over the time course of this experiment. hMVECs treated only with TIMP-2 show increased levels of p27Kip1 protein that remain elevated throughout the time course of these experiments. These findings suggest that treatment with FGF-2 and Ala+TIMP-2 stimulates de novo protein synthesis as evidenced by the increase in total cellular p27Kip1 protein and transcript levels at the early (3-6 h) time points. However, the cellular levels of p27Kip1 present at 24 h (Figure 2A) are the result of balance between de novo synthesis and protein turnover.
To directly examine the contribution of de novo synthesis of p27Kip1 to the TIMP-2-mediated growth arrest of hMVECs we studied TIMP-2-mediated changes in p27Kip1 levels in the presence of the protein synthesis inhibitor cycloheximide (CHX, 10 μM). CHX treatment completely abrogated the TIMP-2-mediated induction of p27Kip1 expression in FGF-2 stimulated hMVECs, Figure 2D. In contrast, the proteasome inhibitor lysyl-lysyl-norleucine (LLnL) prevented the marked decrease in p27Kip1 protein levels normally associated with FGF-2 stimulation, but had little effect on p27Kip1 protein levels in Ala+TIMP-2 treated cells. These findings are entirely consistent with previous reports demonstrating that mitogenic signaling and S phase entry require reduced cellular p27Kip1 levels (21) and that agents that block mitogenic responses enhance p27Kip1 levels either through transcriptional activation and/or inhibition of protein degradation (24). Our findings suggest that the TIMP-2-mediated increase of p27Kip1 levels is predominantly due to de novo protein synthesis, as evidenced by the CHX effect. Furthermore, inhibition of proteasomal processing in cells treated with Ala+TIMP-2 had no detectable effect of p27Kip1 levels. This is in contrast to the effect of the proteasome inhibitor, LLnL, following FGF-2 in which the levels of p27Kip1 protein are markedly increased. These findings suggest that the levels of p27Kip1 in Ala+TIMP-2 treated cells are the result of a balance of de novo protein synthesis and protein turnover by a non-proteasome-dependent pathway.
TIMP-2 increases p27Kip1 association with Cdk complexes and inhibits Cdk activity
To investigate if the increase in p27Kip1 expression observed following Ala+TIMP-2 treatment is involved in cell cycle regulation, we performed p27Kip1 immunoprecipitation and Western blot analysis for Cdks. We also examined the functional consequence of p27Kip1 association with these Cdks. Ala+TIMP-2 treatment enhanced the association of p27Kip1 with both Cdk4 and Cdk2 compared with control and growth factor treated cells, Figure 3A. Furthermore, the kinase activity of both Cdk4 and Cdk2 complexes are reduced in Ala+TIMP-2 treated cells, again compared with both control and growth factor treated cells. Figure 3B. The association of p27Kip1 with Cdk4 and Cdk2 complexes results in a 26 % and 37 % reduction in pRb-C phosphorylation, respectively, when compared with untreated control cells. However, the actual decreases in the kinase activity of these complexes is much greater when compared with FGF-2 stimulation alone, resulting in a 33% and 52 % decrease in activity for the Cdk4 and Cdk2 complexes, respectively. Collectively, these findings demonstrate that TIMP-2-mediated inhibition of hMVEC cell proliferation in response to FGF-2 is due to the loss of Cdk activity that occurs through increased de novo synthesis of the CKI p27Kip1. These findings are consistent with pRb hypo-phosphorylation and decrease in S phase observed in our prior experiments. These observations show that the formation of p27Kip1-inhibitory complexes with specific Cdk/cyclin complexes involved in the G1 to S phase transition are important in TIMP-2-mediated growth inhibition.
Figure 3. Increased Association of p27Kip1 with Cdks Mediates TIMP-2-induced Cell Growth Arrest.
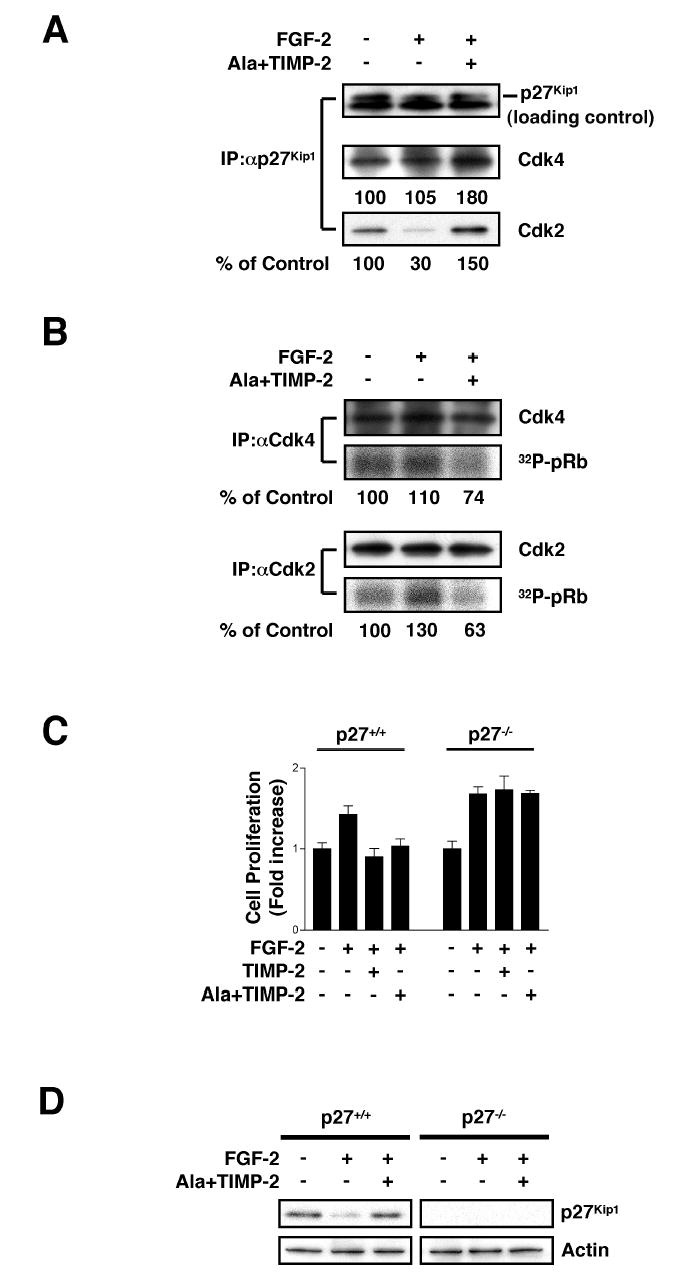
(A) TIMP-2 increases p27Kip1 association with Cdk4 and Cdk2. Following FGF-2 stimulation (24 h) with or without Ala+TIMP-2, anti-p27Kip1 immunoprecipitates were resolved by SDS-PAGE and Western blotted with anti-Cdk4, anti-Cdk2, or anti-p27Kip1 antibodies. The results shown are representative of two independent experiments.
(B) Enhanced association of p27Kip1 with Cdk4 and Cdk2 by Ala+TIMP-2 treatment results in inhibition of pRb kinase activity. Following FGF-2 stimulation for 24 h with or without Ala+TIMP-2, the kinase activity of Cdk4 and Cdk2 immunoprecipitates was measured using Rb-C fusion protein as substrate in the presence of [γ-32P]ATP/ATP and presented as the percentage of unstimulated control. The results shown are representative of two independent experiments.
(C) p27−/− murine embryonic fibroblasts (MEFs) are resistant to TIMP-2 inhibition of cell growth. Both p27+/+ and p27−/− MEFs were treated with FGF-2 (50 ng/ml) in the absence or presence of TIMP-2 (or Ala+TIMP-2) (100 nM). Cell growth assays were conducted following 48 h incubation. Values represent mean ± SD of triplicate determinations.
(D) Ala+TIMP-2 treatment induces p27Kip1 expression in p27+/+ MEFs. Cells were pretreated with Ala+TIMP-2 prior to FGF-2 stimulation for 48 h. The Western blot results are representative of two independent experiments.
TIMP-2 inhibition of cell growth is impaired in cells lacking p27Kip1
To demonstrate the absolute requirement for p27Kip1 in the mechanism of TIMP-2-mediated growth arrest, we examined TIMP-2 effects on growth of p27+/+ and p27−/− immortalized murine embryonic fibroblasts (MEFs), Figure 3C. Both of these cells are Shp-1 positive by Western blot analysis of whole cell lysates (data not shown). TIMP-2 and Ala+TIMP-2 treatment completely inhibits FGF-2-stimulated proliferation in p27+/+ cells. However, p27−/− cells are unresponsive to TIMP-2 growth inhibitory effect. The suppressive effect of TIMP-2 on cell growth in p27+/+, and loss in p27−/− cells are also observed following PDGFs-timulated growth of these MEFs (data not shown). We next performed Western blot analysis of p27Kip1 expression in both p27+/+ and p27−/− cells. The levels of p27Kip1 expression following FGF-2 stimulation in p27+/+ cells are markedly reduced (~80%) compared to levels in untreated cells. Ala+TIMP-2 pretreatment of p27+/+ MEFs induced p27Kip1 expression and prevented the FGF-2-induced reduction, Figure 3D. These findings are identical to previous results using hMVECs and confirm that p27Kip1 is a critical regulator of TIMP-2-mediated cell growth arrest.
TIMP-2 induction of p27Kip1 expression requires α3β1 integrin and PTP activity
We have previously demonstrated that TIMP-2 binding to α3β1 integrin receptor is required for hMVEC growth inhibition (6). To investigate whether α3β1was required for TIMP-2 induction of p27Kip1, we examine the effects of TIMP-2 on p27Kip1 expression levels in β1-null MEFs (GD25 cells) and following siRNA disruption of α3 integrin subunit expression in A549 human tumor cells. A549 cells were utilized in these siRNA experiments because of difficulty transfecting primary endothelial cell cultures. Again, both of these cell lines are Shp-1 positive (data not shown).
First we examined the expression of p27Kip1 following mitogenic stimulation of integrin β1-null MEFs (GD25 cells). We have previously demonstrated that these GD25 cells respond in vitro to PDGF as a mitogen and that PDGF-stimulated growth of these cells is resistant to TIMP-2 inhibition, whereas cells with reconstituted β1 expression (GD25-ß1A cells) are responsive to TIMP-2 inhibition of PDGF-stimulated growth (6). Treatment of β1-null MEFs (GD25 cells) with TIMP-2 prior to PDGF, not only failed to increase, but actually shows a decrease in p27Kip1 expression, Figure 4A. This is in contrast to the >50% increase in p27Kip1 levels observed in MEFs with reconstituted β1 integrin subunit expression (GD25-b1A cells), Figure 4A. Targeted disruption of α3 expression by RNA silencing successfully reduced expression of total α3 subunit > 85% as evidenced by Western blot analysis, Figure 4B. This reduction in integrin α3 subunit expression is sufficient to block Ala+TIMP-2-mediated induction of p27Kip1 expression in A549 human tumor cells, Figure 4B. However, reduced expression of α3 did not prevent the expected FGF-2 reduction in p27Kip1 expression that occurs through enhanced proteasome processing. Together these findings from the β1-null and α3 siRNA-transduced cells clearly demonstrate that α3β1 integrin signaling events are upstream of p27Kip1.
Fig. 4. TIMP-2 Induction of p27Kip1 Expression Requires α3β1 Integrin and PTP Activity.
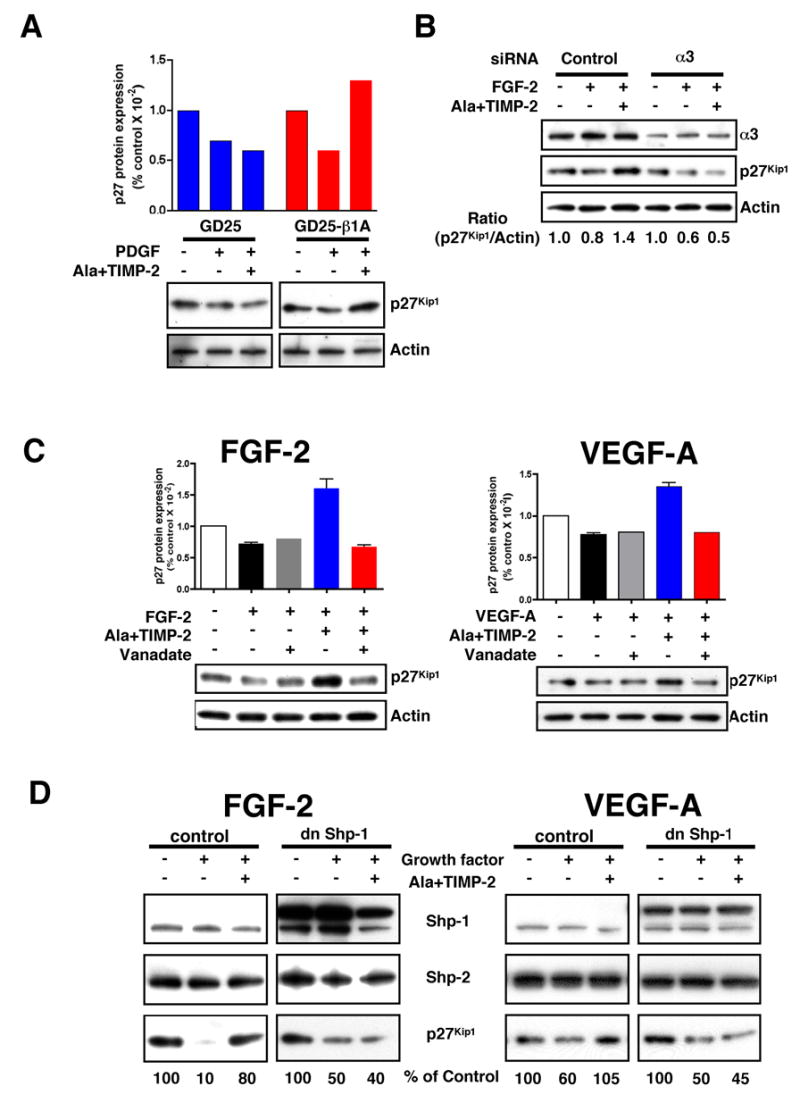
(A) TIMP-2 does not induce the expression of p27Kip1 levels in β1-deficient MEFs. Both GD25 and GD25-β1A MEFs were pretreated with Ala+TIMP-2 (100 nM), followed by PDGF (100 ng/ml) treatment for 24 h. The results shown are representative of two independent experiments with each cell line.
(B) Integrin α3 siRNA abrogates TIMP-2 induction of p27Kip1. A549 lung carcinoma cells were transfected with integrin α3 siRNA or control (scrambled) siRNA. Cells were pretreated with Ala+TIMP-2 (100 nM) and followed by FGF-2 (100 ng/ml) for 24 h. The results shown are representative of two independent experiments with similar results.
(C) The effect of PTP inhibitor orthovanadate on TIMP-2-induced increase in p27Kip1 levels. Orthovanadate (1 μM) reverses TIMP-2 induction of p27Kip1 on FGF-2 and VEGF-A stimulation at 6 h time point. Values represent mean ± SEM from three experiments, and the Western blots are representative.
(D) TIMP-2 induction of p27Kip1 level is dependent on Shp-1 activity. In dn Shp-1 transduced hMVECs, Ala+TIMP-2 fails to induce p27Kip1 protein level in response to FGF-2 and VEGF-A stimulation for 24 h. Vector control and dn mutant Shp-1 cell lysates were Western blotted with anti-Shp-1, anti-Shp-2, or anti-p27Kip1 antibodies. Western blots shown are representative of two independent experiments with similar results.
We next determined if TIMP-2-mediated induction of p27Kip1 also requires PTP activity. Initially, we examined the effect of the PTP inhibitor orthovanadate on p27Kip1 expression following TIMP-2 treatment of hMVECs. Pretreatment of cells with 1 μM orthovanadate completely abrogates the TIMP-2-mediated increase in p27Kip1 expression, Figure 4C, left panel, but has little effect on cells treated with FGF-2 alone. Previously we have demonstrated that TIMP-2 and Ala+TIMP-2 also inhibit hMVEC proliferation in response to VEGF-A mitogenic stimulation (6). In hMVECs stimulated with VEGF-A, Ala+TIMP-2 pretreatment did increase p27Kip1 expression, which was also abrogated by orthovanadate, Figure 4C. These findings are in agreement with our prior observations that TIMP-2-induced suppression of FGFR-1- and VEGFR-2-induced mitogenic responses in hMVECs is PTP-dependent (6). Furthermore, these findings also implicate enhanced p27Kip1 expression in TIMP-2-mediated growth arrest of hMVECs following VEGF-A stimulation. In contrast to observed changes in p27Kip1, no significant changes in expression of Cdk4, cyclin D, cyclin D3, p53 tumor suppressor protein, or p21WAF1/Cip1 are observed following orthovanadate pretreatment (data not shown), suggesting that induction of p27Kip1 levels by TIMP-2 is selectively controlled by PTP activity.
TIMP-2 induction of p27Kip1 is mediated by Shp-1 activity
Shp-1 is an Src-homology 2 (SH2)-containing PTP that is recruited to multiple receptor complexes as a negative regulator of cell growth in many cell types (25). To examine the requirement for Shp-1 in TIMP-2-mediated induction of p27Kip1 and hMVEC growth arrest in vitro we analyzed the effects of a dominant-negative (dn), catalytically inactive Shp-1 mutant on the TIMP-2-mediated increase in p27Kip1 levels. Endogenous Shp-1 protein is expressed in vector control hMVECs, and the transduced dn Shp-1 gene was evaluated by the enhancement in immunoreactive Shp-1 protein that is not associated with an increase in total PTP activity (6). Western blot analysis of control and dn Shp-1 transduced cells demonstrates that expression of dn-Shp-1 did not alter expression of Shp-2, a related PTP that is associated with positive growth regulation, Figure 4D. FGF-2 or VEGF-A stimulation did not alter the expression of Shp-1 or Shp-2 in vector control hMVECs, Figure 4D. The p27Kip1 protein levels in the presence of FGF-2 or VEGF-A alone in vector control hMVECs were reduced to similar levels observed in previous experiments. In control cells, 24 h treatment with Ala+TIMP-2 and FGF-2 results in similar levels of p27Kip1 (Figure 4D, left panel) as observed in wild type hMVECs (Figure 2A). However, in dn Shp-1 transduced hMVECs, Ala+TIMP-2 failed to significantly induce p27Kip1 protein expression levels in either FGF-2 (Figure 4D, left panel) or VEGF-A (Figure 4D, right panel) –stimulated hMVECs. This finding implies that Shp-1 activity is specifically required for TIMP-2-mediated p27Kip1 up-regulation.
TIMP-2 fails to inhibit angiogenesis in Shp-1-deficient mice
The requirements for α3β1 and the putative involvement of Shp-1 in mediating TIMP-2-enhanced de novo expression of p27Kip1 are identical to our previous characterization of the mechanism for TIMP-2 inhibition of angiogenesis in vivo. These findings suggest that Shp-1 is a critical mediator of the anti-angiogenic effects of TIMP-2. To test the requirement for Shp-1 in mediating TIMP-2 inhibition of angiogenesis in vivo we utilized the motheaten viable (mev/mev) mouse model (26). The mev/mev mouse is the result of a spontaneous mutation in the Shp-1 gene that results in severe immunologic defects and autoimmune disorder. These mice express two forms of Shp-1 that have only 20 % of normal Shp-1 activity, but survive to 12 weeks of age in contrast with the motheaten mice which have no detectable levels of Shp-1 activity and a lifespan of only 3 weeks (26).
Angiogenic responses to both FGF-2 and VEGF-A, as well as the ability of TIMP-2 or Ala+TIMP-2 to inhibit these responses were examined in both wild type and m ev/mev strains using the DIVAA system as previously described (6,16). The results of these experiments show that in wild type mice both FGF-2 and VEGF-A induce typical angiogenic responses that are both significantly inhibited (~60%-100% inhibition) by inclusion of either TIMP-2 or Ala+TIMP-2, Figure 5. In homozygous m ev/mev mice the angiogenic response to FGF-2 is similar to that in wild type animals, although this angiogenic response is now resistant to inhibition with TIMP-2 or Ala+TIMP-2, and is actually enhanced by these reagents. This is in contrast to the angiogenic response to VEGF-A in mev/mev which is exaggerated compared with that observed in the wild type animals, but is also resistant to inhibition by Ala+TIMP-2, Figure 5. The lack of sensitivity to the angio-inhibitory activity of TIMP-2 or Ala+TIMP-2 in the mev/mev mice is consistent with our proposed role for Shp-1 as a mediator of negative growth regulation in endothelial cells.
Figure 5. In Vivo Effect of TIMP-2 and Ala+TIMP-2 on Angiogenesis in mev/mev Mice.
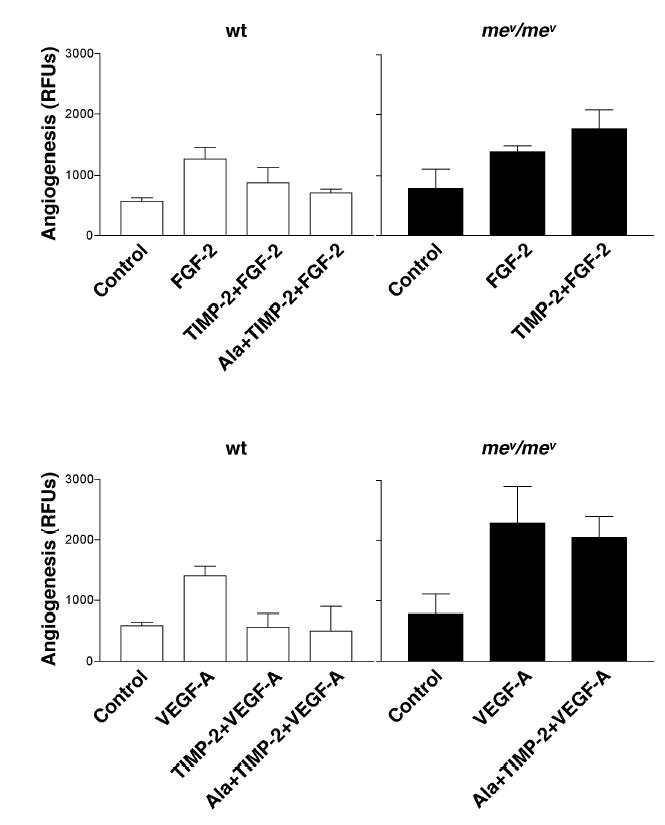
FGF-2 or VEGF-A (each 500 ng/ml) induces significant angiogenic responses in both wild type and homozygous mev/mev mice. However, the suppressive effect of both TIMP-2 and Ala+TIMP-2 are abrogated in homozygous mev/mev mice. Values represent mean of triplicate determinations ± SD.
TIMP-2 induces p27Kip1 nuclear localization
The regulation of p27Kip1 protein expression and localization are complex, but p27Kip1 must be present in the cell nucleus to inhibit the G1 to S phase transition. The levels of p27Kip1 are regulated via transcriptional activation, post-translational modifications (selective phosphorylation) and proteolytic turnover via a proteasome-dependent pathway (21). Nuclear localization of p27Kip1 is controlled by phosphorylation at either residue S10 or T187. Phosphorylation at either site facilitates nuclear export of p27Kip1 to the cytoplasm, resulting in a low level of nuclear p27Kip1. This facilitates the G1 to S phase transition by releasing Cdk activity from p27Kip1 inhibition (27,28). Phosphorylation of p27Kip1 on T187 generates a binding site for Skp2-containing E3 ubiquitin-protein ligase (SCF) that results in subsequent proteasome-dependent degradation (29).
To investigate TIMP-2-mediated regulation of p27Kip1 localization, we first analyzed the phosphorylation levels of total cellular p27Kip1. Following FGF-2 treatment alone there is approximately a 2-3-fold increase in phosphorylation of p27Kip1 on T187 over basal levels, as demonstrated by the increase in the ratio of T187-phospho-p27Kip1 to total p27Kip1, Figure 6A. TIMP-2 pretreatment prior to FGF-2 decreased p27Kip1 phosphorylation levels below those observed in control hMVECs, Figure 6A. Decreased T187 phosphorylation of p27Kip1 following TIMP-2 pretreatment is consistent with enhanced nuclear localization of p27Kip1, as phosphorylation at this site results in increased association with Skp2 and subsequent proteasome-dependent turnover (29).
Figure 6. TIMP-2 Regulation of p27Kip1 Expression and Localization.
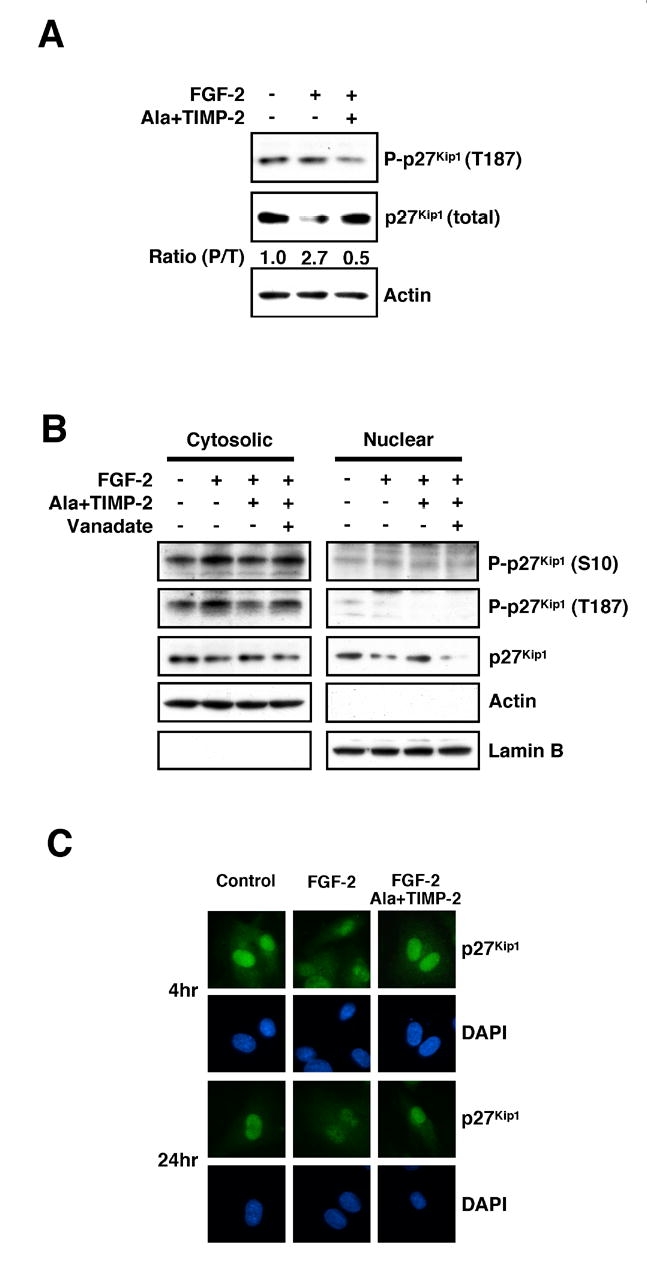
(A) Cell lysates from untreated, FGF-2 alone, or Ala+TIMP-2 and FGF-2-treated hMVECs are Western blotted for phospho-T187-p27Kip1, p27Kip1, or Skp2 at 24 h time point. Integrated density values (and ratios) are determined using NIH Image 1.6 software.
(B) TIMP-2 reduces phosphorylation of p27Kip1 on S10 and T187 in cytosolic fraction and increases nuclear p27Kip1.
(C) TIMP-2 enhances nuclear localization of p27Kip1. The cellular localization of p27Kip1 was detected with anti-p27Kip1 antibody. DNA was stained with DAPI.
p27Kip1 localization in the nuclear compartment was also examined directly by Western blot analysis of nuclear and cytosolic extracts of FGF-2-stimulated hMVECs with and without Ala+TIMP-2 pretreatment. As anticipated, phospho-T187- or phospho-S10-p27Kip1 were not readily detected in the nuclear compartment, Figure 6B, following FGF-2 with or without Ala+TIMP-2 pretreatment. In contrast, Ala+TIMP-2 pretreatment prior to FGF-2 results in a slight decrease in phospho-T187- and phospho-S10-p27Kip1 localization in the cytosolic compartments, Figure 6B. Total p27Kip1 was decreased in the nuclear compartment following FGF-2 alone, but increased with Ala+TIMP-2 pretreatment prior to FGF-2 stimulation, Figure 6B. The increase in nuclear localization of p27Kip1 induced by TIMP-2 pretreatment is abrogated by inclusion of the PTP inhibitor orthovanadate, Figure 6B.
To confirm the changes in nuclear localization of p27Kip1 observed in these experiments we also examined p27Kip1 localization by immunofluorescence staining. In quiescent, untreated hMVECs, anti-p27Kip1 staining reveals strong nuclear localization with weak but detectable cytoplasmic staining at both the 4 and 24 h time points, Figure 6C. In contrast FGF-2 alone shows a slight decrease in the nuclear localization of p27Kip1 at the 4 h time point that is further accentuated at 24 h, Figure 6C. Ala+TIMP-2 pretreatment prior to FGF-2 results in enhanced nuclear localization of the p27Kip1 signal, with weak staining of the cytoplasm at both the 4 and 24 h time points. These data confirm the shift from cytoplasmic to nuclear localization of p27Kip1 following Ala+TIMP-2 treatment observed by Western blotting of cytosolic and nuclear extracts, Figure 6B. Collectively, these findings demonstrate that in addition to inducing de novo synthesis of p27Kip1 protein, TIMP-2 also enhances nuclear localization of p27Kip1.
Discussion
In this study we demonstrate that TIMP-2 prevents endothelial cell growth in vitro by induction of de novo synthesis of p27Kip1. Enhanced levels of p27Kip1 are consistent with the observations that TIMP-2-induced growth arrest of hMVECs is characterized by decreased 3H-thymidine uptake (17), inactivation of growth factor receptors and enhanced PTP activity (6), as well as hypo-phosphorylation of pRb and a decrease in cells entering S phase. Collectively, these data support the concept that TIMP-2 inhibition of endothelial cell growth is the result of G1 cell cycle arrest. Furthermore, TIMP-2 growth arrest and induction of p27Kip1 are both mediated by α3β1 integrin, and inhibited by PTP inhibitors or expression of dn Shp-1. Finally, TIMP-2 anti-angiogenic activities against FGF-2/VEGF-A-mediated responses in vivo are ablated in Shp-1-deficient (mev/mev) mice, demonstrating that Shp-1 is important for regulation of angiogenesis.
The role of integrins in the regulation of cell attachment, migration, proliferation and survival are well established. Recent studies have described several systems in which reciprocal crosstalk between growth factor receptors and integrins may influence cell behavior and stimulate cell growth in a growth factor-independent manner. However, few examples of negative growth regulation or heterologous receptor inactivation (growth suppression) by integrin receptors have been reported. In our previous report we demonstrate that TIMP-2 interaction with α3β1 results in inactivation of both FGFR-1 and VEGFR-2 (6). This inactivation was associated with a decrease in integrin associated PTP activity and an increase in the association of Shp-1 with receptor tyrosine kinases (RTKs). These findings led us to suggest that this mechanism is an example of integrin-mediated heterologous receptor inactivation. A recent report demonstrates the association of the PTP known as T cell PTP, or TCPTP, with the α1 subunit of the collagen-binding integrin α1β1 (30). These authors demonstrate that the cytoplasmic tail of the α1 integrin subunit selectively interacts with TCPTP and activates this PTP activity following cell adhesion to collagen. Integrin-mediated activation of TCPTP results in reduced EGFR phosphorylation following EGF stimulation (30). Thus, integrin initiated, PTP-mediated inactivation of RTK appears to be an emerging concept. However, little is known about down-stream signaling events that may also accompany this new mechanism of RTK suppression.
Few examples of negative regulation of cell growth mediated by integrin receptors have been reported. These include suppression of cell growth following expression of α5β1 in human colorectal carcinoma cells (31), or expression of specific isoforms of the integrin β1 subunits, such as β1c or β1d, in fibroblasts, myoblasts or tumor cells (32,33). Many of these studies were directed at identifying the growth inhibitory domains in the cytoplasmic tail of these integrins and details of the signaling mechanisms involved were not investigated. One report implicates p27Kip1 as a downstream effector of β1c growth inhibition of prostate cells via suppression of cyclin A-dependent kinase activity (34). This study reports a strong correlation between expression of β1c and increased p27Kip1 expression in cells from human prostate carcinoma tissues. However, the mechanism of enhanced p27Kip1 expression was not investigated.
In the present investigation we extend the observations of Matilla et al. (30) and Fornaro et al. (34), as well as those of our previous reports on TIMP-2 suppression of hMVEC growth (6) and inhibition of hMVEC migration (5). We demonstrate a critical role for both α3 and β1 integrin subunits, and Shp-1 activity in mediating the induction of p27Kip1 during hMVEC growth arrest in vitro, and conclude that TIMP-2-mediated suppressive effect on cell cycle progression is exerted through up-regulation of p27Kip1. Enhanced p27Kip1 localization in the nucleus inactivates Cdk4/cyclin D and Cdk2/cyclin E complexes, resulting in pRb hypo-phosphorylation. This effect is mediated by orthovanadate-sensitive PTP activity and is independent of MMP inhibition. Furthermore, studies using expression of dn Shp-1 specifically implicate this PTP activity in mediating TIMP-2 inhibition of mitogenic responses.
Shp-1 is a member of the family of non-transmembrane PTPs that contain SH2 domains known as the Shps. There are two mammalian Shps, Shp-1 and Shp-2, each having two N-terminal SH2 domains, a classic PTP domain and C-terminal tail containing a nuclear localization signal. Both Shp-1 and Shp-2 are expressed in a variety of cell types including hematopoeitic, epithelial and endothelial cells (25). Shp-2 activity is primarily associated with enhanced cell growth, however, Shp-1 activity is associated with specific negative growth regulatory pathways (25). In endothelial cells Shp-1 activation by tumor necrosis factor alpha (TNF-a) inhibits proliferative responses to growth factors such as VEGF and EGF, whereas expression of dn SHP-1 prevented TNF-a inhibition of endothelial cell growth (35). Similarly, our previous report implicates Shp-1 in mediating the observed inactivation of FGFR-1 and VEGFR-2 (6). However, these reports did not examine potential downstream signaling mechanisms by Shp-1 in mediating cell growth arrest. In fact, identification of downstream targets of Shp-1 has been difficult in many systems, although identification of potential targets has been an objective of investigations in this area for several years (25). Furthermore, potential involvement of Shp-1 in integrin-mediated signaling has not been examined in depth. It is reported that bone-marrow macrophages from Shp-1-deficient mice are strongly adherent to ß1- and ß2-integrin ligands, suggesting that Shp-1 is necessary for cell detachment from these substrates (36). Shp-1 deficient macrophages demonstrate a 2-5-fold increase in membrane-associated phosphatidylinositol (PI) 3-kinase activity and inhibition of this activity resulted in dramatic detachment of adherent cells. These studies suggest that Shp-1 regulates PI 3-kinase activity, specifically membrane-associated PI 3-kinase, which in turn modulates integrin-mediated attachment. These findings are consistent with reports demonstrating PI 3-kinase involvement in integrin inside-out signaling that regulates integrin receptor affinity (37), however, this study did not specifically identify the target of Shp-1 activity.
The motheaten viable mouse model (m ev/mev) is deficient in Shp-1 activity, has multiple hematopoietic cell defects that result in inflammatory processes that affect multiple organ systems, and result in premature death of these animals secondary to severe interstitial pneumonia at 9-12 weeks of age (26). However, to our knowledge there are no prior reports on altered angiogenic responses in m ev/mev animals. Furthermore, the failure of m ev/mev to limit angiogenic responses in the presence of TIMP-2 is consistent with the loss of Shp-1 function in these mice.
The results of the present study implicate the TIMP-2 interaction with α3β1 resulting in Shp-1 activation. However, our focus in this report is on the downstream effects following activation of Shp-1 following the binding of TIMP-2 toα3β1. We demonstrate that TIMP-2 interaction with α3β1 results in concomitant induction of p27Kip1 transcriptional activation, de novo synthesis of p27Kip1 and enhanced p27Kip1 nuclear localization. These effects are dependent on PTP activity, and are lost upon forced expression of dn Shp-1. Finally angiogenic responses to VEGF-A and FGF-2 in the Shp-1 deficient mouse model (motheaten viable mice) are resistant to inhibition by TIMP-2, demonstrating that Shp-1 is a principal mediator of the anti-angiogenic activity of TIMP-2 in vivo.
This is the first demonstration that interaction of TIMP-2 with its cognate receptor, α3β1 integrin, mediates the induction of Cdk inhibitor p27Kip1 via an Shp-1-dependent mechanism. Collectively, the data support the concept that TIMP-2 interaction with α3β1 initiates a signaling cascade mediated by the PTP Shp-1 (Figure 7). Activation of this signal cascade results in three distinct mechanisms that function to resist endothelial cell ‘activation’ and preserve endothelial cell homeostasis. These three mechanisms are: 1) Shp-1-mediated inactivation of ‘angiogenic’ receptors such as FGFR-1 and VEGFR-2 (6); 2) Shp-1 activation of Rap1 signaling resulting in enhanced expression of RECK and suppression of endothelial cell migration (5); 3) Shp-1-dependent induction of p27Kip1 which results in pRb hypo-phosphorylation and prevents entry to the S phase of the cell cycle. These findings suggest that TIMP-2 is a key regulator of endothelial cell proliferation, migration and angiogenesis, and that Shp-1 is a new potential therapeutic target in angiogenesis that warrants further investigation.
Figure 7. Proposed TIMP-2 Signaling Pathway.
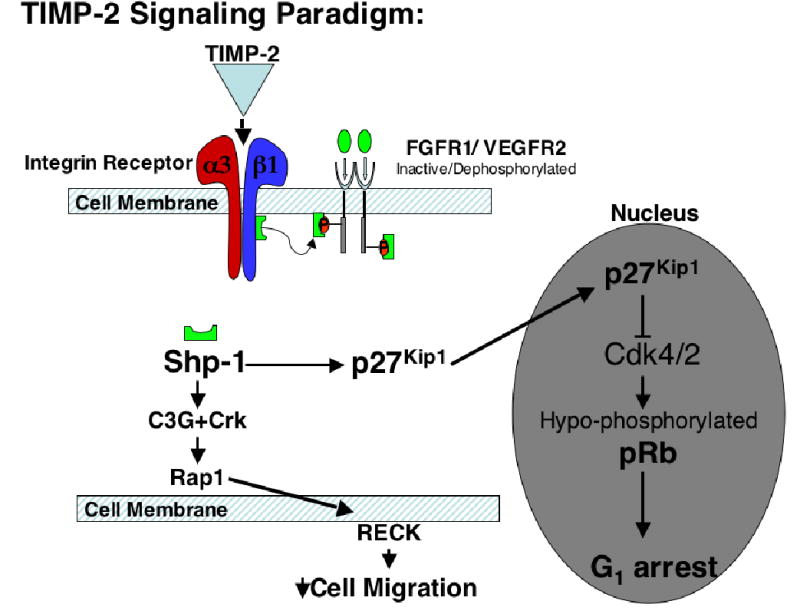
TIMP-2 binds to a3b1 resulting in activation of Shp-1. Shp-1 activation is required for: 1) inhbition of angiogenic receptor activation (6); 2) activation of Rap1 and downstream increase in RECK expression which inhibits hMVEC migration (5); 3) induction of p27Kip1 expression resulting in inhibition of cell cycle progression in hMVECs.
Acknowledgments
The authors thank Gregory A. Jasper and Dr. Maryalice Stetler-Stevenson, NCI, NIH for flow cytometry analysis, Dr. Deane F. Mosher, University of Wisconsin-Madison for the GD25 and GD25-β1A cells, and Drs. Matthew L. Fero and James M. Roberts, Fred Hutchinson Cancer Research Center for p27−/− murine embryonic fibroblasts. We also thank Drs. Manfred Boehm and Elizabeth Nabel, NHLBI, NIH for providing p27Kip1–phospho-S10 antibodies. This work was supported by intramural research funds from the National Cancer Institute, Center for Cancer Research (Project # Z01SC 009179).
References
- 1.Carmeliet P. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 2.Kalluri R. Nat Rev Cancer. 2003;3:422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 3.Chang C, Werb Z. Trends Cell Biol. 2001;11:S37–43. doi: 10.1016/s0962-8924(01)02122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visse R, Nagase H. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 5.Oh J, Seo DW, Diaz T, Wei B, Ward Y, Ray JM, Morioka Y, Shi S, Kitayama H, Takahashi C, Noda M, Stetler-Stevenson WG.(2004) Cancer Research64, 9062–9069, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei BY, Stetler-Stevenson WG. Cell. 2003;114:171–180. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez CA, Butterfield C, Jackson G, Moses MA. J Biol Chem. 2003;278:40989–40995. doi: 10.1074/jbc.M306176200. [DOI] [PubMed] [Google Scholar]
- 8.Liu XW, Bernardo MM, Fridman R, Kim HR. J Biol Chem. 2003;278:40364–40372. doi: 10.1074/jbc.M302999200. [DOI] [PubMed] [Google Scholar]
- 9.Hoegy SE, Oh HR, Corcoran ML, Stetler-Stevenson WG. J Biol Chem. 2001;276:3203–3214. doi: 10.1074/jbc.M008157200. [DOI] [PubMed] [Google Scholar]
- 10.Wingfield PT, Sax JK, Stahl SJ, Kaufman J, Palmer I, Chung V, Corcoran ML, Kleiner DE, Stetler-Stevenson WG. J Biol Chem. 1999;274:21362–21368. doi: 10.1074/jbc.274.30.21362. [DOI] [PubMed] [Google Scholar]
- 11.Guedez L, Stetler-Stevenson WG, Wolff L, Wang J, Fukushima P, Mansoor A, Stetler-Stevenson M. J Clin Invest. 1998;102:2002–2010. doi: 10.1172/JCI2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guedez L, McMarlin AJ, Kingma DW, Bennett TA, Stetler-Stevenson M, Stetler-Stevenson WG. American Journal of Pathology. 2001;158:1207–1215. doi: 10.1016/S0002-9440(10)64070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akahane T, Akahane M, Shah A, Connor CM, Thorgeirsson UP. Experimental Cell Research. 2004;301:158–167. doi: 10.1016/j.yexcr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, Baker A, Anand-Apte B. Nature Medicine. 2003;9:407–415. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 15.Kim CH, Qu CK, Hangoc G, Cooper S, Anzai N, Feng GS, Broxmeyer HE. Journal of Experimental Medicine. 1999;190:681–690. doi: 10.1084/jem.190.5.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guedez L, Rivera AM, Salloum R, Miller ML, Diegmueller JJ, Bungay PM, Stetler-Stevenson WG. Am J Pathol. 2003;162:1431–1439. doi: 10.1016/S0002-9440(10)64276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy AN, Unsworth EJ, Stetler-Stevenson WG. J Cell Physiol. 1993;157:351–358. doi: 10.1002/jcp.1041570219. [DOI] [PubMed] [Google Scholar]
- 18.Malumbres M, Barbacid M. Nat Rev Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 19.Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Mol Cell Biol. 2001;21:4773–4784. doi: 10.1128/MCB.21.14.4773-4784.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vidal A, Koff A. Gene. 2000;247:1–15. doi: 10.1016/s0378-1119(00)00092-5. [DOI] [PubMed] [Google Scholar]
- 21.Servant MJ, Coulombe P, Turgeon B, Meloche S. J Cell Biol. 2000;148:543–556. doi: 10.1083/jcb.148.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato JY, Matsuoka M, Polyak K, Massague J, Sherr CJ. Cell. 1994;79:487–496. doi: 10.1016/0092-8674(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 23.Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, Fusco A, Santoro M. Nat Med. 2002;8:1136–1144. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- 24.Chen F, Harrison LE. Ceullular Signaling. 2005;17:809–816. doi: 10.1016/j.cellsig.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Neel BG, Gu H, Pao L. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 26.Kozlowski M, Mlinaric-Rascan I, Feng GS, Shen R, Pawson T, Siminovitch KA. J Exp Med. 1993;178:2157–2163. doi: 10.1084/jem.178.6.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. Embo J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, Nabel EG. Embo J. 2002;21:3390–3401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrano AC, Eytan E, Hershko A, Pagano M. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 30.Mattila E, Pellinen T, Vuorluoto K, Arjonen A, Ivaska J. Nature Cell Biology. 2005;7:78–85. doi: 10.1038/ncb1209. [DOI] [PubMed] [Google Scholar]
- 31.Varner JA, Emerson DA, Juliano RL. Mol Biol Cell. 1995;6:725–740. doi: 10.1091/mbc.6.6.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belkin AM, Retta SF. J Biol Chem. 1998;273:15234–15240. doi: 10.1074/jbc.273.24.15234. [DOI] [PubMed] [Google Scholar]
- 33.Meredith JE, Jr, Kiosses WB, Takada Y, Schwartz MA. J Biol Chem. 1999;274:8111–8116. doi: 10.1074/jbc.274.12.8111. [DOI] [PubMed] [Google Scholar]
- 34.Fornaro M, Tallini G, Zheng DQ, Flanagan WM, Manzotti M, Languino LR. J Clin Invest. 1999;103:321–329. doi: 10.1172/JCI4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakagami H, Cui TX, Iwai M, Shiuchi T, Takeda-Matsubara Y, Wu L, Horiuchi M. Arterioscler Thromb Vasc Biol. 2002;22:238–242. doi: 10.1161/hq0202.104001. [DOI] [PubMed] [Google Scholar]
- 36.Roach TI, Slater SE, White LS, Zhang X, Majerus PW, Brown EJ, Thomas ML. Current Biology. 1998;8:1035–1038. doi: 10.1016/s0960-9822(07)00426-5. [DOI] [PubMed] [Google Scholar]
- 37.Berrier AL, Mastrangelo AM, Downward J, Ginsberg M, LaFlamme SE. Journal of Cell Biology. 2000;151:1549–1560. doi: 10.1083/jcb.151.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]