

Abstract
Formins induce the nucleation and polymerisation of unbranched actin filaments via the formin-homology domains 1 and 2. Diaphanous-related formins (Drfs) are regulated by a RhoGTPase-binding domain situated in the amino-terminal (N-terminal) region and a carboxy-terminal Diaphanous-autoregulatory domain (DAD), whose interaction stabilises an autoinhibited inactive conformation. Binding of active Rho releases DAD and activates the catalytic activity of mDia. Here, we report on the interaction of DAD with the regulatory N-terminus of mDia1 (mDiaN) and its release by Rho•GTP. We have defined the elements required for tight binding and solved the three-dimensional structure of a complex between an mDiaN construct and DAD by X-ray crystallography. The core DAD region is an α-helical peptide, which binds in the most highly conserved region of mDiaN using mainly hydrophobic interactions. The structure suggests a two-step mechanism for release of autoinhibition whereby Rho•GTP, although having a partially nonoverlapping binding site, displaces DAD by ionic repulsion and steric clashes. We show that Rho•GTP accelerates the dissociation of DAD from the mDiaN•DAD complex.
Keywords: actin polymerisation, Diaphanous, Diaphanous autoregulatory domain, formin, RhoGTPase
Introduction
Cellular processes like cell migration, cytokinesis, maintenance of cell polarity, vesicular trafficking, morphology and phagocytosis are driven by the dynamics of the actin cytoskeleton (Pantaloni et al, 2001; Pollard and Borisy, 2003). Actin filaments are polar structures characterised by a fast-growing barbed and a slow-growing pointed end. They are regulated by extracellular stimuli that activate signalling pathways, including Rho GTP-binding proteins, which in turn, upon activation, can stimulate the actin nucleation and polymerisation machinery (Bishop and Hall, 2000; Pollard and Borisy, 2003; Higgs and Peterson, 2005). So far, three conserved mechanisms of actin nucleation–elongation involving WASP/WAVE-Arp2/3, formins and the recently described Drosophila protein Spire have been identified (Pollard and Borisy, 2003; Quinlan et al, 2005).
Formins generate linear, unbranched actin cables and stress fibres (Pruyne et al, 2002; Zigmond, 2004; Higgs, 2005). They are characterised by formin homology (FH) domains, where the FH1 and FH2 domains are responsible for the actin nucleation and polymerisation activity (Evangelista et al, 2003; Zigmond et al, 2003). Most formins have additional domains mediating the regulation of the polymerisation activity and are grouped into seven subfamilies: Diaphanous (Dia), dishevelled-associated activator of morphogenesis (DAAM), formin-related gene in leukocytes (FRL), formin-homology domain-containing protein (FHOD), inverted formin (INF), formin (FMN) and delphilin (Higgs, 2005). Some of these contain an FH3 domain, which is the structurally and functionally least conserved FH domain (Wallar and Alberts, 2003; Zigmond, 2004) and is believed to mediate the subcellular localisation of mDia proteins (Kato et al, 2001; Ozaki-Kuroda et al, 2001).
The central catalytic element of formins is the FH2 domain, which nucleates actin filament formation and regulates filament elongation (Higgs and Peterson, 2005). The FH2 domain is the catalytically active element of formins and is sufficient in vitro for actin nucleation (Pruyne et al, 2002; Li and Higgs, 2003; Shimada et al, 2004; Xu et al, 2004; Higgs and Peterson, 2005). In contrast to tight capping proteins, which do not allow any actin dynamics to occur at their binding site, the FH2 domain forms a leaky cap at the barbed end and allows processive elongation of actin filaments (Wear et al, 2003; Zigmond et al, 2003; Harris et al, 2004; Kovar and Pollard, 2004; Kovar et al, 2005). The structures of a dimeric FH2 domain construct of the yeast formin Bni1p and a shorter construct of mDia1 (p140mDia) revealed that the core FH2 domain is all helical (Shimada et al, 2004; Xu et al, 2004). The difference between these structures is a short, unstructured region at the N-terminus, which is of high importance for dimerisation and converts the FH2 domain from a micromolar inhibitor to a nanomolar inducer of actin polymerisation (Shimada et al, 2004), in line with previous findings on the role of FH2 domains (Pring et al, 2003; Zigmond et al, 2003; Harris et al, 2004; Xu et al, 2004). The structure of the dimeric FH2 domain with TMR-actin shows that each dimer of FH2 contacts three actin molecules and suggests a mechanism of processive barbed-end elongation in the presence of FH2 (Otomo et al, 2005a).
The proline-rich stretches of the FH1 domain are potential targets for SH3- and WW-domain-containing proteins, but also for profilin (Bedford et al, 1997; Macias et al, 2002; Pring et al, 2003). The actin–profilin complex constitutes the major G-actin pool in the cell and in most cases requires the FH1 domain for efficient polymerisation via the FH2 domain (Otomo et al, 2005a). Binding of the profilin–actin complex to the FH1 domain mediates fast barbed-end elongation by an yet unexplained mechanism that may include high local concentration near the polymerisation site (Zigmond et al, 2003; Romero et al, 2004). It has also been suggested that the increased ATP hydrolysis rate of actin induced by the FH1–FH2 unit decreases the affinity of profilin for actin (Higashida et al, 2004; Romero et al, 2004).
In addition to the FH1 and FH2 domains, Diaphanous-related formins (Drfs) contain an N-terminally located GTPase-binding domain (mDiaN) and a C-terminally located Diaphanous-autoregulatory domain (DAD) (Figure 1A) (Watanabe et al, 1997; Alberts, 2001; Otomo et al, 2005b; Rose et al, 2005b). The Drosophila protein Diaphanous, which plays an important role during cytokinesis (Castrillon and Wasserman, 1994), the Saccharomyces cerevisiae proteins Bni1p and Bnr1p, and the mammalian homologues of Diaphanous, mDia1–3, belong to this subgroup (Wallar and Alberts, 2003). In the absence of an activating signal, Drfs are in an inactive autoinhibited conformation due to the interaction between the N-terminal regulatory region and DAD (Alberts, 2001; Li and Higgs, 2005; Otomo et al, 2005b; Rose et al, 2005b). Lacking the N-terminal RhoGTPase-binding domain (GBD) and/or the C-terminal DAD mDia1 is constitutively active and induces formation of stress fibres and SRF-dependent nuclear transcription (Watanabe et al, 1999; Ishizaki et al, 2001; Copeland and Treisman, 2002; Li and Higgs, 2003). The intrasteric inhibition of Drfs is relieved by binding of active Rho proteins to the regulatory region. Rho proteins are Ras-related GTP-binding proteins, which act as molecular switches that cycle between an inactive GDP- and an active GTP-bound conformation. In the latter state, they interact with downstream effectors defined as proteins, which bind tightly to the GTP-bound conformation only (Vetter and Wittinghofer, 2001; Etienne-Manneville and Hall, 2002). A common mechanism by which Rho proteins activate effectors is the release of an intramolecular inhibitory interaction, which can be found in many effector proteins such as WASP and the Ser/Thr kinases PAK, ROK and PKN (Maesaki et al, 1999; Millard et al, 2004). In the case of PAK, upon binding of active Rac or Cdc42, an intramolecular regulatory domain is displaced from the active site and thus allows substrate binding (Lei et al, 2000, 2005). For WASP, it is the interaction of the N-terminal CRIB domain with parts of the C-terminal VCA motif that is relieved by Cdc42 (Buck et al, 2004; Leung and Rosen, 2005). In vitro, the interaction of the regulatory N-terminus (mDiaN) with DAD of mDia1 is released by active Rho, thus mimicking the formin activation reaction (Li and Higgs, 2005; Rose et al, 2005b), although some studies suggest that Rho•GTP may not be quite sufficient for full relief of autoinhibition (Li and Higgs, 2003, 2005).
Figure 1.
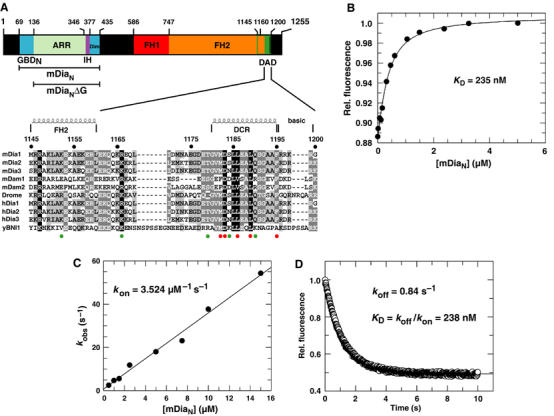
N- and C-terminal constructs of mDia1 and their interaction. (A) Schematic representation of the domain structure of mDia1 and the constructs used. mDiaN, the regulatory region; GBDN, GTPase-binding domain; mDiaNΔG, the regulatory region minus GBDN; ARR, armadillo-repeat region; IH, interdomain helix; Dim, dimerisation domain; FH1-2, formin-homology domains 1, 2; DAD, Diaphanous-autoregulatory domain. Shown below is an alignment of the DAD1145−1200 fragment from different genes and organisms (accession numbers in brackets): mDia1–3: mouse (O08808; Q9Z207; O70566); mDam1–2: mouse (Q8BPM0; Q80U19); Dia: Drosophila (P48608); hDia1–3, human (O60610; O60879; Q9NSV4); yBNI1, Saccharomyces cerevisiae (P41832). Black shaded residues are conserved in 100%, dark grey shaded in 80% and bright grey shaded in 60% of the depicted organisms at the specific position. Furthermore, residues with similar physiochemical properties are combined into groups. Residues 1145–1160 might belong to the FH2 domain and have a helical structure as shown previously (Shimada et al, 2004); the structure of the DCR (residues 1180–1195) was determined here. The red and green circles below the alignment represent residues whose mutations do or do not influence affinity towards mDiaN, respectively. (B) Determination of affinity between F-DAD1145−1200 and mDiaN using fluorescence. F-DAD1145−1200 (100 nM) was titrated with increasing concentration of mDiaN and the change in fluorescence obtained is plotted against the concentration of mDiaN. The data were fitted to a quadratic binding equation. (C) Association kinetics of F-DAD1145−1200 and mDiaN as determined by stopped flow. F-DAD is reacted with increasing concentrations of mDiaN and the observed first-order rate constants (kobs) are plotted against the mDiaN concentration. The slope of the linear fit represents kon. (D) The dissociation rate constant koff is determined by incubating 100 nM of a preformed F-DAD•mDiaN complex in the presence of a large excess of unlabelled DAD peptide. The fluorescence transient is fitted to a single-exponential decay.
Recently, the structures of mDiaN (residues 69–451) alone and in complex with RhoC•GppNHp were solved by X-ray crystallography (Otomo et al 2005b; Rose et al, 2005b), revealing that the all-helical mDiaN consists of the three subdomains: GBDN (the N-terminal part of the Rho-binding domain), the armadillo-repeat region (ARR) and a C-terminal dimerisation domain (Dim). Surprisingly, mutational and NMR studies showed that the DAD and Rho•GTP binding sites on the mDiaN surface were apparently not overlapping, suggesting that the mutual exclusive binding of Rho and DAD is not simply due to steric exclusion (Otomo et al, 2005b; Rose et al, 2005b). In order to more clearly identify the DAD binding site and to clarify the structural basis for regulation of the Drf subfamily of proteins, we solved the three-dimensional structure of the complex between mDiaNΔG (residues 136–451) and DAD encompassing residues 1145–1200. Using a number of biochemical and biophysical measurements, we can propose a two-step binding mechanism for the release of autoinhibition by Rho•GTP.
Results
The mDiaN binds DAD with submicromolar affinity
The N-terminal regulatory domain of mDia1, mDiaN, binds Rho•GTP and the C-terminal DAD in a mutually exclusive manner, where Rho•GTP binds with a low nanomolar affinity to mDiaN (Rose et al, 2005b). In order to compare Rho with DAD binding towards mDiaN, we determined the kinetic and equilibrium constants of DAD binding using isothermal titration calorimetry (ITC) and fluorescence-based equilibrium and kinetic measurements. For these, a fluorescently labelled DAD fragment (F-DAD) was made by coupling 1,5-Iaedans to an extra cysteine on the C-terminus of the DAD fragment encompassing residues 1145–1200 (DAD1145−1200) (Figure 1A). Binding of the fluorescent F-DAD to mDiaN leads to an increase in fluorescence, presumably due to transfer of the fluorescence label to a more hydrophobic environment. This fluorescence change was used for equilibrium binding measurements, by titrating F-DAD with increasing concentrations of mDiaN to obtain a KD of 235 nM (Fig 1B). Furthermore, active site titration using concentrations of reactants above the KD (3 μM; roughly 13-fold above the KD) shows that DAD binds with a clear 1:1 stoichiometry (data not shown).
To analyse the dynamics of the reaction, we measured the association and dissociation rate constants kon and koff by stopped-flow. For association, F-DAD was mixed with increasing concentrations of mDiaN, and the observed rate constants kobs were plotted against the mDiaN concentration. This plot was linear over the concentration range and gives, from the slope, an association rate constant kon of 3.5 μM−1 s−1 (Figure 1C). The dissociation rate constant was obtained by following the fluorescence decrease on incubating the preformed F-DAD•mDiaN complex with an excess of unlabelled DAD1145−1200 to obtain a koff of 0.84 s−1 (Figure 1D). The resulting dissociation equilibrium constant KD (koff/kon) is 238 nM, in good agreement with the equilibrium titration experiment (Figure 1C).
For an independent measurement of affinity and to get more information on the thermodynamics of binding, we measured the interaction of DAD with mDiaN via ITC, which gives an equilibrium dissociation constant (KD) of 109 nM and a stoichiometry of 1:1 (Figure 2A and Table I). The affinity is twofold higher than determined by fluorescence, which is due to the fluorescence label at the C-terminus of DAD, since an ITC experiment using mDiaN and F-DAD resulted in an affinity of 249 nM (data not shown). Quite unusually for a protein–protein interaction, the mDiaN–DAD1145−1200 interaction is highly endothermic, with an enthalpy change ΔH of 10.2 kcal mol−1. The thermodynamically unfavourable enthalpy of the reaction is compensated by the highly favourable (positive) entropic contribution (T ΔS=19.5 kcal mol−1), which is the driving force of the reaction.
Figure 2.
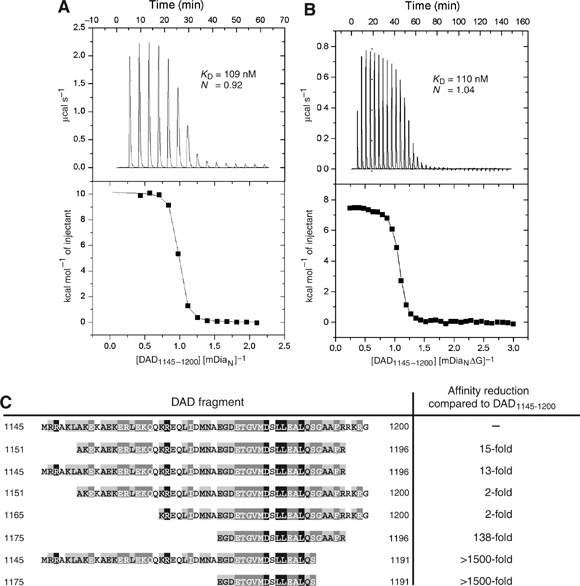
ITC analysis of the DAD–mDiaN interaction. ITC of the interaction between DAD1145−1200 and either mDiaN (A) or mDiaNΔG (B), measured by titrating 40 μM/30 μM mDiaN/mDiaNΔG in the chamber with 940 μM/400 μM DAD1145−1200 in the syringe. Top panels, raw heating power over time; bottom panels, fit of the integrated energy values normalised for injected protein. (C) Result of affinity analysis of different DAD fragments for mDiaN, measured by ITC as in (A, B) and indicated as affinity reduction relative to the ‘full-length' DAD1145−1200 fragment.
Table 1.
Thermodynamic properties of binding of mDiaN to DAD1145–1200, as determined by ITC
| Mutation | KD (nM) | ΔH (kcal mol−1) | T ΔS (kcal mol−1) | N |
|---|---|---|---|---|
| WT | 109±10 | 10.16±0.05 | 19.5 | 0.9 |
| N165D | 130±6 | 10.17±0.03 | 19.4 | 1.0 |
| N217A | 8470±697 | 8.01±0.28 | 14.8 | 1.0 |
| A256D | ⩾1 440 000a | |||
| I259D | ⩾232 000a | |||
| E264K | 91±9 | 10.16±0.05 | 17.9 | 1.1 |
| D366A | 153±6 | 7.81±0.02 | 16.9 | 1.0 |
| R269E | 90±5 | 9.5±0.03 | 18.9 | 1.1 |
| K1152A | 117±7 | 8.69±0.03 | 18.0 | 1.1 |
| R1166A | 288±13 | 7.92±0.03 | 16.7 | 0.9 |
| T1179A | 180±8 | 9.51±0.04 | 18.6 | 0.9 |
| T1179D | 500±19 | 9.85±0.05 | 18.3 | 1.1 |
| M1182A | 88 500±3370 | 17.92±1.22 | 23.3 | 1.0 |
| M1182D | ⩾200 000a | |||
| D1183R | 4100±81 | 9.03±0.04 | 16.3 | 1.5 |
| D1183N | 934±24 | 12.09±0.49 | 20.2 | 1.0 |
| S1184D | 216±17 | 6.77±0.06 | 15.7 | 0.9 |
| L1186A | 1830±91 | 8.10±0.08 | 15.8 | 1.0 |
| L1189A | 9804±865 | 9.06±0.52 | 15.8 | 1.1 |
| Q1190A | 114±7 | 8.19±0.04 | 17.5 | 1.2 |
| F1195A | ⩾150 000a | |||
| mDiaNΔG DADWT |
110±7 |
7.43±0.03 |
16.8 |
1.0 |
| N is the stoichiometry of binding. The last row shows the characteristics concerning the mDiaNΔG–DAD1145–1200WT interaction. | ||||
| a Affinity of A256D, I259D, F1195A and M1182D is too low for accurate measurements of enthalpy, entropy and stoichiometry. | ||||
The core DAD binding site on mDiaN
The N-terminal part of the Rho binding site (GBDN) is detached from the rest of the ARR of mDiaN (see Figure 2 in Rose et al, 2005b). We thus wondered about the contribution of this fragment to DAD binding, expressed mDiaNΔG, and measured its interaction with DAD1145−1200. It binds with an affinity of 110 nM (stoichiometry 1:1), very similar to mDiaN, and the enthalpy is likewise unfavourable, with ΔH 7.4 kcal mol−1 and a compensating entropy term of 16.8 kcal mol−1 (Figure 2B). Thus, the three-helix motif GBDN makes no apparent contribution to the binding of DAD and mDiaNΔG is thus well suited for structural studies on the DAD–mDiaN interaction (see below).
In contrast to mDiaN, DAD binding to mDiaNΔG is not released by RhoA, as shown by a fluorescence polarisation assay. Here, 0.5 μM of a DAD peptide encompassing residues 1175–1196 labelled at the N-terminus with a fluorescent AMCA label (from now: A-DAD) was treated with 2 μM of either mDiaN or mDiaNΔG and the development of polarisation was followed. Due to complex formation and the corresponding increase of mass, the mobility of the fluorophore decreases, which leads to an increase in the polarisation signal (Figure 3). When adding 4 μM of active RhoA•GppNHp to the fluorescent A-DAD–mDiaN complex, polarisation returns to the starting level, whereas addition of RhoA•GDP does not lead to dissociation of the complex, as shown previously under slightly different conditions (Rose et al, 2005b). However, with the A-DAD•mDiaNΔG complex, neither the active nor the inactive Rho can release DAD from the complex, even after addition of a 100-fold excess (data not shown). Using this polarisation assay, the same results could be obtained using the longer F-DAD fragment, which includes the basic residues in the patch 1197–RKRG–1200, which also demonstrates that the nature of the fluorescence label does not influence the results (Supplementary Figure 1). Thus, although part of the Rho binding site is on the ARR region, as shown by the structure of the mDiaN•RhoC complex, the affinity of Rho•GTP for this region is too weak to effectively release DAD, or the mode of binding is inappropriate for such a release.
Figure 3.
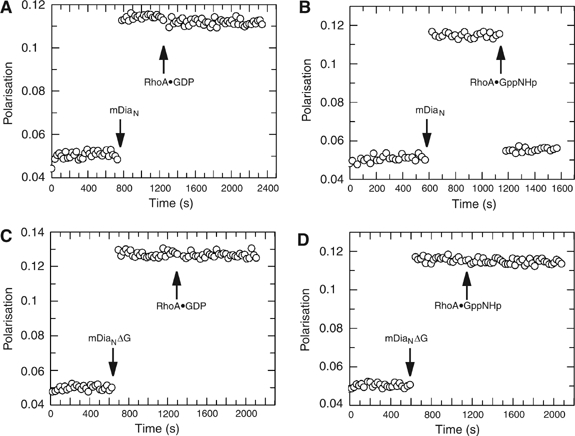
Fluorescence polarisation assay to analyse competitive binding of RhoA or DAD towards mDiaN and mDiaNΔG, respectively. Fluorescently (AMCA)-labelled A-DAD peptide (residues 1175–1196) is incubated with either mDiaN (A, B) or mDiaNΔG (C, D) and leads to increase of the polarisation signal due to the decreased mobility of A-DAD upon complex formation. RhoA•GppNHp (B, D) and RhoA•GDP (A, C) are added as indicated.
Requirements for the DAD–mDiaN interaction
The DAD regions of Drfs can be aligned and show different levels of conservation (Figure 1A), with a central portion that is conserved between Bni1p and mammalian Dia. In UniProt (www.uniprot.org), it is defined as a stretch of 15 amino acids (mDia1: aa 1180–1194) with a large number of identical and conserved residues. In order to sort out the requirements for efficient intramolecular interaction, we screened various DAD constructs and mutants for interaction with mDiaN. Whereas the 17mer DAD1175−1191 was synthesised with and the 22mer DAD1175−1196 peptide with and without a fluorescent label, all the other DAD fragments were recombinantly expressed as GST fusion proteins and cleaved. Using ITC, values for affinity, stoichiometry, enthalpy and entropy of the binding were obtained and are summarised in Table I and Figure 2C. The 22mer containing the highly conserved 15 amino acids of DAD (Figure 1A) has micromolar affinity and is essential for binding. N- and C-terminal elongations of this DAD core lead to an increasing affinity towards mDiaN. The 56mer peptide covering the complete DAD region shows a 138-fold increase in affinity, with the C-terminus having a much larger influence. Comparison of DAD1151−1200 (KD=209 nM), DAD1165−1200 (KD=194 nM) and DAD1145–1196 (KD=1.4 μM) with the fragments DAD1151−1196 (KD=1.6 μM) and DAD1145−1200 (KD=109 nM) demonstrates that the C-terminal residues 1197–RKRG–1200 are mostly responsible for the increased affinities, while the N-terminal residues 1145–1165 have nearly no effect. This is consistent with the findings of Li and Higgs (2005), who found that a 1177–1200 DAD peptide shows a 250 nM affinity towards mDia1 1–548 and 129–548. The synthesised DAD 17mer fragment 1175–1191 and the recombinantly expressed DAD1145−1191 47mer fragment do not show measurable binding affinity in ITC. Even in a qualitative polarisation assay using an AMCA-labelled DAD1175−1191 peptide and F-DAD1145−1191, no increase of polarisation could be detected upon addition of mDiaN, even in a high excess, showing that some of the residues between 1192 and 1200 are essential for binding to mDiaN (data not shown).
Structure of the mDiaNΔG–DAD1145−1200 complex
The mDiaNΔG•DAD1145−1200 complex was purified using a bicistronic expression system in Escherichia coli. The soluble complex could be purified using a GSH-Sepharose column, subsequent cleavage and size exclusion chromatography (S200 16/60). It eluted with an apparent molecular mass of about 80 kDa (data not shown), corresponding to a 2:2 hetero-tetramer, which was crystallised. While no molecular replacement solution could be found for a native data set, the better diffracting Se–Met crystals and the structure of mDiaN in complex with RhoC•GppNHp (Table II; Rose et al, 2005a, 2005b; PDB 1Z2C) were used for phasing by molecular replacement. The anomalous signal of Se–Met1182 was used to identify DAD. A ribbon model of the complete structure shows that the DAD binding site is located on the concave site of the ARR (Figure 4A), as predicted from previous experiments (see Figure 3; Rose et al, 2005b; see also Otomo et al, 2005b). As the site chain density of the DAD peptide corresponding to chain D of the hetero-tetramer was better defined, chain B (mDiaNΔG) and chain D (DAD) will be used for further discussions (Table III).
Table 2.
Data collection statistics
| Wavelength (Å) | 0.950 |
| Resolution (Å) | 3.4 |
| Space group | P6122 |
| Unit-cell parameters | a=138.7, b=138.7, c=210.9 |
| VM (Å3 Da−1) | 3.6 |
| Total measurements | 437 327 |
| Unique reflections | 33 924 |
| Average redundancy | 11.51 |
| I/σ(I) | 21.24 (6.92) |
| Completeness (%) | 99.4 (99.4) |
| Wilson B-factor (Å2) | 77 |
| Rsym (%) | 8.4 |
Figure 4.
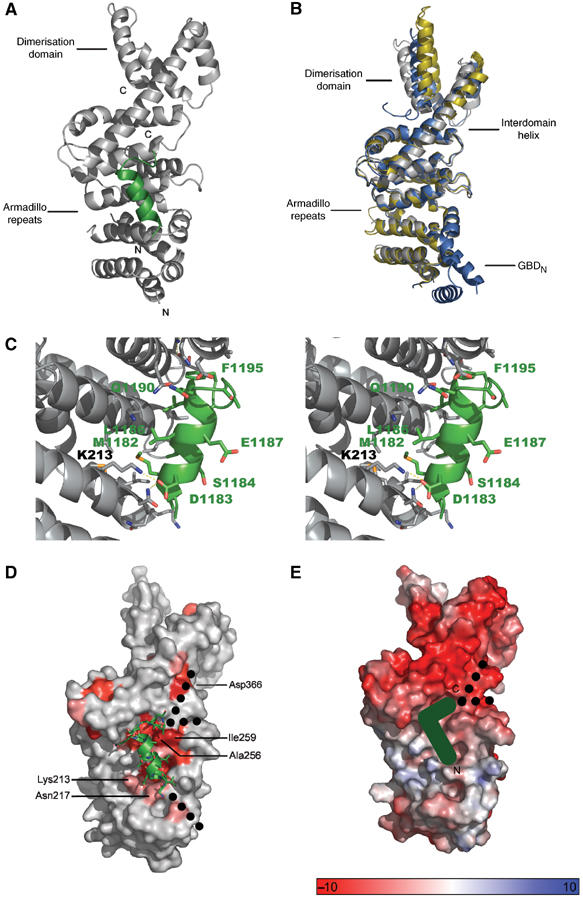
Structure of the mDiaNΔG–DAD complex. (A) Ribbon diagram of the structure, where mDiaNΔG is grey and DAD green. Only residues 1180–1195, the DCR, are visible in the structure. (B) Superimposition of the mDiaNΔG from this structure (grey) and the structures of unbound (yellow) and Rho-bound (blue) mDiaN (Otomo et al, 2005b; Rose et al, 2005b), leaving out RhoC and GBDN. (C) Stereo view of the interface between the ARR and DAD, highlighting residues mentioned in the text. (D) Conservation of residues, where the intensity of red indicates the degree of conservation (accession numbers of compared proteins: mDia1 O08808, mDia2 Q9Z207; mDia3 O70566; hDia1 O60610; hDia2 O60879; DAAM1 human Q9Y4D1; DAAM1 mouse Q8BPM0; DAAM2 human Q86T65; DAAM2 mouse Q80U19; Gallus gallus Dia Q9DEH3; E. histolytica Dia Q514T8; DroMe Dia P48608); dashed lines show the possible paths of the N- and C-terminal extensions of the polypeptide chain; Asp366, Ile259, Ala256, Lys213 and Asn217 are highlighted. (E) Electrostatic potential of the mDiaNΔG surface as calculated with APBS (Baker et al, 2001), position of the DCR and the suspected polypeptide path from N to C as indicated.
Table 3.
Refinement statistics
| Resolution (Å) | 20–3.3 |
| Reflections (working/test set) | 17 565/925 |
| Number of atoms | 4966 |
| Rwork (%)a | 28.8 |
| Rfree (%)b | 36.4 |
| Mean B-value (Å2) | 92.2 |
| R.m.s. deviations from standard geometryc | Counts/r.m.s.d./weight |
| Bond length (Å) | 5031/0.010/0.022 |
| Bond angle (deg) | 6770/1.248/1.990 |
| Torsion angle (deg) | 615/5.907/5.0 |
| Isotropic thermal factor restraints | Counts/r.m.s.d./weight |
| Main-chain bond refined atoms (Å2) | 3192/0.411/1.500 |
| Main-chain angle refined atoms (Å2) | 4974/0.718/2.000 |
| Side-chain bond refined atoms (Å2) | 2018/0.694/3.000 |
| Side-chain angle refined atoms (Å2) |
1796/1.132/4.500 |
| a Rwork=∑∣Fo−Fc∣/∑Fo where Fo and Fc are the observed and calculated structure factor amplitudes. | |
| b Rfree is claculated similarly to Rwork using the test set reflections. | |
| c For definition, see REFMAC5 (www.ysbl.york.ac.uk/~garib/refmac/index.html). | |
The overall structure of mDiaNΔG shows no significant conformational changes for the ARR in comparison to both the structure of mDiaN in complex with RhoC•GppNHp and one subunit of the unliganded form of the protein (Figure 4B and Table IV) (Otomo et al, 2005b; Rose et al, 2005b). The long interdomain helix and the dimerisation domain appear to be more flexible and show the largest differences. In the structure of the unbound form (Otomo et al, 2005b), the long interdomain helix of one of the monomers is interrupted and causes a large distortion of the dimerisation domain, which cannot be observed in the two other structures and the other monomer of the unbound structure (Figure 4B), suggesting that the deviation is due to the extensive crystal contacts at the N-terminus.
Table 4.
RMSD scores of compared mDia-structures
| mDia Molecules compared | Compared | Number of atoms compared | R.m.s.d. (Å) |
|---|---|---|---|
| DAD-bound/Rho-bound | Cα | 246 | 0.87 |
| DAD-bound/unbound | Cα | 226 | 0.7 |
| Rho-bound/unbound | Cα | 241 | 0.8 |
| DAD-bound/Rho-bound | All atoms | 2108 | 1.514 |
| DAD-bound/unbound | All atoms | 2015 | 2.286 |
| Rho-bound/unbound | All atoms | 2064 | 2.078 |
Only 16 (1180–1195) of the 56 residues of the DAD fragment used for crystallisation are visible in the structure (Figure 4C, Supplementary Figure 2). The first 12 of these form an amphipathic helix that makes extensive hydrophobic contacts to the second helices of armadillo-repeats (ARM) two, three and four. At residue 1192, the polypeptide makes a 90° bend and seems to form another helical turn (Figure 4A and C). Apart from the hydrophobic interactions, Asp1183 forms a salt bridge with Lys213 of ARM2 (Figure 4C). The same aspartate is also in contact distance of Asn217 (Table I), which is important for the correct orientation of the ‘arginine wedge' (Arg68) of Rho, being crucial for the binding of the latter (Rose et al, 2005b). Although the precise orientation of the motif SGAA directly C-terminal of the core DAD was difficult to trace at this resolution, Phe1195 following the SGAA motif is clearly defined. It is wedged into the space between the interdomain helix and ARM4 (Figure 4C).
In the structure, DAD is situated on a patch of surface exposed residues with very high conservation between different Drfs down to Entamoeba histolytica (Figure 4D). It is close to, but only marginally overlapping with the Rho binding site (see below), as had been predicted from NMR and biochemical data (Otomo et al, 2005b; Rose et al, 2005b). We have shown above that the basic residues (1196–RRKRG–1200) following the 1191–SGAAF–1195 motif are essential for high-affinity binding (Figure 2C and Table I), but are not visible in the structure of the complex, most likely due to the high salt conditions used in crystallisation, which tend to mask ionic interactions in the crystal. We wondered where this basic stretch of residues could bind and can identify two negatively charged grooves on the interdomain helix or between ARM3 and ARM4, with conserved residues as the possible binding sites (Figure 4D and E). However, since mutation of Asp366 to Ala did not lead to a significant decrease of affinity towards DAD, we are uncertain about the path of the DAD polypeptide on mDiaN (Table I).
The topology of the complex presented here is in analogy to the interactions of catenin with the competing ligands Tcf, cadherin and APC (Graham et al, 2000; Eklof Spink et al, 2001), and to the binding of Ran to the concave side of the helical HEAT repeat motif of its effector β-importin (Chook and Blobel, 1999; Vetter et al 1999), suggesting that the concave side of armadillo or HEAT repeats is a favoured protein–protein interaction surface.
Mutational analysis of the interface
To analyse the interface in more detail, we introduced point mutations into either DAD or mDiaN and measured the affinities using ITC. The hydrophobic residues Phe1195 and Met1182 make major contributions to affinity as their substitution by Ala leads to roughly 800- and 1400-fold reductions in affinity, respectively (Table I). As a control, the mutants T1179A, S1184A and Q1190A with substitutions of residues on the hydrophilic side of the helix have only a minor effect on affinity. The disruption of the salt bridge between Asp1183 and Lys213 led to a 37-fold reduction in affinity, further supporting the notion that hydrophobic interactions provide the major driving force of the interaction between mDiaN and DAD (Table I).
Recently, it was shown structurally and in mutational studies that although Rho•GTP and DAD binding to mDiaN are mutually exclusive, their binding sites do not seem to overlap (Otomo et al, 2005b; Rose et al, 2005b). It was shown that the mutations A256D and I259D in mDiaN affected DAD1145−1200 binding but not RhoA binding, and the mutation N165D affected RhoA binding but not DAD1145−1200 binding (Figure 4D) (Otomo et al, 2005b; Rose et al, 2005b). The affinities of these and additional mDiaN mutants towards DAD1145−1200 were further characterised by ITC measurements. A256D and I259D were drastically reduced indeed to approximately 1.4 mM and 232 μM, respectively, as compared to 109 nM for wild type (Table I). In contrast, mutant N165D, which does affect Rho binding (Rose et al, 2005b), showed wild-type affinity towards DAD1145−1200, with a value 130 nM. While the mutant N217A (KD=8.4 μM) does influence DAD binding, mutations of residues far away from the Rho binding site, such as R269E, E264K and D366A, do not (KD=91, 90 and 153 nM) (Table I).
Rho actively displaces DAD from mDiaN
After localising the DAD binding site by both biochemical and structural analysis, we wondered how Rho•GTP would dissociate the mDiaN•DAD1145−1200 complex, which is a mimic of the autoinhibited full-length mDia1. We and others have shown that Rho•GTP can displace DAD from binding (Figure 3) (Li and Higgs, 2003; Rose et al, 2005b), which is supported by our affinity measurements, that show a 18-fold higher affinity of mDiaN for Rho (KD=6 nM) (Rose et al, 2005b) compared to DAD1145−1200–mDiaN (KD=109 nM). Considering the only slightly overlapping binding sites (see below), the question arises as to why and how Rho•GTP would displace DAD. Does Rho passively compete with DAD for a marginal overlapping binding site on mDiaN and only binds after DAD has dissociated, or does it actively induce DAD dissociation? If the former holds, Rho•GTP binding would be regulated by the kinetics of mDiaN–DAD dissociation, which is 0.84 s−1 for the intermolecular interaction investigated here (Figure 1D), but would be expected to be much slower for the intramolecular interaction of full-length mDia.
Since the structure of the ARR is very similar between the mDiaNΔG•DAD1145−1200 and the mDiaN•RhoC•GppNHp structure (Figure 4B), we can use the superimpositon of the two structures to show that the Rho and the core DAD binding sites are very close to each other, but overlap only slightly (Figure 5A and B). The N-terminal Gly1180 in DAD, the first residue visible in the structure, would probably clash with GBDN upon Rho binding. Thr1179, which is not visible in the structure, would definitely clash with RhoC in any reasonable orientation that we can model into the structure (Figure 5B). In addition, the negative charges of Asp1183 and Glu1187 on DAD1145−1200 would experience electrostatic repulsion from the negative charges of Glu64, Asp65 and Asp67 from Rho. Although this might be sufficient to mediate release of DAD, it might well be that additional residues on the N- or C-terminal extension of the DAD peptide, which we cannot identify in the current structure, would also interfere with Rho. The most likely residues are those on the N-terminal end of DAD, which, although not making any contribution to mDiaN binding (Figure 2), might nevertheless interfere with Rho binding.
Figure 5.
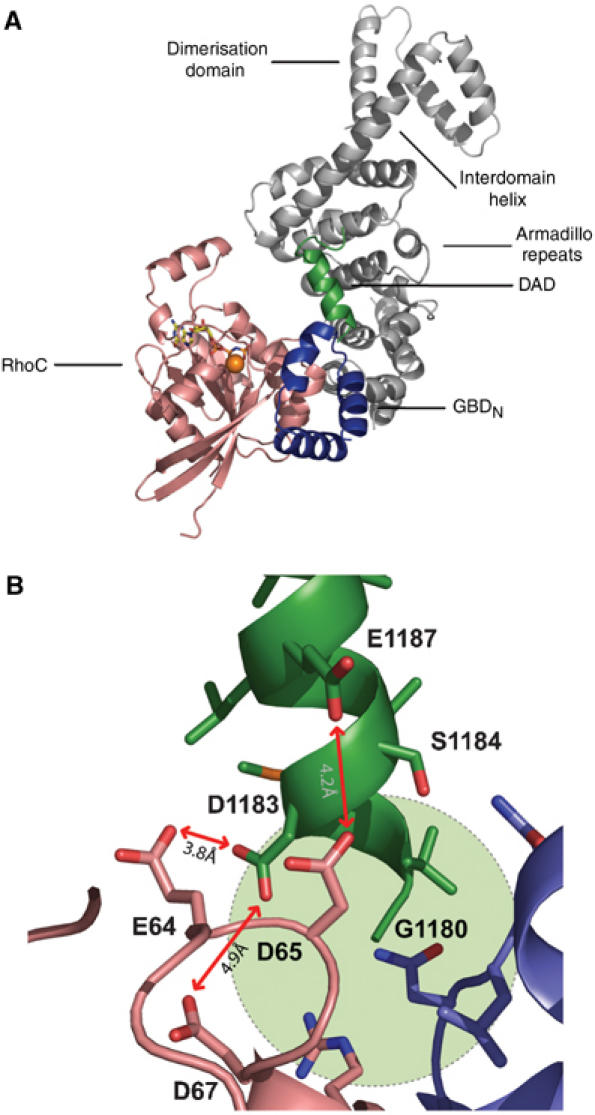
Structural model for release of DAD by Rho binding. (A) Model of a complex of mDiaN with Rho•GTP and the DCR, using the previous RhoC complex structure (Rose et al, 2005b) and the DAD complex structure obtained here and the superimposition shown in Figure 4B. (B) Details of the electrostatic repulsion between DAD and Rho in a proposed ternary mDiaN–Rho–DAD complex; the steric clash on inserting the next DAD residue, Thr1179, is indicated by a green circle.
For a quantitative analysis of competitive binding by ITC, the mDiaN•DAD1145−1200 complex (30 μM mDiaN; 60 μM DAD1145−1200) was titrated with RhoA•GppNHp (Figure 6A). The primary heat changes were fitted using a simple one-site binding model. DAD is released with an apparent affinity IC50 of 29 μM, with a molar ratio of 1:1 for the RhoA•GppNHp mDiaN binding (Figure 6A). The enthalpy of the reaction was –9.8 kcal mol−1 (Figure 6A), which is close to (but with opposite sign) the highly endothermic ΔH (+10.6 kcal mol−1) obtained for the binding of DAD1145−1200 to mDiaN (Figure 2A). Direct ITC measurements of RhoA•GppNHp binding to mDiaN at different concentrations and temperatures did not lead to a measurable signal. This could be due to the binding of RhoA•GppNHp to mDiaN having an endothermic and an exothermic part, which neutralise each other (e.g. due to conformational change), or that the high-affinity binding reaction is indeed solely entropy driven.
Figure 6.
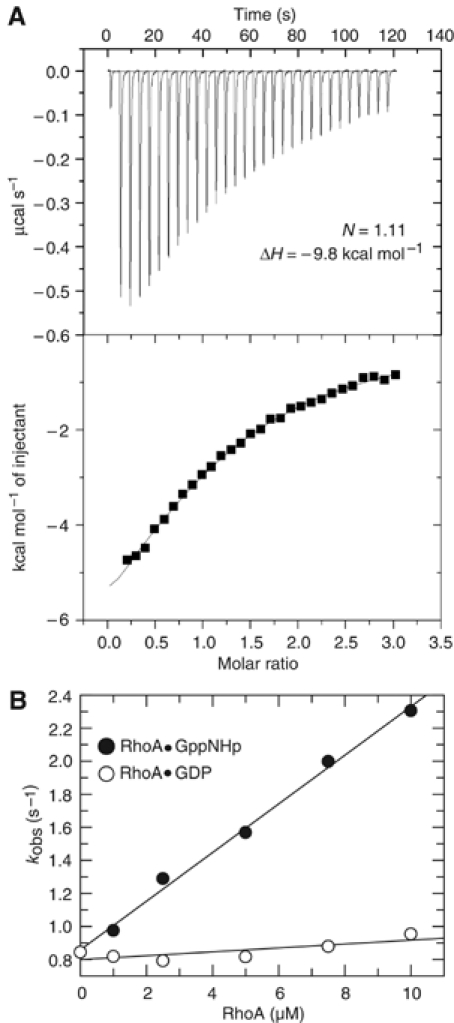
Effect of Rho binding on the mDiaN•DAD1145−1200 complex. (A) ITC analysis of the dissociation mDiaN–DAD1145−1200 complex (30 μM mDiaN; 60 μM DAD1145−1200) by titration with 400 μM RhoA•GppNHp, with upper and lower panels as in Figure 2A and B. (B) Dissociation of DAD from the mDiaN–DAD1145−1200 complex is followed by fluorescence as described in Figure 1D, in the presence of increasing amounts of RhoA•GppNHp (•) or RhoA•GDP (○). The dissociation rate constants kobs are plotted against the concentration of Rho.
To see if Rho has an active dissociating effect on the dissociation of the mDiaN•DAD complex, a preformed complex between mDiaN and F-DAD was mixed with increasing concentrations of either RhoA•GppNHp or RhoA•GDP, in the presence of a 100-fold excess nonlabelled DAD1145−1200 to silence the back reaction. Dissociation was followed by stopped-flow. RhoA•GppNHp does indeed accelerate the dissociation of the mDiaN•F-DAD complex, with a threefold increase over the concentration range used, while RhoA•GDP does not (Figure 6B). This argues for an active displacement mechanism by the binding of RhoA•GTP in the release of autoinhibition.
Discussion
In the absence of a full-length protein structure, it is difficult to delineate the structural and functional boundaries of the FH2 and DAD domains of mDia. Here, we show that there is a DAD core region encompassing residues 1180–1195, which represents the smallest functional and structural unit of autoinhibition. This core region forms a helical structure and binds to a highly conserved patch of residues on the ARR of mDiaN (Figure 4D). No electron density could be detected for the rest of the peptide that was used in crystallisation, suggesting that this part is either partially or fully unstructured and/or does not contribute significantly to binding, or that the high salt crystallisation conditions prevent charge–charge interactions, as observed, that is, for the interaction of the DEDDL motif of Ran with its basic patch (Vetter et al, 1999; Scheffzek et al, 2002).
Our biochemical data (Figure 2C) show that a number of basic residues following Phe1195 do have a pronounced influence since deletion of residues 1197–RKRG–1200 leads to a 13-fold reduction in affinity. Further truncation of a 36mer peptide (residues 1165–1200) to a 22mer (1175–1196) on the C-terminal side and another set of basic residues on the N-terminal side drastically reduces the affinity (36mer: KD=194 nM; 22mer: KD=15 μM). This suggests that there should be additional contacts between DAD and ARR, although the mutation of the conserved N-terminally located Lys1152 on DAD or Asp366 on ARR alone did not decrease affinity (Figure 2C and Table I). One half of the surface of mDiaN is strongly negatively charged, so it could represent a binding site for the two basic patches flanking the DAD-core region (DCR) (Figure 4D and E). The structure of the N-terminal 16 residues of DAD1145−1200 has been solved as part of the FH2 structure and is helical (Shimada et al, 2004). The fact that it is not visible in the structure indicates that there is no significant interaction of this part of the FH2 domain with the ARR.
While the binding of Rho to mDiaN does not involve any apparent enthalpy change, the binding of DAD to mDiaN is highly endothermic, and is compensated by a large favourable entropy change. There is corresponding heat released on binding of Rho to the mDiaN•DAD complex, showing that the release of DAD is at least enthalpically favoured. Conformational changes of GBDN upon binding of DAD1145−1200 to mDiaN can be ruled out as an explanation for the positive ΔH, since three different structures of the N-terminal region in three different binding states superimpose very well. A conformational change of DAD, such as the kink of the helix observed in the structure, might contribute, but no structure of the unbound DAD fragment is available right now. The highly favourable entropy change argues for hydrophobic contacts as the major contribute to binding, and against loss of conformational flexibility on binding, since positive ΔS values indicate exclusion of water molecules from the interface. Thermodynamic analysis of the DAD mutants shows that a weaker binding is a result of both lower enthalpy and entropy, another example of the frequently observed enthalpy–entropy compensation in protein–protein interactions (Table I) (Frisch et al, 1997).
How then does Rho induce the dissociation of the mDiaN•DAD complex? The structure of the mDiaN•RhoC complex has shown that the Rho binding site consists of two subdomains, GBDN and the ARR (Rose et al, 2005b), which are both required for tight binding of Rho•GTP (Figure 5A). GBDN is only loosely connected to the ARR, suggesting that it can easily change its orientation relative to it when not bound to Rho. We would thus propose a model (Figure 7) whereby the GBDN in the autoinhibited state is in a conformation that would allow Rho•GTP binding such that it would not interfere with DAD. Rho binds to the large hydrophobic patch on the GBDN via switch I and a part of switch II, and forms a low-affinity ternary RhoC•DAD–mDiaN complex. In the second step, a conformational change occurs, which positions Rho to make additional contact to the ARR of mDiaN, as observed in the structure of the RhoC•mDiaN complex. The movement of the GBDN towards this conformation would firstly induce long-range electrostatic repulsion by similar charges on DAD (Asp1183, Glu1187) and Rho (Glu64, Asp65, Asp67); secondly, short-range steric clashes would induce the release of DAD from the regulatory region of mDia to form a binary tight complex. This two-step binding model of releasing the mDiaN–DAD interaction by active Rho is comparable to the GEF-catalysed nucleotide exchange on Ras-like proteins, where GEF goes from a weak binding to a strong binding state, thereby inducing a transition from a high- to a low-affinity state of the nucleotide (Vetter and Wittinghofer, 2001).
Figure 7.
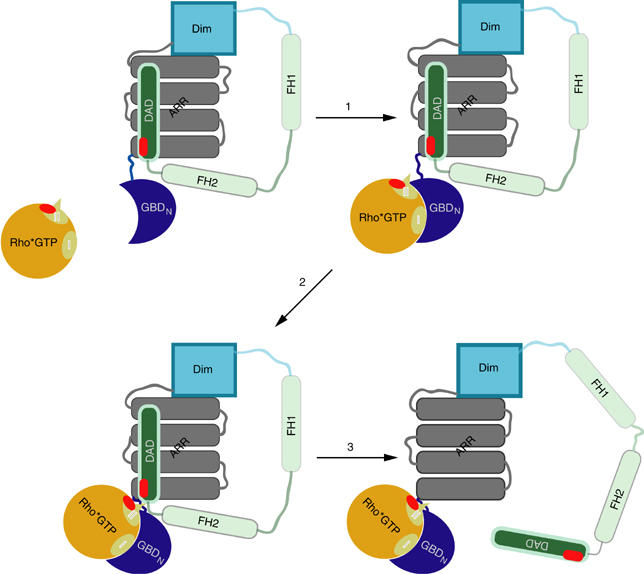
Schematic two-step-binding model of DAD release from the regulatory region of mDia1 by Rho•GTP, as described in the text.
In our studies, the equilibrium of the displacement reaction of the DAD•mDiaN complex by Rho•GTP is mostly on the side of the Rho–mDiaN complex due to a 18-fold higher affinity. In the full-length mDia protein the former interaction is intramolecular, even when one considers that the protein is a dimer and the DAD binding occurs in trans between the subunits. Thus, release of autoinhibition is more complex in this case, and preliminary experiments with constructs containing the FH2 domain in addition to DAD and mDiaN indicate that, in this case, Rho binding does not fully activate the actin polymerisation activity of FH2 (Li and Higgs 2003, 2005). Full activation might thus require an additional step that shifts the equilibrium towards the active state. While our data fully describe the structural requirements of mDia autoinhibition and its release by Rho•GTP, further experiments with full-length mDia are probably required to understand the energetic requirements for activation.
Materials and methods
Recombinant proteins
Proteins were expressed as GST fusions using the vector pGEX-4T1 (Amersham Biociences). The bicistronic mDiaNΔG–DAD1145−1200 construct was generated using an overlapping PCR. For expression, E. coli BL21DE3 cells were grown to an OD600 of 0.7 (37°C, 160 r.p.m.), induced with 100 μM of isopropyl-β-D-thiogalactopyranoside (IPTG) overnight at 20°C. The cells were harvested, suspended in buffer A (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 2 mM β-mercaptoethanol) containing 0.1 mM phenylmethylsulphonyl fluoride (PMSF) and 2 mM EDTA. Cells were lysed by sonication or microfluidizing, the extract applied to a GSH-Sepharose column (Amersham Biosciences) and the column washed with buffer B (50 mM Tris–HCl, pH 7.5, 300 mM NaCl, 2 mM β-mercaptoethanol). The fusion protein was digested with 5 U thrombin (serva) per mg of GST-fusion protein on the column overnight at 4°C. The final purification was carried out with size exclusion chromatography in buffer B using a Superdex S75 for mDiaN or S200 for the DAD fragments. For the mDiaNΔG–DAD1145−1200 complex used for crystallisation, the buffer used for gel filtration contained 20 mM HEPES, pH 7.1, 300 mM NaCl and 2 mM β-mercaptoethanol. For selenomethionine labelling, mDiaNΔG–DAD1145−1200 was expressed in LeMaster medium containing 0.1 g/l Se–Met. The gel filtration buffer contained 20 mM HEPES, pH 7.1, 300 mM NaCl and 10 mM β-mercaptoethanol.
Point mutations were introduced into DAD by the QuickChange protocol (Stratagene) or the PCR mutagenesis protocol of Picard and Bock (1997). Mutant proteins were purified like the wild-type proteins. Concentrations of mDiaN and mDiaNΔG were determined by absorption at 280 nm using the extinction coefficient deduced from the sequence. Rho was purified and loaded with GppNHp as described elsewhere (Rose et al, 2005b).
Synthesised DAD peptides and constructs
DAD peptides comprising residues 1175–1196 and the N-terminally AMCA-labelled (7-amino-4-methylcoumarin-3-acetic acid (AMCA)) peptide were synthesised by Biosynthan, Berlin. The Iaedans labelling of 100 μM of DAD1145−1200 carrying a C-terminal Cys in buffer C (50 mM Tris–HCl, pH 7.5, 300 mM NaCl, 10 mM ascorbic acid) was carried out by incubating it overnight (4°C) with a 3–5-fold excess of the fluorophore (5-((((2-iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid (1.5-Iaedans)) and exchanging the buffer to buffer B. Concentrations of the DAD fragments were determined by the methods of Ehresmann et al (1973) and Ellman (1958).
Stopped-flow kinetics
The experiments were performed at 20°C using a SX18 MW Applied Photophysics apparatus (Leatherhead, UK). The Aedans fluorophore (Molecular Probes) was excited at 336 nm with a bandpass of 6.4 nm. Emission (at 490 nm) was recorded using a 408 nm cut-off filter. All measurements were carried out with a final concentration of 100 nM of F-DAD and mDiaN–F-DAD. Increasing concentrations of mDiaN (1–15 μM) and RhoA•GppNHp/RhoA•GDP (1–10 μM) were used in the experiments using pseudo-first-order conditions. The observed fluorescence transients were fitted to a single-exponential function to obtain kobs. The association rate constant kon was derived from the slope of kobs versus protein concentration. The dissociation rate constants were obtained by mixing a preformed 100 nM mDiaN–F-DAD complex with a 100-fold excess of unlabelled DAD1145−1200. When investigating the dissociation acceleration by RhoA, RhoA•GppNHp or RhoA•GDP was used together with a 100-fold excess of unlabelled DAD1145−1200. All measurements were carried out in buffer B, additionally containing 5 mM MgCl2 in the RhoA experiments. GraFit 3.0–GraFit 5.0 was used for data analysis.
ITC measurements
The interaction of mDiaN and DAD, characterisation of DAD and mDiaN mutants, and analysis of different DAD constructs was performed by ITC based on Wiseman et al (1989) (MicroCal™; VP-ITC microcalorimeter). All measurements were carried out in buffer B. Concentrations of 0.02–3.7 mM DAD or 400 μM RhoA•GppNHp (syringe) were stepwise injected to the 20–280 μM mDiaN or to the 30 μM mDiaN–60 μM DAD1145−1200 complex solution (cell), respectively. The heating power per injection was observed over the reaction time until equilibrium was reached. The data were analysed using the software provided by the manufacturer.
Fluorescence equilibrium titration and polarisation measurements
All experiments were performed by using a FluoroMax II spectrofluorimeter at 20°C. Polarisation experiments were carried out using a polarisation filter (SPEX Instruments, NJ), buffer B plus 5% DMSO plus 5 mM MgCl2 if RhoA was used. The AMCA fluorophore was excited at 353 nm, the Aedans fluorophore at 336 nm. The emission was detected at 442 nm for the AMCA and at 490 nm for the Aedans fluorophore. Fluorescence titration experiments were performed in buffer B plus 0.001% Tween20. The AMCA- or Aedans-labelled DAD fragments (100 nM) were preincubated until the baseline was stable. In addition, mDiaN was added stepwise (0.017–67 μM), mixed rapidly and the fluorescence was followed until the signal did not change anymore. The relative fluorescence yield or polarisation was plotted against the protein concentration and the obtained curve was analysed by fitting a quadratic function to the data as described by Herrmann and Nassar (1996). For data analysis, Grafit 3.0–Grafit 5.0 was used.
Crystallisation and structure determination
Selenomethionine crystals of the GBD136−451–DAD1145−1200 complex were grown in buffer D containing 3.7 M Na formate, pH 7.1, 100 mM HEPES, pH 7.1 and 4.0% PEG5000MME, using the hanging drop/vapour diffusion method. The protein was solved in buffer containing 20 mM HEPES, pH 7.1, 300 mM NaCl and 10 mM β-mercaptoethanol. In all, 1 μl of protein solution of various concentrations (2.5 μg/μl, 5 μg/μl, 10 μg/μl) was mixed with the reservoir solution (buffer D). After 1 day, crystals that had a size of 200 × 100 × 100 μM grew at 20°C. These crystals were flash frozen in liquid nitrogen using buffer D as cryoprotectant.
Data-set collection has been performed at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland (Table II). A Se–Met data set was collected at 100 K on beam line PXII at a wavelength of 0.95 Å, using a Mar225 CCD detector. The detector distance was 350 mm, the oscillation range was 0.5° and 404 frames were collected. Data were indexed, integrated and scaled with the XDS package (Kabsch, 1993).
Crystals belonged to the hexagonal space group P6122 and contained two mDiaNΔG–DAD1145−1200 heterodimers in the asymmetric unit. Initial phases were determined with the program Phaser (McCoy et al, 2005) using residues 132–369 of chain B of the mDiaN•RhoC structure (PDB: 1Z1C) as a template and searching for two molecules. The program Coot (Emsley and Cowtan, 2004) was used to build the model into the 2Fo–Fc and Fo–Fc maps in iterative rounds of NCS refinement with REFMAC (Table III) (Murshudov et al, 1999). The final model has a good geometry with 98% of all residues in the allowed regions of the Ramachandran plot, as judged by the program Procheck (Laskowski et al, 1993). All structure figures were prepared with PyMOL (DeLano, 2002).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We are grateful to the machine and beamline groups, whose outstanding efforts have made these experiments possible. We thank Alex Berndt and Wulf Blankenfeldt for data collection, Michael Weyand for help in X-ray crystallography, Dorothee Kühlmann and Caroline Koerner for expert technical assistance, and Rita Schebaum for expert secretary assistance. The structure factors and coordinates were deposited in the PDB under the ID 2BAP. AW thanks the Deutsche Forschungsgemeinschaft for financial support (SFB 642).
References
- Alberts AS (2001) Identification of a carboxyl-terminal Diaphanous-related formin homology protein autoregulatory domain. J Biol Chem 276: 2824–2830 [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, Chan DC, Leder P (1997) FBP WW domains and the Abl SH3 domain bind to a specific class of proline-rich ligands. EMBO J 16: 2376–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AL, Hall A (2000) Rho GTPases and their effector proteins. Biochem J 348: 241–255 [PMC free article] [PubMed] [Google Scholar]
- Buck M, Xu W, Rosen MK (2004) A two-state allosteric model for autoinhibition rationalizes WASP signal integration and targeting. J Mol Biol 338: 271–285 [DOI] [PubMed] [Google Scholar]
- Castrillon DH, Wasserman SA (1994) Diaphanous is required for cytokinesis in Drosophila and shares domains of similarity with the products of the limb deformity gene. Development 120: 3367–3377 [DOI] [PubMed] [Google Scholar]
- Chook YM, Blobel G (1999) Structure of the nuclear transport complex karyopherin–beta2-Ran xGppNHp. Nature 20: 230–237 [DOI] [PubMed] [Google Scholar]
- Copeland JW, Treisman R (2002) The Diaphanous-related formin mDia1 controls serum response factor activity through its effects on actin polymerization. Mol Biol Cell 13: 4088–4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL User's Manual. San Carlos, CA, USA: DeLano Scientific [Google Scholar]
- Ehresmann B, Imbault P, Weil JH (1973) Spectrophotometric determination of protein concentration in cell extracts containing tRNA's and rRNA's. Anal Biochem 54: 454–463 [DOI] [PubMed] [Google Scholar]
- Eklof Spink K, Fridman SG, Weis WI (2001) Molecular mechanisms of beta-catenin recognition by adenomatous polyposis coli revealed by the structure of an APC–beta-catenin complex. EMBO J 20: 6203–6212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman GL (1958) A colorimetric method for determining low concentrations of mercaptans. Arch Biochem Biophys 74: 443–450 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for Molecular Graphics. Acta Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420: 629–635 [DOI] [PubMed] [Google Scholar]
- Evangelista M, Zigmond S, Boone C (2003) Formins: signaling effectors for assembly and polarization of actin filaments. J Cell Sci 116: 2603–2611 [DOI] [PubMed] [Google Scholar]
- Frisch C, Schreiber G, Johnson CM, Fersht AR (1997) Thermodynamics of the interaction of barnase and barstar: changes in free energy versus changes in enthalpy on mutation. J Mol Biol 267: 696–706 [DOI] [PubMed] [Google Scholar]
- Graham TA, Weaver C, Mao F, Kimelman D, Xu W (2000) Crystal structure of a beta-catenin/Tcf complex. Cell 103: 885–896 [DOI] [PubMed] [Google Scholar]
- Harris ES, Li F, Higgs HN (2004) The mouse formin, FRLalpha, slows actin filament barbed end elongation, competes with capping protein, accelerates polymerization from monomers, and severs filaments. J Biol Chem 279: 20076–20087 [DOI] [PubMed] [Google Scholar]
- Herrmann C, Nassar N (1996) Ras and its effectors. Prog Biophys Mol Biol 66: 1–41 [DOI] [PubMed] [Google Scholar]
- Higashida C, Miyoshi T, Fujita A, Oceguera-Yanez F, Monypenny J, Andou Y, Narumiya S, Watanabe N (2004) Actin polymerization-driven molecular movement of mDia1 in living cells. Science 303: 2007–2010 [DOI] [PubMed] [Google Scholar]
- Higgs HN (2005) Formin proteins: a domain-based approach. Trends Biochem Sci 30: 342–353 [DOI] [PubMed] [Google Scholar]
- Higgs HN, Peterson KJ (2005) Phylogenetic analysis of the formin homology 2 domain. Mol Biol Cell 16: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T, Morishima Y, Okamoto M, Furuyashiki T, Kato T, Narumiya S (2001) Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat Cell Biol 3: 8–14 [DOI] [PubMed] [Google Scholar]
- Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26: 795–800 [Google Scholar]
- Kato T, Watanabe N, Morishima Y, Fujita A, Ishizaki T, Narumiya S (2001) Localization of a mammalian homolog of diaphanous, mDia1, to the mitotic spindle in HeLa cells. J Cell Sci 114: 775–784 [DOI] [PubMed] [Google Scholar]
- Kovar DR, Pollard TD (2004) Progressing actin: formin as a processive elongation machine. Nat Cell Biol 6: 1158–1159 [DOI] [PubMed] [Google Scholar]
- Kovar DR, Wu JQ, Pollard TD (2005) Profilin-mediated competition between capping protein and formin Cdc12p during cytokinesis in fission yeast. Mol Biol Cell 16: 2313–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Moss DS, Thornton JM (1993) Main-chain bond lengths and bond angles in protein structures. J Mol Biol 231: 1049–1067 [DOI] [PubMed] [Google Scholar]
- Lei M, Lu W, Meng W, Parrini MC, Eck MJ, Mayer BJ, Harrison SC (2000) Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell 102: 387–397 [DOI] [PubMed] [Google Scholar]
- Lei M, Robinson MA, Harrison SC (2005) The active conformation of the PAK1 kinase domain. Structure 13: 769–778 [DOI] [PubMed] [Google Scholar]
- Leung DW, Rosen MK (2005) The nucleotide switch in Cdc42 modulates coupling between the GTPase-binding and allosteric equilibria of Wiskott–Aldrich syndrome protein. Proc Natl Acad Sci USA 102: 5685–5690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Higgs HN (2003) The mouse formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr Biol 13: 1335–1340 [DOI] [PubMed] [Google Scholar]
- Li F, Higgs HN (2005) Dissecting requirements for auto-inhibition of actin nucleation by the formin, mDia1. J Biol Chem 280: 6986–6992 [DOI] [PubMed] [Google Scholar]
- Macias MJ, Wiesner S, Sudol M (2002) WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett 513: 30–37 [DOI] [PubMed] [Google Scholar]
- Maesaki R, Ihara K, Shimizu T, Kuroda S, Kaibuchi K, Hakoshima T (1999) The structural basis of Rho effector recognition revealed by the crystal structure of human RhoA complexed with the effector domain of PKN/PRK1. Mol Cell 4: 793–803 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr 61: 458–464 [DOI] [PubMed] [Google Scholar]
- Millard TH, Sharp SJ, Machesky LM (2004) Signalling to actin assembly via the WASP (Wiskott–Aldrich syndrome protein)-family proteins and the Arp2/3 complex. Biochem J 380: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Lebedev A, Vagin AA, Wilson KS, Dodson EJ (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr 55: 247–255 [DOI] [PubMed] [Google Scholar]
- Otomo T, Otomo C, Tomchick DR, Machius M, Rosen MK (2005b) Structural basis of Rho GTPase-mediated activation of the formin mDia1. Mol Cell 18: 273–281 [DOI] [PubMed] [Google Scholar]
- Otomo T, Tomchick DR, Otomo C, Panchal SC, Machius M, Rosen MK (2005a) Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature 433: 488–494 [DOI] [PubMed] [Google Scholar]
- Ozaki-Kuroda K, Yamamoto Y, Nohara H, Kinoshita M, Fujiwara T, Irie K, Takai Y (2001) Dynamic localization and function of Bni1p at the sites of directed growth in Saccharomyces cerevisiae. Mol Cell Biol 21: 827–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni D, Le Clainche C, Carlier MF (2001) Mechanisim of actin-based motility. Science 292: 1502–1506 [DOI] [PubMed] [Google Scholar]
- Picard V, Bock SC (1997) Rapid and efficient one-tube PCR-based mutagenesis method. Methods Mol Biol 67: 183–188 [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112: 453–465 [DOI] [PubMed] [Google Scholar]
- Pring M, Evangelista M, Boone C, Yang C, Zigmond SH (2003) Mechanism of formin-induced nucleation of actin filaments. Biochemistry 42: 486–496 [DOI] [PubMed] [Google Scholar]
- Pruyne D, Evangelista M, Yang C, Bi E, Zigmond S, Bretscher A, Boone C (2002) Role of formins in actin assembly: nucleation and barbed-end association. Science 297: 612–615 [DOI] [PubMed] [Google Scholar]
- Quinlan ME, Heuser JE, Kerkhoff E, Mullins RD (2005) Drosophila Spire is an actin nucleation factor. Nature 433: 382–388 [DOI] [PubMed] [Google Scholar]
- Romero S, Le Clainche C, Didry D, Egile C, Pantaloni D, Carlier MF (2004) Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell 119: 419–429 [DOI] [PubMed] [Google Scholar]
- Rose R, Weyand M, Lammers M, Ishizaki T, Ahmadian MR, Wittinghofer A (2005b) Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature 435: 513–518 [DOI] [PubMed] [Google Scholar]
- Rose R, Wittinghofer A, Weyand M (2005a) The purification and crystallization of mDia1 in complex with RhoC. Acta Crystallogr F 61: 225–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffzek K, Klebe C, Fritz-Wolf K, Kabsch W, Wittinghofer A (2002) Crystal structure of the nuclear Ras-related protein Ran in its GDP-bound form. Nature 374: 378–381 [DOI] [PubMed] [Google Scholar]
- Shimada A, Nyitrai M, Vetter IR, Kuhlmann D, Bugyi B, Narumiya S, Geeves MA, Wittinghofer A (2004) The core FH2 domain of diaphanous-related formins is an elongated actin binding protein that inhibits polymerization. Mol Cell 13: 511–522 [DOI] [PubMed] [Google Scholar]
- Vetter IR, Nowak C, Nishimoto T, Kuhlmann J, Wittinghofer A (1999) Structure of a Ran-binding domain complexed with Ran bound to a GTP analogue: implications for nuclear transport. Nature 398: 39–46 [DOI] [PubMed] [Google Scholar]
- Vetter IR, Wittinghofer A (2001) The guanine nucleotide-binding switch in three dimensions. Science 294: 1299–1304 [DOI] [PubMed] [Google Scholar]
- Wallar BJ, Alberts AS (2003) The formins: active scaffolds that remodel the cytoskeleton. Trends Cell Biol 13: 435–446 [DOI] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S (1999) Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol 1: 136–143 [DOI] [PubMed] [Google Scholar]
- Watanabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, Saito Y, Nakao K, Jockusch BM, Narumiya S (1997) p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J 16: 3044–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wear MA, Yamashita A, Kim K, Maéda Y, Cooper JA (2003) How capping protein binds the barbed end of the actin filament. Curr Biol 13: 1531–1537 [DOI] [PubMed] [Google Scholar]
- Wiseman T, Williston S, Brandts JF, Lin LN (1989) Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem 179: 131–137 [DOI] [PubMed] [Google Scholar]
- Xu Y, Moseley JB, Sagot I, Poy F, Pellman D, Goode BL, Eck MJ (2004) Crystal structures of a formin homology-2 domain reveal a tethered dimer architecture. Cell 116: 711–723 [DOI] [PubMed] [Google Scholar]
- Zigmond SH (2004) Formin-induced nucleation of actin filaments. Curr Opin Cell Biol 16: 99–105 [DOI] [PubMed] [Google Scholar]
- Zigmond SH, Evangelista M, Boone C, Yang C, Dar AC, Sicheri F, Forkey J, Pring M (2003) Formin leaky cap allows elongation in the presence of tight capping proteins. Curr Biol 13: 1820–1823 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2