

Abstract
Endogenous small RNAs function in RNA interference (RNAi) pathways to guide RNA cleavage, translational repression, or methylation of DNA or chromatin. In Tetrahymena thermophila, developmentally regulated DNA elimination is governed by an RNAi mechanism involving ∼27–30-nucleotide (nt) RNAs. Here we characterize the sequence features of the ∼27–30-nt RNAs and a ∼23–24-nt RNA class representing a second RNAi pathway. The ∼23–24-nt RNAs accumulate strain-specifically manner and map to the genome in clusters that are antisense to predicted genes. These findings reveal the existence of distinct endogenous RNAi pathways in the unicellular T. thermophila, a complexity previously demonstrated only in multicellular organisms.
Keywords: Tetrahymena, small RNA, RNAi, Dicer, genome rearrangement
In diverse eukaryotes from parasitic protozoa to humans, RNA interference (RNAi) pathways regulate gene expression, establish heterochromatin, and/or protect the genome from viruses and mobile DNA elements (Matzke and Birchler 2005; Sontheimer and Carthew 2005). Although the biological function of RNAi varies, central to all pathways are ∼21–30-nucleotide (nt) small noncoding RNAs (sRNAs) that provide specificity for RNA or DNA targets. In multicellular organisms, three major classes of endogenous sRNAs have been characterized in detail: micro RNAs (miRNAs), repeat-associated small interfering RNAs (rasiRNAs), and trans-acting small interfering RNAs (ta-siRNAs) (Bartel 2005; Sontheimer and Carthew 2005). The miRNAs and tasiRNAs direct translational repression and/or degradation of messenger RNAs. The rasiRNAs, derived from repetitive DNA elements such as transposons and centromeres, function to promote heterochromatin formation, DNA methylation, and/or RNA degradation. Less-well-characterized sRNAs include those with precise complementarity to protein-coding genes, pseudogenes, and intergenic regions (e.g., see Ambros et al. 2003).
The biogenesis of diverse sRNAs depends on an RNaseIII family nuclease called Dicer (Tomari and Zamore 2005). The Dicer substrates for miRNA production are single-stranded RNAs with stem-loop structures, while precursors to ta-siRNAs and most rasiRNAs are double-stranded RNAs (dsRNAs) resulting from bidirectional transcription or RNA-dependent RNA polymerase activity. Dicer processing of precursors yields short sRNA duplexes of homogeneous length. One strand of each sRNA duplex is stabilized by assembly into an effector ribonucleoprotein (RNP) containing a Piwi/PAZ domain (PPD) protein of the Argonaute family. Multicellular eukaryotes express multiple paralogs of RNAi pathway components that are specialized in function.
In contrast to the diversity of sRNAs in multicellular organisms, unicellular eukaryotes are only known to express rasiRNA-like sRNAs (Djikeng et al. 2001; Reinhart and Bartel 2002; Chicas et al. 2004; Ullu et al. 2005). In the free-living ciliated protozoan Tetrahymena thermophila, RNAs ∼26–31 nt in length direct developmentally programmed DNA elimination (Mochizuki and Gorovsky 2004b). T. thermophila, like other ciliates, has nuclear dualism, with a diploid, germline micronucleus (MIC) that remains phenotypically silent and a polyploid, transcriptionally active, somatic macronucleus (MAC). When starved for nutrients, T. thermophila ceases to divide vegetatively and becomes competent to reproduce sexually by conjugation. In conjugating cells, new MACs are developed from mitotic siblings of the zygotic MIC in a process involving site-specific chromosome fragmentation and deletion of ∼6000 internally eliminated sequences (IESs). The IESs are single-copy elements or moderately repetitive, transposon-like sequences that together account for ∼15% of the MIC genome (Yao and Chao 2005). DNA elimination occurs under epigenetic regulation: Sequences in the parental MAC can protect corresponding sequences in the developing MAC from elimination.
Normal MAC development and the conjugation-induced accumulation of ∼26–31-nt sRNAs require the PPD-containing TWI1 and the Dicer-like DCL1 (Mochizuki et al. 2002; Malone et al. 2005; Mochizuki and Gorovsky 2005). Bidirectional nongenic transcription in the MIC during conjugation (Chalker and Yao 2001) is proposed to provide dsRNA precursors that are processed by Dcl1p into sRNAs (Yao et al. 2003; Mochizuki and Gorovsky 2004b). Northern blot assays have confirmed that a known MIC-limited IES is represented in the conjugation-induced sRNA population (Chalker et al. 2005). In addition, DNA hybridization studies using sRNAs isolated from conjugating cells have suggested that as conjugation progresses, the sRNA population becomes enriched for MIC-limited sequence (Mochizuki and Gorovsky 2004a). To account for this finding and provide a mechanism for the epigenetic influence of the parental MAC, the ∼26–31-nt sRNAs, termed the scan (scn)RNAs, are proposed to enter the parental MAC in association with Twi1p and scan for homologous sequence in a manner that results in degradation of MAC-cognate sRNAs. The sRNAs remaining after parental MAC subtraction are thought to then transit to the developing MAC where they guide the histone H3 Lys 9 (H3K9) methylation of MIC-limited chromatin, which likely marks IESs for subsequent elimination (Taverna et al. 2002; Liu et al. 2004). In this manner, sRNA-guided DNA elimination in T. thermophila is similar to rasiRNA-guided heterochromatin formation in Schizosaccharomyces pombe (Matzke and Birchler 2005).
The recently sequenced MAC genome of T. thermophila encodes multiple Dicer and PPD family members, implying the existence of additional RNAi pathways with roles other than DNA elimination. RasiRNA-like sRNAs derived from MIC centromeres may function in MIC maintenance in a manner dependent on DCL1 during vegetative growth (Mochizuki and Gorovsky 2005), although conflicting results have been reported (Malone et al. 2005). However, the full complexity of sRNAs in T. thermophila has not been examined. Here we present our analysis of sRNAs expressed in vegetatively growing, starving, and conjugating cells. We describe a second class of T. thermophila sRNAs with ubiquitous accumulation throughout the life cycle. These ∼23–24-nt sRNAs have features characteristic of sRNAs from other organisms but with interesting differences that suggest a novel biogenesis pathway distinct from those previously described for miRNAs, rasiRNAs, and ta-siRNAs. Analogous to the diversity of sRNAs found in multicellular organisms, the ∼27–30-nt sRNAs and the ∼23–24-nt sRNAs in T. thermophila represent coexisting yet genetically separable RNAi pathways.
Results and Discussion
The three Dicer-related genes in T. thermophila have distinct expression profiles
Database searches for Dicer homologues in the T. thermophila genome using tBLASTn analysis revealed three loci with homology to known Dicer enzymes. We and others (Malone et al. 2005; Mochizuki and Gorovsky 2005) have used RT–PCR and Northern blot assays to demonstrate that all three Dicer mRNAs are expressed. The domain structures of the T. thermophila Dicer-like proteins are depicted in Figure 1A. DCL1 bears the dual RNaseIII domains and dsRNA-binding motif (dsrm) (Fig. 1A) conserved among Dicers but lacks the canonical N-terminal helicase domain. DCR1 encodes a predicted protein with a conserved Dicer helicase domain and highly divergent RNaseIII domains that seem unlikely to support canonical Dicer activity (Supplementary Figs. S1, S2). The predicted N terminus of DCR1 is a unique ∼750-amino-acid extension lacking known protein motifs. In contrast, DCR2 is highly homologous to other Dicers, encoding a protein with an N-terminal helicase domain and C-terminal RNaseIII domains (Fig. 1A).
Figure 1.

Sequence composition and expression profile of the Dicer-related proteins. (A) Schematic of conserved domains in previously characterized Dicers (top) and the T. thermophila Dicers. The less highly conserved RNaseIII domain of DCR1 is denoted in light gray (see Supplementary Figs. S1, S2). The arrow on DCR1 denotes an N-terminal extension relative to DCR2. Bold lines represent regions used for Northern blot probes. (B) Total RNA was used for Northern blot analysis of Dicer expression during the T. thermophila life cycle. Probes for DCR1 and DCR2 mRNAs were used concurrently, followed by probing of the same blot for DCL1 mRNA.
To further characterize the three Dicer-like genes, we examined their mRNA expression profiles during all stages of the T. thermophila life cycle: vegetative growth, starvation, and conjugation. Northern blot assays revealed that DCL1 is highly expressed in conjugating cells (Fig. 1B). We also detected low levels of a transcript from the DCL1 locus by RT–PCR during vegetative growth and starvation (data not shown), as reported in a concurrent independent study (Mochizuki and Gorovsky 2005). DCR1 and DCR2 mRNAs are expressed ubiquitously, with DCR2 expressed maximally during vegetative growth and DCR1 expressed most highly during the initial stages of starvation (Fig. 1B). The dissimilar life cycle expression profiles of the three Dicer-like proteins suggested that distinct classes of sRNAs and RNAi pathways could exist in T. thermophila.
Three size classes of small RNAs accumulate with distinct expression profiles
To identify sRNAs expressed during the T. thermophila life cycle, total RNA from cultures in vegetative growth, starvation, and conjugation was prepared and enriched for RNAs <125 nt in length using size-selective filtration. The RNA in filtration flow-through and wash fractions was resolved by denaturing gel electrophoresis and visualized directly by SYBR Gold staining (Fig. 2). We observed abundant ∼27–30-nt RNAs in 4 h and 10 h conjugating cells as expected from previous study of scnRNAs (Mochizuki et al. 2002). Similarly sized RNAs were not readily detected in vegetatively growing or starving cells (Fig. 2). In addition to the ∼27–30-nt conjugation-induced RNAs, we identified two additional size classes of RNA. A population of ∼23–24-nt RNAs accumulates throughout the life cycle, and ∼30–35-nt RNAs accumulate specifically during starvation (Fig. 2). The latter class is generated by a non-RNAi-like pathway and is described in a separate report (Lee and Collins 2005). The ∼27–30-nt and ∼23–24-nt RNAs share features with sRNAs from other organisms and are therefore the focus of the rest of this study. To investigate these RNA populations in greater detail, we separately cloned and sequenced RNAs from each size class (see Materials and Methods). In brief, RNAs were size selected by gel fractionation, eluted from gel slices, and cloned using a modified protocol based on previously described methods (Pfeffer et al. 2005).
Figure 2.

Three classes of small RNAs accumulate with distinct life cycle expression profiles. Total RNA was enriched by size filtration for sRNAs from vegetatively growing (3 × 106 cell equivalents), starving (7 h: 3 × 106; overnight: 7.5 × 106), or conjugating (4 h: 1 × 107; 10 h: 7 × 106) cells. The first lane of each triplet set represents column flow-through; the second and third lanes represent first and second washes, respectively. SYBR Gold was used to visualize RNAs.
Sequence characteristics of the ∼27–30-nt sRNAs support a role in DNA elimination
We obtained 125 cDNAs for the ∼27–30-nt RNAs prepared from 10–12-h conjugating cells. The majority of cDNAs not derived from rRNA or tRNA did not match sequence scaffolds representing the MAC genome (Table 1; for sequences, see Supplementary Table S1). This finding suggests that the ∼27–30-nt RNA population is highly enriched for sequences cognate to MIC-limited DNA, which represents only ∼15% of the MIC genome. From this, we infer that these ∼27–30-nt RNAs represent the scnRNAs that function in the late stages of conjugation as sequence-specific guides for DNA elimination.
Table 1.
Summary of sRNA cloning and genomic matches
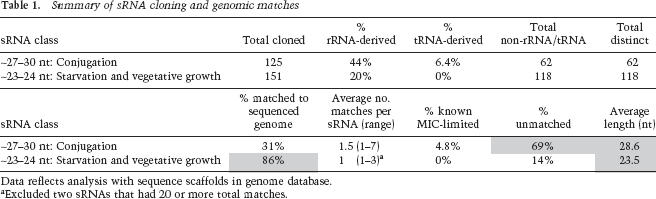
Each ∼27–30-nt sRNA was cloned once (Table 1), suggesting a complexity in the sRNA population consistent with the estimated 20 Mbp of DNA eliminated during MAC development (Yao and Chao 2005). A few MIC-limited elements have been cloned and their sequences deposited in GenBank; three sRNAs matched three known IESs (Supplementary Table S1). These IESs are also present in the MAC genome database, with two mapping to sequence scaffolds <2 kb in length and one to a scaffold <80 kb in length. Such scaffolds are relatively shorter than others in the genome database and are thus likely to represent the low level of MIC contamination anticipated in the MAC preparations used for genomic library construction. Several additional ∼27–30-nt sRNA sequences mapping to scaffolds <7 kb in length or matched unassembled sequence reads likely represent MIC-limited DNA as well (Supplementary Table S1).
The few ∼27–30-nt sRNAs matching long MAC scaffolds likely derive from true MAC loci. Some of these RNAs mapped to the sense strand of predicted protein-coding genes and may be mRNA degradation products. Alternatively, MAC-cognate ∼27–30-nt sRNAs could have escaped parental MAC subtraction or been generated after the window of opportunity for parental MAC scanning had closed.
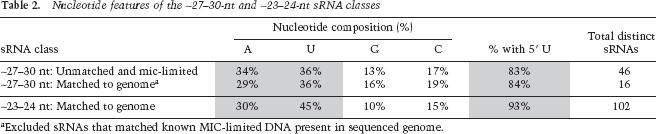
Consistent with genetic evidence linking DNA elimination to RNAi, the ∼27–30-nt sRNAs have sequence features characteristic of sRNAs generated by RNAi pathways in other organisms. Significantly, 83% of the ∼27–30-nt sRNA sequences cloned have a 5′ uridine (U) (Table 2). This 5′ U bias is not an artifact of cloning, as no such bias exists for the rRNA breakdown products cloned in parallel (Supplementary Table S1). A 5′ U bias characterizes miRNAs in plants and metazoans and rasiRNAs in Drosophila melanogaster (Lau et al. 2001; Aravin et al. 2003). The mechanism underlying this bias is unknown. The ∼27–30-nt sRNAs have a nearly 1:1 ratio in A:U frequency that is consistent with accumulation of sRNAs from both strands of dsRNA precursors, similar to rasiRNAs (Table 2). In summary, the sequences of ∼27–30-nt sRNAs that are cognate to MIC-limited DNA support their proposed function in directing DNA elimination and expand existing knowledge of MIC-specific genome content.
Table 2.
Nucleotide features of the ∼27–30-nt and ∼23–24-nt sRNA classes
The ∼23–24-nt sRNAs derive from a second RNAi-related pathway distinct from DNA elimination
We restricted our cloning of ∼23–24-nt sRNAs to vegetatively growing and starving cells to avoid contamination by the conjugation-induced ∼27–30-nt sRNAs. From the isolated RNA, 118 distinct sRNAs not derived from rRNA or tRNA were each cloned a single time, reflecting a high complexity in the ∼23–24-nt sRNA population (Table 1; Supplementary Table S2). In contrast to the ∼27–30-nt sRNAs, the vast majority of ∼23–24-nt sRNA sequences matched the sequenced MAC genome once, mapping to previously uncharacterized loci. A few sRNAs matched two or three loci, and two matched 20 or more positions in the MAC genome. Only 16 sRNAs failed to match the MAC genome; of these, 10 matched rRNA and tRNA of fungal/bacterial origin, likely ingested by T. thermophila cells from the growth media. Two sequences matched the T. thermophila mitochondrial genome.
To verify that the ∼23–24-nt MAC-cognate sRNAs were not degradation products of longer RNAs, we examined sRNA accumulation by Northern blot hybridization. All sRNAs examined accumulated as discrete species (Fig. 3A; data not shown). In addition, the expression levels of individual sRNAs were fairly constant throughout the life cycle (Fig. 3B). These findings are consistent with the observed SYBR Gold staining of the sRNAs in bulk (Fig. 2). Like the T. thermophila ∼27–30-nt sRNAs and sRNAs of other eukaryotes, the ∼23–24-nt sRNAs have a strong bias toward a 5′ U; 93% of the MAC-cognate sRNAs share this feature (Table 2). Together, these findings demonstrate that the ∼23–24-nt sRNAs represent a novel sRNA class in T. thermophila, distinct from the conjugation-induced sRNAs.
Figure 3.

Individual ∼23–24-nt sRNAs accumulate throughout the life cycle with strain-specific expression differences. Total RNA enriched for sRNAs was probed on Northern blots either for an individual sRNA from a single cluster or for sRNAs from all 12 sRNA clusters (sRNA mix) (for actual sRNAs probed, see Supplementary Table 2). The sRNAs 3 and 4 are derived from the same sRNA cluster, while all other sRNAs are derived from distinct clusters. (A) RNA was from SB210 cells in vegetative growth (Veg) or a mix of different time points in starvation (St): 3 h (33%), 6–7 h (58%), and 16–24 h (9%). (B) RNA was from SB210 or CU428 cells in the life cycle stages indicated. Conjugation (Conj) was between SB210 and CU428. (C) RNA was from cells of different strain backgrounds in vegetative growth. Progeny from conjugation were analyzed as a pool before sexual maturity. In B and C, U6 spliceosomal small nuclear RNA served as a loading control.
For roughly half of the ∼23–24-nt sRNAs, the 3′-terminal nucleotide did not match the genomic locus (Supplementary Table S2). Because aberrant 3′ nucleotides were not characteristic of any other RNA population cloned in our study, we suspect that the ∼23–24-nt sRNAs undergo untemplated 3′ nucleotide addition. The only systematic modification reported for sRNAs generated by RNAi pathways is ribose methylation of the 3′ nucleotide by the plant-specific methyltransferase HEN1 (Li et al. 2005). Methylation may influence sRNA stability and reduce the occurrence of a second 3′ end modification: the addition of one to five U residues. Intriguingly, the most common 3′ addition to the T. thermophila ∼23–24-nt sRNAs is a single U (Supplementary Table S2). Identification of a potential role for untemplated 3′ nucleotide addition in the stability or function of the ∼23–24-nt sRNAs awaits further study.
The vast majority of the 118 sRNAs mapped in 12 clusters to the MAC genome, with each cluster on a different sequence scaffold and represented by two to 16 cloned sRNAs. Within a cluster, all sRNAs were encoded on the same strand (Supplementary Tables S2, S3). In addition, in contrast to the near 1:1 ratio in A:U frequency of ∼27–30-nt sRNAs, this ratio in the ∼23–24-nt sRNA population is skewed toward higher U content (Table 2), even if the 3′ untemplated nucleotides are excluded from the analysis. These findings suggest that the sRNAs derive from single-stranded precursors or accumulate in a biased manner from dsRNA substrates. Attempts to model pre-miRNA precursors for individual ∼23–24-nt sRNAs yielded stem-loop structures for only a few sRNAs, even when deviation from canonical pre-miRNA-like structures was allowed (Supplementary Fig. S3). We also found no evidence for more extensive single-stranded fold-back structures similar to that proposed to yield sRNAs cognate to the Caenorhabditis elegans transposon Tc1 (Sijen and Plasterk 2003).
In conjugating ΔDCL1 strains incapable of generating the ∼27–30-nt sRNAs, shorter RNAs ∼24 nt in length accumulate instead (Mochizuki and Gorovsky 2005). This observation suggests that in the absence of Dcl1p, precursors to the ∼27–30-nt sRNAs can be processed by the Dicer normally responsible for biogenesis of the ∼23–24-nt sRNAs. Because the ∼27–30-nt sRNA precursors are thought to be double-stranded, we propose that precursors to the ∼23–24-nt sRNAs are also double-stranded. In agreement with this hypothesis, both sense and antisense transcripts from ∼23–24-nt sRNA genomic clusters were detectable by RT–PCR (data not shown).
Conjugation of MIC-knockout strains of DCL1 and DCR1 but not DCR2 produced viable progeny, suggesting that of the three Dicer-like proteins in T. thermophila, only Dcr2p is essential (Mochizuki and Gorovsky 2005). In vegetatively growing or starving ΔDCL1 and ΔDCR1 cultures, the overall levels of ∼23–24-nt sRNAs were similar (Supplementary Fig. S5). We attempted to deplete Dcr2p during vegetative growth to test the Dcr2p dependence of the ∼23–24-nt sRNAs, but viable strains significantly reduced in DCR2 mRNA could not be generated (data not shown). The ubiquitous expression of both DCR2 and the ∼23–24-nt sRNAs throughout the life cycle suggests that Dcr2p is likely the Dicer nuclease required for biogenesis of the ∼23–24-nt sRNAs. However, we cannot exclude the possibility that these sRNAs are generated by a novel, Dicer-independent pathway.
A complete strand bias in the production of sRNAs unlinked to stem-loop precursors has only been reported for the C. elegans “X cluster” sRNAs of unknown function, which derive from an intergenic region on the X chromosome (Ambros et al. 2003). Some plant ta-siRNA clusters have a substantial but incomplete strand bias that can be accounted for by asymmetry in the internal stability of sRNA duplexes, which influences strand selection for RNP assembly (Vazquez et al. 2004). Thermodynamic asymmetry is also a hallmark of miRNAs and siRNAs derived from exogenous dsRNA substrates (Khvorova et al. 2003). However, such asymmetry is not characteristic of the T. thermophila ∼23–24-nt sRNAs (Supplementary Fig. S4), indicating that another mechanism must account for the extreme strand bias observed.
Accumulation of ta-siRNAs from dsRNA precursors occurs with near perfect ∼21-nt phasing (Bartel 2005). In contrast, we found no support for precise phasing within a ∼23–24-nt sRNA cluster. In fact, ∼10% of the sRNAs overlapped in sequence (Supplementary Table S2). Notably, overlapping sRNAs have also been identified from the C. elegans X cluster (Ambros et al. 2003).
Our findings suggest that the ∼23–24-nt sRNAs and ∼27–30-nt sRNAs are both processed from dsRNA precursors but have otherwise distinct biogenesis pathways. Overall, although the ∼23–24-nt sRNAs share some characteristics with previously described sRNAs, their sequence features and inferred biogenesis pathway resist assignment to any single category of sRNAs yet characterized in detail.
Possible function of ∼23–24-nt sRNAs and their transcripts of origin
The ubiquitous accumulation of individual ∼23–24-nt sRNAs in the T. thermophila strain SB210 suggests that their precursor transcripts are expressed throughout the life cycle. To determine whether the same population of sRNAs is expressed universally in T. thermophila, we examined sRNA accumulation in additional strains. T. thermophila strains are established through extensive vegetative propagation to obtain clonal populations. Differences between wild-type strains have not been extensively studied, although it is known that individual strains belong to one of seven distinct mating types. Conjugation between two compatible mating types produces progeny that are genetically polyclonal, with differences dependent on parental genotypes and alternative DNA rearrangement during macronuclear development (Yao and Chao 2005). To our surprise, individual ∼23–24-nt sRNAs differed in expression between different strains (Fig. 3C), although no correlation was found between mating type and sRNA expression profile. This finding suggests that the population of precursor transcripts giving rise to the ∼23–24-nt sRNAs may be strain-specific. In addition, loci beyond those identified in our sRNA cloning from SB210 may be able to contribute to the ∼23–24-nt sRNA population.
Following our initial analysis of the ∼23–24-nt sRNA clusters in SB210, preliminary gene predictions for the sequenced T. thermophila genome were released. Strikingly, the majority of the 12 sRNA clusters are antisense to the introns and exons of predicted protein-coding genes (Fig. 4; Supplementary Table S3). Interestingly, the majority of these gene predictions are not supported in the existing collection of ESTs, and Northern blot assays for the expression of putative mRNAs did not yield detectable levels of a discrete transcript in any strain examined, regardless of sRNA expression profile (data not shown). No structural homologues could be identified in the protein sequence predictions; in fact, BLASTx analysis of entire sRNA cluster loci failed to reveal substantial primary sequence homology with known proteins. A few sRNA clusters do not overlap predicted genes, and a single cluster overlaps a gene predicted to encode a short, 58-amino-acid protein.
Figure 4.

The majority of sRNA clusters are antisense to predicted protein-coding genes. Alignments of three sRNA clusters with annotated genome scaffolds were generated by Gbrowse (see Materials and Methods). The arrows above the scaffold denote the location and orientation of cloned sRNAs, with the number of sRNAs cloned noted in parentheses. (Top) The sRNAs 3 and 4 in Figure 3 map to the cluster on CH445461. (Bottom) The clusters on CH445618 and CH445681 illustrate that within some sRNA clusters, an additional level of sRNA grouping was observed.
We suggest that the ∼23–24-nt sRNAs represent a pathway that regulates gene expression at a post-transcriptional level. It is unlikely that the sRNAs act similarly to rasiRNAs in promoting H3K9 methylation to direct heterochromatin formation or cytosine methylation in DNA, because these modifications are thought to be absent from the T. thermophila MAC during vegetative growth (Pratt and Hattman 1981; Strahl et al. 1999). Instead, the ∼23–24-nt sRNAs may serve as guides for RNA cleavage, targeting transcripts from the antisense strand of sRNA clusters or related RNAs from other loci. Notably, the ∼23–24-nt sRNAs show a reduced thermodynamic stability of base-pairing in positions 9–12 (Supplementary Fig. S4). This feature is shared by exogenous siRNAs that efficiently silence mRNA targets and has been proposed to reflect requirements for optimal recycling of the nucleolytic effector RNP (Khvorova et al. 2003).
Remarkably, we found that the putative proteins encoded on the antisense strands of the 12 sRNA clusters are highly related. BLASTp searches of the T. thermophila gene predictions revealed that the putative proteins form three distinct families of related genes (Supplementary Table S4). Proteins within a family were more related to each other than to any other predicted protein in the MAC genome. All genes within a single sRNA cluster were part of the same family, and each family was represented by more than one sRNA cluster. Some sRNA clusters shared ∼70 nt to 3.5-kb stretches of nearly identical sequence; however, homology among predicted protein family members extended beyond regions of sequence identity. These findings suggest that the genome regions corresponding to sRNA clusters have undergone gene duplication and divergence.
Other features of the predicted genes within sRNA clusters suggest that the genomic loci may not code for intact, functional proteins and may be more akin to mobile element DNA. Using RT–PCR, we could detect contiguous transcripts linking adjacent predicted genes within a sRNA cluster (data not shown), suggesting that the predicted genes may not be independent transcription units. Also, the genomic loci of some sRNA clusters include tracts of degenerate direct or inverted repeats even within predicted coding regions (Supplementary Table S4). In addition, the sRNA strand of the majority of clusters contains one or more thymidine (T)-rich tracts ranging in length from ∼30–85 nt, with as many as 20 consecutive Ts. On the putative protein-coding strand, these T-rich tracts are polyadenosine tracts located between predicted genes. Taken together, these sequence features suggest a history of DNA rearrangements and/or integration of reverse-transcribed polyadenylated mRNAs. It will be of interest to analyze the sRNA cluster loci, associated transcripts and the T. thermophila genome further to ascertain whether the sRNA clusters and possibly other related loci express only aberrant mRNAs or encode proteins under regulation by RNAi.
To our knowledge, T. thermophila represents the first unicellular organism known to express more than one class of endogenous sRNAs. The T. thermophila ∼23–24-nt sRNAs are distinct from the conjugation-specific ∼27–30-nt sRNAs in size, developmental expression, genomic origin, and putative function. The unique features of the ∼23–24-nt sRNAs reveal the existence of a greater diversity in the biogenesis, function, and regulation of sRNAs than previously known.
Materials and methods
Analysis of T. thermophila Dicer-related genes
Dicer identification and sequence analysis is described in the Supplemental Material. For Northern blots, total RNA isolated with Trizol (GIBCO-BRL) was resolved on agarose/formaldehyde gels and hybridized with hexamer-labeled probes.
sRNA detection and cloning
For sRNA cloning and detection by SYBR Gold (Molecular Probes) or Northern blot, total RNA was enriched for sRNAs using YM50 Microcon columns (Amicon). Northern blots were hybridized with 5′ end-labeled DNA oligonucleotides. RNA cloning was performed according to established methods (Pfeffer et al. 2005), with slight modification. Additional details of sRNA enrichment, cloning, and sequence analysis are described in the Supplemental Material.
Acknowledgments
We thank Ed Orias, Kaz Mochizuki, and Marty Gorovsky for strains; Zasha Weinberg and Larry Ruzzo for genome-wide tRNA prediction; Jacob Kitzman and Chris Burge for discussions; Dan Pollard for bioinformatics assistance; and the Collins laboratory for manuscript discussions. Preliminary sequence data were obtained from The Institute for Genomic Research Web site at http://www.tigr.org. This work was supported by an HHMI predoctoral fellowship to S.R.L.
Supplemental material is available at http://www.genesdev.org.
Article published online ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.1377006.
References
- Ambros V., Lee, R.C., Lavanway, A., Williams, P.T., and Jewell, D. 2003. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr. Biol. 13: 807–818. [DOI] [PubMed] [Google Scholar]
- Aravin A.A., Lagos-Quintana, M., Yalcin, A., Zavolan, M., Marks, D., Snyder, B., Gaasterland, T., Meyer, J., and Tuschl, T. 2003. The small RNA profile during Drosophila melanogaster development. Dev. Cell. 5: 337–350. [DOI] [PubMed] [Google Scholar]
- Bartel B. 2005. MicroRNAs directing siRNA biogenesis. Nat. Struct. Mol. Biol. 12: 569–571. [DOI] [PubMed] [Google Scholar]
- Chalker D.L. and Yao, M.C. 2001. Nongenic, bidirectional transcription precedes and may promote developmental DNA deletion in Tetrahymena thermophila. Genes & Dev. 15: 1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker D.L., Fuller, P., and Yao, M.C. 2005. Communication between parental and developing genomes during Tetrahymena nuclear differentiation is likely mediated by homologous RNAs. Genetics 169: 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicas A., Cogoni, C., and Macino, G. 2004. RNAi-dependent and RNAi-independent mechanisms contribute to the silencing of RIPed sequences in Neurospora crassa. Nucleic Acids Res. 32: 4237–4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A., Shi, H., Tschudi, C., and Ullu, E. 2001. RNA interference in Trypanosoma brucei: Cloning of small interfering RNAs provides evidence for retroposon-derived 24–26-nucleotide RNAs. RNA 7: 1522–1530. [PMC free article] [PubMed] [Google Scholar]
- Khvorova A., Reynolds, A., and Jayasena, S.D. 2003. Functional siRNAs and miRNAs exhibit strand bias. Cell 115: 209–216. [DOI] [PubMed] [Google Scholar]
- Lau N.C., Lim, L.P., Weinstein, E.G., and Bartel, D.P. 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862. [DOI] [PubMed] [Google Scholar]
- Lee S.R. and Collins, K. 2005. Starvation-induced cleavage of the tRNA anticodon loop in Tetrahymena thermophila. J. Biol. Chem. (in press). [DOI: 10.1074/jbc.M510356200.] [DOI] [PubMed]
- Li J., Yang, Z., Yu, B., Liu, J., and Che, X. 2005. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol. 15: 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Mochizuki, K., and Gorovsky, M.A. 2004. Histone H3 lysine 9 methylation is required for DNA elimination in developing macronuclei in Tetrahymena. Proc. Natl. Acad. Sci. 101: 1679–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone C.D., Anderson, A.M., Motl, J.A., Rexer, C.H., and Chalker, D.L. 2005. Germ line transcripts are processed by a Dicer-like protein that is essential for developmentally programmed genome rearrangements of Tetrahymena thermophila. Mol. Cell. Biol. 25: 9151–9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke M.A. and Birchler, J.A. 2005. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 6: 24–35. [DOI] [PubMed] [Google Scholar]
- Mochizuki K. and Gorovsky, M.A. 2004a. Conjugation-specific small RNAs in Tetrahymena have predicted properties of scan (scn) RNAs involved in genome rearrangement. Genes & Dev. 18: 2068–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 2004b. Small RNAs in genome rearrangement in Tetrahymena. Curr. Opin. Genet. Dev. 14: 181–187. [DOI] [PubMed] [Google Scholar]
- ____. 2005. A Dicer-like protein in Tetrahymena has distinct functions in genome rearrangement, chromosome segregation, and meiotic prophase. Genes & Dev. 19: 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K., Fine, N.A., Fujisawa, T., and Gorovsky, M.A. 2002. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in Tetrahymena. Cell 110: 689–699. [DOI] [PubMed] [Google Scholar]
- Pfeffer S., Lagos-Quintana, M., and Tuschl, T. 2005. Cloning of small RNA molecules. In Current protocols in molecular biology (eds. R.B.F.M. Ausubel et al.), pp. 26.4.1–26.4.18. Wiley Interscience, New York. [DOI] [PubMed]
- Pratt K. and Hattman, S. 1981. Deoxyribonucleic acid methylation and chromatin organization in Tetrahymena thermophila. Mol. Cell. Biol. 1: 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart B.J. and Bartel, D.P. 2002. Small RNAs correspond to centromere heterochromatic repeats. Science 297: 1831. [DOI] [PubMed] [Google Scholar]
- Sijen T. and Plasterk, R.H. 2003. Transposon silencing in the Caenorhabditis elegans germ line by natural RNAi. Nature 426: 310–314. [DOI] [PubMed] [Google Scholar]
- Sontheimer E.J. and Carthew, R.W. 2005. Silence from within: Endogenous siRNAs and miRNAs. Cell 122: 9–12. [DOI] [PubMed] [Google Scholar]
- Strahl B.D., Ohba, R., Cook, R.G., and Allis, C.D. 1999. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc. Natl. Acad. Sci. 96: 14967–14972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S.D., Coyne, R.S., and Allis, C.D. 2002. Methylation of histone h3 at lysine 9 targets programmed DNA elimination in Tetrahymena. Cell 110: 701–711. [DOI] [PubMed] [Google Scholar]
- Tomari Y. and Zamore, P.D. 2005. Perspective: Machines for RNAi. Genes & Dev. 19: 517–529. [DOI] [PubMed] [Google Scholar]
- Ullu E., Lujan, H.D., and Tschudi, C. 2005. Small sense and antisense RNAs derived from a telomeric retroposon family in Giardia intestinalis. Eukaryot. Cell 4: 1155–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F., Vaucheret, H., Rajagopalan, R., Lepers, C., Gasciolli, V., Mallory, A.C., Hilbert, J.L., Bartel, D.P., and Crete, P.Y. 2004. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol. Cell 16: 69–79. [DOI] [PubMed] [Google Scholar]
- Yao M.C. and Chao, J.L. 2005. RNA-guided DNA deletion in Tetrahymena: An RNAi-based mechanism for programmed genome rearrangements. Annu. Rev. Genet. 39: 537–559. [DOI] [PubMed] [Google Scholar]
- Yao M.C., Fuller, P., and Xi, X. 2003. Programmed DNA deletion as an RNA-guided system of genome defense. Science 300: 1581–1584. [DOI] [PubMed] [Google Scholar]