

Abstract
Wunen (Wun), a homologue of a lipid phosphate phosphatase (LPP), has a crucial function in the migration and survival of primordial germ cells (PGCs) during Drosophila embryogenesis. Past work has indicated that the LPP isoforms may show functional redundancy in certain systems, and that they have broad-range lipid phosphatase activities in vitro, with little apparent specificity between them. We show here that there are marked differences in biochemical activity between fly Wun and mammalian LPPs, with Wun having a narrower activity range than has been reported for the mammalian LPPs. Furthermore, although it is active on a range of substrates in vitro, mouse Lpp1 has no activity on an endogenous Drosophila germ-cell-specific factor in vivo. Conversely, human LPP3 is active, resulting in aberrant migration and PGC death. These results show an absolute difference in bioactivity among LPP isoforms for the first time in a model organism and may point towards an underlying signalling system that is conserved between flies and humans.
Introduction
Primordial germ cells (PGCs) are the stem-cell progenitors of the germ line. In Drosophila, as in most organisms, the PGCs originate in an area of the developing embryo that is remote from their target tissue, the somatic gonad. During their migration through the embryo in search of the somatic gonad, they are subject to several guidance cues that are essential for a successful journey (Wylie, 1999; Starz-Gaiano & Lehmann, 2001).
The process of PGC migration provides a system for studying the control of cellular migration in general. wunen (wun) and its closely related counterpart wunen 2 (wun2) encode Drosophila homologues of mammalian lipid phosphate phosphatases (LPPs). Wun and Wun2 have dynamic expression patterns, but are most strongly evident in the hindgut primordium at stage 10, becoming localized to the side of the midgut from which the PGCs migrate. In embryos that are mutant for both genes, the PGCs scatter on exiting the midgut at stage 10, and fail to complete migration to the gonads. Ectopic expression of Wun or Wun2 in the mesoderm at stage 10 repels PGCs from its permissive environment, and is concomitant with a marked reduction in the number of PGCs by stage 13. This activity can be prevented by point mutations in the conserved catalytic residues, indicating that these proteins function as enzymes to dephosphorylate an unknown substrate. The observation that misexpression of either protein results in PGC death shows that this substrate, which is presumably an attractive signal for the PGCs, also regulates their survival (K.H., unpublished data; Starz-Gaiano et al., 2001; Zhang et al., 1996, 1997).
The LPPs are grouped into three isoforms, 1, 2 and 3, and the human genes also have splice variants (Waggoner et al., 1999). We analysed sequence alignments of Wun and Wun2 with mammalian LPPs, and conclude that both proteins have the greatest homology with human LPP3.
Past work has indicated that the three known LPP isoforms can dephosphorylate a broad range of lipid phosphates, notably lysophosphatidic acid (LPA), phosphatidic acid (PA), ceramide-1-phosphate (C(1)P), diacylglycerol pyrophosphate (DGPP) and sphingosine-1-phosphate (S(1)P), with relatively little apparent specificity (Dillon et al., 1997; Jasinska et al., 1999; Kai et al., 1997; Roberts et al., 1998; Waggoner et al., 1996). Apart from Wun and Wun2, little is known about the biological functions of these proteins. Dri42 (differentially expressed in rat intestine 42), the rat homologue of LPP3, is upregulated during differentiation of the crypt cells in the small intestine (Barila et al., 1996), whereas the human splice-variant LPP1-α1 is downregulated in human colon-tumour tissue (Leung et al., 1998). In 2000, Zhang et al. reported a homozygous null mutation in murine Lpp2 that produced viable, fertile mice with no detectable phenotype. In addition, Lpp2 is expressed at lower levels than the Lpp1 and Lpp3 isoforms, leading to the proposal that Lpp2 functions redundantly with them (Zhang et al., 2000). Starz-Gaiano et al. reported that null mutations in either wun or wun2 also present no detectable phenotype, with embryonic development and PGC migration occurring normally. By contrast, removal of both genes results in highly perturbed PGC migration, with PGCs scattering widely on exiting the midgut at stage 10. This led to the suggestion that these LPPs function redundantly (Starz-Gaiano et al., 2001).
Given the remarkable bioactivity of the potential lipid substrates for these enzymes, and the specificity of some of their receptors (Takuwa et al., 2002), we found it curious that the separate isoforms present such broad-range activity in biochemical assays and seem to be functionally redundant in vivo. We were interested in examining any potential specificity in substrate recognition among the fly and mammalian isoforms in vitro for a range of known substrates, and in vivo by investigating their ability to dephosphorylate the PGC-specific guidance molecule that functions as a substrate for Wun. We show here that there are marked differences in relative activity between immunopurified Wun and the mammalian isoforms in a biochemical phosphate-release assay, with Wun showing negligible activity for two of the substrates. We created transgenic flies expressing mouse Lpp1 and human LPP3, and show that, although active on all of the tested substrates in vitro, mouse Lpp1 is completely inactive in the germ-cell migration bioassay. Conversely, overexpression of human LPP3 results in aberrant PGC migration and death, with a remarkably similar phenotype to Wun overexpression. This demonstrates a distinct difference in bioactivity between the isoforms for the first time, and may point towards an underlying signalling system that is conserved between flies and humans.
Results
wun and wun2 have the same messenger RNA expression pattern and the same ectopic expression phenotype, indicating a high level of conservation between the two genes. To investigate the relationship between mammalian LPPs and Wun and Wun2, we performed ClustalW alignments (http://www.ebi.ac.uk/clustalw) of the entire amino-acid sequences of each protein alongside those of the human LPPs, LPP1, LPP2 and LPP3 (Fig. 1). This showed almost complete conservation of the phosphatase domains. A phylogenetic tree, as calculated by ClustalW, indicates that Wun and Wun2 are most homologous to human LPP3 (Fig. 2).
Figure 1.

Sequence alignment of lipid phosphate phosphatase proteins. The conserved phosphatase domains are shown in light blue, with those residues thought to be required for catalytic activity shown in dark blue (Brindley & Waggoner, 1998). The position of the WunD:248>T mutation is shown in white. Six transmembrane domains are indicated, as predicted by TMPred (http://www.ch.embnet.org/software/TMPRED_form.html; Hofmann & Stoffel, 1993), and are shown in boxes. Amino acids that are identical between all five sequences are indicated by asterisks, and are shown on a dark pink background or by dark pink letters. Where conserved substitutions have been identified, the amino acids are indicated by a colon, and are shown on a purple background or by purple letters; where semi-conserved substitutions have been identified, amino acids are indicated by a dot, and are shown on a green background or by green letters. The GenBank accession numbers for each sequence are as follows: human lipid phosphate phosphatase 1 (LPP1), AB000888 (Kai et al., 1997); human LPP2, AF047760 (Roberts et al., 1998); human LPP3, AB000889 (Kai et al., 1997); Wunen, U73822 (Zhang et al., 1997); Wunen2, AF331162 (Starz-Gaiano et al., 2001).
Figure 2.
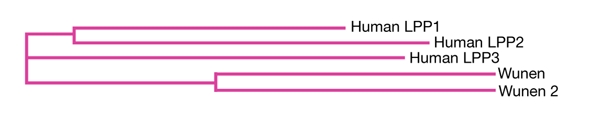
Phylogenetic tree of Wunen and Wunen 2 with the human lipid phosphate phosphatase isoforms. The tree was generated using the sequences shown in Fig. 1. LPP, lipid phosphate phosphatase.
Alongside Wun, we focused on mouse Lpp1 and human LPP3, as they had been previously cloned and characterized (Kai et al., 1996, 1997). We tagged each protein at the carboxyl terminus with green fluorescent protein (GFP). We also tagged WunD:248>T, which has a mutation in the sixth transmembrane domain that disrupts the putative catalytic site (Neuwald, 1997) and should result in a catalytic null (Fig. 3A). Complementary DNAs encoding these proteins were cloned into the Gal4-regulated transformation vector pUAST, and the sequences of the modified inserts were confirmed. To verify that they encoded proteins of the predicted sizes, we transfected them individually into Drosophila S2 cells with the ubiquitous driver Actin5C–Gal4 and analysed them by western blotting (Fig. 3B).
Figure 3.
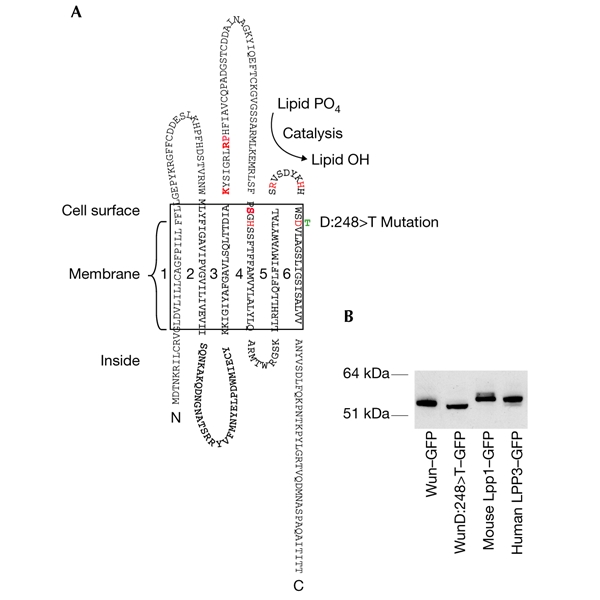
The Wunen protein and confirmation of protein sizes. (A) The Wunen (Wun) protein, indicating the position of the D:248>T point mutation. Conserved residues required for catalysis are shown in red (Neuwald, 1997). (B) Western blot confirming that each protein runs to the correct predicted size. GFP, green fluorescent protein; LPP, lipid phosphate phosphatase.
We investigated the activity of immunopurified Wun and the two mammalian LPPs on PA, LPA, S(1)P and C(1)P in a biochemical assay. The results show that, despite the presence of high levels of D:248>T, it is completely inactive for any of the substrates. Whereas mouse Lpp1 and Wun showed similar levels of activity for LPA, human LPP3 was considerably less active (Table 1; Fig. 4). However, human LPP3 showed ∼1.6 times more activity for PA, than mouse Lpp1 (Table 1), whereas mouse Lpp1 was ∼1.7 times more active on C(1)P than was human LPP3 (Table 1). By comparison, Wun showed negligible activity on C(1)P or PA. We were unable to obtain any results for S(1)P, possibly due to an inability to present the substrate optimally in this system.
Table 1.
Relative activities from the phosphate-release assay and densitometry
| Construct | nmol PO4 released (mean ± s.d.) | nmol PO4 released per unit density (mean ± s.d.) | Relative activity |
|---|---|---|---|
| LPA | |||
| Wun–GFP | 2.819 ± 0.180 | 0.079 ± 0.012 | 1.00 |
| Mouse Lpp1–GFP | 3.056 ± 0.496 | 0.079 ± 0.024 | 1.00 |
| Human LPP3–GFP | 0.359 ± 0.109 | 0.013 ± 0.004 | 0.16 |
| WunD:248>T–GFP | ∼0.000 ± 0.156 | 0.000 ± 0.002 | 0.00 |
| Untransfected S2 | ∼0.000 ± 0.017 | n.a. | 0.00 |
| PA | |||
| Wun–GFP | 0.021 ± 0.016 | 0.002 ± 0.001 | 1.00 |
| Mouse Lpp1–GFP | 0.745 ± 0.070 | 0.024 ± 0.003 | 12.00 |
| Human LPP3–GFP | 0.530 ± 0.018 | 0.038 ± 0.005 | 19.00 |
| WunD:248>T–GFP | ∼0.000 ± 0.029 | 0.000 ± 0.002 | 0.00 |
| Untransfected S2 | ∼0.000 ± 0.015 | n.a. | 0.00 |
| C(1)P | |||
| Wun–GFP | 0.098 ± 0.045 | 0.004 ± 0.001 | 1.00 |
| Mouse Lpp1–GFP | 3.754 ± 0.207 | 0.083 ± 0.020 | 20.75 |
| Human LPP3–GFP | 0.933 ± 0.662 | 0.049 ± 0.034 | 12.25 |
| WunD:248>T–GFP | ∼0.000 ± 0.026 | 0.000 ± 0.003 | 0.00 |
| Untransfected S2 | ∼0.000 ± 0.049 | n.a. | 0.00 |
Each immunocaptured enzyme was incubated in triplicate with lysophosphatidic acid (LPA), phosphatidic acid (PA) or ceramide-1-phosphate (C(1)P), in the PiPer® phosphate-release assay, and the nanomoles of phosphate released for each individual sample were calculated from a standard curve. The means of the three readings for each enzyme are shown. Less than 11% of total substrate was broken down in any of the experiments. The density for each individual sample on a western blot was calculated using BioRad Quantity One, and this was used to calculate the nanomoles of phosphate released per unit density.
Figure 4.
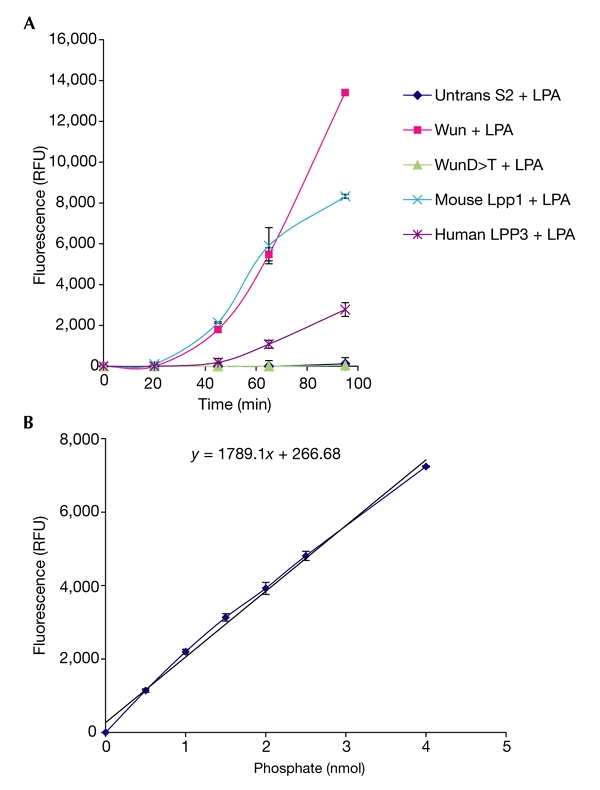
PiPer® phosphate-release assay with lysophosphatidic acid. Fluorescence was measured using a SPECTRAmax™ GEMINI XS Dual Scanning Microplate Spectrafluorometer. (A) Mean fluorescent readout ± s.d. over time from the three experiments for each protein. The sequential action of the enzymes involved in the detection system after exposure to phosphate in solution accounts for the 20-min lag period seen at the start of the assay. Running phosphate standards alone produces the same lag period (see supplementary information online). Plotting phosphate standards against the corresponding fluorescent readout at the chosen timepoint (t = 65 min) gives a straight line in the presence of up to 4 nmol, indicating that the detection system shows first-order kinetics at this timepoint (B). These values are the means ± s.d. of the three samples for each standard. LPA, lysophosphatidic acid; LPP, lipid phosphate phosphatase; RFU, relative fluorescence units; Untrans, untransfected; Wun, Wunen.
We generated transgenic flies for each construct and crossed them to the mesodermal driver Twist–Gal4 (Greig & Akam, 1993). Expression of Wun–GFP disrupted PGC migration and reduced PGC number, as previously reported for untagged Wun. PGCs are seen in tissues other than the gonads, having been repelled from the mesoderm and therefore failing to contact the somatic gonadal precursors (Fig. 5A,F).
Figure 5.
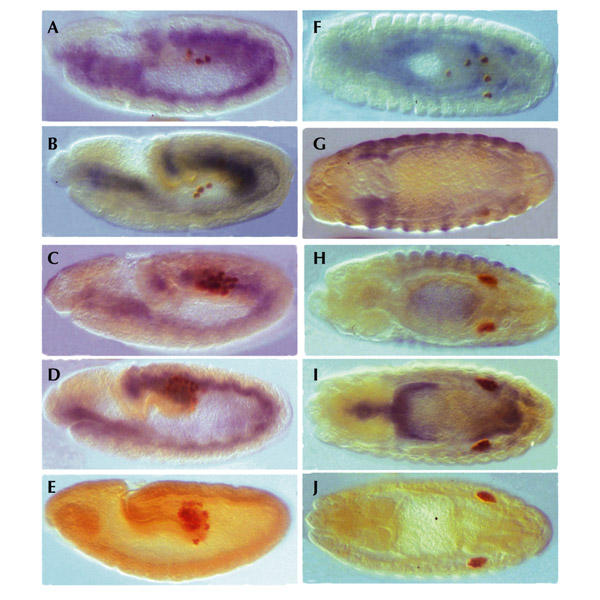
Ectopic expression in the mesoderm. Embryos were immunostained with anti-Vasa antibody to visualize the primordial germ cells (PGCs; brown) and with anti-green-fluorescent-protein (GFP) to visualize protein expression (blue). Embryos in (A–E) are viewed laterally, with the posterior pole to the right; embryos in (F–J) are viewed dorsally. Expression of Wunen (Wun)–GFP (A) or human lipid phosphate phosphatase 3 (LPP3)–GFP (B) at stage 10 results in an early loss of PGCs compared with the ectopic expression of WunD:248>T–GFP (C) or mouse Lpp1–GFP (D). These can be compared with a wild-type embryo (E) with a full complement of PGCs at the same stage. By the end of embryogenesis, those embryos expressing Wun–GFP (F) or LPP3–GFP (G) show a marked loss of PGCs. Embryos expressing WunD:248>T–GFP (H) or mouse Lpp1–GFP (I), however, show no apparent perturbation or loss of PGCs, and clearly form two gonads, as seen in a wild-type embryo at the same stage (J). Wild-type embryos have not been stained with anti-GFP, and consequently show no blue staining.
The Wun catalytic-null D:248>T had no biological activity. PGC migration occurred normally, even in the presence of high levels of the mutant protein in the mesoderm (Fig. 5C,H). Starz-Gaiano et al. (2001) have shown previously that point mutations in the conserved catalytic domain of Wun2 abolish its biological activity in vivo. However, this is the first time that loss of phosphatase activity, as confirmed in a biochemical assay, has also been shown to abolish biological function, and this indicates that converting a single aspartic acid to a threonine at residue 248 is sufficient to remove catalytic activity both in vitro and in vivo in Wun.
Surprisingly, expression of human LPP3–GFP also resulted in highly perturbed PGC migration and a marked reduction in PGC number. In some embryos, only one or two PGCs remain at the end of embryogenesis (Fig. 5B,G). Human LPP3 may therefore recognize and dephosphorylate the same PGC-specific substrate in flies as Drosophila Wun.
Conversely, expression of biochemically active mouse Lpp1–GFP gave no phenotype, with no aberrant migration or apparent PGC loss (Fig. 5D,I). Mouse Lpp1 is therefore incapable of dephosphorylating the same germ-cell-specific factor as Wun in vivo, demonstrating an absolute difference in functional bioactivity between the LPP1 and LPP3 isoforms.
The LPPs have been shown to have ecto-enzymatic activity (Ishikawa et al., 2000; Jasinska et al., 1999; Roberts et al., 1998). Although we do not know if the germ-cell migration phenotype is caused by an extracellular activity of Wun or human LPP3, we wanted to exclude the possibility that mouse Lpp1–GFP fails to be correctly trafficked and to confirm that all of the GFP-tagged proteins are trafficked to the cell surface. We examined embryos that express these proteins in the mesoderm by confocal microscopy. All of the proteins were detected on the mesodermal cell surface (Fig. 6A). None of the proteins localized to intracellular membrane structures when transiently expressed in the embryonic mesoderm by this method. We biochemically analysed the cell surfaces of intact S2 cells that expressed either human LPP3–GFP or mouse Lpp1–GFP by incubating them with N-hydroxysuccinimide–biotin in solution and running the lysates over a monomeric-biotin-binding avidin column. Western blot analysis also confirmed each protein's presence at the cell surface (Fig. 6B).
Figure 6.
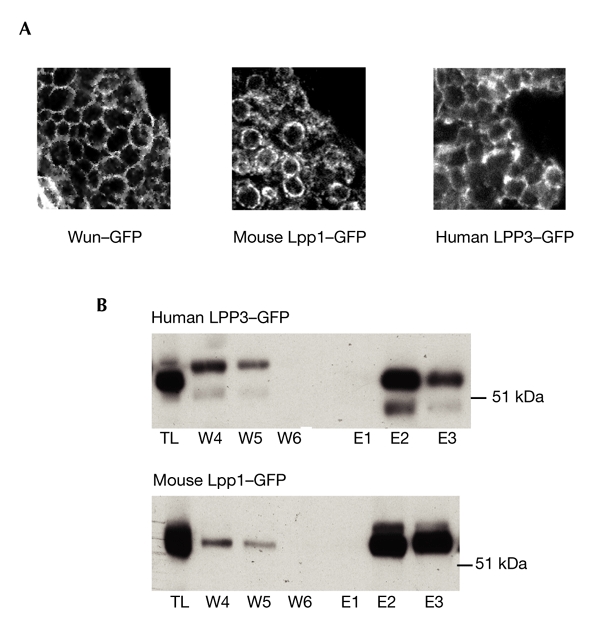
Confirmation of proteins present at the cell surface. (A) Images obtained by confocal microscopy, showing Wunen (Wun)–green-fluorescent-protein (GFP), mouse lipid phosphate phosphatase 1 (Lpp1)–GFP and human LPP3–GFP at the surface of mesodermal cells in Drosophila embryos. Protein expression is driven by Twist–Gal4. (B) Biotinylation of human LPP3–GFP and mouse Lpp1–GFP at the surface of S2 cells, detected with anti-GFP antibody. E, elutions (protein that is biotinylated and has bound to the column); TL, total lysate before capture on the column; W, washes (protein that is not biotinylated and has not bound to the column).
Discussion
Although we have not performed detailed kinetic analyses, we show here that the fly LPP Wun and the mammalian LPP1 and LPP3 isoforms differ in their relative activities on PA, LPA and C(1)P in the PiPer® phosphate-release assay. Although Wun can dephosphorylate LPA with a similar efficiency to mouse Lpp1, it shows negligible activity on both PA and C(1)P. This indicates that there are distinct differences in substrate preference between the fly and mammalian enzymes, with Wun showing a narrower activity range in this assay than was previously reported for the mammalian LPPs. Both mouse Lpp1 and human LPP3 are active on all three substrates. We found that mouse Lpp1 has a 1.7-fold higher activity on C(1)P than human LPP3 in this assay. Human LPP3, however, had a 1.6-fold higher activity than mouse Lpp1 on PA. Previous data on the activity for each isoform on these substrates has tended to vary (Kai et al., 1997; Roberts et al., 1998). The inconsistencies that have been observed for the same isoform on the same substrates may be due to the particular assay conditions or the method of enzyme preparation. This could account for the variation in relative activities between Wun and human LPP3 in vitro, despite their close homology. Alternatively, there may be fundamental differences between the fly and mammalian isoforms that we are unable to uncover without a more detailed analysis of their structures.
We also show, however, that human LPP3 is highly active when ectopically expressed in Drosophila embryos, as assayed by the disruption of PGC migration and survival, which results in a phenotype similar to the ectopic expression of Wun or Wun2. That human LPP3 shows the same phenotype in a bioassay as Drosophila Wun suggests a conserved signalling pathway that regulates germ-cell migration and survival from flies to humans. Conversely, although active in the biochemical assay, mouse Lpp1 is completely inactive in vivo, and has no apparent effect on PGC migration or survival. This shows an absolute difference in functional bioactivity between the mammalian LPP isoforms. That the LPP isoforms present different functional outputs when assayed in vivo has a number of implications. It may be that the Lpp1 isoform simply cannot recognize or catalyse the dephosphorylation of the specific factor acted on by Wun and LPP3. This would demonstrate specificity in substrate choice between the isoforms, and may indicate that the germ-cell-specific factor is not PA, LPA or C(1)P, on which mouse Lpp1 is active in vitro, particularly as Wun shows negligible activity on PA and C(1)P in the same assay. Alternatively, there may be specific components of the pathway that are required for selection and recognition of the factor. It is possible that as yet unidentified conformational or structural differences in mouse Lpp1 preclude its association with these factors, thus inhibiting its enzymatic function in this system. Thus, LPA could be the factor, and the unnatural presentation and high concentration of LPA in the biochemical assay may override the specific selection mechanisms used to regulate activity in vivo, allowing mouse Lpp1 access to this otherwise inaccessible substrate. Primary sequence analyses have not identified any immediate candidates for residues conferring such a difference. We speculate that the observed differences in substrate preference may be related to the proteins' structure, which are as yet unsolved. We note that Barila et al. have already studied the properties of the internal sequences of Dri42 in its trafficking to the cell surface (Barila et al., 1996). The contributions of the termini to biological and biochemical properties are yet to be reported in any detail.
In conclusion, we present evidence of differences in relative activity between the mammalian and fly LPP isoforms on LPA, PA and C(1)P in a biochemical assay. Our results indicate that the fly LPP Wun has a narrower activity range than the mammalian LPPs in this assay. We also present evidence to show that, although active in vitro, mouse Lpp1 cannot dephosphorylate the same endogenous germ-cell-specific factor as Wun in vivo, whereas human LPP3 seems to do so. This demonstrates that despite broad-range activity in Triton-micelle assays, the mammalian LPP isoforms do show distinct differences in bioactivity when assayed in a physiological context. We expect that a combination of biochemical and biological data, such as those presented here, will in time help to identify the physiological substrate for Wun, which is, presumably, a stem-cell control factor.
Methods
Cloning and expression in S2 cells.
Human LPP3–GFP cloning was performed using the construct made in EGFPN3 by I. Wada. The Wun–GFP construct was also made in pEGFPN3 by I. Wada. We transferred sequences to pUAST using conventional restriction enzyme digestion techniques. The mouse Lpp1 and mutant wun constructs were made in our laboratory. Where we used PCR, oligonucleotide mutagenesis or linker insertion, the resulting clones were sequenced through the entire coding region of the modified protein and the sequences were deposited in GenBank. All tags were added to C termini.
Drosophila S2 cells were maintained in HyQCCM3 media (Perbio Science). We used the Effectene Transfection Reagent (Qiagen). Forty-eight hours post-transfection, cells were washed in PBS, spun at 3,000 r.p.m. and lysed on ice in lysis buffer (0.5 M Hepes, 5 M sodium chloride, 1 M sodium fluoride, 0.5 M EDTA, 0.5 M sodium orthovanadate, 0.1% triton X-100, 2 mM N-ethylmaleimide, and Complete protease inhibitors (Roche)). Western blots were performed using standard techniques. We used a mouse monoclonal antibody to GFP (Roche) followed by a horseradish peroxidase (HRP)-conjugated anti-mouse antibody (Jackson ImmunoResearch Laboratories).
Protein immunoprecipitation.
Transfected S2 cells were lysed as before and an equal volume of lysate was added to an aliquot of Fusion Aid GFP resin (Vector laboratories). After incubation for 2 h at 4 °C, the resin was washed extensively and resuspended in a 5× volume of cell lysis buffer plus protease inhibitors before western blot analysis. Lysate from untransfected S2 cells at the same density was also immunocaptured as a control.
Phosphate-release assay.
We used the Molecular Probes PiPer® phosphate-release assay (Cambridge Bioscience). In the presence of inorganic phosphate, maltose phosphorylase converts maltose to glucose-1-phosphate and glucose. Gluconolactone and H2O2 are then formed by the action of glucose oxidase. Using HRP as a catalyst, the H2O2 reacts with the Amplex™ Red reagent to produce the fluorescent product resorufin. The increase in detectable fluorescence/absorbance is therefore proportional to the amount of phosphate present.
Each reaction was performed in triplicate, using 50 μl of the anti-GFP resin containing the immunocaptured proteins. Assays were performed at 37 °C and the resulting fluorescence was measured in a standard plate-reader in a final volume of 100 μl at up to seven timepoints during the assay period. Each substrate control gave a small but constant value, which was subtracted from each of the samples. LPA (Sigma) in 50% ethanol and C(1)P (Affiniti) in 100% ethanol were used at a final concentration of 500 μM. PA (Sigma) in 100% ethanol was used at a final concentration of 70 μM in the supplied buffer with 0.01% phosphate-free triton X-100 (Sigma) and 1 mg ml−1 fatty-acid-free BSA (Sigma). To test the linearity of the detection system, a range of phosphate standards were run simultaneously in triplicate. We examined activity at an arbitrary timepoint that seemed to fall within the linear range for the enzymes for each assay. The PO4 standards were converted to nanomoles of PO4 at this time, and were used to produce a standard curve. This was linear in each case, indicating that the detection system shows first-order kinetics in the presence of up to 4 nmol of PO4. A line of best fit was used to calculate the amount of PO4 released from each protein sample. Each sample was western blotted, and densitometry was performed using BioRad Quantity One software to compare relative protein amounts in each sample on the same blot.
Ectopic expression.
The constructs were microinjected into white118 embryos and transformants were recovered using standard techniques. To visualize the construct in the mesoderm and the PGCs, we used a mouse monoclonal antibody to GFP (Roche) followed by an alkaline-phosphatase-conjugated anti-mouse antibody (Jackson ImmunoResearch Laboratories), and chicken anti-Vasa (K.H.) followed by biotin-conjugated anti-chicken (Jackson ImmunoResearch Laboratories).
Cell-surface biotinylation.
We used Pierce No-Weigh™ Premeasured NHS-PE04 Biotin (Perbio Science). The reaction was quenched with 100 mM ammonium chloride. Cells were washed to remove extraneous unbound biotin, lysed on ice in 1.5 ml cell lysis buffer (1% Triton X-100) and spun for 5 min at 3,000 r.p.m. at 4 °C. The supernatant was applied to a Pierce Immunopure Monomeric Avidin column (Perbio Science) in accordance with the manufacturer's instructions. Fractions were analysed by western blotting.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor900-s1.pdf).
Supplementary Material
Supplementary information
Acknowledgments
We thank A. Harwood, J. Ryves, P. Makridou, A. Morris, D. Cutler, R. Lehmann and M. Raff for help and advice, and T. Kornberg for the Actin5C–Gal4 driver. We also thank H. Kanoh, I. Wada and M. Kai for sharing materials and for help with cloning. This work was supported by the Wellcome Trust, grant number 054375.
References
- Barila D. et al. ( 1996) The Dri 42 gene, whose expression is up-regulated during epithelial differentiation, encodes a novel endoplasmic reticulum resident transmembrane protein. J. Biol. Chem., 271, 29928–29936. [DOI] [PubMed] [Google Scholar]
- Brindley D. & Waggoner W. ( 1998) Mammalian lipid phosphate phosphohydrolases. J. Biol. Chem., 273, 24281–24284. [DOI] [PubMed] [Google Scholar]
- Dillon D.A. et al. ( 1997) Mammalian Mg2+-independent phosphatidate phosphatase (PAP2) displays diacylglycerol pyrophosphate phosphatase activity. J. Biol. Chem., 272, 10361–10366. [DOI] [PubMed] [Google Scholar]
- Greig S. & Akam M. ( 1993) Homeotic genes autonomously specify one aspect of pattern in the Drosophila mesoderm. Nature, 362, 630–632. [DOI] [PubMed] [Google Scholar]
- Hofmann K. & Stoffel W. ( 1993) TMbase—a database of membrane spanning proteins segments. Biol. Chem. Hoppe Seyler, 374, 166. [Google Scholar]
- Ishikawa T., Kai M., Wada I. & Kanoh H. ( 2000) Cell surface activities of the human type 2b phosphatidic acid phosphatase. J. Biochem., 127, 645–651. [DOI] [PubMed] [Google Scholar]
- Jasinska R. et al. ( 1999) Lipid phosphate phosphohydrolase-1 degrades exogenous glycerolipid and sphingolipid phosphate esters. Biochem. J., 340, 677–686. [PMC free article] [PubMed] [Google Scholar]
- Kai M., Wada I., Imai S., Sakane F. & Kanoh H. ( 1996) Identification and cDNA cloning of 35-kDa phosphatidic acid phosphatase (type 2) bound to plasma membranes. Polymerase chain reaction amplification of mouse H2O2-inducible hic53 clone yielded the cDNA encoding phosphatidic acid phosphatase. J. Biol. Chem., 271, 18931–18938. [DOI] [PubMed] [Google Scholar]
- Kai M., Wada I., Imai S., Sakane F. & Kanoh H. ( 1997) Cloning and characterization of two human isozymes of Mg2+-independent phosphatidic acid phosphatase. J. Biol. Chem., 272, 24572–24578. [DOI] [PubMed] [Google Scholar]
- Leung D.W., Tomkins C.K. & White T. ( 1998) Molecular cloning of two alternatively spliced forms of human phosphatidic acid phosphatase cDNAs that are differentially expressed in normal and tumor cells. DNA Cell Biol., 17, 377–385. [DOI] [PubMed] [Google Scholar]
- Neuwald A.F. ( 1997) An unexpected structural relationship between integral membrane phosphatases and soluble haloperoxidases. Protein Sci., 6, 1764–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R., Sciorra V.A. & Morris A.J. ( 1998) Human type 2 phosphatidic acid phosphohydrolases. Substrate specificity of the type 2a, 2b, and 2c enzymes and cell surface activity of the 2a isoform. J. Biol. Chem., 273, 22059–22067. [DOI] [PubMed] [Google Scholar]
- Starz-Gaiano M. & Lehmann R. ( 2001) Moving towards the next generation. Mech. Dev., 105, 5–18. [DOI] [PubMed] [Google Scholar]
- Starz-Gaiano M., Cho N.K., Forbes A. & Lehmann R. ( 2001) Spatially restricted activity of a Drosophila lipid phosphatase guides migrating germ cells. Development, 128, 983–991. [DOI] [PubMed] [Google Scholar]
- Takuwa Y., Takuwa N. & Sugimoto N. ( 2002) The Edg family G protein-coupled receptors for lysophospholipids: their signaling properties and biological activities. J. Biochem. (Tokyo), 131, 767–771. [DOI] [PubMed] [Google Scholar]
- Waggoner D.W., Gomez-Munoz A., Dewald J. & Brindley D.N. ( 1996) Phosphatidate phosphohydrolase catalyzes the hydrolysis of ceramide 1-phosphate, lysophosphatidate, and sphingosine 1-phosphate. J. Biol. Chem., 271, 16506–16509. [DOI] [PubMed] [Google Scholar]
- Waggoner D.W., Xu J., Singh I., Jasinska R., Zhang Q.X. & Brindley D.N. ( 1999) Structural organization of mammalian lipid phosphate phosphatases: implications for signal transduction. Biochim. Biophys. Acta, 1439, 299–316. [DOI] [PubMed] [Google Scholar]
- Wylie C. ( 1999) Germ cells. Cell, 96, 165–174. [DOI] [PubMed] [Google Scholar]
- Zhang N., Zhang J., Cheng Y. & Howard K. ( 1996) Identification and genetic analysis of wunen, a gene guiding Drosophila melanogaster germ cell migration. Genetics, 143, 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Zhang J., Purcell K.J., Cheng Y. & Howard K. ( 1997) The Drosophila protein Wunen repels migrating germ cells. Nature, 385, 64–67. [DOI] [PubMed] [Google Scholar]
- Zhang N., Sundberg J.P. & Gridley T. ( 2000) Mice mutant for Ppap2c, a homolog of the germ cell migration regulator wunen, are viable and fertile. Genesis, 27, 137–140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information