

Abstract
Histone variants play important roles in the maintenance and regulation of the chromatin structure. In order to characterize the biochemical properties of the chromatin structure containing histone variants, we investigated the dynamic status of nucleosome core particles (NCPs) that were assembled with recombinant histones. We found that in the presence of nucleosome assembly protein I (NAP-I), a histone chaperone, H2A-Barr body deficient (H2A.Bbd) confers the most flexible nucleosome structure among the mammalian histone H2A variants known thus far. NAP-I mediated the efficient assembly and disassembly of the H2A.Bbd-H2B dimers from NCPs. This reaction was accomplished more efficiently when the NCPs contained H3.3, a histone H3 variant known to be localized in the active chromatin, than when the NCPs contained the canonical H3. These observations indicate that the histone variants H2A.Bbd and H3.3 are involved in the formation and maintenance of the active chromatin structure. We also observed that acidic histone binding proteins, TAF-I/SET and B23.1, demonstrated dimer assembly and disassembly activity, but the efficiency of their activity was considerably lower than that of NAP-I. Thus, both the acidic nature of NAP-I and its other functional structure(s) may be essential to mediate the assembly and disassembly of the dimers in NCPs.
A nucleosome is the fundamental repeating unit of chromatin and consists of 147 base pairs of DNA wrapped around a histone octamer. Nucleosome assembly and disassembly have a great impact on the regulation of nuclear gene functions, such as transcription, replication, repair, and recombination. The enzymes that are responsible for the posttranslational modifications of histones in combination with chromatin remodeling complexes have been suggested to be important for mediating the assembly and disassembly of the chromatin structure. Recent experiments that used the fluorescent recovery after photobleaching technique have clearly demonstrated that the H2A-H2B dimers are highly dynamic and are rapidly exchanged in living cells (21). This dynamic exchange of the H2A-H2B dimers was also confirmed by several biochemical studies. The H2A-H2B dimers in the nucleosomes are removed by chromatin remodeling complexes (6), transcription elongation complexes (5), and a histone binding protein (40). Therefore, the assembly and disassembly of the H2A-H2B dimers from a nucleosome are indicated to be crucial steps during chromatin remodeling.
Histones that are expressed before and during DNA replication are utilized for the packaging of the newly synthesized DNA into nucleosomes during the S phase. In contrast, histone variants are expressed throughout the cell cycle. To date, four histone H2A variants, namely, H2A.X, H2A.Z, macroH2A, and H2A-Barr body deficient (H2A.Bbd) and two histone H3 variants, namely, H3.3 and CENP-A, have been identified in mammalian somatic cells so far (45). Genetic studies have clearly demonstrated that the histone variants, H2A.Z (9) and CENP-A (14), are encoded by essential genes. H2A.X also plays a crucial role in the DNA repair and recombination pathways, although H2A.X is not essential (7). These genetic studies have suggested that the histone variants are crucial for the formation of a specialized chromatin structure. Despite the presence of significant sequence similarities, each histone variant shows a specific localization pattern. For instance, macroH2A and CENP-A are enriched in the inactive X chromosome and the centromere chromatin, respectively. However, the mechanisms by which these proteins are recruited to the specific chromosome loci and the functions of these proteins at these specialized chromosome regions are largely unknown.
When mixed directly under physiological conditions, histones and DNA form insoluble aggregates. Acidic histone-binding proteins bind to histones and maintain their solubility within the cell. Nucleoplasmin was the first acidic protein to be discovered as a functional histone-binding protein in Xenopus egg extracts (24). Nucleoplasmin decondenses sperm chromatin by stripping the sperm-specific basic proteins and depositing the H2A-H2B dimers on chromatin (43). Several acidic histone-binding proteins having properties similar to those of nucleoplasmin have been identified from mammalian cells (2, 42). We have identified acidic proteins termed template activating factors that are involved in the remodeling of adenovirus chromatin (19, 27, 36). Three acidic proteins, namely, template activating factor I (TAF-I)/SET, TAF-II/NAP-I, and TAF-III/nucleophosmin/B23, were shown to remodel the structure of viral chromatin in order to stimulate the replication and transcription. These proteins bind to histones and mediate nucleosome assembly in vitro in a similar manner. Although the bona fide functions of these proteins in the cell are unclear, several biochemical studies on these proteins strongly indicate that they function as histone chaperones.
Here, we investigated the histone chaperone-mediated dynamic nature of the nucleosome core particles (NCPs) containing various histone variants. An NCP comprises the first-order packaging of DNA in eukaryotic cells and is the best substrate for studying the stability of the chromatin structure. In order to simplify the assay system, a well-characterized 5S rRNA gene fragment from L. variegatus sea urchin was used as a nucleosome positioning sequence for the purpose of NCP assembly. All the histones were prepared as recombinant proteins from bacteria in order to exclude the effect of posttranslational modifications. We observed that NCPs that contains the histone H2A variant, H2A.Bbd, are unstable, and nucleosome assembly protein I (NAP-I) efficiently removes the H2A.Bbd-H2B dimers from the NCPs. By systematically comparing the stability of the NCPs that contain various mammalian histone H2A variants, including canonical H2A, H2A.X, H2A.Z, the histone fold domain of macroH2A1.2, and H2A.Bbd, it was found that H2A.Bbd confers exceptional flexibility to the NCP structure. Furthermore, our data demonstrated that NAP-I mediates the reversible assembly and disassembly of the dimers. These results gave rise to the hypothesis that NAP-I is involved in the exchange between the dimers containing H2A variants and those containing canonical H2A during chromatin remodeling. Further, the activity of NAP-I was found to be significantly higher than those of the other acidic histone binding proteins, TAF-I/SET and B23. Thus, the acidic nature of NAP-I, though essential, is not the sole criteria for nucleosome assembly and disassembly.
MATERIALS AND METHODS
Plasmid DNA.
To express the human histones H2A, H2B, H3, and H4, pET22b-H2A, pET22b-H2B, pET22b-H3, and pET22b-H4, respectively, were used (47). The mouse H3.3 cDNA was amplified from cDNA prepared from NIH 3T3 cells by PCR and subcloned into the NcoI and BamHI sites of pET14b (Novagen). The human H2A.Z, H2A.X, macroH2A1.2, and H2A.Bbd cDNAs were amplified from cDNA prepared from HeLa cells by PCR and subcloned into the NdeI and BamHI sites of pET14b. To prepare the histone fold domain of macroH2A1.2, the original BamHI site in the macroH2A1.2 cDNA was used. The fragment subcloned into the NdeI and BamHI sites of pET14b encodes the histone fold domain (amino acids 1 to 119) of macroH2A1.2. The human NAP-I cDNA was amplified by PCR using cDNA prepared from HeLa cells as a template and subcloned into the NdeI and XhoI sites of pET14b. In this report, the nomenclature NAP-I is used for the human NAP1-like 1 protein. In order to express NAP-I without a hexahistidine tag (His tag), pET21a-NAP-I was constructed by inserting the cDNA encoding NAP-I into the NdeI and BamHI sites of pET21a (Novagen). The sequences of all cDNAs were confirmed by ABI Prism BigDye terminator cycle sequencing (PE Applied Biosystems) using appropriate primers. The sequences of oligonucleotides used are available upon request.
Expression and purification of recombinant proteins.
Recombinant proteins were expressed in BL21(DE3) CodonPlus RIL-pLys S cells (Stratagene). For H2B, H3, H4, and H3.3, cells expressing histones were disrupted by sonication and purified as described previously (26). To purify H2A variants, cells were disrupted by sonication, and the soluble proteins were removed by centrifugation. His-tagged recombinant histones were purified from the insoluble fractions using metal chelating resins (SIGMA) under denaturing condition. Purified His-H2A variant proteins were mixed with purified H2B in 20 mM sodium acetate, pH 5.2, 5 mM β-mercaptoethanol, and 1 mM phenylmethanesulfonyl fluoride (PMSF) containing 8 M urea and then dialyzed against TE buffer (10 mM Tris, pH 7.4, 1 mM EDTA, 5 mM β-mercaptoethanol, and 1 mM PMSF) containing 2 M NaCl for 12 h. To prepare histone H2A-H2B and H2A.Bbd-H2B dimers without the His tag, the refolded dimers (1 mg of total proteins) were dialyzed stepwise with TE buffer containing 1.5, 1.0, 0.5, and 0.1 M NaCl for 3 h at each step and then treated with 3 units of thrombin (Nacalai Tesque) on ice overnight. Histone H2A and its variant proteins contain three additional amino acids (Gly-Ser-His) before the first methionine of the original proteins after thrombin digestion. Thrombin-treated dimers were loaded on a Superdex 200 column (Amersham-Pharmacia) in TE buffer containing 2 M NaCl to remove the His-tag peptide. The histone H2A-H2B and H2A variant-H2B dimers were concentrated by double-stranded DNA-Sepharose column chromatography (Sigma).
Recombinant human NAP-I proteins with or without the His tag were purified from Escherichia coli soluble extracts. His-tagged NAP-I was purified using metal-chelating resins (Sigma) according to the manufacture's protocol. To purify nontagged NAP-I, soluble extracts were fractionated by ammonium sulfate. The soluble proteins in 35% saturation of ammonium sulfate were dialyzed against buffer A (20 mM HEPES-NaOH, pH 7.9, 0.5 mM dithiothreitol [DTT], 0.5 mM PMSF, and 10% glycerol) containing 100 mM NaCl and then loaded on a Mono Q column (1 ml; Amersham-Pharmacia). After extensive washing with the same buffer, the bound proteins were eluted with a linear salt gradient from 100 to 600 mM NaCl. Peak fractions containing NAP-I were dialyzed against buffer A containing 200 mM NaCl and then loaded on a Mini Q column (240 μl; Amersham-Pharmacia). The bound proteins were eluted with a linear salt gradient from 200 to 500 mM NaCl. Peak fractions were collected, and the protein concentration was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) that was stained with Coomassie brilliant blue R250 (CBB).
Nucleosome reconstitution and DNA analyses.
NCPs were assembled with the salt dilution method as described previously (46). Briefly, recombinant histones (2 μg) were mixed with the 5S rRNA gene fragment (2 μg) in 10 μl of 10 mM Tris, pH 7.4, 1 mM EDTA, 0.1 mg/ml bovine serum albumin, 1 mM DTT, and 0.1 mM PMSF in the presence of 2 M NaCl and incubated at 37°C for 10 min. The reaction was serially diluted to 1.5, 1, 0.8, 0.7, 0.6, 0.5, 0.4, 0.25, and 0.2 M NaCl by adding 50 mM HEPES (pH 7.5), 1 mM EDTA, 5 mM DTT, and 0.5 mM PMSF, with 15-min incubations at 30°C for each dilution step. The salt concentration was brought to 0.1 M by adding 100 μl of 10 mM Tris (pH 7.5), 1 mM EDTA, 5 mM DTT, 0.5 mM PMSF, 10% glycerol, and 0.1 mg/ml bovine serum albumin and incubated for 15 min at 30°C. The reconstitutions were confirmed by the nucleoprotein gel analysis.
Nucleoprotein gel analyses, DNase I footprinting, and ExoIII mapping were carried out as described previously (39).
Western blotting.
After the electrophoresis of the NCPs on a nucleoprotein gel, DNA was visualized by ethidium bromide (EtBr) staining. After a brief wash with water, the proteins and DNA on the gel were transferred to a polyvinylidene difluoride (PVDF) membrane at 90 V for 3 h in Tris-glycine buffer (25 mM Tris and 192 mM glycine) containing 20% methanol. Histone H3 and His-tagged H2A proteins were detected by anti-histone H3 (Abcam) and antipolyhistidine (Sigma) antibodies, respectively. To detect H2A.Bbd, antiserum against H2A.Bbd was raised in rabbits by immunizing recombinant full-length His-tagged H2A.Bbd. Recombinant human NAP-I was detected by a monoclonal antibody against NAP-I (a generous gift from A. Kikuchi, Nagoya University).
RESULTS
Reconstitution of NCP containing histone variants.
In order to investigate the stability of NCPs that contained histone variants, recombinant human histones were expressed in bacteria and purified as described in Materials and Methods. Since it has been suggested that the histone variants H3.3 and H2A.Bbd are incorporated into active chromatin, we wanted to clarify the effect of these variant histones on the stability of a nucleosome core particle. We used the expression vector encoding mouse H3.3, the amino acid sequence of which is identical to that of the human H3.3 protein, for the expression of the histone H3.3 protein. Histone H2A and its variants were expressed as N-terminal His-tagged proteins, and the His tag was removed by thrombin digestion (Fig. 1A, lanes 3 and 4). These recombinant histones were first refolded into either H2A-H2B dimers or H3-H4 tetramers and then assembled into histone octamers (Fig. 1A, lanes 7 to 10). As reported previously (4), gel filtration analyses revealed that octamer formation by combination of the H2A.Bbd-H2B dimers with the H3-H4 tetramers was not observed even under high salt concentrations (data not shown). Nevertheless, they were incorporated into the NCPs in the presence of DNA under the condition employed here (see below). The salt dilution method (46) was used to assemble the NCPs on the 196-bp DNA fragment containing the sea urchin 5S rRNA gene with the refolded recombinant histone octamers (Fig. 1B). This 5S rRNA gene-derived sequence was chosen because of its ability to position nucleosomes (10). The assembled NCPs were hereafter designated NCP1, NCP2, NCP3, and NCP4 for NCPs assembled with H2A/H2B/H3/H4, H2A/H2B/H3.3/H4, H2A.Bbd/H2B/H3/H4, and H2A.Bbd/H2B/H3.3/H4, respectively. Nucleoprotein gel analysis revealed that the recombinant histones were assembled into NCPs, and different NCP species, which were designated N1, N2, and N3, were detected (Fig. 1B). Two NCP bands (N1 and N2) were formed in NCP1 and NCP2 due to the presence of a distinct NCP species in which histone octamers occupied distinct positions along the DNA fragments (Fig. 2C) (39). No differences between NCP1 and NCP2 were observed on the nucleoprotein gel, and a single NCP band, N3, was detected in NCP3 and NCP4, which were assembled with histone octamers containing H2A.Bbd (Fig. 1B). Subsequently, the NCPs assembled with recombinant histones on the 5′-end-labeled DNA (Fig. 2A) were subjected to DNase I footprinting analysis (Fig. 2B). DNase I digestion of NCP1 and NCP3 revealed a ladder having the prominent cutting sites that are characteristic for NCPs, whereas DNase I digested naked DNA randomly. It is reported that DNase I digestion of the nonnucleosomal DNA-histone complex also gave rise to a similar ladder (20). To further verify the NCPs assembled with the salt dilution method, the MNase digestion assay was carried out (Fig. 2C). The MNase digestion of NCP1 and NCP3 generated protected DNA fragments of 145 bp and 110 bp, respectively. The size of the DNA fragment generated by the MNase digestion of NCP1 is almost similar to the 147-bp DNA, which is required for the assembly of a mononucleosome. Although a histone octamer containing H2A.Bbd was reported to occupy DNA fragments of approximately 118 bp (4), a slightly shorter DNA fragment of 110 bp was protected from MNase digestion of NCP3 under our experimental conditions (Fig. 2C, bottom panel, lanes 2 to 4). Subsequently, we mapped the positioning of the NCPs along the 5S rRNA gene fragment by digestion with MNase in combination with restriction endonuclease (Fig. 2C). NCP1 and NCP3 were subjected to MNase digestion, and DNA was purified and incubated in the presence of the restriction endonuclease, DraI. As indicated in Fig. 2C, DraI digests the 5S rRNA gene fragment at position +41 and generates DNA fragments of 131 bp and 65 bp (Fig. 2C, left panels). In contrast, DraI digestion of the 5S rRNA gene from NCP1 that was treated with MNase gave rise to mainly two combinations of DNA fragments, 111 bp and 34 bp as well as 96 bp and 49 bp, suggesting that the N1 and N2 nucleosomes, which were assembled with canonical histones, are present between −70 and +75 and between −55 and +90 along the 5S rRNA gene. These findings are harmonious with the results obtained from the ExoIII digestion analysis (see Fig. 4) (39). On the other hand, DraI digestion of the 5S rRNA gene in NCP3 that was treated with MNase mainly gave rise to two DNA fragments of 76 bp and 34 bp, suggesting that NCP3 is located between −35 and +75 along the 5S rRNA gene fragment.
FIG. 1.

Purification of recombinant histones and assembly of the nucleosome core particles. A. Expression, purification, and refolding of recombinant histones. Refolded histone H3-H4, H3.3-H4, His-H2A-H2B, His-H2A.Bbd-H2B (lanes 1 to 4, respectively), histone octamers purified from HeLa cells (lanes 5 and 6), refolded histone octamers containing H2A, H2B, H3, and H4 (lane 7), H2A, H2B, H3.3, and H4 (lane 8), H2A.Bbd, H2B, H3, and H4 (lane 9), or H2A.Bbd, H2B, H3.3, and H4 (lane 10) were separated by 15% SDS-PAGE and visualized with CBB staining. Lanes M indicate the molecular weight markers. Positions of histones are indicated at the left side of the panels. B. Assembly of NCPs by the recombinant histone octamers. NCP1, NCP2, NCP3, and NCP4 were assembled with the recombinant histone octamers containing H2A/H2B/H3/H4, H2A/H2B/H3.3/H4, H2A.Bbd/H2B/H3/H4, or H2A.Bbd/H2B/H3.3/H4, respectively, on the 196-bp-5S rRNA gene fragment by the salt dilution method. Naked DNA (lane 1) and NCP1, NCP2, NCP3, and NCP4 (lanes 2 to 5, respectively) were separated on a 6% nucleoprotein gel in 0.5× Tris-borate-EDTA and visualized with EtBr staining. Positions of NCPs and free DNA are indicated to the right of the panel. N1 and N2 indicate two NCPs appeared in NCP1 and NCP2, whereas N3 indicates NCPs appeared in NCP3 and NCP4 (see Fig. 2C).
FIG. 2.


Characterization of the NCPs assembled with recombinant histone octamers. A. Nucleoprotein gel analysis of the NCPs. NCPs were assembled on the 5S rRNA gene fragment without (lane 1) or with histones H2A, H2B, H3, and H4 for NCP1 (lane 2) and H2A.Bbd, H2B, H3, and H4 for NCP3 (lane 3), loaded on a 6% polyacrylamide gel in 0.5× Tris-borate-EDTA, and visualized with autoradiography. The 5′ end of the sense strand relative to the transcription direction was labeled with 32P. Positions of the nucleosome (N1, N2, and N3) and the free DNA are indicated at the right side of the panel. B. DNase I footprinting of NCPs. Naked DNA and NCPs (NCP1 and NCP3) (lanes 1 to 3, respectively) were treated with increasing amounts of DNase I. DNA was purified, separated by electrophoresis on a 6% polyacrylamide gel containing 8 M urea in 1× Tris-borate-EDTA, and visualized with autoradiography. The 10-bp periodicity of DNase I-sensitive sites in the nucleosomal DNA is shown by bullets. DNA size markers are indicated at the left side of the panel. C. MNase digestion of NCPs and mapping of the nucleosome positioning. NPC1 and NCP3 (top and bottom panels, respectively; 200 ng DNA) were incubated with 0.1 unit of MNase at 37°C for 0, 2, 5, or 10 min (lanes 1 to 4, respectively). DNA was purified, separated on a 6% polyacrylamide gel in 0.5× Tris-borate-EDTA, and visualized with staining with EtBr (lanes 1 to 4). After MNase digestion, DNA was purified and digested with 1 unit of DraI at 37°C for 1 h. MNase- and DraI-digested DNA was purified, separated by 6% PAGE, and visualized with EtBr staining (lanes 5 to 8). Positions of DNA fragments generated by digestion of the full-length 5S rRNA gene with DraI and by digestion of the MNase-treated N1, N2, and N3 nucleosomal DNA with DraI were indicated by filled circles, filled triangles, filled squares, and blank triangles, respectively. Lane M indicates DNA size markers produced by digestion of the 196-bp 5S rRNA gene fragment with either DraI, ScaI, or MspI. Nucleosome positioning along the 5S rRNA gene fragment obtained from the MNase and restriction enzyme digestion assay is schematically summarized to the right of the panels.
FIG. 4.


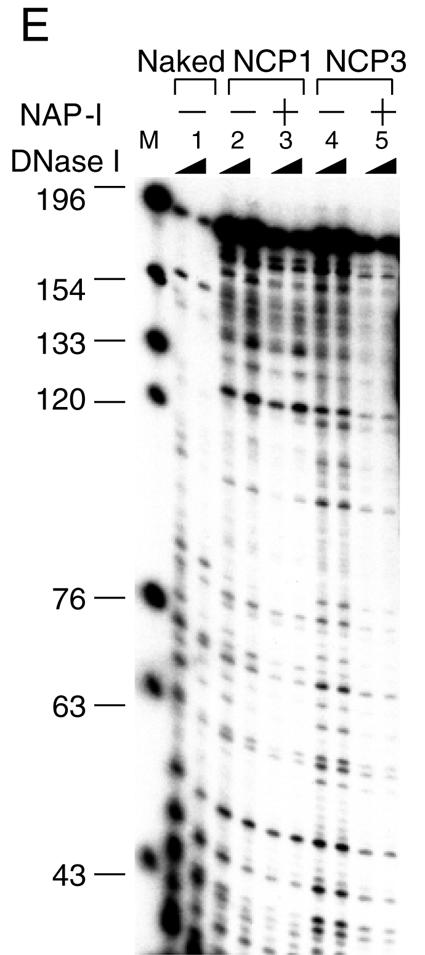
NAP-I removes the histone H2A.Bbd-H2B dimers from the NCPs. A. NAP-I does not induce nucleosome sliding. NCP1 and NCP3 were assembled on the 5′-[32P]-labeled 5S rRNA gene fragment as shown in Fig. 2A. Naked DNA (lanes 1 and 2), NCP1 (lanes 3 and 4), and NCP3 (lanes 5 and 6) (100 ng, 0.8 pmol of DNA) were incubated in the absence (lanes 1, 3, and 5) or presence (lanes 2, 4, and 6) of excess amounts of NAP-I (500 ng, 6 pmol) followed by digestion with ExoIII. DNA was purified and analyzed by 6% PAGE with 8 M urea in 1× Tris-borate-EDTA. Positions of the major ExoIII posing sites that correspond to the nucleosome border are indicated by bullets. DNA size markers (lane M) and nucleosome positioning were shown at the right of the panel. B. Western blotting of the NCPs. NCP1 and NCP3 (100 ng, 0.8 pmol of DNA) were incubated at 30°C for 30 min without (lanes 1, 5, and 9) or with increasing amounts of NAP-I (100 ng [1.2 pmol] for lanes 2, 6, and 10, 200 ng [2.4 pmol] for lanes 3, 7, and 11, and 500 ng [6 pmol] for lanes 4, 8, and 12) followed by electrophoresis on a native 6% polyacrylamide gel. DNA was visualized with EtBr staining (lanes 1 to 4). DNA and proteins were transferred to a PVDF membrane, followed by Western blotting with anti-histone H3 and anti-histone H2A.Bbd antibodies (lanes 5 to 8 and 9 to 12, respectively). Positions of the NCPs (N1, N2, and N3), free DNA, and the free dimer-NAP-I complexes are indicated at the right side of the panel. C. NAP-I forms a complex with the H2A.Bbd-H2B dimers dissociated from the NCPs. Recombinant NAP-I (100 ng [1.2 pmol], 200 ng [2.4 pmol], and 500 ng [6 pmol]) for lanes 1 to 4, 5 to 8, and 9 to 12, respectively) were incubated in the absence (lanes 1, 5, and 9) or presence of the free H2A.Bbd-H2B dimers (1.5 pmol, lanes 2, 6, and 10), NCP3 (0.64 pmol of DNA, lanes 3, 7, and 11), or NCP4 (0.64 pmol of DNA, lanes 4, 8, and 12) and loaded on a 6% polyacrylamide gel in 0.5× Tris-borate-EDTA. DNA was visualized by EtBr staining (top panel), and then protein and DNA on the gel were transferred to a PVDF membrane followed by Western blotting with an anti-NAP-I antibody (middle panel). The same membrane was washed and proved with an anti-H2A.Bbd antibody (bottom panel). Positions of nucleosome N3 and N3*, DNA, the free dimer and NAP-I complexes, and free NAP-I were indicated to the right of the panels. D. Two-dimensional electrophoresis of NCPs treated with NAP-I. NCP3 (200 ng, 1.6 pmol) incubated in the presence of an excess of NAP-I (1,000 ng, 12 pmol) was incubated at 30°C for 30 min and separated on a native 6% polyacrylamide gel. DNA was visualized with EtBr staining (top panel). The lane containing NCPs was cut out and analyzed by 15% SDS-PAGE. Proteins and DNA were visualized with silver staining. Directions of electrophoresis are indicated by arrows. Positions of histones and DNA are indicated at the left of and under the panels. The band intensities of DNA and histones in the N3 and N3* positions were quantified by using NIH Image (bottom table). Right columns (ratio) of N3 and N3* indicate the amounts of histones when the amount of DNA was set as 1. E. NAP-I-mediated structural change of NCPs enhances the nuclease sensitivity. Naked DNA (lanes 1), NCP1 (lanes 2 and 3), and NCP3 (lanes 4 and 5) (0.8 pmol of DNA) preincubated without (lanes 1, 2, and 4) or with (6 pmol, lanes 3 and 5) NAP-I were subjected to DNase I digestion at 37°C for 15 s and 60 s. DNA was purified, separated on a 6% polyacrylamide gel containing 8 M urea in 1× Tris-borate-EDTA, and visualized with autoradiography. DNA size markers are indicated to the left of the panel.
Effect of acidic histone chaperones on the stability of NCPs.
Since histone chaperones are suggested to be responsible for the assembly and disassembly of nucleosomes, we attempted to assess the role of these proteins in the stability of NCPs containing histone variants. We chose three acidic histone chaperones, TAF-I/SET, NAP-I, and B23.1 (Fig. 3A). These proteins have been identified as remodeling factors of adenovirus chromatin, and they show different localization patterns in the cell. TAF-I/SET is present throughout the nucleoplasm, and B23.1 is present in the nucleolus. NAP-I is mainly localized in the cytoplasm but has been demonstrated to shuttle between the cytoplasm and the nucleus (16, 30, 44). The acidic regions of these proteins were shown to be critical for their adenovirus chromatin-remodeling and histone-binding activities (19, 33, 36).
FIG. 3.

Disassembly of NCPs by acidic histone chaperones. A. Purified acidic histone chaperones. His-tagged recombinant acidic histone chaperones, TAF-I/SET, NAP-I, and B23.1 (lanes 1 to 3, respectively; 200 ng each) were separated by a 10% SDS-PAGE and visualized with CBB staining. Lane M indicates molecular weight markers. B. NAP-I specifically alters the structure of the NCPs containing H2A.Bbd. NCPs assembled with histone octamers as shown in Fig. 1 (100 ng, 0.8 pmol of DNA) were incubated at 30°C for 30 min without (lanes 1, 3, 6, 9, and 12) or with increasing amounts of histone chaperones (100 ng for lanes 4, 7, 10, and 13 and 500 ng for lanes 2, 5, 8, 11, and 14) followed by electrophoresis on a 6% polyacrylamide gel in 0.5× TBE. DNA was visualized with EtBr staining. TAF-I, NAP-I, and B23.1 were used as acidic histone chaperones for the top, middle, and bottom panels, respectively. TAF-I and NAP-I form dimers (28, 31), and B23.1 forms a pentamer (34) in solution, so that 500 ng of TAF-I, NAP-I, and B23.1 corresponds to 8, 6, and 3 pmol of oligomers, respectively. Positions of nucleosome and free DNA are indicated to the right of the panels.
NCPs that were assembled, as shown in Fig. 1, were incubated in either the absence or presence of increasing amounts of acidic histone chaperones, and the reaction was followed by nucleoprotein gel analyses. NCP1 and NCP2, which were assembled with canonical histone octamers and octamers containing H3.3, respectively, were stable even in the presence of excess amounts of TAF-I/SET and B23.1 under our assay conditions (Fig. 3B, lanes 3 to 8). Further, although yeast NAP-I was reported to mediate the nucleosome sliding (40), NAP-I did not affect the stability of NCP1 and NCP2. TAF-I/SET and B23.1 did not significantly affect the structure of NCP3 and NCP4 containing H2A.Bbd (Fig. 3B, lanes 9 to 14). In contrast, the structures of NCP3 and NCP4 were drastically changed after incubation with NAP-I (middle panel of Fig. 3B, lanes 9 to 14). The effect of NAP-I on the structure of NCP4 was slightly greater than that on NCP3 (compare lanes 9 to 11 with lanes 12 to 14 in Fig. 3B). Although TAF-I/SET, NAP-I, and B23.1 bind to histones and transfer them to DNA in a similar manner, only NAP-I induced structural change in NCPs containing H2A.Bbd. The molar ratio of B23.1 relative to DNA used here was lower than that of TAF-I/SET or NAP-I, as shown in Fig. 3B, because B23 presumably functions as a pentamer or decamer in solution (34). However, no significant structural change in NCPs was observed in the presence of B23.1 at the same dose used for Fig. 3B (data not shown). These results prompted us to further analyze the structural change of NCPs containing H2A.Bbd by NAP-I.
Removal of the H2A.Bbd-H2B dimers from NCPs by NAP-I.
In order to investigate the particulars of the NAP-I-induced structural change of the NCPs containing H2A.Bbd, two possibilities were addressed. The first possibility was that NAP-I mediated nucleosome sliding, since rotational positioning of NCPs along a DNA fragment often changes the mobility on the nucleoprotein gel. To test this possibility, we made use of ExoIII mapping analysis to map the positioning of NCPs along the DNA fragment. ExoIII progressively cuts the DNA from the 3′ to the 5′ direction so that when the enzyme reaches the 3′ border of the nucleosome, the digestion of DNA is blocked and strong posing sites are observed. The NCPs were assembled with the 5S rRNA gene fragment in which the 5′ end of the sense strand relative to the direction of transcription was labeled with 32P and subjected to the ExoIII digestion assay. Consistent with the results shown in Fig. 2C, ExoIII posing sites appeared at positions +75 and +90 and at position +75 when NCP1 and NCP3, respectively, were subjected to ExoIII digestion. If NAP-I mediates nucleosome sliding, novel ExoIII posing sites should appear on incubation with NAP-I. However, as shown in Fig. 4A, the ExoIII digestion patterns of NCP1 and NCP3 that were preincubated without or with NAP-I were not different from each other. The ExoIII posing sites that were observed at positions +75 and +90 for NCP1 and at +75 for NCP3, indicated by bullets in Fig. 4A, were detected regardless of whether NCPs were incubated with or without NAP-I. This suggests that NAP-I did not mediate nucleosome sliding.
Since yeast NAP-I has been reported to transiently remove the H2A-H2B dimers from NCPs, which results in an active exchange of the H2A-H2B dimers (40), we addressed the other possibility that human NAP-I stripped the histone H2A.Bbd-H2B dimers from NCPs. NCPs containing canonical H2A-H2B or H2A.Bbd-H2B dimers (NCP1 and NCP3, respectively) were incubated in the absence or presence of NAP-I, and the reaction was followed by nucleoprotein gel analysis (Fig. 4B). DNA was visualized by EtBr staining, the proteins and DNA fragments were transferred to a PVDF membrane, and histone H3 and H2A.Bbd were detected by Western blotting. When the NCP1 that was assembled with canonical histones was incubated in the absence or presence of increasing amounts of NAP-I, the electrophoretic patterns of DNA and histone H3 did not change significantly (Fig. 4B, top panels). In sharp contrast, as shown in Fig. 3B, NCP3 containing H2A.Bbd was perturbed and a slower-mobility band designated N3* appeared on incubation with increasing amounts of NAP-I. As can be observed in the bottom panel of Fig. 4B, both the N3* and the N3 species contained DNA, H3, and H2A.Bbd. A prominent new band that contained H2A.Bbd appeared on incubation with NAP-I. Since this band did not contain any detectable DNA, it could correspond to either free H2A.Bbd-H2B dimers or a ternary complex having NAP-I. Since H2A.Bbd-H2B dimers cannot enter the gel due to their positive charge, it is likely that the band migrating faster than the NCPs corresponds to a ternary complex having NAP-I. To demonstrate this, NAP-I that was incubated in the absence or presence of free H2A.Bbd-H2B dimers or NCPs was separated on the nucleoprotein gel and analyzed by Western blotting (Fig. 4C). NAP-I alone was distributed throughout the lanes (lanes 1, 5, and 9). This may be because of the possibility that NAP-I alone cannot form a stable conformation under a nondenaturing condition. In fact, it has been reported that yeast NAP-I forms a dimer, and each NAP-I dimer further forms complex oligomers under physiological salt concentrations (28). However, NAP-I incubated with free H2A.Bbd-H2B was concentrated mainly in two bands: one of these bands migrated faster and the other migrated slower than the NCPs. Since these two bands that were detected by an anti-NAP-I antibody also contained H2A.Bbd (Fig. 4C, bottom panel), both correspond to a ternary complex between the H2A.Bbd-H2B dimers and NAP-I. These two different ternary complexes possibly were generated due to the different stoichiometry between the H2A.Bbd-H2B dimers and NAP-I. In addition, the bands migrating faster than the NCPs appeared with incubation with NCP3 and NCP4, which were assembled with histone octamers containing H2A.Bbd. These observations support the idea that the band containing H2A.Bbd and migrating faster than NCPs corresponded to a ternary complex having NAP-I rather than only a free H2A.Bbd-H2B dimer.
Since we prepared the NCPs by the salt dilution method and the assembled NCPs were not purified, it was possible that the free H2A.Bbd-H2B dimers that were not assembled into the NCPs corresponded to the band that migrated faster than the NCP on incubation with NAP-I. However, the same band containing H2A.Bbd appeared when NCP3 purified through a sucrose density gradient was incubated with increasing amounts of NAP-I (see Fig. S1 in the supplemental material). Therefore, it is unlikely that the free H2A.Bbd-H2B dimers that were not assembled into the NCPs cause the band to migrate faster than the NCPs.
We also noted that the ratio of DNA to histone H3 in N3 was similar to that in N3*; however, the amount of H2A.Bbd in N3* was significantly lower than that in N3. Thus, we assumed that N3* is a product that is generated by the removal of one H2A.Bbd-H2B dimer from the NCPs by NAP-I. To test this assumption, we assessed the stoichiometry of histones in N3 and N3* species by using nondenaturing PAGE in combination with SDS-PAGE. The NCPs containing H2A.Bbd incubated with NAP-I were separated by 6% PAGE under nondenaturing conditions, and the lane was cut out and subjected to SDS-PAGE (Fig. 4D). Subsequently, the proteins and DNA were visualized by silver staining. Although an excess of NAP-I was broadly distributed and not detected as a distinct band, the silver-stained gel clearly demonstrated that the H2A.Bbd-H2B dimers were partially stripped on incubation with NAP-I and that the N3* nucleosome appeared simultaneously. The amounts of histones and DNA present in N3 and N3* were quantitatively measured by using NIH Image (Fig. 4D, table). The amounts of the H2A.Bbd-H2B dimers in N3* were distinctly low, and N3* had almost half the amount of dimers of H3 and H4. Therefore, we concluded that N3* contains one H2A.Bbd-H2B dimer and one H3-H4 tetramer. These results revealed that NAP-I mediates removal of the H2A.Bbd-H2B dimers from the NCPs rather than nucleosome sliding.
Since NAP-I removed the H2A.Bbd-H2B dimer from the NCPs, we assumed that this structural change of the NCPs makes the trans-acting factors accessible to nucleosomal DNA. To test this assumption, the NCPs that were preincubated in the presence or absence of NAP-I were subjected to a DNase I digestion assay. The accessibility of DNase I to nucleosomal DNA was much lower than that of naked DNA (Fig. 4E, compare lane 1 with lanes 2 and 4), and the periodic digestion pattern for NCP1 and NCP3 was observed as shown in Fig. 2B. In contrast, the accessibility of DNase I significantly increased when NCP3 was preincubated in the presence of NAP-I (Fig. 4E, lanes 4 and 5). Although distinct changes in the electrophoretic pattern of NCP1 were not observed even in the presence of an excess of NAP-I (Fig. 3 and 4), NAP-I increased the accessibility of DNase I to DNA in NCP1 (Fig. 4D, lanes 2 and 3). Thus, NAP-I may also mediate the transient disassembly of the H2A-H2B dimers from the NCPs, or incubation of NCP1 with NAP-I may result in uncharacterized structural changes of NCPs without removing histone octamers. This could also be true in the cases of TAF-I/SET and B23.1. TAF-I/SET increases the nuclease sensitivity of nucleosomal DNA and stimulates transcription from the chromatin template, whereas significant structural changes of NCPs were not observed on the nucleoprotein gel (Fig. 3B) (12, 38).
Stability of NCPs containing various H2A variants.
It has been reported that a histone variant, H2A.Z, confers rigidity to the nucleosome structure by enhancing the interaction between the H2A.Z-H2B dimers and an H3-H4 tetramer within an NCP (41). On the other hand, H2A.Bbd is suggested to be involved in the formation of a more flexible nucleosome structure than that formed by H2A (Fig. 3 and 4) (4, 13). By examining the stability of NCPs containing various histone variants, using the same biochemical assay system, the effect of histone H2A variants on the stability of NCPs could be investigated in a holistic manner. Therefore, we tested the effect of the histone H2A variants known thus far on the stability of NCPs in the presence of NAP-I. In order to detect the histone H2A variant proteins easily by Western blotting, we used N-terminal His-tagged H2A variant proteins to assemble the NCPs, and His-tagged histone H2A variants were detected by an anti-His-tag antibody. His-tagged H2A, H2A.X, H2A.Z, the histone fold domain of macroH2A1.2 (mH2AN), and H2A.Bbd were expressed in E. coli, purified, and refolded into the dimers with H2B (Fig. 5A); subsequently, histone octamers were prepared. Using these octamers, the NCPs were assembled on the 5S rRNA gene fragment (Fig. 5B). NCPs containing each H2A variant demonstrated differences in electrophoretic mobility in nucleoprotein gels. These differences could be due to the difference in either size or charge among the H2A variants or due to the differential effects of the variants on the position of the octamer along the DNA. The assembled NCPs were incubated in the absence or presence of NAP-I and subjected to nucleoprotein gel analysis and Western blotting with anti-His-tag or anti-histone H3 antibodies (Fig. 5C). As shown in the middle panel of Fig. 5C, each of the His-tagged histone variants was incorporated into the NCPs. The electrophoretic patterns of the NCPs in the absence or presence of NAP-I did not significantly differ from each other except for the NCPs containing H2A.Bbd (lanes 17 to 20). Thus, we concluded that among various H2A variants tested, H2A.Bbd was the most susceptible to dissociation in the presence of NAP-I.
FIG. 5.

Stability of NCPs containing H2A variants. A. Purification and refolding of the dimers containing histone H2A variants. His-tagged histone H2A, H2A.X, H2A.Z, the histone fold domain of macroH2A1.2 (mH2AN), and H2A.Bbd (lanes 1 to 5, respectively) were expressed in E. coli and purified as described in Materials and Methods. Purified histone variants refolded into dimers with H2B (lanes 1 to 5) and a H3-H4 tetramer (lane 6) were separated on a 15% SDS-PAGE and visualized with CBB staining. Lane M indicates molecular weight markers. B. Assembly of the NCPs containing histone H2A variants. Refolded dimers as shown in A were mixed with H3-H4 tetramers and used for assembly of NCPs. The NCPs assembled with histone octamers containing His-tagged H2A variants (indicated above each lane) were separated by 6% PAGE and visualized with EtBr staining. Positions of nucleosome and free DNA are indicated at the right of the panel. C. Stability of NCPs containing histone H2A variants in the presence of NAP-I. NCPs (100 ng, 0.8 pmol of DNA) containing His-tagged histone variants (indicated above the panel) were incubated in the absence (lanes 1, 5, 9, 13, and 17) or presence of increasing amounts of NAP-I (100 ng [1.2 pmol], 200 ng [2.4 pmol], and 500 ng [6 pmol] for lanes 2, 6, 10, 14, and 18; 3, 7, 11, 15, and 19; and 4, 8, 12, 16, and 20, respectively), followed by a nucleoprotein gel analysis. DNA was visualized with EtBr staining (top panel), and His-tagged histones and histone H3 were detected by Western blotting using anti-His (middle panel) and anti-histone H3 (bottom panel) antibodies, respectively. Positions of nucleosome (Nuc), free dimer-NAP-I complexes, and DNA are shown to the right of the panels.
Preferential removal of the H2A.Bbd-H2B dimers from NCPs containing H3.3 rather than those containing canonical H3.
Since the effect of NAP-I on the structure of NCP4 containing H3.3 was slightly more pronounced than that on NCP3 containing canonical H3 (Fig. 3B, middle panel), it is reasonable to hypothesize that the H2A.Bbd-H2B dimers were removed more efficiently from NCP4 than from NCP3. To be more precise, the NCPs assembled with histone octamers containing H2A.Bbd, H2B, H3, and H4 (NCP3) or H2A.Bbd, H2B, H3.3, and H4 (NCP4) were incubated in the absence or presence of increasing amounts of NAP-I and separated on the nucleoprotein gel, and this was followed by EtBr staining and Western blotting with an anti-H2A.Bbd antibody (Fig. 6A). As shown in Fig. 3 and 4, the free H2A.Bbd-H2B dimers appeared on incubation with NAP-I, and NAP-I stripped the H2A.Bbd-H2B dimers more efficiently from NCP4 than from NCP3 (compare lanes 2 to 4 with lanes 6 to 8 in Fig. 6A, bottom panel). Similar results were obtained when the reactions were carried out at 4°C (Fig. 6B). The amounts of H2A.Bbd present at the free dimer position were measured and plotted as a function of increasing amounts of NAP-I (Fig. 6B). As much as 40% and 25% of the H2A.Bbd-H2B dimers were stripped from NCP3 at 37°C and 4°C, respectively, whereas approximately 75% and 50% of the H2A.Bbd-H2B dimers were stripped from NCP4 at 37°C and 4°C, respectively. Further, a new NCP band designated N3** appeared on incubation of NCP4 in the presence of an excess of NAP-I (Fig. 6A, top panel). Since this band contained DNA but not H2A.Bbd and was detected by an anti-H3 antibody (data not shown), it is likely to correspond to the nucleoprotein complexes containing an H3-H4 tetramer and DNA. From these observations, we propose that H2A.Bbd and H3.3 work in combination to make the NCPs flexible.
FIG. 6.

Effect of the H3 variant, H3.3, on NAP-I-mediated removal of the H2A.Bbd-H2B dimers from NCPs. A. Nucleoprotein gel analysis of NCP3 and NCP4 preincubated with NAP-I. NCPs were assembled with histone octamers containing H2A.Bbd, H2B, H3, and H4 (NCP3, lanes 1 to 4) or H2A.Bbd, H2B, H3.3, and H4 (NCP4, lanes 5 to 8). NCPs (100 ng, 0.8 pmol of DNA) incubated without (lanes 1 and 5) or with increasing amounts of NAP-I (100 ng [1.2 pmol, lanes 2 and 6], 200 ng ′2.4 pmol, lanes 3 and 7], and 500 ng [6 pmol, lanes 4 and 8]) were analyzed by nucleoprotein gel analysis and Western blotting with an anti-H2A.Bbd antibody (top and bottom panels, respectively). B. Quantitative analysis of the dimer removal by NAP-I from NCP3 and NCP4 under different temperature conditions. NCP3 and NCP4 (0.4 pmol of DNA) were incubated with increasing amounts of NAP-I (1.2, 2.4, 4.8, and 12 pmol) at 4°C or 37°C for 1 h and subjected to the nucleoprotein gel analysis followed by Western blotting with an anti-H2A.Bbd antibody. The ratios of the H2A.Bbd-H2B dimers removed from the NCPs were quantitatively analyzed by using NIH Image and plotted as a function of the amounts of NAP-I. The amounts of H2A.Bbd derived from NCP3 at 4°C, NCP3 at 37°C, NCP4 at 4°C, and NCP4 at 37°C are indicated by filled circles, filled squares, blank circles, and blank squares, respectively. Data shown were from the averages for two independent experiments.
Reversible assembly and disassembly of the H2A.Bbd-H2B dimers by NAP-I.
Since NAP-I efficiently stripped the H2A.Bbd-H2B dimers from NCPs (Fig. 4 to 6) and yeast NAP-I was reported to mediate the assembly and disassembly of the nucleosome (40), it was assumed that human NAP-I could mediate the reversible assembly and disassembly of the H2A-H2B and H2A.Bbd-H2B dimers. To test this assumption, the His-H2A-H2B or His-H2A.Bbd-H2B dimers that were preincubated with NAP-I were mixed with NCPs assembled with nontagged histones. The NCPs incubated with the His-tagged dimer-NAP-I complexes were analyzed by nucleoprotein gels (Fig. 7A). If NAP-I mediates an exchange of the dimers, the His-tagged dimers would be incorporated into the NCPs and detected at the positions of the NCPs on the nucleoprotein gel. As expected, the nontagged H2A.Bbd-H2B dimers were replaced by the His-H2A-H2B dimers as well as the His-H2A.Bbd-H2B dimers in a NAP-I-dependent manner, and these dimers were detected at the positions corresponding to the N1-, N2-, and N3-NCPs on the nucleoprotein gel (Fig. 7A, lanes 17 to 32). Consistent with the observation shown in Fig. 6, the replacement of the nontagged H2A.Bbd-H2B dimers with the His-tagged dimers in the presence of NAP-I (Fig. 7A, compare lanes 18 to 24 with lanes 27 to 32 of the middle and bottom panels) was higher in NCP4 than in NCP3. In addition, a low but distinct level of the His-tagged H2A-H2B dimers was also detected at the NCP positions when NCP1 and NCP2 were incubated with the His-H2A-H2B dimer-NAP-I complexes (Fig. 7A, lanes 3 to 5 and 11 to 13), suggesting that NAP-I mediates the disassembly and assembly of the H2A-H2B dimers in canonical NCPs. As indicated by the bullets in the top and middle panels of Fig. 7A, the bands having slow mobility appeared during the exchange of H2A-H2B or H2A.Bbd-H2B dimers with His-H2A-H2B dimers. These bands contained DNA and were recognized by an anti-His-tag antibody, thus suggesting that the bands are intermediate NCPs containing one H2A-H2B dimer (with or without His tag) and an H3-H4 tetramer. During the disassembly of the H2A-H2B dimers, the DNA in the NCPs could be transiently exposed, and this structural change in the NCPs could enable the trans-acting factors to access the DNA. Indeed, NAP-I increased the sensitivity of DNase I to nucleosomal DNA in NCP1 (Fig. 4E). It should also be noted that the H2A-H2B dimers in NCP1 and NCP2 were not efficiently replaced by the H2A.Bbd-H2B dimers under these experimental conditions (Fig. 7A, lanes 6 to 8 and 14 to 16), whereas the H2A.Bbd-H2B dimers in NCP3 and NCP4 were efficiently replaced by the His-H2A-H2B dimers in the presence of an excess of NAP-I (Fig. 7A, lanes 19 to 21 and 27 to 29). Once the H2A.Bbd-H2B dimers were replaced by the H2A-H2B dimers, the efficiency of the NAP-I-mediated dimer exchange was significantly decreased. Therefore, the free H2A.Bbd-H2B dimer-NAP-I complexes were accumulated when the H2A.Bbd-H2B dimers were replaced by the H2A-H2B dimers (Fig. 7A, bottom panel, lanes 19 to 21 and 27 to 29). Hence, it is suggested that NAP-I preferentially mediates the replacement of thermodynamically unstable dimers with stable dimers. In order to verify the exchange reaction mediated by NAP-I, we performed the dimer exchange reaction of NCPs containing His-tagged H2A.Bbd-H2B dimers with nontagged H2A-H2B dimers or H2A.Bbd-H2B dimers, and the His-tagged dimers that were removed by NAP-I were quantitatively analyzed (Fig. 7B). Although excess amounts of NAP-I resulted in the efficient removal of the His-tagged H2A.Bbd-H2B dimers from NCP4 in the absence of the free dimers (Fig. 7B, lane 20), the His-tagged dimers were more efficiently accumulated in the presence of the nontagged H2A-H2B dimers than in the absence or presence of nontagged H2A.Bbd-H2B dimers (bottom graphs in Fig. 7B). The replacement of the His-tagged H2A.Bbd-H2B dimers with the nontagged H2A-H2B dimers were evidenced by the appearance of the N1 and N2 nucleosome species on the nucleoprotein gel stained with EtBr (Fig. 7B, top panel, lanes 6 to 10 and 21 to 25). These observations support the idea that NAP-I preferentially mediates the exchange of unstable dimers in NCPs for stable dimers.
FIG. 7.

Reversible assembly and disassembly of the H2A.Bbd-H2B dimers by NAP-I. A. Removal of the nontagged dimers from the NCPs and replacement with the His-tagged dimers by NAP-I. His-tagged H2A-H2B dimers (lanes 3 to 5, 11 to 13, 19 to 21, and 27 to 29) or His-tagged H2A.Bbd-H2B dimers (lanes 6 to 8, 14 to 16, 22 to 24, and 30 to 32) (50 ng, 2 pmol of the dimers each) were preincubated with increasing amounts of NAP-I (200 ng [2.4 pmol], 400 ng [4.8 pmol], 1,000 ng [12 pmol]) as indicated at the top of the panel. Then, the dimer-NAP-I complexes were mixed with the nontagged NCPs (80 ng, 0.6 pmol of DNA) and further incubated at 30°C for 1 h followed by nucleoprotein gel analysis. DNA was visualized with EtBr staining (top panel), and proteins were analyzed by Western blotting with anti-His (middle panel) and anti-H2A.Bbd (bottom panel) antibodies. Positions of DNA and NCPs are indicated to the right of the panels. Bullets shown at the right side of lanes 5, 13, 21, and 29 indicate what could be intermediate NCPs (see the text). B. Removal of the His-tagged dimers from the NCPs and replacement with the nontagged dimers by NAP-I. Increasing amounts of NAP-I (0, 1.2, 2.4, 4.8, and 12 pmol) were preincubated without (lanes 1 to 5 and 16 to 20) or with nontagged H2A-H2B dimers (1.5 pmol, lanes 6 to 10 and 21 to 25) or nontagged H2A.Bbd-H2B dimers (1.5 pmol, lanes 11 to 15 and 26 to 30). The complexes were mixed and incubated with NCP3 (lanes 1 to 15) or NCP4 (lanes 16 to 30) (0.4 pmol of DNA each) that was assembled with histones containing His-tagged H2A.Bbd at 30°C for 1 h, followed by analysis with 6% PAGE in 0.5× Tris-borate-EDTA. DNA was visualized with EtBr staining (top panel), and the protein and DNA were transferred to a PVDF membrane, followed by Western blotting with an anti-His-tag antibody (bottom panel). The amounts of His-tagged H2A.Bbd in the N3 nucleosome position were quantitatively analyzed by using NIH Image and plotted as a function of the amounts of NAP-I (bottom graphs). Several independent experiments showed similar results, and the graphs shown below the panels were from the results shown in this figure.
DISCUSSION
In this report, we examined the stability of NCPs containing histone variants. Among the various histone H2A variants tested thus far, H2A.Bbd was shown to generate the most flexible NCP structure in the presence of human NAP-I (Fig. 5). Interestingly, the NCPs containing H2A.Bbd were more flexible in combination with the histone H3.3 (Fig. 6 and 7), which is a histone H3 variant reported to be preferentially incorporated in the active chromatin (1). These results increase the possibility of the histone variants H2A.Bbd and H3.3 being involved in the formation of the active chromatin structure without the involvement of posttranslational modifications. Interestingly, TAF-I/SET and B23.1, which demonstrate significant similarities in sequence and function to NAP-I and nucleoplasmin, respectively, did not show efficient assembly and disassembly activity of the H2A.Bbd-H2B dimers (Fig. 3). Thus, the acidic nature and the histone-binding activity of NAP-I are not sufficient to explain the mechanism of NAP-I-mediated assembly and disassembly of the NCPs.
Several genetic studies have clearly demonstrated that histone variant proteins play important roles during mammalian development and various cellular processes (7, 9, 14). However, the function of each histone variant in NCPs has not been studied extensively. H2A.Bbd is the most recently identified histone variant and shows the lowest sequence similarity to the canonical histone H2A among the various histone H2A variants known thus far (8). The exogenously expressed H2A.Bbd is preferentially incorporated at the active chromosome loci and almost excluded from the “Barr Body” that is formed by the inactive X chromosome (8). Bao et al. demonstrated that the NCPs containing H2A.Bbd are less rigid than canonical NCPs and the docking domain of the H2A.Bbd is responsible for this effect (4). In agreement with this finding, the NCPs containing H2A.Bbd were exceptionally flexible among the NCPs containing various H2A variants in the presence of NAP-I, as shown in Fig. 5. DNase I digestion assays of NCPs containing H2A.Bbd (NCP3) in the absence of NAP-I also demonstrated that in addition to the periodic cutting sites with about 10-bp distances, spontaneous cutting sites were generated (Fig. 2B and 4E). This suggests that H2A.Bbd weakens the interaction not only between the H2A.Bbd-H2B dimers and H3-H4 tetramers but also between the histone octamers and DNA. The exposure of DNA sites is proposed to occur via the spontaneous transient dissociation of short stretches of DNA from the surface of the histone octamer beginning at one end and extending progressively inwards (3). This exposure is likely to occur more efficiently in the NCPs containing H2A.Bbd than in the canonical NCPs, thereby allowing DNase I to access the DNA in the NCPs containing H2A.Bbd.
When incubated with NAP-I, H3.3, a histone H3 variant, generated a more-flexible nucleosome structure in combination with H2A.Bbd (Fig. 6 and 7). Evidence from several reports demonstrated that H3.3 is preferentially deposited at the active chromatin independently of DNA synthesis (1, 18). It is well established that once H3.3 is deposited at the active chromatin, histone modification enzymes mark the specific amino acids, such as lysine 4 and lysine 9, in order to maintain the active chromatin structure (29). However, it is unclear whether H3.3 generates a less-rigid chromatin structure than canonical H3 without these modifications. H3.3 and H3 differ from each other with regard to only four amino acids, and three of these amino acids are located at the α2 helix of H3, where the solvent-accessible site are suggested to be located (25). At this point in time, it is unknown whether these amino acids alter the interaction between H2A.Bbd-H2B dimers and H3.3-H4 tetramers or between NAP-I and H2A.Bbd-H2B dimers in the NCPs. The biological relevance of NCPs containing H2A.Bbd-H2B dimers and an H3.3-H4 tetramer in vivo is an important issue to be addressed.
A previous report demonstrated that yeast NAP-I mediates the exchange of the histone H2A-H2B dimers for variant dimers (40). Our results clearly demonstrated that NAP-I preferentially mediated the exchange of H2A.Bbd-H2B dimers for H2A-H2B dimers, and this efficient exchange reaction mediated by NAP-I was not observed when the exchange reaction was reversed (Fig. 7). This suggests that the disassembly of the dimer from NCPs is a rate-limiting step of the dimer exchange reaction. Since the NCPs containing the H2A-H2B dimers are more stable than those containing H2A.Bbd-H2B dimers, the disassembly of the H2A-H2B dimers by NAP-I is much slower than that of H2A.Bbd-H2B dimers. Assembly of the dimers by NAP-I would be equally efficient for both canonical dimers and dimers containing H2A.Bbd. This assumption explains the efficient exchange of the H2A.Bbd-H2B dimers for H2A-H2B dimers (Fig. 7). Since NAP-I alone cannot efficiently remove the H2A-H2B dimers from the NCPs by NAP-I alone, other factors, such as the ATP-dependent chromatin remodeling machineries, could be required for this process. Indeed, ATP-dependent histone exchange complexes (22, 32) have been shown to mediate the exchange of the dimers. The stability of the NCPs is also presumed to be regulated by posttranslational modifications, including the acetylation, of histones. In fact, the Tip60 acetyltransferase activity and an ATP-dependent chromatin remodeling protein, Domino, were demonstrated to be required for the efficient execution of a dimer exchange reaction (22). The p300-mediated acetylation of histones in the nucleosome has also been reported to facilitate the transfer of the H2A-H2B dimers from the nucleosome to NAP-I (17). The effect of histone modifications on the stability of NCPs is, therefore, a matter of concern to be investigated in the future.
NAP-I was originally identified as a factor that facilitates nucleosome formation in vitro (15). NAP-I is conserved from yeast to humans, although the biological function of NAP-I has not been completely established. Several lines of genetic evidence revealed that NAP-I is involved in the regulation of a distinct set of genes (23, 35). From these observations, one can conclude that NAP-I, at least in part, is likely to be involved in chromatin remodeling by the disassembly of histone H2A-H2B dimers in vivo. In addition to NAP-I, several acidic histone-binding proteins have been identified. In a manner similar to that of NAP-I, TAF-I/SET and B23.1 bind to histones and transfer them to DNA to assemble the nucleosome (19, 37). Unlike NAP-I, however, TAF-I/SET and B23.1 did not demonstrate efficient dimer stripping activity (Fig. 3). The carboxyl-terminal acidic region of yeast NAP-I was shown to be critical for the stripping of the H2A-H2B dimers and for remodeling of the adenovirus chromatin (19, 40); however, this region is dispensable for histone binding and nucleosome assembly (11). Therefore, the C-terminal acidic region may be required to compete with DNA for the removal of the basic proteins from DNA. However, the mechanism of dimer stripping by NAP-I is more complex, because TAF-I/SET, which has a similar, long acidic stretch at its C terminus, did not demonstrate efficient dimer stripping activity. Thus, the acidic region and the other functional domain(s) yet to be identified are important in order for NAP-I to demonstrate its complete histone chaperone activity.
Supplementary Material
Acknowledgments
We thank A. Verreault for critical reading and helpful comments on the manuscript and A. Kikuchi for an anti-NAP-I antibody.
This work was supported by a grant-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (M.O. and K.N.) and grants from the Bioarchitect Research Program (RIKEN) and the project of Tsukuba Advanced Research Alliance (K.N.).
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Ahmad, K., and S. Henikoff. 2002. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 9:1191-1200. [DOI] [PubMed] [Google Scholar]
- 2.Akey, C. W., and K. Luger. 2003. Histone chaperones and nucleosome assembly. Curr. Opin. Struct. Biol. 13:6-14. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, J. D., A. Thastrom, and J. Widom. 2002. Spontaneous access of proteins to buried nucleosomal DNA target sites occurs via a mechanism that is distinct from nucleosome translocation. Mol. Cell. Biol. 22:7147-7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bao, Y., K. Konesky, Y. J. Park, S. Rosu, P. N. Dyer, D. Rangasamy, D. J. Tremethick, P. J. Laybourn, and K. Luger. 2004. Nucleosomes containing the histone variant H2A.Bbd organize only 118 base pairs of DNA. EMBO J. 23:3314-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belotserkovskaya, R., S. Oh, V. A. Bondarenko, G. Orphanides, V. M. Studitsky, and D. Reinberg. 2003. FACT facilitates transcription-dependent nucleosome alteration. Science 301:1090-1093. [DOI] [PubMed] [Google Scholar]
- 6.Bruno, M., A. Flaus, C. Stockdale, C. Rencurel, H. Ferreira, and T. Owen-Hughes. 2003. Histone H2A/H2B dimer exchange by ATP-dependent chromatin remodeling activities. Mol. Cell 12:1599-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Celeste, A., S. Petersen, P. J. Romanienko, O. Fernandez-Capetillo, H. T. Chen, O. A. Sedelnikova, B. Reina-San-Martin, V. Coppola, E. Meffre, M. J. Difilippantonio, C. Redon, D. R. Pilch, A. Olaru, M. Eckhaus, R. D. Camerini-Otero, L. Tessarollo, F. Livak, K. Manova, W. M. Bonner, M. C. Nussenzweig, and A. Nussenzweig. 2002. Genomic instability in mice lacking histone H2AX. Science 296:922-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chadwick, B. P., and H. F. Willard. 2001. A novel chromatin protein, distantly related to histone H2A, is largely excluded from the inactive X chromosome. J. Cell Biol. 152:375-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faast, R., V. Thonglairoam, T. C. Schulz, J. Beall, J. R. Wells, H. Taylor, K. Matthaei, P. D. Rathjen, D. J. Tremethick, and I. Lyons. 2001. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 11:1183-1187. [DOI] [PubMed] [Google Scholar]
- 10.Flaus, A., K. Luger, S. Tan, and T. J. Richmond. 1996. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc. Natl. Acad. Sci. USA 93:1370-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujii-Nakata, T., Y. Ishimi, A. Okuda, and A. Kikuchi. 1992. Functional analysis of nucleosome assembly protein, NAP-1. The negatively charged COOH-terminal region is not necessary for the intrinsic assembly activity. J. Biol. Chem. 267:20980-20986. [PubMed] [Google Scholar]
- 12.Gamble, M. J., H. Erdjument-Bromage, P. Tempst, L. P. Freedman, and R. P. Fisher. 2005. The histone chaperone TAF-I/SET/INHAT is required for transcription in vitro of chromatin templates. Mol. Cell. Biol. 25:797-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gautier, T., D. W. Abbott, A. Molla, A. Verdel, J. Ausio, and S. Dimitrov. 2004. Histone variant H2ABbd confers lower stability to the nucleosome. EMBO Rep. 5:715-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howman, E. V., K. J. Fowler, A. J. Newson, S. Redward, A. C. MacDonald, P. Kalitsis, and K. H. Choo. 2000. Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc. Natl. Acad. Sci. USA 97:1148-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishimi, Y., J. Hirosumi, W. Sato, K. Sugasawa, S. Yokota, F. Hanaoka, and M. Yamada. 1984. Purification and initial characterization of a protein which facilitates assembly of nucleosome-like structure from mammalian cells. Eur. J. Biochem. 142:431-439. [DOI] [PubMed] [Google Scholar]
- 16.Ito, T., M. Bulger, R. Kobayashi, and J. T. Kadonaga. 1996. Drosophila NAP-1 is a core histone chaperone that functions in ATP-facilitated assembly of regularly spaced nucleosomal arrays. Mol. Cell. Biol. 16:3112-3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ito, T., T. Ikehara, T. Nakagawa, W. L. Kraus, and M. Muramatsu. 2000. p300-mediated acetylation facilitates the transfer of histone H2A-H2B dimers from nucleosomes to a histone chaperone. Genes Dev. 14:1899-1907. [PMC free article] [PubMed] [Google Scholar]
- 18.Janicki, S. M., T. Tsukamoto, S. E. Salghetti, W. P. Tansey, R. Sachidanandam, K. V. Prasanth, T. Ried, Y. Shav-Tal, E. Bertrand, R. H. Singer, and D. L. Spector. 2004. From silencing to gene expression: real-time analysis in single cells. Cell 116:683-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawase, H., M. Okuwaki, M. Miyaji, R. Ohba, H. Handa, Y. Ishimi, T. Fujii-Nakata, A. Kikuchi, and K. Nagata. 1996. NAP-I is a functional homologue of TAF-I that is required for replication and transcription of the adenovirus genome in a chromatin-like structure. Genes Cells 1:1045-1056. [DOI] [PubMed] [Google Scholar]
- 20.Kerrigan, L. A., and J. T. Kadonaga. 1992. Periodic binding of individual core histones to DNA: inadvertent purification of the core histone H2B as a putative enhancer-binding factor. Nucleic Acids Res. 20:6673-6680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura, H., and P. R. Cook. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kusch, T., L. Florens, W. H. Macdonald, S. K. Swanson, R. L. Glaser, J. R. Yates III, S. M. Abmayr, M. P. Washburn, and J. L. Workman. 2004. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306:2084-2087. [DOI] [PubMed] [Google Scholar]
- 23.Lankenau, S., T. Barnickel, J. Marhold, F. Lyko, B. M. Mechler, and D. H. Lankenau. 2003. Knockout targeting of the Drosophila nap1 gene and examination of DNA repair tracts in the recombination products. Genetics 163:611-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laskey, R. A., B. M. Honda, A. D. Mills, and J. T. Finch. 1978. Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 275:416-420. [DOI] [PubMed] [Google Scholar]
- 25.Luger, K., A. W. Mader, R. K. Richmond, D. F. Sargent, and T. J. Richmond. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251-260. [DOI] [PubMed] [Google Scholar]
- 26.Luger, K., T. J. Rechsteiner, and T. J. Richmond. 1999. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 119:1-16. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto, K., K. Nagata, M. Ui, and F. Hanaoka. 1993. Template activating factor I, a novel host factor required to stimulate the adenovirus core DNA replication. J. Biol. Chem. 268:10582-10587. [PubMed] [Google Scholar]
- 28.McBryant, S. J., and O. B. Peersen. 2004. Self-association of the yeast nucleosome assembly protein 1. Biochemistry 43:10592-10599. [DOI] [PubMed] [Google Scholar]
- 29.McKittrick, E., P. R. Gafken, K. Ahmad, and S. Henikoff. 2004. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. USA 101:1525-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyaji-Yamaguchi, M., K. Kato, R. Nakano, T. Akashi, A. Kikuchi, and K. Nagata. 2003. Involvement of nucleocytoplasmic shuttling of yeast Nap1 in mitotic progression. Mol. Cell. Biol. 23:6672-6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyaji-Yamaguchi, M., M. Okuwaki, and K. Nagata. 1999. Coiled-coil structure-mediated dimerization of template activating factor-I is critical for its chromatin remodeling activity. J. Mol. Biol. 290:547-557. [DOI] [PubMed] [Google Scholar]
- 32.Mizuguchi, G., X. Shen, J. Landry, W. H. Wu, S. Sen, and C. Wu. 2004. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303:343-348. [DOI] [PubMed] [Google Scholar]
- 33.Nagata, K., H. Kawase, H. Handa, K. Yano, M. Yamasaki, Y. Ishimi, A. Okuda, A. Kikuchi, and K. Matsumoto. 1995. Replication factor encoded by a putative oncogene, set, associated with myeloid leukemogenesis. Proc. Natl. Acad. Sci. USA 92:4279-4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Namboodiri, V. M., I. V. Akey, M. S. Schmidt-Zachmann, J. F. Head, and C. W. Akey. 2004. The structure and function of Xenopus NO38-core, a histone chaperone in the nucleolus. Structure (Camb). 12:2149-2160. [DOI] [PubMed] [Google Scholar]
- 35.Ohkuni, K., K. Shirahige, and A. Kikuchi. 2003. Genome-wide expression analysis of NAP1 in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 306:5-9. [DOI] [PubMed] [Google Scholar]
- 36.Okuwaki, M., A. Iwamatsu, M. Tsujimoto, and K. Nagata. 2001. Identification of nucleophosmin/B23, an acidic nucleolar protein, as a stimulatory factor for in vitro replication of adenovirus DNA complexed with viral basic core proteins. J. Mol. Biol. 311:41-55. [DOI] [PubMed] [Google Scholar]
- 37.Okuwaki, M., K. Matsumoto, M. Tsujimoto, and K. Nagata. 2001. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 506:272-276. [DOI] [PubMed] [Google Scholar]
- 38.Okuwaki, M., and K. Nagata. 1998. Template activating factor-I remodels the chromatin structure and stimulates transcription from the chromatin template. J. Biol. Chem. 273:34511-34518. [DOI] [PubMed] [Google Scholar]
- 39.Okuwaki, M., and A. Verreault. 2004. Maintenance DNA methylation of nucleosome core particles. J. Biol. Chem. 279:2904-2912. [DOI] [PubMed] [Google Scholar]
- 40.Park, Y. J., J. V. Chodaparambil, Y. Bao, S. J. McBryant, and K. Luger. 2005. Nucleosome assembly protein 1 exchanges histone H2A-H2B dimers and assists nucleosome sliding. J. Biol. Chem. 280:1817-1825. [DOI] [PubMed] [Google Scholar]
- 41.Park, Y. J., P. N. Dyer, D. J. Tremethick, and K. Luger. 2004. A new fluorescence resonance energy transfer approach demonstrates that the histone variant H2AZ stabilizes the histone octamer within the nucleosome. J. Biol. Chem. 279:24274-24282. [DOI] [PubMed] [Google Scholar]
- 42.Philpott, A., T. Krude, and R. A. Laskey. 2000. Nuclear chaperones. Semin. Cell Dev. Biol. 11:7-14. [DOI] [PubMed] [Google Scholar]
- 43.Prado, A., I. Ramos, L. J. Frehlick, A. Muga, and J. Ausio. 2004. Nucleoplasmin: a nuclear chaperone. Biochem. Cell Biol. 82:437-445. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez, P., J. Pelletier, G. B. Price, and M. Zannis-Hadjopoulos. 2000. NAP-2: histone chaperone function and phosphorylation state through the cell cycle. J. Mol. Biol. 298:225-238. [DOI] [PubMed] [Google Scholar]
- 45.Sarma, K., and D. Reinberg. 2005. Histone variants meet their match. Nat. Rev. Mol. Cell Biol. 6:139-149. [DOI] [PubMed] [Google Scholar]
- 46.Steger, D. J., and J. L. Workman. 1999. Transcriptional analysis of purified histone acetyltransferase complexes. Methods 19:410-416. [DOI] [PubMed] [Google Scholar]
- 47.Tsunaka, Y., N. Kajimura, S. Tate, and K. Morikawa. 2005. Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle. Nucleic Acids Res. 33:3424-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.