

Abstract
The Rb/E2F complex represses S-phase genes both in cycling cells and in cells that have permanently exited from the cell cycle and entered a terminal differentiation pathway. Here we show that S-phase gene repression, which involves histone-modifying enzymes, occurs through distinct mechanisms in these two situations. We used chromatin immunoprecipitation to show that methylation of histone H3 lysine 9 (H3K9) occurs at several Rb/E2F target promoters in differentiating cells but not in cycling cells. Furthermore, phenotypic knock-down experiments using siRNAs showed that the histone methyltransferase Suv39h is required for histone H3K9 methylation and subsequent repression of S-phase gene promoters in differentiating cells, but not in cycling cells. These results indicate that the E2F target gene permanent silencing mechanism that is triggered upon terminal differentiation is distinct from the transient repression mechanism in cycling cells. Finally, Suv39h-depleted myoblasts were unable to express early or late muscle differentiation markers. Thus, appropriately timed H3K9 methylation by Suv39h seems to be part of the control switch for exiting the cell cycle and entering differentiation.
Keywords: E2F, histone H3K9 methylation, muscle differentiation, Rb, Suv39h
Introduction
The balance between cell proliferation and differentiation is controlled during early G1, before the restriction point (Planassilva and Weinberg, 1997), and the Rb/E2F pathway plays an essential role in this control (Chan et al, 2001). The E2F family of transcription factors drives the G1/S transition by activating S-phase genes, whose products are involved in either DNA synthesis, such as DNA pol α or DHFR (Nevins, 1998), or cell cycle control, such as cyclin E or cyclin D1 (Ohtani et al, 1995; Watanabe et al, 1998; Lee et al, 2000). Rb, the founding member of the pocket protein family, represses E2F activity. Rb inactivation by hyperphosphorylation triggers the G1/S transition. In addition to being a key element in the control of S-phase genes in cycling cells, Rb/E2F plays a major role in the permanent cell cycle exit that precedes terminal differentiation in a number of tissues including muscle.
Muscle cell terminal differentiation is a multistep genetic program, orchestrated by bHLH transcription factors (Buckingham, 1996), that begins with the permanent withdrawal of myoblast precursor cells from the cell cycle. In this model tissue, proliferation and differentiation are mutually exclusive: on the one hand, when myoblasts enter the differentiation program, they become unresponsive to mitogenic signals, and S-phase genes become permanently repressed (Walsh and Perlman, 1997); on the other hand, cell cycle exit is required for the genetic program of muscle differentiation. Myoblasts that are forced to proliferate, for example by the expression of an oncogenic protein, cannot enter terminal differentiation (Lassar et al, 1989). Cell cycle withdrawal involves induction of p21, a cdk inhibitor that activates the Rb family (Mal et al, 2000), resulting in repression of the transcription factor E2F (Novitch et al, 1999). The next steps in the muscle genetic program involve the sequential activation of muscle-specific markers driven by myogenic bHLH transcription factors in conjunction with various coactivators including histone acetyltransferases (Polesskaya et al, 2000, 2001; Puri et al, 1997a, 1997b) and histone methyltransferases (Chen et al, 2002). Rb family members also participate in this process by acting as coactivators for muscle-specific genes (Novitch et al, 1996; Sellers et al, 1998).
Thus, Rb proteins are essential in the control of both cell proliferation and differentiation. They exert their functions, at least partly, through the recruitment of various chromatin-associated proteins, in particular histone deacetylases (Brehm et al, 1998; Ferreira et al, 1998, 2001; Magnaghi-Jaulin et al, 1998). Histone acetylation is a reversible phenomenon that is generally associated with transcriptional activation, whereas deacetylation is associated with transcriptional repression. In addition, Rb proteins also associate with Dnmt1, a DNA methyltransferase (Robertson et al, 2000). Finally, a physical and functional interaction has been described between Rb proteins and Suv39h (Nielsen et al, 2001; Vandel et al, 2001; Nicolas et al, 2003), a family of pericentromeric proteins (Aagaard et al, 1999, 2000; Melcher et al, 2000; O'Carroll et al, 2000) with intrinsic histone methyltransferase activity specific for histone H3 lysine 9 (H3K9) (Rea et al, 2000). This modification is associated with the transcriptionally silenced heterochromatin compartment (Noma et al, 2001). Histone H3 methylated on K9 specifically binds proteins of the heterochromatin protein 1 (HP1) family, thereby controlling the subcellular localization of these proteins (Bannister et al, 2001; Lachner et al, 2001). Histone H3K9 methylation has been linked to both DNA methylation (Tamaru and Selker, 2001; Fuks et al, 2003) and X-chromosome inactivation (Heard et al, 2001). In mice, the absence of functional Suv39h results in elevated embryonic lethality and a high frequency of leukemia (Peters et al, 2001), which is accompanied by chromosomal instability and illegitimate chromosome association during meiosis, as also observed in a variety of other organisms (Ekwall et al, 1996; Peters et al, 2001). Thus, both histone deacetylases and histone methyltransferases specific for H3K9 may be involved in Rb/E2F function.
In order to gain knowledge about the mechanisms involved in S-phase gene repression—whether transient (cycling cells) or permanent (differentiating cells)—we have addressed the involvement of histone-modifying enzymes in the two cell models. We analyzed histone modifications on E2F target promoters using chromatin immunoprecipitation (ChIP) experiments (Ferreira et al, 2001) and found that histone acetylation at the E2F target promoter DHFR varies depending on the activity of the promoter, in both cycling cells and differentiating muscle cells. In contrast, we found no changes in histone H3 methylation on lysine 9 in cycling cells, but a marked increase of lysine 9 methylation in differentiating cells, suggesting that histone H3K9 methylation is specifically associated with differentiation. Methylation of H3K9 was also observed in differentiating cells at other S-phase gene promoters including B-Myb, Cyclin-E and Cyclin-D1. Phenotypic knock-down of Suv39h protein with siRNAs (Elbashir et al, 2001) did not affect the expression level of cyclin D1 or cyclin A2, two E2F target proteins, in cycling cells. Furthermore, inhibiting Suv39h did not perturb cell cycle distribution. In contrast, inhibiting Suv39h markedly perturbed the silencing of Cyclin-D1 and Cyclin-A2 genes under differentiation conditions: myoblastic cells transfected with Suv39 siRNA expressed low levels of cyclin D1 or cyclin A2 in the absence of serum; however, in contrast to normal differentiating myoblasts, re-introduction of serum induced high levels of both cyclins in these cells. Normal gene silencing was substantially restored by ectopic expression of an siRNA-resistant conservative mutant of Suv39h, indicating that the effect was indeed due to decreased Suv39h levels and not simply due to triggering the siRNA system. Depletion of Suv39h caused a marked decrease in H3K9 methylation at the cyclin D1 promoter, as well as at the promoters of other S-phase genes. Finally, inhibition of Suv39h markedly affected the differentiation program, altering expression of early and late muscle marker proteins, in a myoblastic cell line as well as in primary myoblasts. Normal expression of these proteins was restored by ectopic expression of the siRNA-resistant mutant of Suv39h. These data establish that the pericentromeric protein Suv39h is required for both the silencing of proliferation-associated genes and the activation of muscle differentiation markers. More importantly, they indicate that two distinct mechanisms are used for S-phase gene control: Suv39h-independent transient repression in cycling cells, and Suv39h-dependent permanent silencing in differentiating cells.
Results
Histone modifications at S-phase gene promoters
We analyzed histone modifications at the promoter of DHFR, an E2F target gene, using ChIP experiments. These experiments were performed with cycling cells (NIH3T3 fibroblasts) and terminally differentiating cells (mouse myoblastic cell line C2C12). Our results (Figure 1A) demonstrate that histone acetylation varies with the activity of the promoter in both situations: histone H3 acetylation was low in cycling cells during early G0 (Figure 1A, DHFR/fibroblast), as we have previously shown for histone H4 ((Ferreira et al, 2001), and was also low in differentiating myoblasts (Figure 1A, DHFR/myoblast). Acetylation of both histone H3 (Figure 1A) and histone H4 (data not shown) is higher when the promoter is activated, during the G1/S transition in cycling fibroblasts, or in proliferating myoblasts. A GAPDH control sequence that is constitutively expressed did not show any variation in histone acetylation in either cycling cells (data not shown; Ferreira et al, 2001) or differentiating cells (Figure 1A, GAPDH/myoblast).
Figure 1.
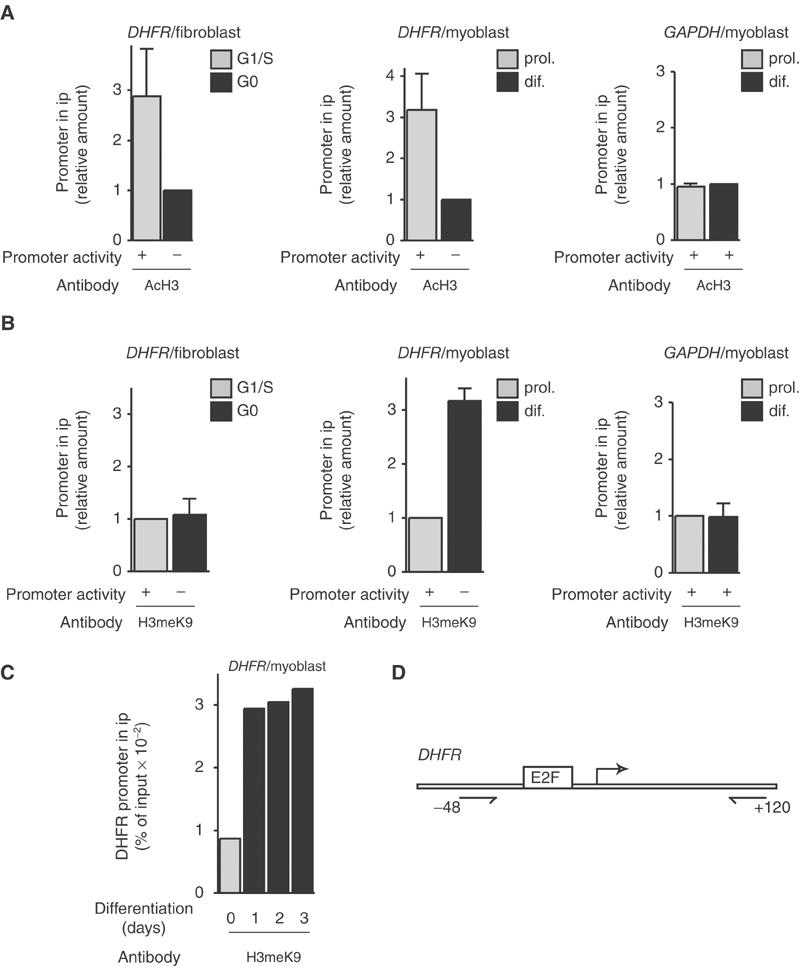
Histone modifications at the DHFR promoter in fibroblasts or myoblasts as indicated. Chromatin was prepared from NIH3T3 fibroblasts at different stages of the cell cycle (G0 or G1/S as indicated, see Materials and methods) or from C2C12 myoblastic cells, either proliferating (prol.) or after either 2 days (dif.) (A, B) or indicated period of time (C) in differentiation medium. Chromatin was immunoprecipitated with antibodies directed against pan-acetylated H3 (AcH3 (A)), or methylated K9 histone H3 (H3meK9 (B, C)) as indicated, and analyzed by Q-PCR to quantify the DHFR promoter copy number, or the GAPDH gene (negative control) copy number. H3 acetylation results (mean±s.d., n=3) are shown as the ratio between values from cells in G1/S and G0 phase (for fibroblasts) or as the ratio of data for proliferating versus differentiating cells (for myoblasts). H3K9 methylation results (mean±s.d., n=3) are shown as the ratio between values from cells in G0 and G1/S phase (for fibroblasts), as the ratio of data for differentiating versus proliferating cells (for myoblasts), or as the percent of input chromatin immunoprecipitated using anti-H3meK9 antibodies (C). Promoter activity was assessed by Northern blot (see Ferreira et al, 2001). (D) Schematic representation of the DHFR promoter showing the positions of the E2F site, transcription start site (bent arrow) and primers used for PCR (with reference to the transcription start site).
In contrast, histone H3 methylation on lysine 9 showed no significant variation during the cell cycle in fibroblasts (Figure 1B, DHFR/fibroblast). In differentiating myoblasts, however, it increased markedly compared to proliferating cells (Figure 1B, DHFR/myoblast), whereas no increase was observed for the constitutively expressed GAPDH sequence (Figure 1B, GAPDH/myoblast). A time course analysis (Figure 1C) revealed that methylation of histone H3K9 at the DHFR promoter was an early event that was detected after 1 day of differentiation. These data indicate that whereas histone acetylation increases following DHFR gene activation both in cycling fibroblasts and in proliferating myoblasts, H3K9 methylation increases only in differentiating myoblasts, concomitant with the silencing of the gene. In order to test whether methylation on H3K9 occurred on other S-phase genes, we performed ChIP analysis of B-Myb, Cyclin-E and Cyclin-D1 promoters. In all three cases, methylation increased in differentiating myoblasts, compared to proliferating cells (Figure 2). These results strongly suggest that H3K9 methylation is a general feature of S-phase gene promoters in differentiating cells.
Figure 2.
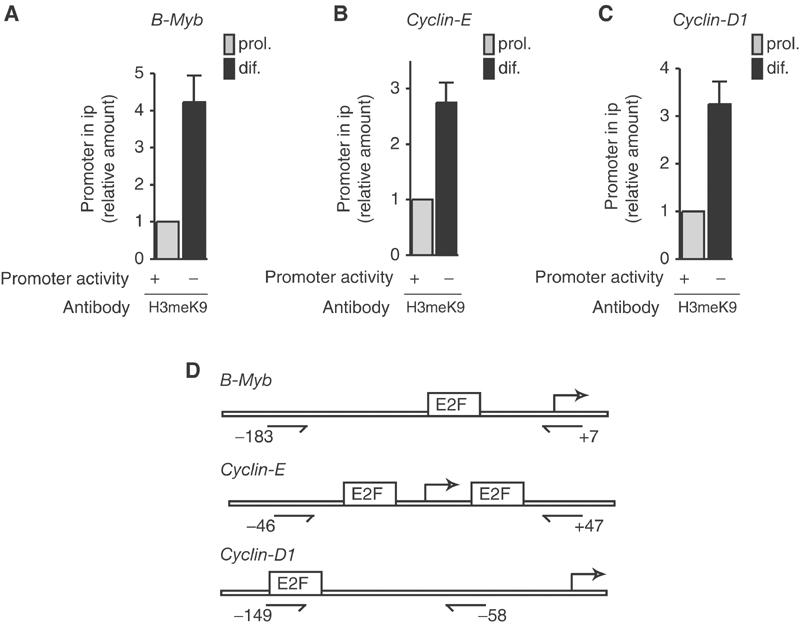
Methylation of H3K9 at various S-phase gene promoters. ChIP experiments were performed as described in Figure 1 and assayed for B-Myb (A), Cyclin-E (B) and Cyclin-D1 (C) promoters (mean±s.d., n=4). (D) Schematic representations of the promoters and positions of the primers as in Figure 1.
Suv39h is responsible for S-phase gene silencing in differentiating cells
Several mammalian enzymes have been shown to methylate histone H3 on lysine 9, including the heterochromatin enzyme Suv39h and the G9A enzyme (Tachibana et al, 2001) that preferentially acts on euchromatin (Tachibana et al, 2002). Suv39h, however, has been previously connected to E2F target gene control (Nielsen et al, 2001; Vandel et al, 2001), and thus seems to be a more likely candidate for S-phase gene inhibition. Quantitative RT–PCR experiments showed that Suv39h mRNA is expressed in proliferating myoblasts (Figure 3). Its expression decreases during the first hours of differentiation to around 50% of the original level and remains stable for 3 days, after which it drops to undetectable levels.
Figure 3.
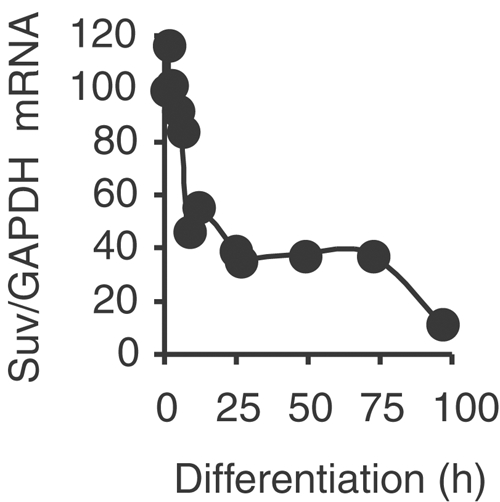
Expression of Suv39h1 mRNA in muscle cells. RNA from C2C12 cells either proliferating (0 h) or after different periods of differentiation were analyzed by Q-RT–PCR. The results are shown after standardization on GAPDH mRNAs quantified in the same experiments.
In order to address the role of Suv39h in S-phase gene control, we designed an siRNA corresponding to a sequence shared by the two murine as well as human isoforms of Suv39h, Suv39h1 and h2, as depicted in Figure 4A (Suv39 siRNA). A scrambled sequence was used as a negative control (control siRNA). A search in sequence libraries indicated that our Suv39h siRNA sequence is restricted to Suv39h1 and Suv39h2, and the control siRNA sequence is absent altogether. The siRNA was transfected into a high proportion of myoblastic cells (Figure 4B). In a stable transfectant cell line derived from HeLa cells (Figure 4C), as well as when transiently expressed in myoblastic cells (Figure 4D), it almost completely inhibited the expression of Myc-tagged Suv39h1.
Figure 4.
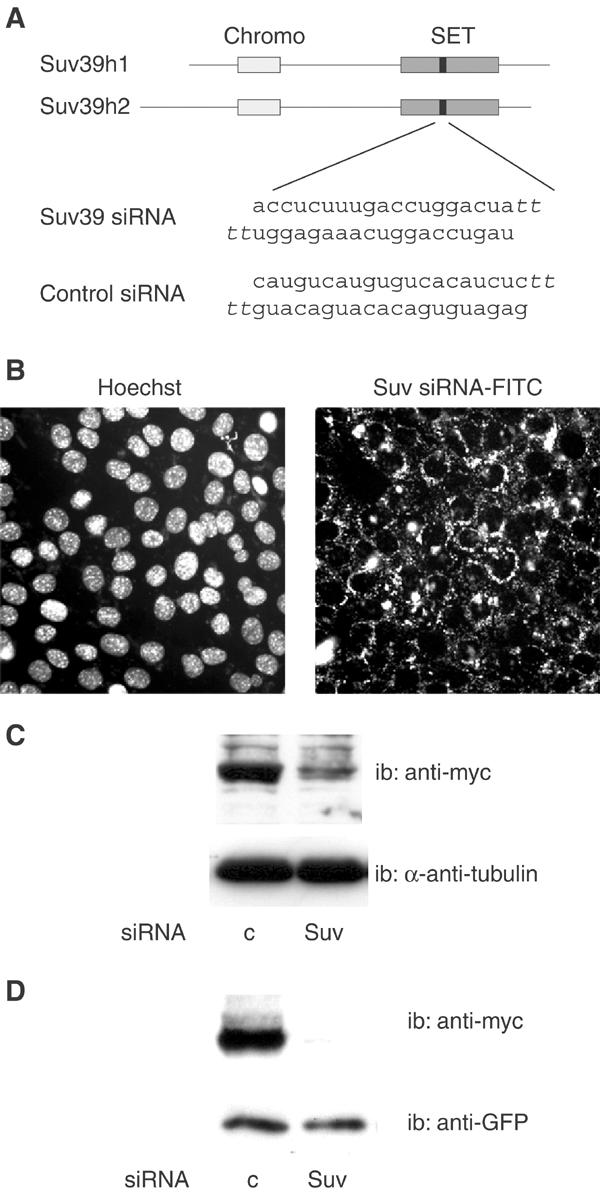
Inhibition of Suv39h expression by siRNAs. (A) Sequence of Suv39h siRNA and position of its target in the mRNAs (black boxes) corresponding to positions 889–908 for Suv39h1 and 1111–1130 for Suv39h2 (chromo: chromodomain; SET: SET domain). The scrambled sequence used as a control is also shown (Control siRNA). SiRNAs contain two 3′ overhanging T's. (B) Penetration of Suv39h siRNA into myoblast. Proliferating C2C12 cells were transfected with FITC-labeled Suv39h siRNA, and examined 18 h later. (C) Inhibition of Suv39h1 protein in HeLa cells. A HeLa-derived human cell line stably expressing a myc-tagged version of Suv39h1 (see Materials and methods) was transfected with Suv39h (Suv) or control (c) siRNAs, and analyzed 40 h later by Western blotting using anti-myc antibodies, or anti-α-tubulin antibodies as a control. (D) Inhibition of Suv39h protein in myoblasts. C2C12 cells were cotransfected with Suv39 siRNA or control siRNA, together with the myc-tagged Suv39h1 expression vector and a GFP expression vector used as a transfection control. At 48 h post-transfection, cell lysates were analyzed by Western blotting with anti-myc or anti-GFP.
We used these siRNAs to investigate the role of Suv39h in S-phase gene control, in both cycling and differentiating cells. Due to the lack of a reliable antibody recognizing the Suv39h protein, the efficiency of the siRNA was routinely monitored at the level of the target mRNA, using real-time RT–PCR, and results can be found in the legends of the corresponding figures. NIH3T3 fibroblasts were transfected with control or Suv39h siRNAs, prior to synchronization by serum deprivation. We monitored the expression of cyclin D1, whose gene promoter is, at least in part, controlled by E2F (Watanabe et al, 1998; Lee et al, 2000), at different time points as described in Figure 5A. Inhibition of Suv39h did not significantly affect the expression of cyclin D1, nor did it affect the cell cycle as assayed by FACS analysis (data not shown). Thus, Suv39h is not involved in the regulation of the Cyclin-D1 gene in cycling cells. To explore the role of Suv39h in differentiating cells, C2C12 cells were transfected with siRNAs, cultured in differentiation medium for 4 days, and then treated with serum for 10 h, as described in Figure 5B. Under normal conditions, differentiated myoblasts are unresponsive to mitogenic signals and indeed differentiating cells treated with control siRNA expressed very little cyclin D1 in response to serum, the residual level of cyclin D1 detected being most likely due to cells that are refractory to differentiation. Similarly, differentiating cells treated with Suv39 siRNA expressed little cyclin D1 in the absence of serum. However, treatment of these cells with serum induced a much higher level of cyclin D1 than in control cells (Figure 5B). This result suggests that Suv39h is involved in the silencing of the Cyclin-D1 gene in differentiating cells. To demonstrate that the absence of Suv39h was responsible for the inappropriate cyclin D1 expression in these cells, we performed rescue experiments using an siRNA-resistant conservative mutant. Conservative mutations that do not affect the protein sequence were introduced into five codons located in the siRNA target sequence (Figure 6A). The resulting mSuv39h1 protein was entirely resistant to inhibition by Suv39 siRNA, as shown in a transient cotransfection assay (Figure 6B). Transfection of the expression vector for mSuv39h1 together with Suv39 siRNA restored the silencing of cyclin D1 protein expression in differentiating myoblasts, whereas transfection of the empty vector did not have any effect (Figure 6C). Thus, taken together, these data demonstrate that Suv39h protein is essential for the silencing of the Cyclin-D1 gene during muscle terminal differentiation. Similar results were obtained with the S-phase cyclin, cyclin A2, which is strongly reduced in differentiating cells. Cyclin A2 expression in response to serum was much higher in cells depleted of Suv39h than in cells treated with the control siRNA. Normal silencing was restored upon expression of the siRNA-resistant Suv39h expression vector (mSuv, Figure 6C).
Figure 5.
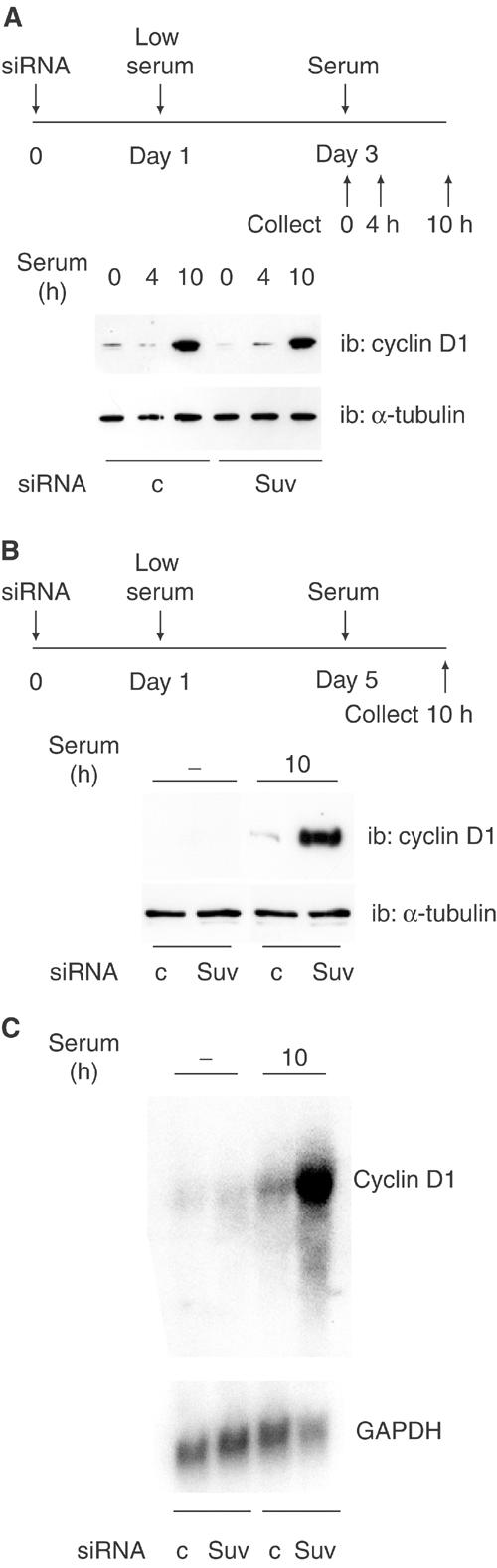
Suv39h inhibition affects differentiating cells but not cycling cells. (A) NIH3T3 cells were treated with control siRNA or Suv39h siRNA, synchronized and treated with serum as described in the upper panel; cyclin D1 expression was monitored by Western blotting. Suv39h1 mRNA was downregulated by 84% in the experiment shown. (B) C2C12 cells were treated with control siRNA or Suv39h siRNA, placed in differentiation medium and treated with serum as indicated in the upper panel, and analyzed as in (A). Suv39h1 mRNA was downregulated by 86% in the experiment shown. (C) C2C12 cells were treated with control siRNA or Suv39h siRNA as in (B), and RNA was extracted and analyzed by Northern blot using indicated probes. Inhibition of Suv39h was 79% as measured by RT–PCR. These experiments were repeated 2–4 times.
Figure 6.
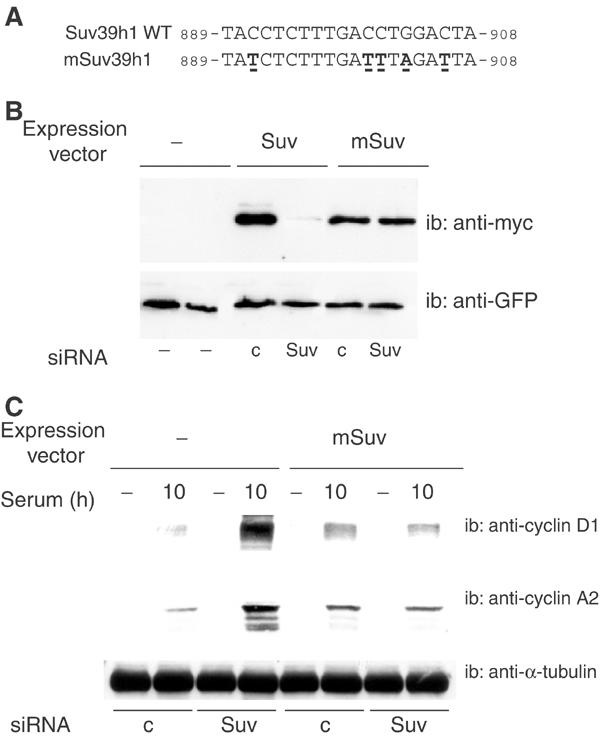
Rescue experiments using a Suv39h siRNA-resistant mutant. (A) Sequences of wild type and mutant Suv39h; conservative mutations are underlined. (B) C2C12 cells were cotransfected with Suv39 siRNA or control siRNA, together with the indicated expression vectors: myc-tagged Suv39h1 wild type (Suv), myc-tagged Suv39h siRNA-resistant mutant (mSuv) or the empty vehicle vector (−); a GFP expression vector was used as a transfection control. At 48 h post-transfection, cell lysates were analyzed by Western blotting with anti-myc or anti-GFP. (C) C2C12 cells were cotransfected with Suv39h siRNA (Suv) or control siRNA (c) together with either the Suv39h siRNA-resistant mutant (mSuv) expression vector or the empty vehicle vector (−), and treated as in Figure 5B; extracts were analyzed by Western blotting using anti-cyclin D1 or -cyclin A2 antibodies, as indicated. Suv39h1 mRNA was downregulated by 80% in the experiment shown. These experiments were repeated twice.
Suv39h is required for H3K9 methylation at S-phase gene promoters in differentiating cells
Our data show that silencing of both Cyclin-D1 and Cyclin-A2 genes requires normal expression of Suv39h. It thus appears that silencing is most likely due to methylation of histone H3K9 by Suv39h at the corresponding loci. To test this hypothesis, we monitored methylation of H3K9 by ChIP experiments in Suv39h-depleted myoblasts (Figure 7): depletion of Suv39h resulted in low levels of H3K9 methylation at the Cyclin-D1 promoter, as well as at the promoters of DHFR, B-Myb and Cyclin-E, but did not affect GAPDH used as a control. These experiments demonstrate that Suv39h is the enzyme responsible for H3K9 methylation at these promoters in differentiating cells. Interestingly, depletion of Suv39h did not result in a general demethylation of H3K9 in myoblast nuclei (data not shown), in contrast to results in mouse embryonic fibroblasts (MEFs) from double knockout mice (Peters et al, 2001). This could suggest that Suv39h is not responsible for the majority of H3K9 methylation in the myoblastic lineage. Methylation, however, is thought to be a stable modification since no histone demethylase has been identified so far; therefore, demethylation most likely would require several cell divisions and would not be observed within the time frame of these experiments (2–3 days).
Figure 7.
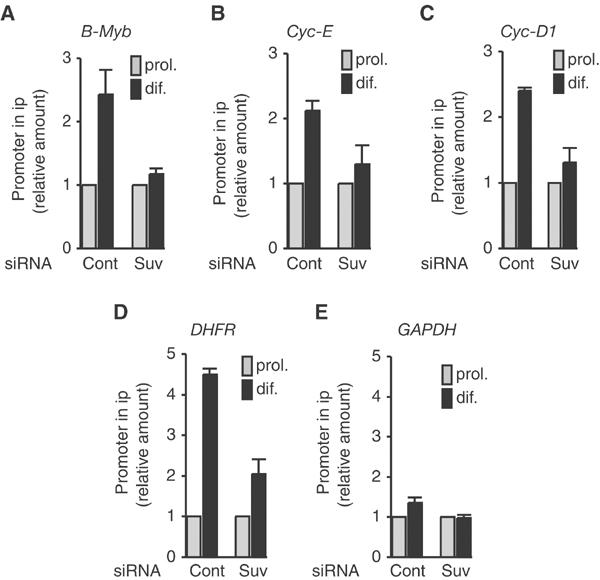
Suv39h depletion affects H3K9 methylation at S-phase gene promoters. C2C12 cells were transfected with control siRNA (cont) or Suv39h siRNA (Suv) as indicated. At 48 h post-transfection, cells were placed in differentiation medium for 72 h and chromatin was prepared and immunoprecipitated as in Figure 1. S-phase gene promoters were detected in immunoprecipitates as described in Figure 2 (mean±s.d., n=3–4).
Suv39h is required for myoblastic differentiation
We next investigated the effect of inhibiting Suv39h on the muscle-specific genetic program. Indeed, forcing proliferation of myoblastic cells markedly affects their differentiation potential (Konieczny et al, 1989; Caruso et al, 1993; La Rocca et al, 1994; Tiainen et al, 1996)—for example, when cyclin D1 is overexpressed (Skapek et al, 1995). Under conditions of low Suv39h expression, cyclin D1 protein is low in the absence of serum, and is only induced by mitogenic signals (Figure 5B and C). Nevertheless, Suv39h-depleted cells show a poor differentiation capacity: the proportion of cells fusing into myotubes was much lower in Suv39h-depleted cells (Figure 8A), and the expression of muscle markers was greatly impaired. In particular, myogenin, an early marker of the cell differentiation program, was expressed much less in cells treated with Suv39 siRNA than in control cells (Figure 8B). Myogenin expression was partially restored by ectopic expression of the siRNA-resistant mutant (mSuv, Figure 8C). The expression of late markers was also affected by inhibition of Suv39h (Figure 8D): the expression of myosin heavy chain (MHC), as well as muscle creatine kinase (MCK), was lower in cells treated with Suv39 siRNA than in control cells. Furthermore, similar results were obtained in primary myoblasts in which the knock-down of Suv39h dramatically affected the expression of the late muscle-specific marker MCK (Figure 8E). Taken together, these results indicate that Suv39h inhibition affects the differentiation program at both early and late stages.
Figure 8.
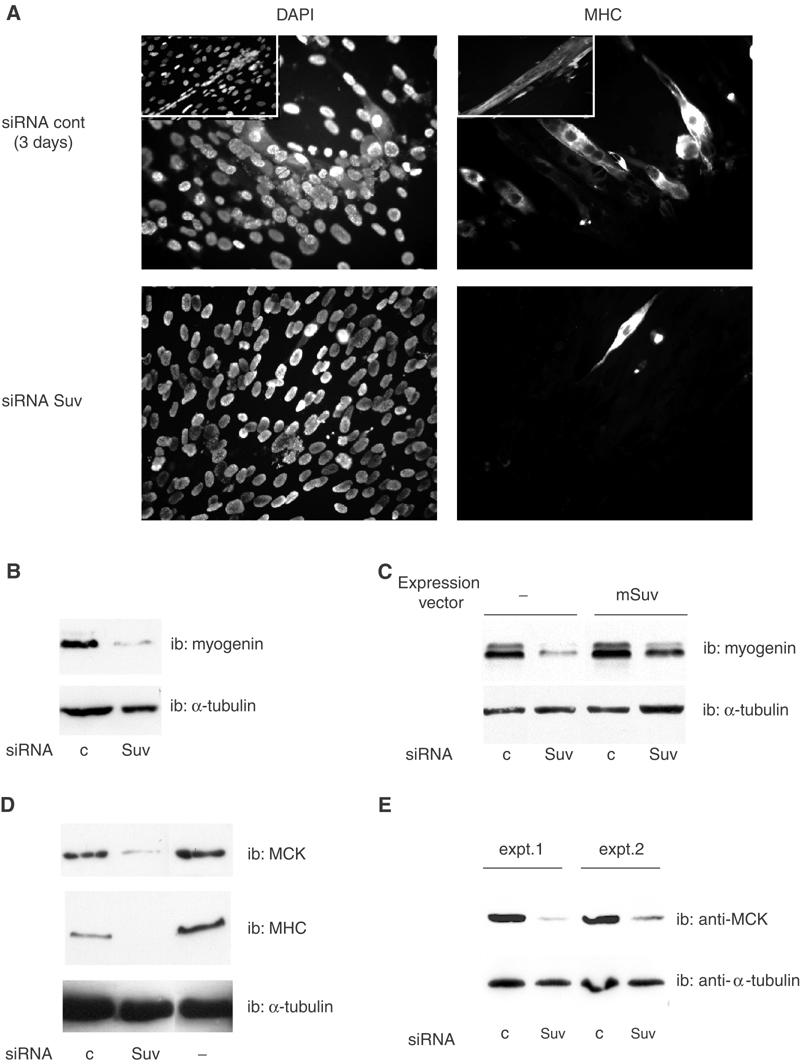
Suv39h depletion affects the muscle differentiation program. (A, B) C2C12 cells were transfected with control siRNA or Suv39h siRNA as indicated. At 48 h post-transfection, cells were placed in differentiation medium for 72 h (or 96 h for control siRNA-transfected cells in the insets; no mature myotubes were observed in Suv39 siRNA-transfected cells under the same conditions) and labeled by immunofluorescence using an anti-MHC antibody (A) or analyzed by Western blotting with anti-myogenin (B). (C) C2C12 cells were transfected as in (A), together with the expression vector for the siRNA-resistant form of Suv39h (mSuv) or the empty vehicle vector for the control; differentiation was induced 24 h after transfection. (D) C2C12 cells were transfected as in (A), and extracts were analyzed by Western blotting with anti-MHC and -MCK antibodies. Suv39h mRNA was downregulated by 80–86% in these experiments. These experiments were repeated 2–3 times. (E) Primary myoblasts were transfected with siRNAs using Lipofectamine (experiment 1) or Lipofectamine 2000 (experiment 2); inhibition of Suv39h was 74.1 and 73.9%, respectively. Cells were placed in differentiation medium for 2 days, and assayed for MCK expression.
Discussion
Two distinct mechanisms for S-phase genes repression/silencing in cycling and differentiating cells
We have investigated histone modifications at S-phase gene promoters in cycling or differentiating cells. Whereas histone acetylation varied as a function of gene activity, histone H3K9 methylation remained low throughout the cell cycle in proliferating cells, but increased significantly in terminally differentiating myoblasts (Figures 1 and 2). In good agreement with these results, depletion of the histone methyltransferase Suv39h affected methylation of histone H3 as well as S-phase gene control in differentiating but not in cycling cells. These results indicate that two distinct mechanisms based on histone modifications are used for S-phase gene control in cycling and in terminally differentiating cells.
H3K9 methylation at S-phase gene promoters in cycling cells?
Our findings that H3K9 is not methylated in cycling fibroblasts contradict those of Nicolas et al (2003), who found H3K9 methylation at the DHFR promoter in growth-arrested fibroblasts. This discrepancy is probably due to the antibodies used: they used an antibody directed against a regular linear peptide, which gives a low signal to noise ratio in ChIP experiments (data not shown). We, on the other hand, have used an antibody raised against a ‘branched peptide', designed to mimic condensed heterochromatin (Peters et al, 2001), that has been thoroughly characterized: it preferentially recognizes trimethylated H3K9, but also binds to dimethylated H3K9 and shows a mild reactivity with H3 methylated on other lysines (in our experiments, the binding that we see is most likely due to methylated H3K9 since most of the binding is sensitive to Suv39h depletion, see Figure 7). A major difference between the two antibodies is that the anti-‘branched' antibody used here clearly labels heterochromatin whereas the anti-‘linear' antibody shows a widespread labeling of the nucleus (data not shown). Thus, an interesting hypothesis is that, in cycling cells, H3K9 methylation at S-phase gene promoters does occur, but in the euchromatin compartment, without chromatin condensation and possibly in a qualitatively different manner. However, if methylation of H3K9 does occur in cycling cells, it does so independently of the heterochromatin-associated protein Suv39h, as inhibition of Suv39h does not perturb S-phase gene repression in fibroblasts (Figure 5). This result is in good agreement with data obtained with Suv39h−/− fibroblasts, which do not show any abnormal phenotype in G1 phase (Peters et al, 2001), indicating that histone H3K9 methylation by Suv39h is not involved in cycling cells. In contrast, in differentiating cells, H3K9 methylation is due to Suv39h since it is sensitive to Suv39h depletion. Furthermore, it is detected using the anti-‘branched' antibody and thus is likely to be associated with condensation and local heterochromatinization.
Silencing, but not repression, of S-phase genes is impaired in Suv39h-low myoblasts
Under normal conditions, serum removal triggers terminal differentiation in myoblasts. The differentiating cells become refractory to mitogenic signals: they do not express S-phase genes; moreover, these genes cannot be re-expressed upon serum addition. Differentiating C2C12 cells with reduced levels of Suv39h showed a low level of cyclin D1 or cyclin A2 expression—comparable to control cells—in the absence of mitogenic signals; however, in contrast to control cells, they expressed high levels of cyclins in response to serum. In rescue experiments, a normal inhibition was restored by ectopic expression of a Suv39h form that resists knock-down by siRNA, indicating that the abnormal responsiveness to serum of Suv39h-deficient cells was indeed due to the inhibition of Suv39h, and not to other nonspecific effects. Notably, the above result indicates that the corresponding genes are not constitutively de-repressed in the absence of Suv39h, analogous to the results obtained in cycling fibroblasts, where low levels of expression were observed in early G1. Thus, in both cell types, S-phase genes are normally repressed in the absence of mitogenic signal. Inhibiting Suv39h perturbs an additional control mechanism that is triggered only in differentiating cells: Suv39h is required to lock S-phase genes into a silenced state. Suv39h is probably not required, however, for the maintenance of this state, as its expression decreases at late times of differentiation.
Suv39h is a pericentromeric protein, and it is unclear how it could directly regulate—by locally modifying histone H3—gene promoters that are not located in the heterochromatin compartment. It is possible that some signals of unknown origin activate a heterochromatinization pathway for S-phase gene promoters in differentiating cells. The results of ChIP experiments with antibodies directed against HP1 proteins, as well as phenotypic knock-down of these proteins with siRNAs, indicate that some of the HP1 proteins are recruited to S-phase gene promoters upon differentiation (S Ait-Si-Ali, L Fritsch and A Harel-Bellan, unpublished observations), supporting the heterochromatinization hypothesis. Furthermore, our preliminary data suggest that, indeed, an S-phase gene promoter can migrate from the euchromatin to the heterochromatin compartment upon myogenic terminal differentiation (V Guasconi, S Ait-Si-Ali and A Harel-Bellan, unpublished observations).
Muscle marker expression is perturbed in Suv39h-low myoblasts
Cells with reduced levels of Suv39h did not differentiate, and early (myogenin) or late (MHC, MCK) muscle differentiation markers were negatively affected. These data indicate that Suv39h is required for activation of the muscle differentiation program. It is noteworthy that Rb proteins, with which Suv39h forms complexes, are coactivators for various transcription factors including C/EBP (Chen et al, 1996) and myogenic bHLHs such as MyoD (Novitch et al, 1996). This function of Rb can be distinguished from E2F repression by mutational analysis (Sellers et al, 1998), although its precise mechanism is unknown. It is tempting to speculate that Suv39h is also involved in the coactivating functions of Rb independent of its effect on S-phase genes. In that case, the effects on S-phase genes would be an indirect consequence of the failure to differentiate properly. A more likely hypothesis, however, is that inhibiting Suv39h affects the ‘main control switch' of cell proliferation and differentiation in muscle. Indeed, activation of muscle-specific genes requires permanent cell cycle exit (Wang et al, 1996). Thus, the absence of muscle marker proteins could be an indirect effect of S-phase gene deregulation (mis-silencing) in Suv39 siRNA treated cells.
Both early (myogenin) and late (MHC, MCK) muscle genes are inhibited in the absence of Suv39h, even though, under normal conditions, Suv39h mRNA levels decrease as cells progress along the differentiation pathway. It is likely that inhibition of late genes is an indirect consequence of the inhibition of early genes, and in particular of myogenin, which plays a central role in late gene activation (Hasty et al, 1993). This hypothesis is consistent with the kinetics of Suv39h expression in differentiating cells (Figure 3), which suggests a function in early differentiation; we believe that this function is the silencing of S-phase genes, muscle gene inhibition in Suv39h-low cells being due to the mis-silencing of S-phase genes.
Is Suv39h-dependent silencing of S-phase genes restricted to permanent cell cycle exit?
A silencing mechanism similar to the one we describe here has been reported for S-phase genes during senescence (Narita et al, 2003). Interestingly, in both senescent and differentiating cells, exit from the cell cycle becomes permanent. Thus, our data support a model in which two distinct mechanisms are used for regulating S-phase genes. In cycling cells, S-phase genes are repressed by a reversible mechanism that involves local histone deacetylation but not H3K9 methylation. In cells permanently exiting from the cell cycle, a silencing process that requires histone methylation occurs in addition to repression by deacetylation. Thus H3K9 seems to be part of the ‘main switch' that controls cell proliferation and differentiation. The molecular relationship between the silencing process and the ability of cells to enter the terminal differentiation pathway is a key issue for understanding the balance between cell proliferation and differentiation.
Materials and methods
Cell culture and transfections
C2C12 skeletal myoblasts, NIH3T3 fibroblasts and stably transfected HeLa cells were cultured under standard conditions. Mouse primary myoblasts were prepared as previously described (Polesskaya et al, 2003). To induce terminal differentiation, C2C12 cells were placed in differentiation medium (DMEM supplemented with 0.5% fetal calf serum) as in Polesskaya et al (2001), and primary myoblasts were treated as in Polesskaya et al (2003). NIH3T3 cells were synchronized by placing them in DMEM supplemented with 0.5% fetal calf serum, and then treated with 20% FCS for 0 h (G0) or 12 h (G1/S). Cell cycle position was monitored by FACS analysis: in the G0 sample, 80–85% of the cells were in G0/G1 and 10–15% in S; in the G1/S sample, 60–65% of the cells were in G1 and 30–35% in S. Transfections were carried out with a Polyfect kit (Qiagen) or a Lipofectamine kit (Invitrogen). For fluorescence analysis, cells were grown and transfected on glass coverslips, and fixed using paraformaldehyde.
Formaldehyde crosslinking and chromatin immunoprecipitation
Chromatin preparation and immunoprecipitation were performed as described in Ferreira et al (2001), with a crosslinking time of 10 min at 37°C and sonication to an average length of 300–800 bp. Extracts were standardized by nondenaturing gel electrophoresis, and standardization was verified by quantitative PCR (Q-PCR) using a LightCycler (Roche Diagnostics).
Rabbit anti-methyl-K9 histone H3 (H3K9) antibodies raised against a ‘branched' peptide corresponding to dimethylated H3K9 were kindly provided by T Jenuwein (Vienna, Austria). This antibody preferentially recognizes trimethylated H3K9, but also binds to dimethylated H3K9 and mildly crossreacts with other methylated lysines in histone H3; this antibody labels preferentially the heterochromatin dots of interphasic nuclei as shown by immunofluorescence experiments (data not shown). The association that we observe with S-phase gene promoters, however, is most likely due to H3K9 methylation since it depends on the proper expression of Suv39h in cells (Figure 7). Anti-acetyl histone H3, which recognizes pan-acetylated histone H3 (AcH3), and anti-acetyl histone H4 (AcH4) were purchased from Upstate Biotech. In all, 50 μg of sonicated chromatin was used for anti-AcH3 or anti-AcH4, and 150 μg for anti-H3meK9. Oligonucleotides used in the Q-PCR experiments are as follows: B-Myb: Fw: TAGGCCCCTCC TAGGGTTCT; Rev: AGGTCTGGTCGCACGTTC; cyclin E: Fw: TGAGGGGCTCGCAGCCCTCG; Rev: CCCGGCTTCGAGCGGGACAT; cyclin D1: Fw: TCACTGCTCCCGAGCC; Rev: CCGTGTGACGT TACTGTTGT. Oligonucleotides used for DHFR and GAPDH were previously described (Ferreira et al, 2001).
Samples were analyzed by Q-PCR on a LightCycler, and copy numbers were calculated as described in Ferreira et al (2001). Two different dilutions of each sample were analyzed independently.
RT–PCR
mRNAs were purified using a GenElute RNA kit (Sigma). Quantitative RT–PCR (Q-RT–PCR) of Suv39h was carried out in hybridization format using a kit from Roche (details available upon request). Primers were: external: forward: ACCTGTGCCGACTAGC CAAG; reverse: CCACGCCACTTAACCAGGTA; internal probes: forward: TGCCCTTGGTGTTTCT-3′-fluorescein; reverse: red640-5′-GAAGAATCTGTATGAC. Q-RT–PCR of GAPDH has been performed in SYBR Green format. Primers were: forward: CCAATGTGTC CGTCGTGGATCT; reverse: GTTGAAGTCGCAGGAGACAACC. Results from Q-RT–PCR are presented as the ratio of Suv39h mRNA to GAPDH mRNA values.
siRNAs
Synthetic siRNAs were purchased from Genset Proligo (France) or MWG Biotech (Germany).
Western and Northern blotting
Monoclonal anti-myogenin antibody (F.5D, kind gift of WE Wright), anti-α-tubulin antibody (Sigma), anti-cyclin A2 (Santa Cruz), rabbit anti-MCK (kindly provided by H Ito) and anti-MHC (MY-32; Sigma) antibodies were used as described in Polesskaya et al (2001). Anti-cyclin D1 antibodies were either a rabbit polyclonal (Santa Cruz Biotechnology) used at 1:500 dilution or a mouse monoclonal (Zymed) used at 2.5 μg/ml. Northern blots were performed using standard procedures.
Acknowledgments
We thank JB Weitzman, LL Pritchard, M Ameyar-Zazoua, A Hamiche and C Francastel for critical reading of the manuscript, Thomas Jenuwein, Idenori Ito, Luis Martinez and Maurizia Caruso for the kind gift of antibodies and probes, Claude Sardet for sharing information, Zohair Mishal and Arlette Vervisch for technical assistance, and François Dautry for helpful discussions, with special thanks to Thomas Jenuwein for the kind gift of the Suv39h1 expression vector, Suv39h1 expressing cell line and anti-methylated H3 antibodies, materials that were instrumental for this study. This work was supported by grants from the Association Française contre les Myopathies, from the Association pour la Recherche sur le Cancer and from the European 5th PCRDT grant no. QLG1-1999-00866. VG was supported by a fellowship from the Société Française du Cancer and from the Ligue Nationale contre le Cancer, RS is a recipient of a fellowship from the Ministère de la Recherche et de la Technologie, and HY was supported by a fellowship from the Société Française du Cancer.
References
- Aagaard L, Laible G, Selenko P, Schmid M, Dorn R, Schotta G, Kuhfittig S, Wolf A, Lebersorger A, Singh PB, Reuter G, Jenuwein T (1999) Functional mammalian homologues of the Drosophila PEV-modifier Su(var)3-9 encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO J 18: 1923–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aagaard L, Schmid M, Warburton P, Jenuwein T (2000) Mitotic phosphorylation of SUV39H1, a novel component of active centromeres, coincides with transient accumulation at mammalian centromeres. J Cell Sci 113: 817–829 [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124 [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391: 597–601 [DOI] [PubMed] [Google Scholar]
- Buckingham M (1996) Skeletal muscle development and the role of the myogenic regulatory factors. Biochem Soc Trans 24: 506–509 [DOI] [PubMed] [Google Scholar]
- Caruso M, Martelli F, Giordano A, Felsani A (1993) Regulation of MyoD gene transcription and protein function by the transforming domains of the adenovirus E1A oncoprotein. Oncogene 8: 267–278 [PubMed] [Google Scholar]
- Chan HM, Shikama N, La Thangue NB (2001) Control of gene expression and the cell cycle. Essays Biochem 37: 87–96 [DOI] [PubMed] [Google Scholar]
- Chen PL, Riley DJ, Chen Y, Lee WH (1996) Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev 10: 2794–2804 [DOI] [PubMed] [Google Scholar]
- Chen SL, Loffler KA, Chen D, Stallcup MR, Muscat GE (2002) The coactivator-associated arginine methyltransferase is necessary for muscle differentiation: CARM1 coactivates myocyte enhancer factor-2. J Biol Chem 277: 4324–4333 [DOI] [PubMed] [Google Scholar]
- Ekwall K, Nimmo ER, Javerzat JP, Borgstrom B, Egel R, Cranston G, Allshire R (1996) Mutations in the fission yeast silencing factors clr4+ and rik1+ disrupt the localisation of the chromo domain protein Swi6p and impair centromere function. J Cell Sci 109: 2637–2648 [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D (1998) The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc Natl Acad Sci USA 95: 10493–10498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R, Naguibneva I, Mathieu M, Ait-Si-Ali S, Robin P, Pritchard LL, Harel-Bellan A (2001) Cell cycle-dependent recruitment of HDAC-1 correlates with deacetylation of histone H4 on an Rb-E2F target promoter. EMBO Rep 2: 794–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T (2003) The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 278: 4035–4040 [DOI] [PubMed] [Google Scholar]
- Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH (1993) Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364: 501–506 [DOI] [PubMed] [Google Scholar]
- Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD, Spector DL (2001) Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 107: 727–738 [DOI] [PubMed] [Google Scholar]
- Konieczny SK, Drobes BL, Menke SL, Taparowsky EJ (1989) Inhibition of myogenic differentiation by the H-ras oncogene is associated with the down regulation of the MyoD1 gene. Oncogene 4: 473–481 [PubMed] [Google Scholar]
- La Rocca SA, Crouch DH, Gillespie DA (1994) c-Myc inhibits myogenic differentiation and myoD expression by a mechanism which can be dissociated from cell transformation. Oncogene 9: 3499–3508 [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120 [DOI] [PubMed] [Google Scholar]
- Lassar AB, Thayer MJ, Overell RW, Weintraub H (1989) Transformation by activated ras or fos prevents myogenesis by inhibiting expression of MyoD1. Cell 58: 659–667 [DOI] [PubMed] [Google Scholar]
- Lee RJ, Albanese C, Fu M, D'Amico M, Lin B, Watanabe G, Haines GK III, Siegel PM, Hung MC, Yarden Y, Horowitz JM, Muller WJ, Pestell RG (2000) Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol 20: 672–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A (1998) Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391: 601–605 [DOI] [PubMed] [Google Scholar]
- Mal A, Chattopadhyay D, Ghosh MK, Poon RY, Hunter T, Harter ML (2000) p21 and retinoblastoma protein control the absence of DNA replication in terminally differentiated muscle cells. J Cell Biol 149: 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher M, Schmid M, Aagaard L, Selenko P, Laible G, Jenuwein T (2000) Structure–function analysis of SUV39H1 reveals a dominant role in heterochromatin organization, chromosome segregation, and mitotic progression. Mol Cell Biol 20: 3728–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716 [DOI] [PubMed] [Google Scholar]
- Nevins JR (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ 9: 585–593 [PubMed] [Google Scholar]
- Nicolas E, Roumillac C, Trouche D (2003) Balance between acetylation and methylation of histone H3 lysine 9 on the E2F-responsive dihydrofolate reductase promoter. Mol Cell Biol 23: 1614–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T (2001) Rb targets histone H3 methylation and HP1 to promoters. Nature 412: 561–565 [DOI] [PubMed] [Google Scholar]
- Noma K, Allis CD, Grewal SI (2001) Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293: 1150–1155 [DOI] [PubMed] [Google Scholar]
- Novitch BG, Mulligan GJ, Jacks T, Lassar AB (1996) Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J Cell Biol 135: 441–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch BG, Spicer DB, Kim PS, Cheung WL, Lassar AB (1999) pRb is required for MEF2-dependent gene expression as well as cell-cycle arrest during skeletal muscle differentiation. Curr Biol 9: 449–459 [DOI] [PubMed] [Google Scholar]
- O'Carroll D, Scherthan H, Peters AH, Opravil S, Haynes AR, Laible G, Rea S, Schmid M, Lebersorger A, Jerratsch M, Sattler L, Mattei MG, Denny P, Brown SD, Schweizer D, Jenuwein T (2000) Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol 20: 9423–9433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR (1995) Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci USA 92: 12146–12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T (2001) Loss of the suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107: 323–337 [DOI] [PubMed] [Google Scholar]
- Planassilva MD, Weinberg RA (1997) The restriction point and control of cell proliferation. Curr Opin Cell Biol 9: 768–772 [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Duquet A, Naguibneva I, Weise C, Vervisch A, Bengal E, Hucho F, Robin P, Harel-Bellan A (2000) CREB-binding protein/p300 activates MyoD by acetylation. J Biol Chem 275: 34359–34364 [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Naguibneva I, Fritsch L, Duquet A, Ait-Si-Ali S, Robin P, Vervisch A, Pritchard LL, Cole P, Harel-Bellan A (2001) CBP/p300 and muscle differentiation: no HAT, no muscle. EMBO J 20: 6816–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesskaya A, Seale P, Rudnicki MA (2003) Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell 113: 841–852 [DOI] [PubMed] [Google Scholar]
- Puri PL, Avantaggiati ML, Balsano C, Sang N, Graessmann A, Giordano A, Levrero M (1997a) p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J 16: 369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri PL, Sartorelli V, Yang XJ, Hamamori Y, Ogryzko VV, Howard BH, Kedes L, Wang JY, Graessmann A, Nakatani Y, Levrero M (1997b) Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol Cell 1: 35–45 [DOI] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP (2000) DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet 25: 338–342 [DOI] [PubMed] [Google Scholar]
- Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, Lassar AB, Kaelin WG (1998) Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Gene Dev 12: 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skapek SX, Rhee J, Spicer DB, Lassar AB (1995) Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase [see comments]. Science 267: 1022–1024 [DOI] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Fukushima T, Shinkai Y (2001) Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem 276: 25309–25317 [DOI] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16: 1779–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaru H, Selker EU (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 414: 277–283 [DOI] [PubMed] [Google Scholar]
- Tiainen M, Spitkovsky D, Jansen-Durr P, Sacchi A, Crescenzi M (1996) Expression of E1A in terminally differentiated muscle cells reactivates the cell cycle and suppresses tissue-specific genes by separable mechanisms. Mol Cell Biol 16: 5302–5312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D (2001) Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol Cell Biol 21: 6484–6494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh K, Perlman H (1997) Cell cycle exit upon myogenic differentiation. Curr Opin Genet Dev 7: 597–602 [DOI] [PubMed] [Google Scholar]
- Wang J, Huang Q, Tang W, Nadal-Ginard B (1996) E2F1 inhibition of transcription activation by myogenic basic helix–loop–helix regulators. J Cell Biochem 62: 405–410 [DOI] [PubMed] [Google Scholar]
- Watanabe G, Albanese C, Lee RJ, Reutens A, Vairo G, Henglein B, Pestell RG (1998) Inhibition of cyclin D1 kinase activity is associated with E2F-mediated inhibition of cyclin D1 promoter activity through E2F and Sp1. Mol Cell Biol 18: 3212–3222 [DOI] [PMC free article] [PubMed] [Google Scholar]