

Abstract
Acute myeloid leukemia (AML) is a heterogeneous group of genetically defined diseases. Their classification is important with regard to prognosis and treatment. We performed microarray analyses for gene expression profiling on bone marrow samples of 37 patients with newly diagnosed AML. All cases had either of the distinct subtypes AML M2 with t(8;21), AML M3 or M3v with t(15;17), or AML M4eo with inv(16). Diagnosis was established by cytomorphology, cytogenetics, fluorescence in situ hybridization, and reverse transcriptase–PCR in every sample. By using two different strategies for microarray data analyses, this study revealed a unique correlation between AML-specific cytogenetic aberrations and gene expression profiles.
Acute myeloid leukemia (AML) is a heterogeneous group of diseases with respect to biology and clinical course. Since the introduction of the French-American-British (FAB) classification in 1976, diagnosis and classification have been based on cytomorphology and cytochemistry (1). As other techniques like immunophenotyping, cytogenetics, and molecular genetics contributed to the definition of AML subtypes the FAB classification was updated. In 1999 the World Health Organization classification for tumors of hematopoietic and lymphoid tissues was proposed. In an attempt to define biologically homogeneous entities that have clinical relevance, morphologic, immunophenotypic, genetic, and clinical features were incorporated (2, 3).
For optimal treatment approaches both a precise diagnosis and prognostic parameters that determine response to therapy and survival are needed. So far, the karyotype of the AML blasts is the most important independent prognostic factor. A favorable outcome under currently used treatment regimens with cure rates from 50% to 85% was observed in several studies in patients with (i) t(8;21)(q22;q22) occurring mostly in FAB subtype AML M2, (ii) inv(16)(p13q22) associated with AML M4eo, and (iii) t(15;17)(q22;q11–12) associated with AML M3 and AML M3v (4–6). In contrast, chromosome aberrations with an unfavorable clinical course are −5/del(5q), −7/del(7q), inv(3)/t(3;3), and complex aberrant karyotypes with cure rates of less than 10% (7, 8). The remainder of AML patients are assigned to a prognostically intermediate group. This latter group is very heterogeneous because it includes patients with a normal karyotype as well as those with rare chromosome aberrations and yet-unknown prognostic impact.
Besides their prognostic impact genetic aberrations are involved in the pathogenesis of leukemia. Although for unbalanced cytogenetic aberrations the heterogeneous pathogenetic mechanisms have not yet conclusively been determined, several studies provide strong evidence for the central pathogenetic role of leukemia-specific fusion genes that are generated by the above-mentioned balanced abnormalities (9–12). Therefore it can be postulated that AML with balanced abnormalities most probably display a homogeneous gene expression profile and thus are promising candidates for microarray analyses.
In a pivotal study, gene expression profiles were analyzed in bone marrow samples of 27 acute lymphoblastic leukemia (ALL) and 11 AML patients. A set of 50 genes of 6,817 analyzed genes was sufficient to discriminate ALL and AML. By leave-one-out cross-validation it was possible to correctly classify 36 of 38 acute leukemia cases. A class predictor could automatically determine new leukemia cases out of an independent test set as belonging to the myeloid or the lymphoid lineage. Thus, these results demonstrated the possibility of cancer classification based on gene expression profiling (13). In a further approach comparing AML with trisomy 8 and AML with normal karyotype expression profiling revealed fundamental biological differences in AML with isolated trisomy 8 and normal cytogenetics (14). More recently, ALL with translocations involving the MLL gene could be separated from ALL cases without MLL translocations and from cases with AML by gene expression profiling (15).
The aim of our investigation was to answer the question of whether a leukemia-specific genotype is associated with a distinct gene expression profile. Therefore, we analyzed three distinct genetic subtypes of AML: t(8;21)(q22;q22), inv(16)(p13q22), and t(15;17)(q22;q12), which lead to subtype-specific fusion genes AML1-ETO, CBFB-MYH11, and PML-RARA, respectively. They are specifically associated with four distinct morphological subtypes according to the FAB classification: AML M2, AML M4eo, AML M3, and AML M3v (16–18). We performed microarray analyses on a cohort of leukemia samples (n = 37) and applied several methodologies to evaluate genes that allowed an assignment to the corresponding type of cytogenetic aberration for classification. We have shown that AML-specific cytogenetic aberrations can be correlated with corresponding gene expression profiles and vice versa.
Methods
Selection and Characterization of Leukemia Samples.
We selected bone marrow samples from 37 AML patients representing four morphological and three underlying cytogenetic subgroups. All samples were newly diagnosed de novo AML and were characterized by cytomorphology, cytogenetics, fluorescence in situ hybridization, and molecular genetics using standard procedures (1, 3, 18–24). Samples used for gene expression analyses had been lysed immediately, frozen, and stored at −80°C from 1 to 34 months. The targets for GeneChip analyses were prepared according to the current Expression Analysis Technical Manual (Affymetrix, Santa Clara, CA). Detailed procedures are described in additional Methods, which are published as supporting information on the PNAS web site, www.pnas.org.
Class Separation by Principal Component Analysis.
Potential clusters corresponding to the genetic subgroups were visualized with a two-step approach. The data were scaled from each array to a target intensity value of 50 (Affymetrix MICROARRAY SUITE 4.0.1) to be able to perform interarray comparisons. All data were permutated 100 cycles by using the multiclass response parameter of the Significance Analysis of Microarrays algorithm (SAM) (25) (http://www-stat.stanford.edu/∼tibs/SAM/index.html). The total set of 12,600 genes was reduced to the significant differentially expressed genes. In a second step, the reduced set of genes was prepared for principal component analysis and analyzed with j-express (26) (http://www.molmine.com/). For visualization in a two-dimensional plot we chose the first two principal components as they captured most of the variation in the original data set.
Class Prediction by Weighted Voting.
We adapted a previously described method to reduce the number of candidate genes that could distinguish between the three different cytogenetic AML subgroups (13). Briefly, to avoid division by zero or negative numbers as occurs because of the expression algorithm (Affymetrix MICROARRAY SUITE 4.0.1) we set all average fluorescence intensities of 1 or less to 1. Then, gene expression levels were log-transformed. Performing pairwise comparisons (A vs. B), for each gene g P(g,c) values and votes [defined by: P(g,c) = (m1(g) − m2(g))/(s1(g) + s2(g))] were calculated based on mean expression levels (m) and standard deviations (s) in the respective cytogenetic subgroup. Subsequently, votes were summed and prediction strength values reflected the margin of victory in the direction of either cytogenetic group A or B of the pairwise comparison. Prediction strength values range between 0 and 1, values >0.45 demonstrate significance (according to the permutation test). The relevance of selected genes was assessed by performing leave-one-out cross-validation. Only those genes that were contained in all cross-validation classifiers were considered important. To determine a random association between genes we performed a permutation test (100 cycles). Because the number of informative genes, which are required to discriminate between samples, is unknown, we applied this method for different numbers of informative genes (range: 2 to 200). The minimal set of genes that provided optimal classification accuracy together with the highest prediction strength was selected to avoid overfitting. To visualize the identified genes and check their suitability for class separation a hierarchical cluster analysis was performed by using j-express (26) (cluster method: average linkage; distance metric: euclidean). The accuracy of this class prediction model was validated on an independent test set of five cases of AML not fulfilling the cRNA high-quality criterion.
Multiple-Tree Classifier.
As basic units in this classifier, classification trees are used (27–29). The optimal number of trees has been determined to be 15 (data not shown). Class votes of these trees are aggregated by a vote-by-majority rule. The classifier was fed with gene expression intensity values from a set of 973 genes that had been chosen based on their r statistic:
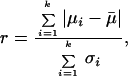 |
where μi refers to the class averages, μ̄ to the overall average, σi, to the within-class standard deviation, and summation is carried out over all k classes. The threshold was set to r > 0.75. Classification trees were constructed as follows: tree building was performed while restricting trees to contain no more than n-1 nodes to discriminate between n classes. The C5.0 algorithm was used (28). The variables (gene expression intensities) used for tree construction were eliminated from the data set, and a new tree was calculated based on the truncated data set. This procedure was iterated until the predetermined number of trees had been reached. The accuracy of the multiple-tree classifier was estimated by 10-fold cross-validation (30) and on an independent test set of data from five bone marrow aspirates, where the quality of the corresponding cRNA preparation was slightly lower than the high-quality standards required for the training set.
Results
Characterization of Leukemia Samples.
We investigated 37 AML cases representing three defined cytogenetic aberrations corresponding to four FAB subtypes: t(8;21)(q22;q22)/AML M2 (n = 9), t(15;17)(q22;q12)/AML M3 or AML M3v (n = 10, n = 8), and inv(16)(p13q22)/AML M4eo (n = 10). All cases were characterized by cytomorphology, cytogenetics, fluorescence in situ hybridization, and reverse transcription–PCR (see Fig. 4, which is published as supporting information on the PNAS web site). All cases with AML and t(8;21) had AML M2, all with AML and inv(16) had AML M4eo, 10 cases with AML and t(15;17) had AML M3, and eight cases with AML and t(15;17) had AML M3v. All patients showed these balanced abnormalities as the sole karyotype change. Using fluorescence in situ hybridization analysis, more than 65% of cells demonstrated the specific signal constellation. The respective fusion transcripts were detected by reverse transcription–PCR in all samples. The median age of all patients was 53 years (range, 19–82 years; male/female = 15:22) and did not differ between the respective groups. AML subtypes M3 and M3v both carry the same chromosomal aberration but differ in morphological aspects like nuclear configuration, granulation, and clinical aspects like white blood cell count. The median white blood cell count was 20,000/μl (range, 800–168,000/μl) and was strikingly lower in patients with AML M3 as compared with all other patients (median, 6,200 vs. 36,500/μl, P = 0.0002).
Microarray Analyses.
The gene expression profiles of 37 AML samples were evaluated. Thirty-two hybridization cocktails demonstrated high-quality cRNA characteristics (Test3 probe arrays: 3′/5′ ratio of glyceraldehyde-3-phosphate dehydrogenase probe sets ≤3.0) and were selected for building class prediction models: t(8;21)/AML M2 (n = 7), t(15;17)/AML M3 or M3v (n = 9, n = 7), and inv(16)/AML M4eo (n = 9). Five cases were primarily excluded (3′/5′ ratios ranging between 3.9 and 5.4) and were used for subsequent validations of the class prediction models: t(8;21)/AML M2 (n = 2), t(15;17)/AML M3 or M3v (n = 1, n = 1), and inv(16)/AML M4eo (n = 1).
Class Separation by Principal Component Analysis.
To visualize clusters corresponding to the three underlying genetic subgroups we applied a two-step approach. Based on a permutation test (100 permutations) we correlated our expression data to the three different cytogenetic parameters (25). We obtained 1,000 significant genes. By principal component analysis we were able to clearly separate the three distinct chromosomal aberrations t(8;21), t(15;17), and inv(16) (Fig. 1) (26). These data suggest that genetically defined AML subtypes can be specified and identified based on their gene expression profiles.
Figure 1.

Three cytogenetically defined AML subtypes with t(15;17), t(8;21), or inv(16) can be separated based on their gene expression profiles of 1,000 preselected genes. The three different subgroups form distinct clusters. For visualization in a two-dimensional plot the first two principal components were chosen as they captured most of the variation in the original data set. The subgroups are colored according to their chromosomal aberrations.
Class Prediction by Weighted Voting.
To identify the genes that enable the accurate discrimination of these subgroups, we applied the data analysis methodology introduced by Golub et al. (13). We selected the minimal set of genes that provided optimal classification accuracy together with the highest prediction strength to avoid overfitting. Thirteen genes were sufficient to separate these AML subtypes with high precision (Table 1). GenBank accession numbers and detailed descriptions of the genes are given in Table 2.
Table 1.
A minimal set of 13 genes (GenBank accession nos. are given) is sufficient for accurate class prediction with optimal classification accuracy and highest prediction strength
| Classes | t(15;17) vs. t(8;21) | t(15;17) vs. inv(16) | inv(16) vs. t(8;21) | inv(16) vs. remainder | t(8;21) vs. remainder | t(15;17) vs. remainder |
|---|---|---|---|---|---|---|
| Accuracy | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Prediction strength | 0.91 | 0.96 | 0.93 | 0.95 | 0.98 | 0.91 |
| P(g,c) | ||||||
| M65066 | −1.52 | |||||
| AL049933 | −2.12 | |||||
| AF010310 | 1.89 | |||||
| N90866 | −2.34 | |||||
| M26326 | 2.85 | −2.56 | ||||
| N99340 | 8.43 | |||||
| M25915 | 1.63 | |||||
| AF013570 | −6.84 | 7.78 | 6.99 | |||
| AI207842 | 3.08 | 3.08 | 3.08 | |||
| X16665 | 6.56 | 6.56 | ||||
| X96719 | −2.36 | |||||
| AF013611 | 2.68 | |||||
| W72424 | −2.05 |
Comparisons (A vs. B) were performed either between two distinct subtypes or between one distinct subtype and all other subtypes (= remainder), respectively. As calculated from pairwise comparisons, positive P(g,c) values indicate a higher expression in the first class listed, negative P(g,c) values a higher expression in the second class listed, respectively.
Table 2.
Thirty-six genes separate accurately three distinct cytogenetic AML subtypes
| GenBank accession no. | Approved UCL/HGNC/HUGO database symbol | Description | Identified according to Golub et al. | Identified by using multiple-tree classifiers |
|---|---|---|---|---|
| M65066 | PRKAR1B | cAMP-dependent protein kinase regulatory subunit RI-beta | X | |
| AL049933 | GNAI1 | Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 | X | |
| AF010310 | PIG6* | Proline oxidase homolog | X | |
| N90866 | CDW52 | CDW52 antigen (CAMPATH-1 antigen) | X | |
| M26326 | KRT18 | Keratin, type i cytoskeletal 18 | X | X |
| N99340 | DKFZP586N1922* | DKFZP586N1922 protein | X | X |
| M25915 | CLU | Clusterin precursor | X | |
| AI207842 | PTGDS | Prostaglandin-H2 D-isomerase precursor | X | |
| X16665 | HOXB2 | Homeobox protein hox-b2 | X | X |
| X96719 | CLECSF2 | C-type (calcium-dependent, carbohydrate-recognition domain) lectin, superfamily member 2 (activation induced) | X | X |
| AF013611 | CTSW | Cathepsin w (lymphopain) precursor | X | X |
| W72424 | S100A9 | Calgranulin b (migration inhibitory factor-related protein 14) | X | |
| AF013570 | MYH11 | Myosin heavy chain, smooth muscle isoform | X | X |
| AF001548 | MYH11 | Myosin heavy chain, smooth muscle isoform | X | |
| X53742 | FBLN1 | Fibulin-1 | X | |
| U37122 | ADD3 | Gamma adducin | X | |
| J03853 | ADRA2C | Alpha-2c-1 adrenergic receptor | X | |
| Y10183 | ALCAM | CD166 antigen precursor (activated leukocyte cell adhesion molecule) | X | |
| AB002313 | PLXNB2 | Plexin B2 | X | |
| X78817 | ARHGAP4 | Rho GTPase activating protein 4 | X | |
| X54486 | SERPING1 | Plasma protease c1 inhibitor precursor | X | |
| L19872 | AHR | Aryl hydrocarbon receptor | X | |
| M15395 | ITGB2 | CD18, integrin beta-2 precursor | X | |
| AF045229 | RGS10 | Regulator of g-protein signaling 10 | X | |
| D43638 | CBFA2T1 | MTG8 protein (ETO protein) | X | |
| M25280 | SELL | I-selectin precursor (lymph node homing receptor) | X | |
| W25986 | DKFZP564K0822* | Hypothetical protein DKFZp564K0822 | X | |
| M36035 | BZRP | Peripheral-type benzodiazepine receptor | X | |
| X64624 | POU4F1 | Brain-specific homeobox/pou domain protein 3a | X | |
| M18728 | CEACAM6 | Carcinoembryonic antigen-related cell adhesion molecule 6 (nonspecific cross-reacting antigen) | X | |
| M77349 | TGFBI | Transforming growth factor-beta induced protein ig-h3 precursor | X | |
| M80899 | AHNAK | Neuroblast differentiation associated protein ahnak | X | |
| M13560 | CD74 | CD74 antigen, (invariant polypeptide of major histocompatibility complex, class II antigen-associated) | X | |
| X62744 | HLA-DMA | Major histocompatibility complex, class II, DM alpha, RING6 | X | |
| M32578 | HLA-DRB1 | HLA class II histocompatibility antigen, dr-1(dw14) beta chain precursor | X | |
| X00457 | HLA-DPA1 | HLA class II histocompatibility antigen, dp alpha chain precursor | X | |
| J00194 | HLA-DRA | HLA class II histocompatibility antigen, dr alpha chain precursor | X |
GenBank accession numbers, approved human gene nomenclature symbol (
= not approved), and description of the function are presented. Six genes are included in the minimal set of both weighted voting according to Golub et al. (13) (total = 13) and multiple-tree classifiers (total = 29).
All 32 clinical samples could be assigned to their corresponding cytogenetic subtype with best accuracy in leave-one-out cross-validation (1.0). Prediction strength values ranged from 0.91 to 0.98 (Table 1). To illustrate these results we applied hierarchical clustering (31). The resulting dendrogram clearly demonstrates the capacity of this subset of genes to separate all AML cases according to their cytogenetic aberration (Fig. 2). This finding demonstrates that class prediction of a chromosomal aberration in AML is feasible solely based on gene expression data.
Figure 2.

Hierarchical cluster analysis of the gene expression pattern of the set of 13 predictor genes as identified according to the adapted class prediction methodology introduced by Golub et al. (13). The three distinct cytogenetic AML subgroups can clearly be separated based on their gene expression profiles. Each row represents a leukemia sample and each column a gene. GenBank accession numbers are shown on the top. Varying expression levels are shown on a scale from black (no gene expression) to bright red (highest expression). The subgroups are colored according to their chromosomal aberrations.
For external validation, we tested whether primarily excluded samples could also be accurately assigned to their specific cytogenetic category. Despite their nonoptimal cRNA quality, all five cases were correctly classified with high prediction strength (0.76, 1.00, 1.00, 1.00, 1.00).
Class Prediction by Multiple-Tree Models.
As a second and independent methodological approach we developed a multiple-tree classifier to separate the three genetically defined subtypes based on the expression level of a minimal set of genes. In short, we computed classification trees to discriminate between the different AML subclasses. To avoid overfitting of a singular tree model, we computed a multiple-tree model by using an iteratively reduced set of genes. For each tree, we used only those genes that have not been used by the previously computed classification tree. The procedure is stopped when a predetermined number of trees has been reached. For this study, the optimal number of trees was calculated to be 15. The votes of the 15 trees were aggregated by a vote-by-majority rule. Equal votes for two of the three classes were counted as misclassification.
The classifier used the expression values of 29 genes (MYH11 was identified twice by two different probe sets; Table 2) to discriminate between three classes, namely samples displaying t(15;17), t(8;21), and inv(16) (Fig. 3). The accuracy on the training set (n = 32) was 100% and on the independent test set (n = 5) 100%. The average accuracy in 10-fold cross-validation was 94%.
Figure 3.

Schematic representation of the 15 decision trees (a–o) used in the multiple-tree classifier. Arrows indicate high (arrow up) or low (arrow down) expression, 0 and + denote absence or presence of a gene, respectively [e.g., in a the low expression of X96719 indicates AML with t(15;17) whereas the high expression of X96719 indicates AML with inv(16) or AML with t(8;21); the latter two entities are distinguished by X53742: lack of expression identifies AML with inv(16) and positive expression predicts AML with t(8;21)]. Accession numbers are given for relevant genes. Nodes are represented as ovals and leaves as rectangles. Classes are referred to as t(15;17), t(8;21), or inv(16).
In summary, we identified 36 genes by using two independent methodologies for class prediction in AML (Table 2). Six genes were described in both calculations, seven were found exclusively in the minimal set according to Golub et al. (13), and another 23 genes by using multiple-tree classifiers.
Correlation of Phenotype and Gene Expression Profile.
We were able to demonstrate striking correlations between genotype and gene expression profiles in three genetically defined subgroups of AML. In addition, we answered the question of whether the cytogenetically identical AML with t(15;17) but appearing with two different phenotypes, AML M3 or AML M3v (see Fig. 4), can also be separated by different gene expression patterns. We used 100-fold permutation of M3 (n = 10) and M3v (n = 8) data followed by principal component analysis and hierarchical cluster analysis based on 82 informative genes (data not shown). Separation into the corresponding two morphologically defined FAB subtypes M3 and M3v was possible in all cases (see Fig. 5, which is published as supporting information on the PNAS web site) and suggests also a close correlation between phenotype and gene expression profile.
Discussion
We have demonstrated an unequivocal association between disease-specific genetic alterations and distinct gene expression profiles in AML. For each of the three analyzed clearly defined subtypes of AML [t(8;21), t(15;17), inv(16)] patterns of gene expression were identified that were homogeneous within all samples of the respective subgroups but clearly differed between these three subgroups. The analyzed samples represent disease subtypes that are specifically defined on the genetic and the phenotypic level by conventional diagnostics including cytomorphology, cytogenetics, and molecular genetics.
By applying two independent approaches for the analysis of microarray data, the present study demonstrates that AML samples from previously defined subtypes (3) can be classified adequately on the basis of gene expression profiles. It is intriguing that there is both sufficient coherence in gene expression within and difference between these subtypes to classify them with high accuracy even though the samples derive from the same myeloid cell lineage.
To correlate gene expression with cytogenetics Virtaneva et al. (14) compared the expression status of 6,606 genes of AML blasts with normal cytogenetics and trisomy 8 as the sole abnormality. While in this study normal CD34+ cells clustered into a distinct group, AML with trisomy 8 and AML with normal karyotype intercalated with each other. Microarray analyses showed an overall increased gene expression of genes located on chromosome 8, suggesting a gene-dosage effect (14). AML with trisomy 8 is heterogeneous on the phenotypic level as it occurs in different FAB subtypes. In contrast, AML with t(15;17), inv(16), and t(8;21) show a very close correlation to distinct morphological subtypes. Furthermore, trisomy 8 is probably not a primary, disease-defining aberration leading to AML as it also occurs in addition to a variety of different cytogenetic and molecular genetic abnormalities (32, 33). In contrast to this study, Armstrong et al. (15) compared samples of the more homogeneous group of ALL with MLL translocations to ALL without MLL translocations and to AML. They demonstrated that ALL with MLL translocations comprises a distinct disease that can be classified robustly by gene expression profiling.
The main focus of the present analyses was the assessment of the differences between three highly characterized subgroups of AML defined by specific primary chromosome aberrations. As anticipated, it was shown that AML with t(8;21) and AML with inv(16), which both involve alterations of the core binding factor complex, are more related to each other as compared with AML with t(15;17) (34). Both phenotypically different subtypes of AML with t(15;17), AML M3 and AML M3v, cluster within one area. In an additional analysis, the latter two subtypes were separated from each other based on their gene expression profiles. These data suggest the existence of further genetic and not-yet-identified alterations leading to the different phenotypes of AML M3 and AML M3v. One possible candidate gene is FLT3, which is mutated more frequently in AML M3v than in AML M3 (67% vs. 19%, P = 0.001) (35).
Several studies confirmed that gene expression profiles can be used for class prediction. This has been shown for acute leukemias, round blue cell tumors, and malignant melanomas (13, 36–38) as well as for different types of solid tumors by using multiclass cancer classification (39). Whereas the selection of different subgroups in these studies was performed by using exclusively phenotypic criteria, other studies were based on genetically defined entities (40, 41). In the present study not only the discrimination of the three genetically defined AML subgroups was accomplished but also all of these cases of AML were separated from normal bone marrow (data not shown).‖
To develop a classifier two independent approaches were applied. Whereas classification by weighted voting according to Golub et al. (13) allows the discrimination between the three classes based on a minimal set of 13 genes, the multiple-tree classifier uses 30 genes. As indicated by cross-validation, generalization properties are excellent for the multiple-tree classifier, i.e., it is likely to perform equally well on new, unseen samples. Furthermore, it can be easily extended to more than the three subclasses described in the present study.
Our classifiers contained genes already known to be primarily involved in the pathogenesis of the respective entities, namely MYH11 (43) and ETO (44). Presumably, the detection of overexpression of MYH11 in inv(16) cases and ETO in t(8;21) cases relates to the detection of the fusion gene transcripts rather than of the wild-type transcripts. The other genes identified belong to various functional categories. Their potential pathogenetic significance in AML has yet to be clarified.
It is expected that the extension of the present analyses to currently less well-defined AML will identify additional subgroups of AML with clinical relevance based on their gene expression profiles. The feasibility of such an approach has been demonstrated for diffuse large B-cell lymphoma (45). Alizadeh et al. have subdivided an entity previously considered homogeneous by various pathological methods into two not only new but also prognostically highly relevant subgroups. In two recent studies, gene expression profiling also in breast cancer revealed subgroups significantly differing in their prognosis (46, 47). With regard to AML, this approach may be most promising in AML with normal karyotype. This subgroup cannot be further defined on the cytogenetic level and is characterized by an intermediate prognosis possibly masking poor and favorable subgroups.
In addition, the current data may have major implications with regard to delineating aberrant gene expression pathways underlying the pathogenesis of AML. As has been shown in mantle cell lymphoma and medulloblastoma (42, 48) the extension of our analyses to all subgroups of AML should enable us to define the deregulated genes important for the initiation and the progression of AML. Finally, these analyses will promote the identification of new targets for specific treatment approaches.
Supplementary Material
Acknowledgments
This study was supported by a grant from the Deutsche José Carreras Leukämie-Stiftung (DJCLS-R00/13).
Abbreviations
- AML
acute myeloid leukemia
- FAB
French-American-British
- ALL
acute lymphoblastic leukemia
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Kohlmann, A., Dugas, M., Schoch, C., Schnittger, S., Mergenthaler, S., Kern, W., Haforlach, T. & Hiddemann, W. (2001) Blood 98, 91a (abstr.).
References
- 1.Bennett J M, Catovsky D, Daniel M T, Flandrin G, Galton D A, Gralnick H R, Sultan C. Br J Haematol. 1976;33:451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 2.Harris N L, Jaffe E S, Diebold J, Flandrin G, Muller Hermelink H K, Vardiman J, Lister T A, Bloomfield C D. J Clin Oncol. 1999;17:3835–3849. doi: 10.1200/JCO.1999.17.12.3835. [DOI] [PubMed] [Google Scholar]
- 3.Jaffe E S, Harris N L, Stein H, Vardiman J W. World Health Organization Classification of Tumors: Pathology and Genetics of Tumors of Hematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2001. [Google Scholar]
- 4.Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, et al. Blood. 1998;92:2322–2333. [PubMed] [Google Scholar]
- 5.Bloomfield C D, Shuma C, Regal L, Philip P P, Hossfeld D K, Hagemeijer A M, Garson O M, Peterson B A, Sakurai M, Alimena G, et al. Cancer. 1997;80:2191–2198. [PubMed] [Google Scholar]
- 6.Büchner T, Hiddemann W, Wörmann B, Löffler H, Gassmann W, Haferlach T, Fonatsch C, Haase D, Schoch C, Hossfeld D, et al. Blood. 1999;93:4116–4124. [PubMed] [Google Scholar]
- 7.Schoch C, Haferlach T, Haase D, Fonatsch C, Löffler H, Schlegelberger B, Staib P, Sauerland M C, Heinecke A, Büchner T, et al. Br J Haematol. 2001;112:118–126. doi: 10.1046/j.1365-2141.2001.02511.x. [DOI] [PubMed] [Google Scholar]
- 8.Schoch C, Kern W, Krawitz P, Dugas M, Schnittger S, Haferlach T, Hiddemann W. Blood. 2001;98:3500. doi: 10.1182/blood.v98.12.3500. [DOI] [PubMed] [Google Scholar]
- 9.Pabst T, Mueller B U, Harakawa N, Schoch C, Haferlach T, Behre G, Hiddemann W, Zhang D E, Tenen D G. Nat Med. 2001;7:444–451. doi: 10.1038/86515. [DOI] [PubMed] [Google Scholar]
- 10.Yuan Y, Zhou L, Miyamoto T, Iwasaki H, Harakawa N, Hetherington C J, Burel S A, Lagasse E, Weissman I L, Akashi K, et al. Proc Natl Acad Sci USA. 2001;98:10398–10403. doi: 10.1073/pnas.171321298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castilla L H, Wijmenga C, Wang Q, Stacy T, Speck N A, Eckhaus M, Marin Padilla M, Collins F S, Wynshaw Boris A, Liu P P. Cell. 1996;87:687–696. doi: 10.1016/s0092-8674(00)81388-4. [DOI] [PubMed] [Google Scholar]
- 12.Brown D, Kogan S, Lagasse E, Weissman I, Alcalay M, Pelicci P G, Atwater S, Bishop J M. Proc Natl Acad Sci USA. 1997;94:2551–2556. doi: 10.1073/pnas.94.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golub T R, Slonim D K, Tamayo P, Huard C, Gaasenbeek M, Mesirov J P, Coller H, Loh M L, Downing J R, Caligiuri M A, et al. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 14.Virtaneva K, Wright F A, Tanner S M, Yuan B, Lemon W J, Caligiuri M A, Bloomfield C D, de la Chapelle A, Krahe R. Proc Natl Acad Sci USA. 2001;98:1124–1129. doi: 10.1073/pnas.98.3.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armstrong S A, Staunton J E, Silverman L B, Pieters R, den Boer M L, Minden M D, Sallan S E, Lander E S, Golub T R, Korsmeyer S J. Nat Genet. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 16.Haferlach T, Bennett J M, Löffler H, Gassmann W, Andersen J W, Tuzuner N, Cassileth P A, Fonatsch C, Schoch C, Schlegelberger B, et al. Leuk Lymphoma. 1996;23:227–234. doi: 10.3109/10428199609054825. [DOI] [PubMed] [Google Scholar]
- 17.Haferlach T, Winkemann M, Löffler H, Schoch R, Gassmann W, Fonatsch C, Schoch C, Poetsch M, Weber Matthiesen K, Schlegelberger B. Blood. 1996;87:2459–2463. [PubMed] [Google Scholar]
- 18.Bennett J M, Catovsky D, Daniel M T, Flandrin G, Galton D A, Gralnick H R, Sultan C. Ann Intern Med. 1985;103:620–625. doi: 10.7326/0003-4819-103-4-620. [DOI] [PubMed] [Google Scholar]
- 19.Dugas M, Schoch C, Schnittger S, Haferlach T, Danhauser-Riedl S, Hiddemann W, Messerer D, Uberla K. Leukemia. 2001;15:1805–1810. doi: 10.1038/sj.leu.2402301. [DOI] [PubMed] [Google Scholar]
- 20.Löffler H, Rastetter J. Atlas of Clinical Hematology. Berlin: Springer; 1999. [Google Scholar]
- 21.Stollmann B, Fonatsch C, Havers W. Br J Haematol. 1985;60:183–196. doi: 10.1111/j.1365-2141.1985.tb07399.x. [DOI] [PubMed] [Google Scholar]
- 22.Fonatsch C, Schaadt M, Kirchner H, Diehl V. Int J Cancer. 1980;26:749–756. doi: 10.1002/ijc.2910260608. [DOI] [PubMed] [Google Scholar]
- 23.Mitelman F, editor. Guidelines for Cancer Cytogenetics: An International System for Human Cytogenetic Nomenclature. Basel: Karger; 1995. [Google Scholar]
- 24.Evans P, Jack A, Short M, Haynes A, Shiach C, Owen R, Johnson R, Morgan G J. Leukemia. 1995;9:1285–1286. [PubMed] [Google Scholar]
- 25.Tusher V G, Tibshirani R, Chu G. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dysvik B, Jonassen I. Bioinformatics. 2001;17:369–370. doi: 10.1093/bioinformatics/17.4.369. [DOI] [PubMed] [Google Scholar]
- 27.Breiman L, Friedman J H, Olshen R A, Stone C J. Classification and Regression Trees. Monterey, CA: Wadsworth & Brooks; 1984. [Google Scholar]
- 28.Quinlan J R. Programs for Machine Learning. San Mateo, CA: Morgan Kaufmann; 1993. [Google Scholar]
- 29.Berrar D, Granzow M, Dubitzky W, Lichter P, Eils R. In: New Insights in Clinical Impact of Molecular Genetic Data by Knowledge-Driven Data Mining, Proceedings of the Second International Conference on Systems Biology. Yi T-M, Hucka M, Morohashi M, Kitano H, editors. Madison, WI: Omnipress; 2001. pp. 275–281. [Google Scholar]
- 30.Efron B, Tibshirani R J. An Introduction to the Bootstrap. New York: Chapman & Hall; 1981. [Google Scholar]
- 31.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoch C, Haase D, Fonatsch C, Haferlach T, Löffler H, Schlegelberger B, Hossfeld D K, Becher R, Sauerland M C, Heinecke A, et al. Br J Haematol. 1997;99:605–611. doi: 10.1046/j.1365-2141.1997.4473257.x. [DOI] [PubMed] [Google Scholar]
- 33.Schnittger S, Kinkelin U, Schoch C, Heinecke A, Haase D, Haferlach T, Büchner T, Wörmann B, Hiddemann W, Griesinger F. Leukemia. 2000;14:796–804. doi: 10.1038/sj.leu.2401773. [DOI] [PubMed] [Google Scholar]
- 34.Friedman A D. Leukemia. 1999;13:1932–1942. doi: 10.1038/sj.leu.2401590. [DOI] [PubMed] [Google Scholar]
- 35.Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, Löffler H, Sauerland M C, Serve H, Büchner T, et al. Blood. 2002;100:159–166. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- 36.Miyazato A, Ueno S, Ohmine K, Ueda M, Yoshida K, Yamashita Y, Kaneko T, Mori M, Kirito K, Toshima M, et al. Blood. 2001;98:422–427. doi: 10.1182/blood.v98.2.422. [DOI] [PubMed] [Google Scholar]
- 37.Khan J, Wei J S, Ringner M, Saal L H, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu C R, Peterson C, et al. Nat Med. 2001;7:673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben Dor A, et al. Nature (London) 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 39.Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang C H, Angelo M, Ladd C, Reich M, Latulippe E, Mesirov J P, et al. Proc Natl Acad Sci USA. 2001;98:15149–15154. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi O P, et al. N Engl J Med. 2001;344:539–548. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- 41.Perou C M, Sorlie T, Eisen M B, van de Rijn M, Jeffrey S S, Rees C A, Pollack J R, Ross D T, Johnsen H, Akslen L A, et al. Nature (London) 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 42.MacDonald T J, Brown K M, LaFleur B, Peterson K, Lawlor C, Chen Y, Packer R J, Cogen P, Stephan D A. Nat Genet. 2001;29:143–152. doi: 10.1038/ng731. . . [DOI] [PubMed] [Google Scholar]
- 43.Miller J D, Stacy T, Liu P P, Speck N A. Blood. 2001;97:2248–2256. doi: 10.1182/blood.v97.8.2248. [DOI] [PubMed] [Google Scholar]
- 44.Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci P G, Lazar M A. Mol Cell Biol. 1998;18:7185–7191. doi: 10.1128/mcb.18.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alizadeh A A, Eisen M B, Davis R E, Ma C, Lossos I S, Rosenwald A, Boldrick J C, Sabet H, Tran T, Yu X, et al. Nature (London) 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 46.Sorlie T, Perou C M, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen M B, van de Rijn M, Jeffrey S S, et al. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.West M, Blanchette C, Dressman H, Huang E, Ishida S, Spang R, Zuzan H, Olson J A, Jr, Marks J R, Nevins J R. Proc Natl Acad Sci USA. 2001;98:11462–11467. doi: 10.1073/pnas.201162998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hofmann W K, de Vos S, Tsukasaki K, Wachsman W, Pinkus G S, Said J W, Koeffler H P. Blood. 2001;98:787–794. doi: 10.1182/blood.v98.3.787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.