

Abstract
Synaptotagmins constitute a large family of membrane proteins implicated in Ca2+-triggered exocytosis. Structurally similar synaptotagmins are differentially localized either to secretory vesicles or to plasma membranes, suggesting distinct functions. Using measurements of the Ca2+ affinities of synaptotagmin C2-domains in a complex with phospholipids, we now show that different synaptotagmins exhibit distinct Ca2+ affinities, with plasma membrane synaptotagmins binding Ca2+ with a 5- to 10-fold higher affinity than vesicular synaptotagmins. To test whether these differences in Ca2+ affinities are functionally important, we examined the effects of synaptotagmin C2-domains on Ca2+-triggered exocytosis in permeabilized PC12 cells. A precise correlation was observed between the apparent Ca2+ affinities of synaptotagmins in the presence of phospholipids and their action in PC12 cell exocytosis. This was extended to PC12 cell exocytosis triggered by Sr2+, which was also selectively affected by high-affinity C2-domains of synaptotagmins. Together, our results suggest that Ca2+ triggering of exocytosis involves tandem Ca2+ sensors provided by distinct plasma membrane and vesicular synaptotagmins. According to this hypothesis, plasma membrane synaptotagmins represent high-affinity Ca2+ sensors involved in slow Ca2+-dependent exocytosis, whereas vesicular synaptotagmins function as low-affinity Ca2+ sensors specialized for fast Ca2+-dependent exocytosis.
Keywords: C2-domain/Ca2+ binding protein/exocytosis/neurotransmitter release/synaptic plasticity
Introduction
When an action potential invades a presynaptic nerve terminal, Ca2+ influx triggers neurotransmitter release (Katz, 1969). Ca2+ activates two components of release: a fast synchronous component that accounts for >90% of total release, and a slow asynchronous component that mediates <10% of total release (Geppert et al., 1994; Goda and Stevens, 1994; Atluri and Regehr, 1998). Both components exhibit a high degree of Ca2+ cooperativity but different apparent Ca2+ affinities (Dodge and Rahamimoff, 1967; Goda and Stevens, 1994). In addition to stimulating release, Ca2+ modulates release during short-term synaptic plasticity (Katz and Miledi, 1968; Dittman et al., 2000). Short-term synaptic plasticity is at least partly induced by accumulation of residual Ca2+ during repetitive stimulation (Delaney and Tank, 1994; Kamiya and Zucker, 1994) and is an important determinant of the properties of neuronal networks (Dobrunz and Stevens, 1999; Gil et al., 1999).
Ca2+ is thought to trigger release and regulate synaptic plasticity by binding to Ca2+ sensors. Although many Ca2+ binding proteins have been identified that could potentially serve as synaptic Ca2+ sensors, the best current candidates at the synapse are synaptotagmins 1 and 2, abundant synaptic vesicle proteins that bind Ca2+. Synaptotagmins 1 and 2 are highly homologous but differentially distributed, with synaptotagmin 1 primarily expressed in forebrain and synaptotagmin 2 in the brain stem and spinal cord (Ullrich et al., 1994; Marqueze et al., 1995). However, at least 11 other synaptotagmins are present in brain besides synaptotagmins 1 and 2. Of these ‘other’ synaptotagmins, synaptotagmins 3 and 7 are the most abundant (reviewed in Südhof, 2002). Different from synaptotagmins 1 and 2, synaptotagmins 3 and 7 are uniformly co-distributed throughout the brain (Ullrich and Südhof, 1995) and are localized to plasma membranes instead of to synaptic vesicles (Butz et al., 1999; Sugita et al., 2001).
All synaptotagmins are composed of an N-terminal transmembrane region, a linker sequence and two C-terminal C2-domains (referred to as the C2A- and C2B-domains). In synaptotagmin 1, the C2A-domain binds three Ca2+ ions, while the C2B-domain binds only two Ca2+ ions (Südhof and Rizo, 1996; Ubach et al., 1998; Fernandez et al., 2001). Both C2-domains bind Ca2+ ions with a low intrinsic affinity (>500 µM Ca2+). The apparent Ca2+ affinity of the synaptotagmin 1 C2-domains is dramatically increased in the presence of phospholipid membranes (∼5–50 µM Ca2+ depending on the lipid composition), presumably because the phospholipid headgroups provide additional coordination sites for the bound Ca2+ ions (Zhang et al., 1998; Fernandez et al., 2001; Fernandez-Chacon et al., 2001). In the ternary C2-domain– Ca2+–phospholipid complex, the C2A-domain probably contains three Ca2+ ions, but the C2B-domain contains only two Ca2+ ions. As a result, the C2B-domain complex is more labile, and Ca2+-dependent phospholipid binding to the synaptotagmin 1 C2B-domain was only detected recently by methods in which binding was studied in solution (Fernandez et al., 2001). The sequences of most C2-domains of other synaptotagmins are very similar to those of synaptotagmin 1, suggesting that most synaptotagmins also form Ca2+-dependent phospholipid complexes via both C2-domains (Li et al., 1995a,b). Ca2+-dependent phospholipid interactions probably constitute an intrinsic component of synaptotagmin function since phospholipid binding is the only confirmed function of C2-domains, as exemplified by enzymes such as phospholipase A2 or protein kinase C in which the C2-domain mediates the Ca2+-dependent recruitment of the enzymes to the membrane (reviewed in Nalefski and Falke, 1996; Newton and Johnson, 1998).
Knockout mice revealed that, in hippocampal synapses, synaptotagmin 1 is essential for fast but not for slow Ca2+-stimulated neurotransmitter release, suggesting that synaptotagmin 1 is an essential Ca2+ sensor for fast exocytosis (Geppert et al., 1994). This hypothesis is supported by mutant mice in which a point mutation, R233Q, was introduced into synaptotagmin 1 by homologous recombination (Fernandez-Chacon et al., 2001). The R233Q mutation caused an ∼2-fold reduction in the overall Ca2+ affinity of synaptotagmin 1, and a similar decrease in the apparent Ca2+ affinity of transmitter release, suggesting that the Ca2+ affinity of synaptotagmin 1 determines the Ca2+ affinity of synaptic exocytosis (Fernandez-Chacon et al., 2001). In contrast to synapses, however, deletion of synaptotagmin 1 in neuroendocrine cells had a surprisingly small effect on exocytosis. In chromaffin cells, the synaptotagmin 1 knockout caused only a minor decrease in fast exocytosis (Voets et al., 2001). Similarly, in PC12 cells lacking synaptotagmins 1 and 2, Ca2+ still induced robust secretion (Shoji-Kasai et al., 1992). The lack of a requirement for synaptotagmin 1 for most large dense-core vesicle exocytosis, but its necessity for synaptic vesicle exocytosis, suggested that other Ca2+ sensors may be more important for large dense-core vesicle exocytosis. Indeed, studies in permeabilized PC12 cells demonstrated that the C2A- and C2B-domains of synaptotagmin 7 potently inhibited exocytosis, whereas the C2A- and C2B-domains of synaptotagmin 1 were without significant effect, indicating that synaptotagmin 7 may constitute a Ca2+ sensor for exocytosis in these cells (Sugita et al., 2001). However, since synaptotagmin 7 is also present in synapses, this raises the question of why synaptotagmin 7 can substitute for synaptotagmin 1 in PC12 cells but not in synapses.
One possible explanation for this conundrum is that in synapses and endocrine cells, vesicular synaptotagmins may be responsible for fast exocytosis triggered at higher Ca2+ concentrations, and plasma membrane synaptotagmins for slower exocytosis stimulated at lower Ca2+ concentrations. The more severe phenotype of the synaptotagmin 1 knockout at synapses, and the less severe phenotype in endocrine cells, would then be due to the fact that most synaptic exocytosis, but only a small part of endocrine exocytosis, are mediated by the fast component. However, it is unclear whether plasma membrane and vesicular synaptotagmins have intrinsic functional differences as predicted by this hypothesis, and also whether these differences apply to other synaptotagmins, especially synaptotagmin 3, which is co-localized with synaptotagmin 7 on synaptic plasma membranes but whose location in neuroendocrine cells is unclear. In the present study, we set out to address these issues. Our results demonstrate an unsuspected functional specialization of synaptotagmins whereby plasma membrane synaptotagmins exhibit a higher Ca2+ affinity than vesicular synaptotagmins, and even vesicular synaptotagmins are heterogeneous with respect to Ca2+ affinity. These findings indicate that at central synapses, a series of Ca2+ sensors with distinct affinities may operate in triggering fast release. In large dense-core vesicle exocytosis, by contrast, fusion is probably largely driven by high-affinity synaptotagmins that operate more slowly but require lower Ca2+ levels.
Results
Relative Ca2+ affinities of the synaptotagmin C2A-domains
We compared, in the same experiment, the apparent Ca2+ affinities of the C2A-domains of synaptotagmins 1, 2, 3, 5, 7 and 10. These synaptotagmins were chosen because synaptotagmins 3 and 7 are the most abundant synaptotagmins after 1 and 2 (Butz et al., 1999; Sugita et al., 2001), and because synaptotagmins 3, 5, 6 and 10 form a class of closely related synaptotagmins (Fukuda et al., 1999). Synaptotagmin 6 was not studied because we were unable to produce soluble properly folded C2-domains from this isoform. We estimated the apparent Ca2+ affinities of the C2A-domains by Ca2+-dependent phospholipid binding, and compared two independent methods and three different buffers to control for potential artifacts. Ca2+-dependent phospholipid binding assays were chosen because synaptotagmins bind Ca2+ at physiological concentrations only in the presence of phospholipids and because Ca2+-dependent phospholipid binding most likely constitutes part of their physiological function (reviewed in Südhof, 2002).
We first measured the apparent Ca2+ affinity of the C2A-domains with a standard resin-based assay in which immobilized glutathione S-transferase (GST) fusion proteins of the synaptotagmin C2A-domains were incubated with radiolabeled liposomes at different Ca2+ concentrations (Davletov and Südhof, 1993). Figure 1 demonstrates that each synaptotagmin is characterized by a distinct Ca2+ affinity, as measured by this assay. The vesicular synaptotagmins 1 and 2 consistently had the lowest Ca2+ affinities [EC50 ≈ 10–20 µM Ca2+ with liposomes composed of 25% phosphatidylserine (PS)/75% phosphatidylcholine (PC); Figure 1A and B]. By contrast, the plasma membrane synaptotagmins 3 and 7 exhibited the highest Ca2+ affinities (EC50 ≈ 1–2 µM Ca2+; Figure 1C and E). Synaptotagmins 5 and 10 (whose localizations are unknown, but which are most closely related to the plasma membrane synaptotagmins 3 and 6) also displayed relatively high Ca2+ affinities (EC50 ≈ 3 µM Ca2+; Figure 1D and F). Moreover, we observed that synaptotagmins 1 and 2 (which are both localized to synaptic vesicles but differentially distributed in brain; Ullrich et al., 1994; Marqueze et al., 1995) also differ in Ca2+ affinity (Figure 1A and B), with synaptotagmin 2 reproducibly exhibiting an ∼2-fold lower Ca2+ affinity than synaptotagmin 1. The specificity of the phospholipid binding reaction was confirmed by a point mutant in the predicted Ca2+ binding site of the synaptotagmin 3 C2A-domain (D333N), which abolished Ca2+-dependent phospholipid binding (Figure 1C).

Fig. 1. Phospholipid binding to the C2A-domains of synaptotagmins 1, 2, 3, 5, 7 and 10 studied by Ca2+-dependent GST pull-downs of radiolabeled liposomes. GST fusion proteins containing the indicated C2A-domains (Syt, synaptotagmin) were immobilized on glutathione–agarose and incubated at increasing concentrations of free Ca2+ with radiolabeled liposomes composed of 25% PS/75% PC. Ca2+ concentrations were clamped by Ca2+/EGTA buffers using the standard NaCl-based buffer (Gerber et al., 2001). Agarose beads were washed three times in the respective incubation buffers and bound liposomes were quantified by liquid scintillation counting. Binding was normalized to 100% for the maximal point. Data shown are means ± SEMs from two experiments performed in triplicate. The binding curve for the synaptotagmin 1 C2A-domain in (A) is repeated in open squares (B–F) as an internal reference point to facilitate comparisons. (C) Gray symbols display binding observed for the synaptotagmin 3 C2A-domain (Syt 3-C2A) containing a point mutation in a Ca2+ binding loop (D333N).
The distinct Ca2+ affinities of synaptotagmins could have important implications for the Ca2+-dependent regulation of neurotransmitter release. This is illustrated by the R233Q point mutation in synaptotagmin 1, which causes an ∼2-fold decrease in the Ca2+ affinity of synaptotagmin 1 and a similar decrease in the Ca2+ responsiveness of synapses (Fernandez-Chacon et al., 2001), suggesting that the Ca2+ affinity of vesicular synaptotagmins controls synaptic responses. In view of the potential importance of differences in Ca2+ affinity, we sought to confirm the measured apparent Ca2+ affinities of synaptotagmins with an independent assay. For this purpose we incubated soluble C2A-domain–GST fusion proteins with liposomes at different Ca2+ concentrations, isolated the liposomes by centrifugation, and determined the amount of bound synaptotagmin C2A-domains by Coomassie Blue staining of SDS gels. This assay, referred to as the liposome centrifugation assay, utilizes C2A-domains in solution and thus avoids possible immobilization artifacts of the standard resin-based assay. Figure 2 shows that the liposome centrifugation assay produced results similar to the resin-based assay, revealing an ∼2-fold higher Ca2+ affinity of synaptotagmin 1 than of synaptotagmin 2, and an ∼5- to 10-fold higher affinity of the plasma membrane synaptotagmins than of vesicular synaptotagmins. A major difference between the two assays was that in the resin-based assay, a bell-shaped Ca2+ concentration dependence was observed for synaptotagmins 3 and 7 (Figure 1), whereas the liposome centrifugation assay exhibited no decrease in phospholipid binding at higher Ca2+ concentrations (Figure 2). This difference may reflect the more stringent washes used in the resin-based assay, which would also explain the inability of the resin-based assay to detect Ca2+ binding to the C2B-domain (see below; Fernandez et al., 2001). The concurrence of the apparent Ca2+ affinities determined with the two assays confirms that the Ca2+ affinity is not dependent on whether the C2A-domains are immobilized or in solution.
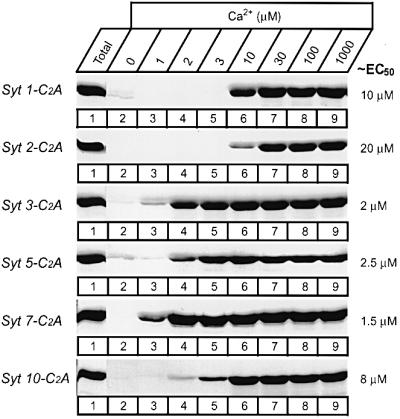
Fig. 2. Phospholipid binding by the C2A-domains of synaptotagmins 1, 2, 3, 5, 7 and 10 studied by co-sedimentation with liposomes. Soluble purified GST fusion proteins containing the indicated C2A-domains were incubated with liposomes composed of 25% PS/75% PC in the presence of free Ca2+ at the concentrations shown, clamped by Ca2+/EGTA buffers. Liposomes were centrifuged and washed, and bound proteins were estimated by SDS–PAGE. Data shown are Coomassie Blue-stained gels from a single representative experiment repeated multiple times. Numbers on the right indicate approximate Ca2+ concentrations required for half-maximal binding as estimated from multiple experiments.
Synaptotagmin C2B-domains exhibit similar differences in Ca2+ affinity
Synaptotagmins have two Ca2+ binding domains: the C2A- and C2B-domains. Initially the C2B-domains were thought to be unable to bind to phospholipids as a function of Ca2+ because the resin-based assay did not detect such binding (reviewed in Südhof and Rizo, 1996). Recently, however, less stringent assays revealed that the C2B-domain of synaptotagmin 1 specifically binds to phospholipids in a Ca2+-dependent manner with an apparent Ca2+ affinity that resembles that of the C2A-domain (Fernandez et al., 2001). The C2B-domain probably forms a less tight Ca2+–phospholipid complex than the C2A-domain because the C2B-domain contains only two Ca2+ binding sites as opposed to the three Ca2+ binding sites of the C2A-domain. To test whether the C2B-domains of synaptotagmins 3 and 7 also bind phospholipids in response to Ca2+ and whether the various C2B-domains exhibit similar differences in Ca2+ affinity as the C2A-domains, we examined the C2B-domains using the liposome centrifugation assay (Figure 2). Figure 3 shows that the synaptotagmin 1 and 7 C2B-domains, but not the synaptotagmin 3 C2B-domain, bind phospholipids as a function of Ca2+. The apparent Ca2+ affinity of the synaptotagmin 7 C2B-domain was ∼10-fold higher than that of the synaptotagmin 1 C2B-domain (Figure 3), demonstrating that the C2A- and C2B-domains of these synaptotagmins exhibit the same difference in Ca2+ affinity. The lack of Ca2+-dependent phospholipid binding by the synaptotagmin 3 C2B-domain, as judged by this assay, is somewhat surprising considering its sequence similarity to other C2B-domains. It may be due either to a lack of Ca2+ binding by this domain (as indicated by the crystal structure; Sutton et al., 1999) or to a selective inability to bind phospholipids as a function of Ca2+.

Fig. 3. Phospholipid binding by the C2B-domains of synaptotagmins 1, 3 and 7 studied by co-sedimentation with liposomes. Purified GST–C2B-domain fusion proteins were incubated in solution with liposomes composed of 25% PS/75% PC in the presence of the indicated concentrations of free Ca2+ and binding was measured as described in Figure 2. Data shown are from a single representative experiment repeated multiple times; numbers on the right indicate approximate Ca2+ concentrations required for half-maximal binding as estimated from multiple experiments.
The relative Ca2+ affinities of synaptotagmin C2A-domains are independent of Ca2+ buffers
The apparent Ca2+ affinities for synaptotagmin C2A-domains determined above and in previous studies (Fernandez-Chacon et al., 2001) were determined with the use of Ca2+/EGTA buffers, which may introduce systematic errors. Furthermore, although Ca2+ binding assays and secretion measurements in permeabilized PC12 cells both employ Ca2+/EGTA buffers, these buffers have distinct compositions (Gerber et al., 2001; Sugita et al., 2001). To better relate Ca2+-dependent phospholipid binding to Ca2+-induced secretion in PC12 cells and to evaluate the accuracy of the Ca2+/EGTA buffers, we directly compared three different Ca2+ buffers: the traditional NaCl-based Ca2+/EGTA buffer (Gerber et al., 2001); the K-glutamate Ca2+/EGTA buffer containing 2 mM Mg2+, 2 mM ATP and 0.1% bovine serum albumin (BSA), which is used for permeabilized PC12 cell experiments (Sugita et al., 2001); and a NaCl-based buffer, which contained only Ca2+ but no EGTA (see Materials and methods). All three buffer systems generally gave similar results, suggesting that the Ca2+/EGTA buffers are reliable (data not shown). The only major difference noted was that the synaptotagmin 1 C2A-domain displayed a lower apparent Ca2+ affinity in the K-glutamate Ca2+/EGTA buffer than in the traditional Ca2+/EGTA buffer or the Ca2+-only buffer, probably because the ATP in the K-glutamate buffer inhibits phospholipid binding to the C2A-domain of synaptotagmin 1 but not to that of the other synaptotagmins (data not shown).
Synaptotagmins in PC12 cells
As a first approach to testing the functional consequences of the different Ca2+ affinities of synaptotagmins, we employed neuroendocrine PC12 cells as a model system. These cells were chosen because they have been productively used in studying Ca2+-triggered exocytosis (Ahnert-Hilger et al., 1987; Walent et al., 1992; McFerran et al., 1998; Avery et al., 2000) and are known to express at least synaptotagmins 1, 3 and 7 (Shoji-Kasai et al., 1992; Mizuta et al., 1994; Sugita et al., 2001). Previous studies have shown that synaptotagmin 1 is present on secretory vesicles in neuroendocrine cells, while synaptotagmin 7 is localized to plasma membranes (Perin et al., 1991; Sugita et al., 2001). The localization of synaptotagmin 3, however, is unclear. Some studies in pancreatic β-cells (the only endocrine cell where it has been studied) reported synaptotagmin 3 on secretory vesicles (Mizuta et al., 1997; Brown et al., 2000; Gao et al., 2000), although a more recent study detected it on the plasma membrane (Gut et al., 2001), similar to its localization in brain (Butz et al., 1999). To address this discrepancy with an independent approach, we examined in PC12 cells the localization of transfected synaptotagmin 3–EYFP fusion protein in comparison with other synaptotagmins. This approach was employed because the low abundance of synaptotagmin 3 made detection of the endogenous protein difficult, and because the direct fluorescence of EYFP avoids possible artifacts due to indirect immunofluorescence procedures. Figure 4 shows that synaptotagmin 3 is quantitatively deposited into the plasma membrane, which is labeled with the fluorescent dye FM4-64. The localization of synaptotagmin 3 is identical to that of synaptotagmin 7–EYFP, whereas synaptotagmin 1–EYFP is exclusively present in intracellular vesicles (Figure 4). These findings suggest that synaptotagmins 3 and 7 are general plasma membrane proteins in neurons and endocrine cells.

Fig. 4. Localization of synaptotagmins 1, 3 and 7 in PC12 cells analyzed with transfected EYFP fusion proteins. PC12 cells were transfected with expression vectors encoding synaptotagmins 1, 3 or 7 as indicated; all synaptotagmins are expressed as C-terminal fusion proteins with EYFP. Unfixed PC12 cells were incubated with the fluorescent dye FM4-64 to stain the plasma membrane and viewed in a confocal microscope. Synaptotagmin 3 is shown in two examples to document reproducibility. Transfected cells are highlighted by a white arrow. The scale bar (2 µm) applies to all panels.
Inhibition of Ca2+-dependent norepinephrine secretion from permeabilized PC12 cells by synaptotagmin C2-domains
We loaded PC12 cells with radioactive norepinephrine, permeabilized the cells by freeze–thawing and triggered exocytosis by addition of Ca2+ at increasing concentrations. In the absence of protein additions or in the presence of only GST, we observed a bell-shaped Ca2+ response curve for exocytosis from the permeabilized PC12 cells (control in Figure 5A). This curve was similar to the Ca2+-concentration dependence of phospholipid binding by synaptotagmins 3 and 7 as measured by the resin assay (Figure 1; Sugita et al., 2001). Addition of the synaptotagmin 1 GST–C2A-domain fusion protein to the PC12 cells caused a small but significant inhibition of exocytosis at high Ca2+ concentrations (Figure 5A). A similar but lower amount of inhibition was also observed for the C2A-domain of synaptotagmin 2 (Figure 5B), consistent with its lower apparent Ca2+ affinity (Figures 1 and 2). The C2A-domain of synaptotagmin 3, however, almost abolished Ca2+-triggered exocytosis at all Ca2+ concentrations (Figure 5C). This inhibition is similar to the effect we previously observed with the C2A-domain of synaptotagmin 7 (Sugita et al., 2001). Inhibition by the C2A-domain of synaptotagmin 3 required Ca2+ binding to the C2A-domain because the Ca2+ binding site mutant of the C2A-domain (D333N), which is unable to mediate Ca2+-dependent phospholipid binding (Figure 1C), had no effect on exocytosis (Figure 5D). Finally, the C2A-domains of synaptotagmins 5 and 10 had an intermediate effect and significantly inhibited Ca2+-triggered exocytosis only at higher Ca2+ concentrations (Figure 5E and F).

Fig. 5. Inhibition of Ca2+-triggered exocytosis in permeabilized PC12 cells by the C2A-domains of synaptotagmins 1, 2, 3, 5 and 10. PC12 cells were loaded with 3H-labeled norepinephrine, permeabilized and pre-incubated with the indicated purified GST fusion proteins of wild-type synaptotagmin C2A-domains (A–C, E and F) or mutant synaptotagmin 3 C2A-domain containing a single amino acid substitution in the Ca2+ binding site (D333N) (D). Exocytosis was triggered by addition of Ca2+ clamped at the concentrations shown with Ca2+/EGTA buffers. Norepinephrine release was normalized to 100% for the maximal release observed under control conditions run in parallel for each experiment with GST (gray symbols). Synaptotagmin C2A-domains and GST were added at 6 and 9 µM, respectively. Data are means ± SEMs from three experiments performed in duplicate and repeated multiple times. Note that the control traces in each graph (amount of secretion in the same assays observed with addition of 9 µM GST alone) differ slightly between graphs because each assay was performed with its own separate controls.
The PC12 cell experiments suggest that the Ca2+ sensor responsible for triggering PC12 cell exocytosis is activated by low micromolar Ca2+ concentrations, which precisely match the apparent Ca2+ affinities of synaptotagmins 3 and 7, and is inhibited by the C2A-domains from these synaptotagmins. The Ca2+ binding measurements described above showed that the C2B-domains of synaptotagmins 1 and 7 exhibited similar differences in Ca2+ affinities, raising the question of whether this also applies to the PC12 cell inhibitions. To test this, we measured the effects of the synaptotagmin 1, 3 and 7 C2B-domains on PC12 cell exocytosis. Figure 6 shows that the synaptotagmin 1 and 3 C2B-domains had little effect on Ca2+-triggered exocytosis, whereas the synaptotagmin 7 C2B-domain effectively inhibited exocytosis at all Ca2+ concentrations. These results again agree well with the Ca2+-dependent phospholipid binding measurements (Figure 3), indicating that there is an overall correlation for synaptotagmins in their apparent Ca2+ affinities as measured by Ca2+-dependent phospholipid binding and their effects in PC12 cells.

Fig. 6. Inhibition of Ca2+-triggered exocytosis in permeabilized PC12 cells by the C2B-domains of synaptotagmins 1 (A), 3 (B) and 7 (C). PC12 cells were loaded with 3H-labeled norepinephrine, permeabilized, pre-incubated with the indicated purified GST fusion proteins of synaptotagmin C2B-domains or GST alone, and stimulated for exocytosis as described in Figure 5. Data are means ± SEMs from two experiments performed in duplicate.
We had previously found that the synaptotagmin 7 C2A-domain inhibits Ca2+-triggered exocytosis at all Ca2+ concentrations except very low ones (Sugita et al., 2001), and a similar trend was observed for the synaptotagmin 3 C2A-domain (Figure 5C). Interestingly, the synaptotagmin 7 C2B-domain exhibited the opposite behavior, in that inhibition of exocytosis was most effective at low Ca2+ concentrations (Figure 6C), suggesting that exocytosis at very low Ca2+ concentrations is primarily mediated by this C2-domain, and is not due to calmodulin. This suggestion is also supported by the finding that Sr2+, a divalent cation that is unable to activate calmodulin (Chao et al., 1984) but does bind to synaptotagmins, strongly activates exocytosis in PC12 cells (see below) and that calmodulin antagonists had no effect on exocytosis under our assay conditions (data not shown).
Sr2+ as a Ca2+ mimetic for synaptotagmins and PC12 cell exocytosis
As an independent approach to test the potential Ca2+ sensor function of synaptotagmins in exocytosis, we examined the ability of Sr2+ to substitute for Ca2+. Differently to previous studies (Li et al., 1995b), the current experiments were performed using Sr2+/EGTA buffers to control the free Sr2+ concentration more precisely, to eliminate potential interference by contaminating Ca2+ and to allow a direct comparison of Ca2+ binding and of Ca2+ triggered exocytosis in PC12 cells. Figure 7 shows that at all Sr2+ concentrations examined, the C2A-domains of vesicular synaptotagmins 1 and 2 did not bind to phospholipids, whereas synaptotagmins 3 and 7 exhibited similar apparent Sr2+ affinities in both buffers (EC50 ≈ 30–50 µM). The resin-based and the liposome centrifugation assays for measuring the apparent Sr2+ affinity gave identical results (Figure 7). The specificity of the reactions was confirmed with the synaptotagmin 3 mutant D333N, which, similarly to Ca2+-dependent phospholipid binding (Figure 1C), was unable to support Sr2+-dependent phospholipid binding (data not shown).
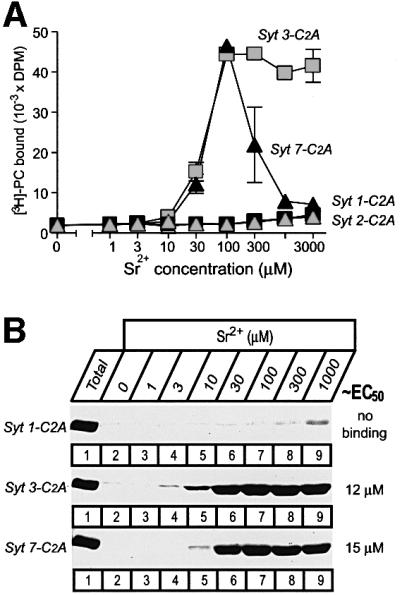
Fig. 7. Sr2+-dependent phospholipid binding by synaptotagmin C2A-domains measured with the resin-based assay (A) or the liposome centrifugation assay (B). (A) Liposome binding to immobilized GST fusion proteins containing the C2A-domains shown was measured as described in Figure 1, except that binding was performed at the indicated concentrations of free Sr2+, which were clamped by Sr2+/EGTA buffers. Data shown are means ± SEMs from two representative experiments performed in triplicate. (B) Liposome binding of GST fusion proteins containing the indicated C2A-domains was measured as described in Figure 2, except that binding was performed at the indicated concentrations of free Sr2+ clamped by Sr2+/EGTA buffers.
We next measured the response of permeabilized PC12 cells to increasing Sr2+ concentrations and examined the effects of recombinant synaptotagmin C2A-domains on this response. As before, GST alone was employed in all experiments as a negative control analyzed in parallel with the test proteins. Under control conditions, Sr2+ was as effective as Ca2+ in triggering exocytosis (Figure 8). A bell-shaped Sr2+ concentration curve was measured similarly to the Ca2+ concentration curve, although much higher Sr2+ concentrations (100–300 µM) were required for maximal effect. Similarly to Ca2+-triggered exocytosis, the synaptotagmin 1 C2A-domain had only a limited effect on Sr2+-triggered exocytosis, with a moderate inhibition at high Sr2+ concentrations (Figure 8A). In contrast, the wild-type synaptotagmin 3 C2A-domain completely abolished Sr2+-triggered exocytosis (Figure 8B), but the mutant synaptotagmin 3 C2A-domain (which is unable to form Sr2+-dependent phospholipid complexes; see Figure 7) had no effect (Figure 8C). The synaptotagmin 7 C2A-domain was even more effective in inhibiting exocytosis than the synaptotagmin 3 C2A-domain, and depressed release below non-stimulated control levels (Figure 8D).

Fig. 8. Inhibition of Sr2+-triggered exocytosis in permeabilized PC12 cells by the C2A-domains of synaptotagmins 1 (A), 3 (B and C) and 7 (D). PC12 cells were loaded with 3H-labeled norepinephrine, permeabilized and pre-incubated with the indicated purified GST fusion proteins of synaptotagmin C2A-domains. In the case of the synaptotagmin 3 C2A-domain, a wild-type (B) and a mutant C2A-domain (C) containing a single amino acid substitution in the Ca2+ binding site (D333N) were tested. Exocytosis was triggered by addition of the indicated concentrations of Sr2+ clamped with Sr2+/EGTA buffers, and normalized to 100% for the maximal release observed under control conditions run in parallel (gray symbols). Data are means ± SEMs from two experiments performed in duplicate.
Discussion
Synaptotagmins constitute a large family of membrane proteins that are preferentially expressed in brain and are composed of an N-terminal transmembrane region, a linker sequence and two C-terminal C2-domains. Synaptotagmin 1, the first synaptotagmin studied, is an abundant synaptic vesicle protein that binds Ca2+ and phospholipids via both of its C2-domains and is essential for fast Ca2+-triggered neurotransmitter release (reviewed in Südhof and Rizo, 1996). A large body of evidence supports a role for synaptotagmin 1 and its close homolog synaptotagmin 2 as a Ca2+ sensor in exocytosis, especially the observation that changes in the Ca2+ affinity of synaptotagmin 1 produce a coordinate change in the Ca2+ affinity of neurotransmitter release (Fernandez-Chacon et al., 2001). Nevertheless, multiple observations indicate that neurotransmitter release is not triggered by synaptotagmins 1 and 2 alone. These include the findings that some Ca2+-triggered release remains in synapses lacking synaptotagmins 1 and 2 (Geppert et al., 1994), chromaffin cells deficient in synaptotagmin 1 exhibit a selective impairment in the minor fast component of Ca2+-triggered exocytosis (Voets et al., 2001) and PC12 cells without synaptotagmins 1 or 2 are capable of relatively normal Ca2+-stimulated exocytosis (Shoji-Kasai et al., 1992). Although other synaptotagmins, such as synaptotagmins 3 and 7, are present at synapses and in neuroendocrine cells in addition to synaptotagmin 1, these synaptotagmins are localized to the plasma membrane instead of to synaptic vesicles (Butz et al., 1999; Sugita et al., 2001; Figure 4). At synapses, the plasma membrane synaptotagmins are clearly not redundant with synaptotagmin 1 because the synaptotagmin 1 knockout has a strong synaptic phenotype, but the other synaptotagmins may mediate Ca2+-triggered exocytosis in endocrine cells that do not require synaptotagmin 1 (Shoji-Kasai et al., 1992; Sugita et al., 2001). Viewed together, these findings raised the possibility that different synaptotagmins are specialized for distinct Ca2+-regulated functions in exocytosis that correlate with their subcellular localizations. We have tested this hypothesis by examining the apparent Ca2+ affinities of the most abundant synaptotagmins and by relating these affinities to Ca2+-triggered exocytosis in neuroendocrine PC12 cells.
Our major finding is that different synaptotagmins exhibit distinct apparent Ca2+ affinities (Figures 1–3). Affinities were measured by Ca2+-dependent phospholipid binding, which most likely represents the physiological Ca2+ binding activity of C2-domains, because phospholipid binding is the best functionally validated general property of C2-domains and because the apparent Ca2+ affinity of the C2-domains of synaptotagmins in the presence of phospholipids (<0.05 mM Ca2+), but not in their absence (>0.4 mM Ca2+), is in the physiological range (Ubach et al., 1998; Zhang et al., 1998; Fernandez et al., 2001; Fernandez-Chacon et al., 2001). We have examined both C2-domains of the major synaptotagmins. To rule out potential artifacts created, for example, by the use of Ca2+/EGTA buffers or of immobilized GST fusion proteins, we have confirmed the results by independent methods in multiple buffer systems. Our measurements showed that synaptotagmin 2 has an ∼2-fold lower Ca2+ affinity than synaptotagmin 1, providing the first evidence for a functional divergence between these two differentially distributed isoforms of vesicular synaptotagmins. More importantly, our data demonstrated that synaptotagmins 3 and 7 have an ∼5- to 20-fold higher Ca2+ affinity than synaptotagmins 1 and 2, suggesting that the plasma membrane synaptotagmins are activated at lower Ca2+ concentrations than the vesicular synaptotagmins. Moreover, synaptotagmins 5 and 10 also exhibited relatively high Ca2+ affinities, whose significance is difficult to evaluate because the localizations of these synaptotagmins are unknown. It should be noted that the Ca2+ affinities described here are not absolute but depend on the composition of the phospholipid membranes and on the ionic strength (Zhang et al., 1998; Fernandez-Chacon et al., 2001). Our numbers therefore provide only general figures for the relative Ca2+ affinities of the various synaptotagmins. The ‘true’ affinities may vary by a factor of 2–4, depending on the precise lipid composition of the sites of exocytosis, which is unknown. Furthermore, double C2-domain fragments probably have higher apparent Ca2+ affinities than individual C2-domains.
Our second major finding is that the apparent Ca2+ affinities of synaptotagmin C2-domains correlate with their ability to inhibit Ca2+-stimulated exocytosis in PC12 cells (Figures 65 and ). Among others, we show that both of the currently known synaptic plasma membrane synaptotagmins, synaptotagmins 3 and 7, are also present on plasma membranes in PC12 cells (Figure 4) and that interference with their functions severely impairs exocytosis, suggesting that these two synaptotagmins together serve as plasma membrane Ca2+ sensors for exocytosis (Figure 5). The mechanism by which the C2-domains of these synaptotagmins block exocytosis is unknown, but the inhibition is clearly specific, as shown by the differential effects observed with various phospholipid-binding C2-domains (Figure 5) and confirmed in control experiments with C2-domains from another synaptic protein, rabphilin (data not shown). The functional analysis of C2-domains was extended to PC12 cells where exocytosis was stimulated with Sr2+ instead of Ca2+: Sr2+-triggered exocytosis was also abolished by the C2A-domains of synaptotagmins 3 and 7, but not by synaptotagmin 1 (Figure 8). Based on these observations, we propose an overall model whereby two classes of synaptotagmins, vesicular synaptotagmins 1 and 2 and plasma membrane synaptotagmins 3 and 7, execute distinct functions in exocytosis (Figure 9). The central tenet of this working model is that the two classes of synaptotagmins cooperate in exocytosis, with the high-affinity plasma membrane synaptotagmins mediating Ca2+-triggered exocytosis in neuroendocrine cells and the slow component of synaptic vesicle exocytosis, whereas the low-affinity vesicular synaptotagmins are largely dispensable for slow neuroendocrine exocytosis but are required for fast synaptic vesicle exocytosis. It has been suggested that calmodulin may be a major Ca2+ sensor for exocytosis (Chen et al., 1999; Quetglas et al., 2000) instead of modulating release as originally proposed (Chamberlain et al., 1995; Kibble and Burgoyne, 1996). Our data demonstrating that Sr2+ is fully capable of triggering release rule out an essential role for calmodulin as a Ca2+ trigger for exocytosis in PC12 cells.

Fig. 9. Diagram of the symmetrical localizations, but asymmetrical Ca2+ affinities, of synaptotagmins on secretory vesicles and plasma membranes. The structures of synaptotagmins are schematically indicated with extracytoplasmic sequences (yellow; note that vesicular synaptotagmins are glycosylated, as indicated by ‘CHO’), transmembrane regions (white), linker sequences (blue; break in the plasma membrane synaptotagmins indicates alternative splicing in synaptotagmin 7), C2-domains (red) and C-terminal sequences (green). The C2-domains of synaptotagmins are thought to bind to phospholipids as a function of Ca2+ and possibly also to SNARE proteins. The apparent affinities of the C2-domains in the presence of negatively charged phospholipids are decribed next to the C2-domains. Ca2+ affinities shown apply to single C2-domains in the presence of negatively charged phospholipids. Because of cooperativity between C2-domains, the double C2-domain fragments have an ~3-fold higher Ca2+ affinity.
Our findings are at odds with some of the observations on synaptotagmins reported in the literature. In pancreatic β-cells, synaptotagmin 3 was found on secretory vesicles instead of the plasma membrane (Mizuta et al., 1997; Brown et al., 2000; Gao et al., 2000). However, these studies used antibodies that recognized either no synaptotagmin 3 protein in brain, or a protein of different size. Since synaptotagmin 3 is primarily expressed in brain and not subject to alternative splicing (Li et al., 1995a; Südhof, 2002), this result raises the possibility that the antibodies did not react with synaptotagmin 3 with high affinity. Indeed, this possibility was confirmed by Gut et al. (2001), who showed with affinity-purified antibodies that synaptotagmin 3 is on the plasma membrane in pancreatic β-cells. Another discrepancy with published data (Desai et al., 2000) is the fact that we observe no significant inhibition of exocytosis by the C2A- or C2B-domain of synaptotagmin 1 at Ca2+ concentrations <10 µM, but do see inhibition at concentrations >0.1 mM Ca2+. However, our results demonstrate that the overall Ca2+ affinity of synaptotagmin 1, both of the C2A- and the C2B-domain, is much lower than the Ca2+ response curve of exocytosis (compare Figure 1A with 6A). These data are in perfect agreement with previous analyses of cells lacking synaptotagmin 1, showing that this synaptotagmin is not required for the vast majority of neuroendocrine exocytosis (Shoji-Kasai et al., 1992; Voets et al., 2001).
The finding that different synaptotagmins exhibit distinct apparent Ca2+ affinities and subcellular localizations may have significant functional implications. The difference in Ca2+ affinities between synaptotagmins 1 and 2 suggests that these highly homologous synaptotagmins, which are differentially distributed but otherwise similar in subcellular localization (Geppert et al., 1991; Ullrich et al., 1994), could contribute to the Ca2+ responsiveness of a synapse. Our data thus predict that synapses expressing synaptotagmin 2 require higher Ca2+ concentrations for release than those expressing synaptotagmin 1. Furthermore, vesicular and plasma membrane synaptotagmins with distinct Ca2+ affinities appear to be co-expressed on opposing membranes in synapses. Since Ca2+ influx during an action potential causes a rapid increase, but slower decline in the synaptic Ca2+ concentration, plasma membrane and vesicular synaptotagmins are presumably activated in parallel. However, as the Ca2+ concentration declines, the plasma membrane synaptotagmins may remain active, whereas vesicular synaptotagmins are shut off. This would suggest that the remaining slow component in synaptotagmin 1 knockout mice is at least partly mediated by plasma membrane synaptotagmins. Moreover, if the relative abundance of various synaptotagmins differed between synapses, this could potentially explain the differences in release probabilities between nerve terminals. For example, layer 2/3 pyramidal cells in the cortex form synapses with a high probability of release with bitufted interneurons, and synapses with a low probability of release with multipolar interneurons (Rozov et al., 2001). Other examples are the parallel fiber and the climbing fiber synapses in cerebellum, which have similar sizes and numbers of docked vesicles, but which exhibit an up to 10-fold difference in release probability (Xu-Friedman et al., 2001). One factor that might contribute to these differences is the mix of synaptotagmins that is expressed at these synapses. Again, future experiments will have to test whether such differences are, at least in part, caused by variations in synaptotagmins.
Materials and methods
Expression and purification of recombinant proteins
All recombinant proteins were expressed as bacterial GST fusion proteins in pGEX-KG (Guan and Dixon, 1991). Most vectors have been described previously (Sugita et al., 2001) except for the synaptotagmin 3 plasmids, which were constructed by PCR using standard methods (Sambrook et al., 1989). GST fusion proteins were purified essentially as described (Guan and Dixon, 1991; Sugita and Südhof, 2000; Sugita et al., 2001) and C2B-domain proteins were additionally treated to remove the bacterial contaminants that stick to these domains (Ubach et al., 2001). For this purpose, extracted proteins were incubated with 1500 U/l benzonase (Novagen Inc.) for 1 h at room temperature before binding to glutathione–agarose. The agarose was packed into a polypropylene column with a paper disc column (Quick-Sep, Isolab) and additionally washed five times with 5 ml of phosphate-buffered saline (PBS), 1 M NaCl, 1% Triton X-100, 1 mM EDTA and 0.1 g/l phenylmethylsulfonyl fluoride (PMSF), followed by six washes with 5 ml of PBS, 1 M NaCl, 1 mM EDTA and 0.1 g/l PMSF.
Phospholipid binding assays
These were performed essentially as described using GST fusion proteins either immobilized on glutathione–agarose in a resin assay (Davletov and Südhof, 1993; Li et al., 1995a,b) or in solution with a centrifugation assay (Fernandez et al., 2001) with three different buffers. Buffer A: NaCl-based Ca2+/EGTA buffer (50 mM HEPES–NaOH pH 6.8, 0.1 M NaCl, 4 mM Na2EGTA, and total Ca2+ or Sr2+ to produce the indicated concentrations of free Ca2+ or Sr2+); buffer B: K-glutamate-based Ca2+/EGTA buffer (120 mM K-glutamate, 20 mM K-acetate, 2 mM EGTA, 20 mM HEPES–NaOH pH 7.2, and total Ca2+ or Sr2+ to produce the indicated concentrations of free Ca2+ or Sr2+); buffer C: NaCl-based Ca2+-only buffer [50 mM HEPES–NaOH pH 6.8; 0.1 M NaCl passed through AG MP-50 resin (Bio-Rad) to eliminate Ca2+ contamination after resin ion conversion to Na+ from H+, with Ca2+ or Sr2+ added directly to the buffer from 1 M stock solution (Fluka Chemical Corp., NY) to achieve the indicated total Ca2+ or Sr2+ concentration]. For composition of Ca2+/EGTA and Sr2+/EGTA buffers, the respective amounts of total Ca2+ or Sr2+ needed to achieve a defined free Ca2+ or Sr2+ concentration were calculated with EqCal software (Biosoft, Ferguson, MI) using buffer constants given by the program or published by Martell and Smith (1974). All buffers were prepared in high-resistance MilliQ water using a 1 M Ca2+ standard solution (Fluka Chemical Corp.).
Resin assay. PS and PC (1.75 mg total; Avanti Polar Lipids, AL) were dissolved in chloroform, mixed in a 1:3 weight ratio including <0.01% 3H-PC (Amersham Pharmacia Biotech, NJ) and dried under a stream of nitrogen. Dried lipids were resuspended in Ca2+-free buffer A or B (10 ml) by vigorous vortexing for 1 min, sonicated for 5 min in a waterbath sonicator (model G112PIG; Laboratory Supply Co. Inc., NJ; output: 80 kc, 80 W) and centrifuged (15 min at ∼5000 g) to remove aggregates. Binding assays contained ∼25 µg of recombinant protein, with 1 g protein/l wet glutathione beads. Beads were equilibrated in 0.1 ml of the respective binding buffers (buffer A or B with 8.75 µg of phospholipids and 0.025 µCi of 3H-PC). Binding reactions were incubated for 10 min at 30°C with vigorous shaking, briefly centrifuged, washed three times with 0.8 ml of the respective binding buffers and phospholipid binding quantified by scintillation counting of the beads (Beckman LS6000SC; Beckman Instruments Inc., CA).
Centrifugation assay. Phospholipids (PS/PC = 25/75, w/w) in chloroform were dried as a thin layer under a stream of nitrogen. Buffers A, B or C containing 0.5 M sucrose were added to the dried phospholipid layer, vortexed for 20 min and sonicated for 5 min in a bath sonicator (model G112SP1G; Laboratory Supply Co. Inc.). After liposome formation, 4 vols of each buffer without sucrose were added and centrifuged to separate heavy liposome from free phospholipids (100 000 g for 30 min). Heavy liposomes were washed once and re-pelleted (13 000 r.p.m. for 10 min). Liposomes were resuspended in each buffer with various concentrations of free Ca2+ and used within 1 h. Purified soluble recombinant GST–synaptotagmin C2-domains (6 µg) and liposomes (100 µg of phospholipids; total volume = 1 ml) were incubated for 10 min on an Eppendorf thermal mixer at 30°C and 1400 r.p.m., liposomes were re-isolated by centrifugation (20 800 g for 10 min) and washed three times with 0.5 ml of the corresponding buffers. Chloroform:methanol (1:2, v/v) solution was added into the pelleted liposomes to denature protein and dissolve phospholipids. After centrifugation (20 800 g for 15 min), the protein precipitate was resuspended in 30 µl of 2× SDS sample buffer and analyzed by SDS–PAGE and Coomassie Blue staining.
Permeabilized PC12 cell secretion assay
Secretion assays were conducted using freeze–thaw permeabilization of PC12 cells (Klenchin et al., 1998; Sugita et al., 2001). PC12 cells were maintained in 75 cm2 flasks (uncoated) in RPMI1640 with 5% fetal bovine serum (heat inactivated), 10% horse serum (heat inactivated), penicillin (50 U/ml) and streptomycin (50 U/ml) at 37°C in 5% CO2. For each assay, PC12 cells were plated in 10 cm2 dishes (Costar) at 40–50% confluency. Two to three days after plating (70% confluency), PC12 cells fresh medium (total 10 ml) containing 4 µl of [3H]norepinephrine (NEN; 1 µCi/µl in stock) and 0.5 mM ascorbic acid was added. After 24 h, cells were washed with physiological saline solution (145 mM NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, 15 mM HEPES–NaOH pH 7.4), detached from the wells by pipetting a stream of Ca2+-free ice-cold buffer B, washed twice with 6 ml of ice-cold buffer B and resuspended in 6 ml of buffer B in a 15 ml cone tube. For permeabilization, cells were frozen overnight at –80°C, thawed by warming the tube at room temperature, 10 mM EGTA was added and the thawed cells were left on ice for 1–5 h to allow efficient extraction of soluble proteins. Resulting cell ghosts were washed three times with 3 ml of ice-cold Ca2+-free buffer B containing 1 g/l BSA, with centrifugations at 1000 g for 5 min. About 18–36 secretion reactions were conducted with each preparation with standard reaction mixes containing in 1.5 ml microcentrifuge tubes (total volume 0.1 ml): washed cell ghosts, 2 mM ATP, 2 mM MgCl2, 10 µl rat brain cytosol (10 mg/ml) in buffer B with various concentrations of CaCl2 and recombinant proteins. Reactions were incubated for 30 min at 30°C, terminated by chilling to 0°C and samples were centrifuged at 4°C for 3 min at 20 800 g. Supernatants and the pellets solubilized in 1% Triton X-100 were analyzed by liquid scintillation counting.
PC12 cell transfections
Vectors encoding EYFP fusion proteins were constructed in a CMV-based expression plasmid using standard procedures (Sugita et al., 2001). For transfection, PC12 cells were plated on polylysine-coated coverslips in RPMI medium supplemented with 10% fetal bovine serum and 5% horse serum. After 1 day in culture, cells were transfected with appropriate vectors using Tfx-50 (Promega). Forty-eight to 72 h after transfection, culture medium was replaced with 0.2 ml of minimal essential medium containing 10 µM FM4-64 (Molecular Probes Inc.) and cells were incubated at 37°C for 5–10 min. FM4-64-labeled coverslips were mounted on slides and imaged using a Bio-Rad MRC-1024 confocal microscope with a 100× (NA1.3) objective.
Miscellaneous procedures
SDS–PAGE and immunoblotting were performed using standard procedures (Laemmli, 1970; Johnston et al., 1989). Immunoblots were developed by enhanced chemiluminescence (Amersham).
Acknowledgments
Acknowledgements
We thank Ms I.Leznicki, A.Roth and E.Borowicz for technical assistance. The initial phase of this study was supported by a fellowship from the Muscular Dystrophy Association to S.S.
References
- Ahnert-Hilger G., Brautigam,M. and Gratzl,M. (1987) Ca2+-stimulated catecholamine release from α-toxin-permeabilized PC12 cells: biochemical evidence for exocytosis and its modulation by protein kinase C and G proteins. Biochemistry, 26, 7842–7848. [DOI] [PubMed] [Google Scholar]
- Atluri P.P. and Regehr,W.G. (1998) Delayed release of neurotransmitter from cerebellar granule cells. J. Neurosci., 18, 8214–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery J., Ellis,D.J., Lang,T., Holroyd,P., Riedel,D., Henderson,R.M., Edwardson,J.M. and Jahn,R. (2000) A cell-free system for regulated exocytosis in PC12 cells. J. Cell Biol., 148, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown H. et al. (2000) Synaptotagmin III isoform is compartmentalized in pancreatic β-cells and has a functional role in exocytosis. Diabetes, 49, 383–391. [DOI] [PubMed] [Google Scholar]
- Butz S., Fernandez-Chacon,R., Schmitz,F., Jahn,R. and Südhof,T.C. (1999) The subcellular localizations of atypical synaptotagmins: synaptotagmin III is enriched in synapses and synaptic plasma membranes but not in synaptic vesicles. J. Biol. Chem., 274, 18290–18296. [DOI] [PubMed] [Google Scholar]
- Chamberlain L.H., Roth,D., Morgan,A. and Burgoyne,R.D. (1995) Distinct effects of α-SNAP, 14-3-3 proteins, and calmodulin on priming and triggering of regulated exocytosis. J. Cell Biol., 130, 1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao S.H., Suzuki,Y., Zysk,J.R. and Cheung,W.Y. (1984) Activation of calmodulin by various metal cations as a function of ionic radius. Mol. Pharmacol., 26, 75–82. [PubMed] [Google Scholar]
- Chen Y.A., Duvvuri,V., Schulman,H. and Scheller,R.H. (1999) Calmodulin and protein kinase C increase Ca2+-stimulated secretion by modulating membrane-attached exocytic machinery. J. Biol. Chem., 274, 26469–26476. [DOI] [PubMed] [Google Scholar]
- Davletov B. and Südhof,T.C. (1993) A single C2-domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid-binding. J. Biol. Chem., 268, 26386–26390. [PubMed] [Google Scholar]
- Delaney K.R. and Tank,D.W. (1994) A quantitative measurement of the dependence of short-term synaptic enhancement on presynaptic residual calcium. J. Neurosci., 14, 5885–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai R.C., Vyas,B., Earles,C.A., Littleton,J.T., Kowalchyck,J.A., Martin,T.F. and Chapman,E.R. (2000) The C2B domain of synaptotagmin is a Ca2+-sensing module essential for exocytosis. J. Cell Biol., 150, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman J.S., Kreitzer,A.C. and Regehr,W.G. (2000) Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J. Neurosci., 20, 1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz L.E. and Stevens,C.F. (1999) Response of hippocampal synapses to natural stimulus patterns. Neuron, 22, 157–166. [DOI] [PubMed] [Google Scholar]
- Dodge F.A. Jr and Rahamimoff,R. (1967) Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J. Physiol., 193, 419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez I., Arac,D., Ubach,J., Gerber,S.H., Shin,O., Gao,Y., Anderson,R.G.W., Südhof,T.C. and Rizo,J. (2001) Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid-binding module. Neuron, 32, 1057–1069. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón R. et al. (2001) Synaptotagmin I functions as a Ca2+-regulator of release probability. Nature, 410, 41–49. [DOI] [PubMed] [Google Scholar]
- Fukuda M., Kanno,E. and Mikoshiba,K. (1999) Conserved N-terminal cysteine motif is essential for homo- and heterodimer formation of synaptotagmins III, V, VI, and X. J. Biol. Chem., 274, 31421–31427. [DOI] [PubMed] [Google Scholar]
- Gao Z., Reavey-Cantwell,J., Young,R.A., Jegier,P. and Wolf,B.A. (2000) Synaptotagmin III/VII isoforms mediate Ca2+-induced insulin secretion in pancreatic islet β-cells. J. Biol. Chem., 275, 36079–36085. [DOI] [PubMed] [Google Scholar]
- Geppert M., Archer,B.T.,III and Südhof,T.C. (1991) Synaptotagmin II: a novel differentially distributed form of synaptotagmin. J. Biol. Chem., 266, 13548–13552. [PubMed] [Google Scholar]
- Geppert M., Goda,Y., Hammer,R.E., Li,C., Rosahl,T.W., Stevens,C.F. and Südhof,T.C. (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell, 79, 717–727. [DOI] [PubMed] [Google Scholar]
- Gerber S.H., Rizo,J. and Südhof,T.C. (2001) The top loops of the C2 domains from synaptotagmin and phospholipase A2 control function specificity. J. Biol. Chem., 276, 32288–32292. [DOI] [PubMed] [Google Scholar]
- Gil Z., Connors,B.W. and Amitai,Y. (1999) Efficacy of thalamocortical and intracortical synaptic connections: quanta, innervation, and reliability. Neuron, 23, 385–397. [DOI] [PubMed] [Google Scholar]
- Goda Y. and Stevens,C.F. (1994) Two components of transmitter release at a central synapse. Proc. Natl Acad. Sci. USA, 91, 12942–12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan K.L. and Dixon,J.E. (1991) Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem., 192, 262–267. [DOI] [PubMed] [Google Scholar]
- Gut A., Kiraly,C.E., Fukuda,M., Mikoshiba,K., Wollheim,C.B. and Lang,J. (2001) Expression and localisation of synaptotagmin isoforms in endocrine β-cells: their function in insulin exocytosis. J. Cell Sci., 114, 1709–1716. [DOI] [PubMed] [Google Scholar]
- Johnston P.A., Jahn,R. and Südhof,T.C. (1989) Transmembrane topography and evolutionary conservation of synaptophysin. J. Biol. Chem., 264, 1268–1273. [PubMed] [Google Scholar]
- Kamiya H. and Zucker,R.S. (1994) Residual Ca2+ and short-term synaptic plasticity. Nature, 371, 603–606. [DOI] [PubMed] [Google Scholar]
- Katz B. (1969) The Release of Neural Transmitter Substances. Liverpool University Press, Liverpool, UK.
- Katz B. and Miledi,R. (1968) The role of calcium in neuromuscular facilitation. J. Physiol., 195, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibble A.V. and Burgoyne,R.D. (1996) Calmodulin increases the initial rate of exocytosis in adrenal chromaffin cells. Pflügers Arch., 431, 464–466. [DOI] [PubMed] [Google Scholar]
- Klenchin V.A., Kowalshyk,J.A. and Martin,T.F.J. (1998) Large dense-core vesicle exocytosis in PC12 cells. Methods, 18, 204–208. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Li C., Ullrich,B., Zhang,J.Z., Anderson,R.G.W., Brose,N. and Südhof,T.C. (1995a) Ca2+-dependent and Ca2+-independent activities of neural and nonneural synaptotagmins. Nature, 375, 594–599. [DOI] [PubMed] [Google Scholar]
- Li C., Davletov,B.A. and Südhof,T.C. (1995b) Distinct Ca2+ and Sr2+-binding properties of synaptotagmins: definition of candidate Ca2+ sensors for the fast and slow components of neurotransmitter release. J. Biol. Chem., 270, 24898–24902. [DOI] [PubMed] [Google Scholar]
- Marqueze B., Boudier,J.A., Mizuta,M., Inagaki,N., Seino,S. and Seagar,M. (1995) Cellular localization of synaptotagmin I, II, and III mRNAs in the central nervous system and pituitary and adrenal glands of the rat. J. Neurosci., 15, 4906–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell A.E. and Smith,R.M. (1974) In Critical Stability Constants. Plenum Press, New York, NY.
- McFerran B.W., Graham,M.E. and Burgoyne,R.D. (1998) Neuronal Ca2+ sensor 1, the mammalian homologue of frequenin, is expressed in chromaffin and PC12 cells and regulates neurosecretion from dense-core granules. J. Biol. Chem., 273, 22768–22772. [DOI] [PubMed] [Google Scholar]
- Mizuta M., Inagaki,N., Nemoto,Y., Matsukura,S., Takahashi,M. and Seino,S. (1994) Synaptotagmin III is a novel isoform of rat synaptotagmin expressed in endocrine and neuronal cells. J. Biol. Chem., 269, 11675–11678. [PubMed] [Google Scholar]
- Mizuta M., Kurose,T., Miki,T., Shoji-Kasai,Y., Takahashi,M., Seino,S. and Matsukura,S. (1997) Localization and functional role of synaptotagmin III in insulin secretory vesicles in pancreatic β-cells. Diabetes, 46, 2002–2006. [DOI] [PubMed] [Google Scholar]
- Nalefski E.A. and Falke,J.J. (1996) The C2-domain calcium-binding motif: structural and functional diversity. Protein Sci., 5, 2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton A.C. and Johnson,J.E. (1998) Protein kinase C: a paradigm for regulation of protein function by two membrane-targeting modules. Biochim. Biophys. Acta, 1376, 155–172. [DOI] [PubMed] [Google Scholar]
- Perin M.S., Brose,N., Jahn,R. and Südhof,T.C. (1991) Domain structure of synaptotagmin (p65). J. Biol. Chem., 266, 623–629. [PubMed] [Google Scholar]
- Quetglas S., Leveque,C., Miquelis,R., Sato,K. and Seagar,M. (2000) Ca2+-dependent regulation of synaptic SNARE complex assembly via a calmodulin- and phospholipid-binding domain of synaptobrevin. Proc. Natl Acad. Sci. USA, 97, 9695–9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A., Burnashev,N., Sakmann,B. and Neher,E. (2001) Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J. Physiol., 531, 807–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Shoji-Kasai Y. et al. (1992) Neurotransmitter release from synaptotagmin-deficient clonal variants of PC12 cells. Science, 256, 1821–1823. [DOI] [PubMed] [Google Scholar]
- Südhof T.C. (2002) Synaptotagmins: why so many? J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- Südhof T.C. and Rizo,J. (1996) Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron, 17, 379–388. [DOI] [PubMed] [Google Scholar]
- Sugita S. and Südhof,T.C. (2000) Specificity of Ca2+-dependent protein interactions mediated by the C2A-domains of synaptotagmins. Biochemistry, 39, 2940–2949. [DOI] [PubMed] [Google Scholar]
- Sugita S., Han,W., Butz,S., Liu,X., Fernandez-Chacon,R., Lao,Y. and Südhof,T.C. (2001) Synaptotagmin VII as a plasma membrane Ca2+-sensor in exocytosis. Neuron, 30, 459–473. [DOI] [PubMed] [Google Scholar]
- Sutton R.B., Ernst,J.A. and Brünger,A.T. (1999) Crystal structure of the cytosolic C2A-C2B-domains of synaptotagmin III: Implications for Ca2+-independent SNARE complex interaction. J. Cell Biol., 147, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubach J., Zhang,X., Shao,X., Südhof,T.C. and Rizo,J. (1998) Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J., 17, 3921–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubach J., Lao,Y., Fernandez,I., Arac,D., Südhof,T.C. and Rizo,J. (2001) The C2B-domain of synaptotagmin 1 is a Ca2+-binding module. Biochemistry, 40, 5854–5860. [DOI] [PubMed] [Google Scholar]
- Ullrich B. and Südhof,T.C. (1995) Differential distributions of novel synaptotagmins: comparison to synapsins. Neuropharmacology, 34, 1371–1377. [DOI] [PubMed] [Google Scholar]
- Ullrich B., Li,C., Zhang,J.Z., McMahon,H., Anderson,R.G.W., Geppert,M. and Südhof,T.C. (1994) Functional properties of multiple synaptotagmins in brain. Neuron, 13, 1281–1291. [DOI] [PubMed] [Google Scholar]
- Voets T., Moser,T., Lund,P.E., Chow,R.H., Geppert,M., Südhof,T.C. and Neher,E. (2001) Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin 1. Proc. Natl Acad. Sci. USA, 98, 11680–11685.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walent J.H., Porter,B.W. and Martin,T.F. (1992) A novel 145 kd brain cytosolic protein reconstitutes Ca2+-regulated secretion in permeable neuroendocrine cells. Cell, 70, 765–775. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman M.A., Harris,K.M. and Regehr,W.G. (2001) Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J. Neurosci., 21, 6666–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Rizo,J. and Südhof,T.C. (1998) Mechanism of phospholipid binding by the C2A-domain of synaptotagmin I. Biochemistry, 37, 12395–12403. [DOI] [PubMed] [Google Scholar]