

Abstract
Translation initiation of the picornavirus genome is regulated by an internal ribosome entry site (IRES). The IRES of a neurovirulent picornavirus, the GDVII strain of Theiler’s murine encephalomyelitis virus, requires polypyrimidine tract-binding protein (PTB) for its function. Although neural cells are deficient in PTB, they express a neural-specific homologue of PTB (nPTB). We now show that nPTB and PTB bind similarly to multiple sites in the GDVII IRES, rendering it competent for efficient translation initiation. Mutation of a PTB or nPTB site results in a more prominent decrease in nPTB than PTB binding, a decrease in activity of nPTB compared with PTB in promoting translation initiation, and attenuation of the neurovirulence of the virus without a marked effect on virus growth in non-neural cells. The addition of a second-site mutation in the mutant IRES generates a new PTB (nPTB) binding site, and restores nPTB binding, translation initiation and neurovirulence. We conclude that the tissue-specific expression and differential RNA-binding properties of PTB and nPTB are important determinants of cell-specific translational control and viral neurovirulence.
Keywords: IRES/PTB forms/RNA binding/translation initiation/virus-induced disease
Introduction
Factors related to both the host and the infectious agent determine the pathogenesis of virus-induced disease. In the case of picornaviruses, the efficiency of viral translation has been implicated as a major determinant of neurovirulence and disease pathogenesis (Svitkin et al., 1985). Picornaviruses have a complex structure in the 5′-untranslated region (5′ UTR) of the viral RNA called the internal ribosome entry site (IRES). The IRES regulates the initiation of translation of the viral genome by coordinating binding of canonical initiation factors and IRES-specific trans-acting factors (reviewed in Jackson and Kaminski, 1995; Pestova et al., 2001). Host-cell proteins that bind specifically to certain picornavirus IRESs and regulate their function include polypyrimidine tract-binding protein (PTB; Hellen et al., 1993; Hunt et al., 1999; Pilipenko et al., 2000), La autoantigen (Meerovitch et al., 1993; Svitkin et al., 1994), poly(rC)-binding protein 2 (PCBP-2; Blyn et al., 1996, 1997), upstream of N-ras protein (unr; Hunt et al., 1999) and IRES trans-acting factor 45 (ITAF45; Pilipenko et al., 2000). Mutations in the IRES as well as the substitution of one virus IRES for another can impair viral RNA translation (Svitkin et al., 1988; La Monica and Racaniello, 1989; Haller et al., 1996) and virus growth (Evans et al., 1985; Pilipenko et al., 1995, 1999, 2000; Gromeier et al., 1996) in particular cell types, suggesting the involvement of cell-specific IRES-binding proteins in virus tropism and pathogenicity.
To clarify the involvement of cell-specific IRES-binding proteins in translational control and virus pathogenicity, we used a neurovirulent GDVII strain of Theiler’s murine encephalomyelitis virus (TMEV), a member of the cardiovirus genus of Picornaviridae. We demonstrated previously that PTB is necessary for efficient translation initiation of GDVII RNA (Pilipenko et al., 2000). PTB has four non-conventional RNA-recognition motifs (RRMs; Ghetti et al., 1992) and exists as a homodimer in solution (Perez et al., 1997b). PTB is assumed to function as an RNA chaperone, fostering and maintaining a functionally competent folding of the IRES in order to carry out efficient translation initiation. However, central nervous system (CNS) cells including motor neurons (a major host cell for GDVII) are deficient in PTB and contain a neural or brain-enriched homologue of PTB, nPTB (Kikuchi et al., 2000; Markovtsov et al., 2000; Polydorides et al., 2000; Lillevali et al., 2001). nPTB has >70% amino acid identity with PTB and is physically associated with two other neuron-specific proteins, Nova-1 and -2 (Polydorides et al., 2000), which play a role in paraneoplastic CNS disease (reviewed in Darnell, 1996). These findings led us to hypothesize that nPTB in motor neurons (possibly complexed with Nova-1) binds to the GDVII IRES and thereby regulates translation of the viral RNA in a cell-specific manner and controls GDVII-induced CNS disease.
We now demonstrate that PTB and nPTB bind to the same regions of the GDVII IRES, and stimulate viral RNA translation initiation equally, as judged by the assembly of the 48S complex (i.e. the ternary complex of a 40S ribosome, initiator tRNA and template mRNA). Mutation of a PTB (nPTB)-binding site in the IRES, which leads to attenuation of the virus, preferentially impairs nPTB compared with PTB binding and decreases activity of nPTB more significantly than PTB in promoting translation initiation, providing an explanation for the decreased neurovirulence of the mutant virus. A second-site mutation that generates a new PTB (nPTB)-binding site leads to efficient translation initiation and restoration of neurovirulence to wild-type levels. These studies show that binding of cell-specific proteins that regulate translation is critical to virus pathogenicity. The data also show that PTB and nPTB act as RNA chaperones by changing the structure of the IRES into one that is optimal for translation initiation.
Results
PTB and nPTB stimulate 48S complex formation on the GDVII IRES equally
mRNA template activity largely depends on the efficiency of 48S complex formation since the assembly of the 48S pre-initiation complex (i.e. the 40S ribosomal subunit and initiator tRNA at the start codon of the mRNA) precedes the joining of the 60S subunit and formation of a translationally active 80S ribosome. The major, if not the only, function of the IRES appears to be to promote the generation of the competent 48S complex. The IRES activity may vary significantly in different cell types due to the differential expression of specific host cell proteins that are essential for 48S complex formation. A comparison of the amount of 48S complex formed in the presence of different host cell-specific proteins allows one to estimate the efficiency of mRNA translation in different cell types. We have used an in vitro translation initiation system and toeprint analysis (Pestova et al., 1996) to investigate the efficiency of 48S complex formation and to assess the fidelity of the complexes formed (i.e. the localization of the complexes on the initiator AUG). The adequacy of this assay as a measure of the efficiency of translation initiation on picornavirus RNA is supported by experiments showing conversion of the 48S complex formed in this system into the 80S initiation complex (T.V.Pestova, personal communication). In this in vitro system, translation initiation is reconstituted to the stage of 48S complex formation with pure components. The in vitro system allows one to investigate directly the factors required for the assembly of 48S complex on an mRNA of interest, to simulate the cell type-specific translation machinery by supplementing the system with host cell-specific proteins and to study the effect of a mutation of a particular cis-element on the efficiency of 48S complex formation in the presence of different host cell proteins.
We took advantage of this in vitro translation system in order to clarify the role of PTB, nPTB and Nova-1 in GDVII IRES function. We found that nPTB and PTB stimulate 48S complex formation on GDVII RNA equally (Figure 1). The efficiency of 48S complex formation was proportional to the amount of PTB and nPTB, and reached a plateau when the PTB- or nPTB-dimers were added in equal molar amounts with the GDVII RNA. Thus, PTB and nPTB act stoichiometrically rather than catalytically in stimulating GDVII IRES-mediated translation initiation, a finding consistent with their presumed function as RNA chaperones, in which their binding fosters and maintains the IRES structure optimal for 48S complex formation. Relevant to this function is the finding that addition of either PTB or nPTB resulted in significant changes in the intensity of some reverse transcriptase (RT) stops on the GDVII RNA (Figure 1), suggesting a global rearrangement of the IRES conformation following their binding. Nova-1 did not affect 48S complex formation when added by itself or along with nPTB (not shown).
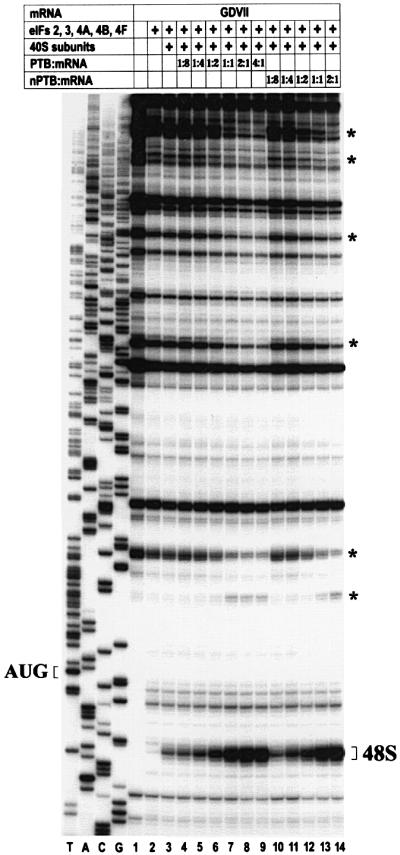
Fig. 1. Toeprint analysis of 48S complexes assembled on wt GDVII RNA in the presence of increasing amounts of PTB and nPTB. Reactions were assembled with translation components as indicated in the table above the gel; molar ratios of PTB- and nPTB-dimers to mRNA are listed. Reference lanes T, A, C and G depict the negative-strand sequence and the position of the initiator AUG is indicated to the left. Positions of cDNA products terminated due to 48S complex formation are labeled as 48S to the right. Other RT stops that changed in intensity in the presence of PTB and nPTB are labeled by an asterisk on the right.
PTB and nPTB have the same binding sites in the GDVII IRES
We modified a model of the structure of the GDVII IRES (Pilipenko et al., 1989; Duke et al., 1992) on the basis of comparative sequence analysis and biochemical probing (Figure 2). The refined model contains several novel features: a small hairpin (H′), a pseudoknot (pk), and a long stem arising from the stacking of helices F, G and the pseudoknot. Despite a significant variation in the nucleotide sequence, these features are conserved among all cardioviruses and aphthoviruses (except for H′, which is present in all cardioviruses but not aphthoviruses).

Fig. 2. A model of the structure of the GDVII IRES showing the location of PTB- and nPTB-binding sites. Stem–loops are named according to Duke et al. (1992). A pseudoknot is designated as pk. Thin lines with arrows connect different structural domains. Some regions of the IRES that are not detailed are shown with a bold line connecting the number of the downstream and upstream nucleotides of the region. The initiator AUG is boxed. Nucleotides susceptible to DMS and CMCT are marked by circles and squares, respectively, while cleavages produced by RNase ONE and RNase V1 are marked by arrowheads and arrows, respectively. Nucleotides and bonds with no change in susceptibility to chemicals and RNases following PTB or nPTB incubation are marked by black symbols. Nucleotides and bonds protected by PTB or nPTB are marked by red symbols. The names of the PTB- and nPTB-binding sites are shown near the protected RNA segments (see text). Nucleotides and bonds in which susceptibility to RNases and chemicals was enhanced following PTB or nPTB binding are marked by green symbols.
To investigate and localize binding sites of PTB and nPTB in the GDVII IRES, we tested the protection of RNA in the presence of an equal molar amount of PTB- or nPTB-dimers from cleavage or modification with RNase ONE, RNase V1, dimethyl sulfate (DMS) and N-cyclo hexyl-N′-(2-morpholinoethyl)-carbodiimide methyl-p-toluene sulfonate (CMCT). Footprints demonstrated six single-stranded (ss) RNA segments well protected by PTB and nPTB from RNase ONE cleavage (and, in some cases, from chemical modification), designated 1–4ss, 6ss and 7ss (Figures 2 and 3, lanes 8–10). Site 5ss is not shown in Figure 2 since it is only utilized in second-site revertants of mutant GDVII (see later). Footprints also demonstrated five double-stranded (ds) sites well protected by PTB and nPTB from RNase V1 treatment, designated 2ss–6ss (Figures 2 and 3, lanes 11–13). The double-stranded sites are adjacent to the single-stranded sites and are named with the same numbers as given to the adjacent single-stranded sites. RNase V1 cleaves a double-stranded segment on each of the two strands of stem H, which are designated 4dsa for the upstream part and 4dsb for the downstream one. Footprints demonstrated no protection of the IRES in the presence of Nova-1 alone and addition of Nova-1 to nPTB did not affect the nPTB protection pattern (data not shown), suggesting that nPTB alone or in a complex with Nova has similar binding properties to the GDVII IRES. Several regions of the GDVII IRES had enhanced cleavage or modification in the presence of PTB (nPTB) following RNase or chemical treatment (Figure 2, green symbols), again suggesting that PTB and nPTB act as RNA chaperones that bind to the IRES and cause a global effect on RNA conformation.
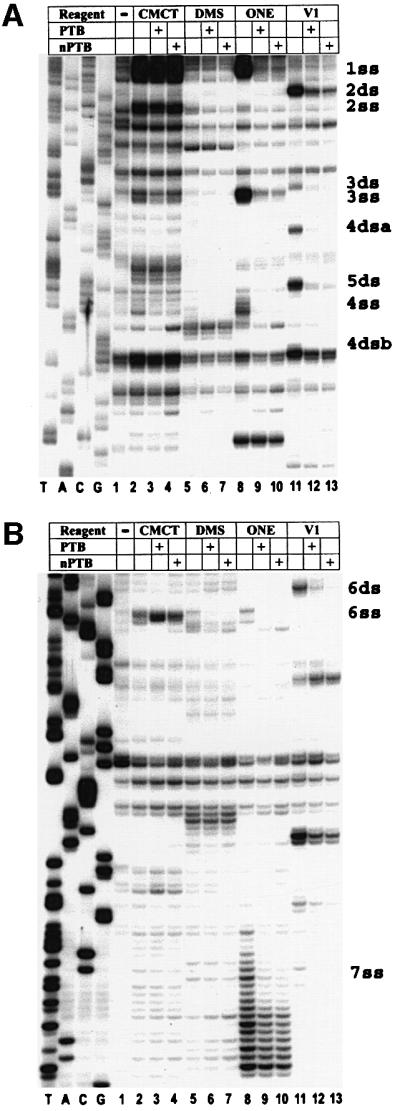
Fig. 3. Chemical and enzymatic footprints of wt GDVII IRES alone and in complexes formed with PTB or nPTB (as noted in the table above the gel). RNA probes were extended from a primer complementary to nt 740–758 (A) or nt 1107–1121 (B). Reference lanes T, A, C and G depict the negative-strand sequence derived using the relevant primer with GDVII plasmid DNA. Positions of PTB- and nPTB-binding sites are indicated on the right.
Mutations in PTB- and nPTB-binding sites affect virus neurovirulence but not virus growth in non-neural cells
In order to determine the importance of PTB and nPTB binding to the biological activities of GDVII, we generated a number of mutations in the PTB- and nPTB-binding sites: GD-91 [with an A to C base change at nucleotide (nt) 649 in site 4dsa in stem–loop H], GD-105 (with two substitutions in 6ss) and GD-121 [with eight substitutions in 7ss—the same changes as in a previously published mutant GD-21 (Pilipenko et al., 1995; Table I; see also Figure 2)]. All of these mutants were attenuated in mice, but grew relatively well in BHK-21 cells, with a plaque size only slightly smaller than wild-type (wt) GDVII (Table I). In contrast to the wt GDVII 50% paralytic dose (PD50) of 0.2 (log10 PFU), GD-91 had a PD50 of 3.3 (Table I); in addition, high doses of GD-91 caused paralysis of mice with relatively few deaths [with a 50% lethal dose (LD50) >10-fold greater than the PD50], while the wt GDVII PD50 was identical to the LD50 and indicative of the extreme neurovirulence. In the case of GD-105 and GD-121, no mice died at the highest dose of virus (Table I).
Table I. GDVII viruses and their biological properties.
| Virus | Sequence of PTB (nPTB)-binding sitesa |
Plaque size (mm) | PD50 (log10PFU) | LD50 (log10PFU) | |||
|---|---|---|---|---|---|---|---|
| 4dsa | 5ssb | 6ss | 7ss | ||||
| GDVII | 635ACACG639 | 648UUUUU653 | 981CUUUA985 | 1042UUUCUCUUUUU1052 | 4.0 | 0.2 | 0.2 |
| GD-91 | 635ACcCG639 | 648UUUUU653 | 981CUUUA985 | 1042UUUCUCUUUUU1052 | 3.0 | 3.3 | 4.5c |
| GD-91Rev A, Bd | 635ACcCG639 | 648UcUUU653 | 981CUUUA985 | 1042UUUCUCUUUUU1052 | 4.0 | <0.3 | <0.3 |
| GD-98 | 635ACcCG639 | 648UcUUU653 | 981CUUUA985 | 1042UUUCUCUUUUU1052 | 3.9 | 0.3 | 0.3 |
| GD-105 | 635ACACG639 | 648UUUUU653 | 981aaUUA985 | 1042UUUCUCUUUUU1052 | 2.7 | 5.8 | >6.5 |
| GD-106 | 635ACACG639 | 648UcUUU653 | 981aaUUA985 | 1042UUUCUCUUUUU1052 | 3.1 | 1.5 | 1.5 |
| GD-121 | 635ACACG639 | 648UUUUU653 | 981CUUUA985 | 1042UagacgcagUU1052 | 3.1 | >5.9 | >5.9 |
| GD-122 | 635ACACG639 | 648UcUUU653 | 981CUUUA985 | 1042UagacgcagUU1052 | 3.0 | 1.5 | 3.0 |
| GD-121Rev A, B, Ce | 635ACACG639 | 648UcUUU653 | 981CUUUA985 | 1042UagacgcagUU1052 | N/D | N/D | N/D |
| GD-100 | 635ACACG639 | 648UcUUU653 | 981CUUUA985 | 1042UUUCUCUUUUU1052 | 4.0 | 1.0 | 1.0 |
aEngineered and acquired mutations in GDVII viruses are shown in bold lower case.
bPTB-binding site 5ss is present only in viruses that have a second-site mutation (see text for further details).
cDeath seen with GD-91 was most likely caused by the appearance of pseudo-revertants with a second-site mutation in 5ss in the CNS, as shown for GD-91Rev A, B.
dRev A and B were isolated from the CNS of SJL/J mice inoculated with 3.6 × 104 (A) and 102 PFU (B) of GD-91 that died on day 8 and 16, respectively. The plaque size, PD50 and LD50 were determined for Rev B.
eRev A, B and C were isolated from the CNS of 129S-Rag1™1Mom mice inoculated with 105 (A) and 102 (B and C) PFU of GD-121 that died on days 14, 28 and 32, respectively.
Two viruses, GD-91 revertants (Rev) A and B, were isolated from the CNS of two animals that died following inoculation with GD-91. These isolates were found to be neurovirulent with an LD50 of <0.3 (Table I). Both revertants had an identical second-site mutation, U649C, in the apical loop of domain H (Table I, see also Figure 2) with no change in the original engineered mutation at nt 637. This second-site mutation resulted in a 648UCUUU653 sequence, which is a consensus PTB-binding motif (Perez et al., 1997a) found in all aphthoviruses and cardioviruses (except for GDVII) and has been shown to be protected by PTB in another cardiovirus, encephalomyocarditis virus (EMCV), and in an aphthovirus, foot-and-mouth disease virus (FMDV; Kolupaeva et al., 1996). We confirmed the phenotypic importance of this second-site mutation by demonstrating that the introduction of the U to C mutation at nt 649 in GD-91, GD-105 and GD-121 (resulting in GD-98, GD-106 and GD-122) led to a significant restoration of neurovirulence (Table I). We hypothesized that this second-site mutation at nt 649 generated a new PTB- and nPTB-binding site in GDVII, designated 5ss (Table I), and that the generation of an additional PTB (nPTB)-binding site restored the neurovirulence that had been attenuated by destruction of different PTB (nPTB)-binding sites. The presence of the U to C mutation at nt 649 (with no other mutations) in the background of wt GDVII did not affect either virus growth in BHK cells or virus neurovirulence (see GD-100 in Table I).
In order to clarify further the importance of PTB and nPTB binding to disease phenotype, we infected immunocompromised 129S-Rag1™1Mom mice, which lack mature B and T cells, with 105 or 102 PFU of GD-121. The mice showed no clinical signs for at least 2 weeks post-infection (PI), at which time they became paralyzed and died. Three viruses isolated from the CNS of moribund mice (GD-121Rev A-C) had the original engineered mutations in site 7ss, but also demonstrated the same U to C second-site mutation at nt 649 that generates 5ss (Table I), providing additional support for the importance of this new putative PTB (nPTB)-binding site in determining neurovirulence. The failure of the GD-121 mutant by itself to cause any disease in immunocompromised mice suggests a severe defect in virus replication in neurons, most likely due to a failure of nPTB to stimulate translation initiation on mutant RNA with a destroyed nPTB-binding site. These data suggest that cell-specific translation rather than the immune system is the major factor affecting the neurovirulence of GD-121 (and presumably GD-91 and GD–105).
Mutations in PTB-binding sites differentially affect binding of PTB and nPTB
We hypothesized that mutations in PTB-binding sites in the attenuated GDVII mutants differentially affect binding of PTB and nPTB to the IRES, decreasing the efficiency of translation and growth in neural cells more dramatically than in non-neural cells. We therefore tested the mutant viral RNAs in protection assays with conditions simulating those in non-neural cells (in the presence of PTB) and neural cells (in the presence of nPTB–Nova-1). The susceptibility of single-stranded and double-stranded segments of the mutant RNA to the RNases and chemical reagents (in the absence of added PTB and nPTB) was not changed, suggesting that the mutations did not affect the IRES structure.
Footprints of the IRES of attenuated GD-105 showed that interactions of PTB and nPTB–Nova-1 were disrupted with the mutated 6ss site as well as the adjacent site 6ds (Figure 4C). In addition, there was a loss in protection following incubation of PTB and nPTB at a number of distant sites, e.g. 4ss, 4dsa, 4dsb and 5ds in domain H (Figure 4A). Of special interest was a specific decrease in protection at site 7ss with nPTB but not PTB (Figure 4C, compare lane 9 with 10). Thus, PTB binds more sites than nPTB in the GD-105 IRES, suggesting that the GD-105 IRES acquires a different and presumably more functional conformation in BHK-21 cells (expressing PTB) compared with that in neurons (expressing nPTB–Nova); these differences in binding may account for the more efficient replication of this mutant virus in BHK-21 cells than in neurons. Footprints of GD-106, the neurovirulent revertant of GD-105, showed that the second-site mutation at nt 649 generated a new site, 5ss, which was fully protected by both PTB and nPTB–Nova against RNase ONE cleavage and chemical modification (Figure 4B, lanes 8–10 and 2–7, respectively). Interestingly, this new interaction of PTB and nPTB with site 5ss facilitated binding to other sites that had been lost with GD-105, restoring protection in domain H by both PTB and nPTB–Nova (Figure 4B) and restoring protection of 7ss by nPTB (Figure 4D, compare lane 10 for GD-106 and GD-105 in Figure 4D and C, respectively). Thus, binding of PTB and nPTB to GD-106 IRES was similar and comparable with that seen for wt GDVII rather than with GD-105, presumably explaining the efficient growth of this double-mutant virus in both BHK-21 cells and neurons in the CNS.

Fig. 4. Chemical and enzymatic footprints of the mutant GD-105 (A and C) and GD-106 (B and D) IRES. RNA was incubated with protein(s) and chemical(s) in reactions as listed in the tables above the gels. RNA probes were extended from a primer complementary to nt 740–758 (A and B) or nt 1107–1121 (C and D). Positions of PTB- and nPTB-binding sites in wt GDVII IRES are indicated on the right. Note that compared with wt GDVII (Figure 3A) and GD-105 (A), there is now protection with PTB and nPTB at a new site, 5ss, in GD-106 (B). In GD-105, there is decreased protection with PTB and nPTB at sites 4ss, 4ds and 5ds, as well as decreased protection with nPTB at site 7ss, while in GD-106 protection of the above sites with PTB and nPTB had been restored. Reference lanes T, A, C and G depict the negative-strand sequence derived using the relevant primer with GD-105 plasmid DNA.
Footprints of the GD-121 and GD-122 pair of mutants showed that the mutation of site 7ss abrogated interactions of PTB and nPTB–Nova-1 with this region, but did not affect their binding to sites 6ss and 6ds (Figure 5A for GD-121; data not shown for GD-122). In GD-121, there was a loss in protection following incubation with nPTB at sites 2ds and 3ds and a significant decrease in protection at sites 3ss, 4dsa and 4dsb by nPTB, while PTB binding was only slightly affected at site 4dsa (Figure 5B). Thus, PTB binds more sites than nPTB in the GD-121 IRES, as was also the case with the attenuated mutant GD-105. This finding suggests that the GD-121 IRES acquires a more functional conformation in BHK-21 cells (which express PTB) than in neurons (which express nPTB–Nova). Footprints of the IRES of the neurovirulent revertant GD-122 demonstrated that the second-site mutation generated a binding site 5ss for PTB and nPTB (as was the case with the neurovirulent revertant GD-106). In GD-122, this new interaction with site 5ss stabilized nPTB binding to other sites that had been lost in GD-121, restoring protection of sites 2ds, 3ss, 3ds and 4ds (Figure 5C). This stabilization presumably accounts for the efficient growth of the double-mutant GD-122 virus in CNS neurons.

Fig. 5. Chemical and enzymatic footprints of the mutant GD-121 (A and B) and GD-122 (C) IRES. RNA probes were extended from a primer complementary to nt 740–758 (B and C) or nt 1107–1121 (A). For more details, see the legends to Figures 3 and 4. Note that compared with wt GDVII (Figure 3) and GD-122 (C), there is decreased protection with nPTB at sites 2ds, 3ss, 3ds and 4ds in GD-121 (B). In GD-122, PTB and nPTB protect a new site, 5ss, and there is now restoration of protection with nPTB that had been perturbed in GD-121. Reference lanes T, A, C and G depict the negative-strand sequence derived using the relevant primer with GD-121 plasmid DNA.
Footprints of the remaining pair of mutants, GD-91 and GD-98, demonstrated similar findings to those seen with the previous pairs (data not shown). Of interest, mutation of PTB (nPTB)-binding site 4dsa in GD-91 destabilized the nPTB, but not PTB, interaction with 7ss.
In summary, the IRES of the attenuated mutant viruses had a more severe binding deficit with nPTB than PTB and the generation of a new PTB (nPTB)-binding site in the neurovirulent second-site revertant viruses led to restoration of these deficits.
PTB and nPTB–Nova-1 differentially affect the efficiency of translation initiation on mutant RNA
We hypothesized that mutations in PTB- and nPTB-binding sites in the attenuated GDVII mutants differentially affect the stimulation of IRES-mediated translation initiation by PTB and nPTB, allowing for better growth in non-neural than neural cells. We therefore tested the mutant RNAs in an in vitro reconstituted translation system (Pestova et al., 1996) with conditions simulating those in non-neural cells (in the presence of PTB) and neural cells (in the presence of nPTB–Nova-1). As expected, the amount of 48S complexes formed on wt GDVII RNA was similar in the presence of PTB and nPTB–Nova-1 (Figure 6, lanes 5 and 6), but the efficiency of 48S complex formation on mutant RNA from attenuated viruses (GD-91, GD-105 and GD-121) was significantly greater in the presence of PTB than nPTB–Nova (Figure 6, compare lane 9 with 10). The amount of 48S complexes formed on RNA from second-site neurovirulent revertants (GD-98, GD-106 and GD-122) was similar in the presence of either PTB or nPTB–Nova (Figure 6, lanes 13 and 14) and significantly greater in the presence of nPTB–Nova than that formed on the attenuated mutants’ RNA (Figure 6, compare lane 10 with 14). Thus, the stimulation of 48S complex formation on a mutant IRES by PTB and nPTB–Nova-1 correlated with PTB and nPTB–Nova-1 binding properties to the IRES. Likewise, the efficiency of translation initiation on the mutant RNA in the presence of PTB and nPTB–Nova-1 correlated with the mutant virus growth in cells expressing either of the proteins.
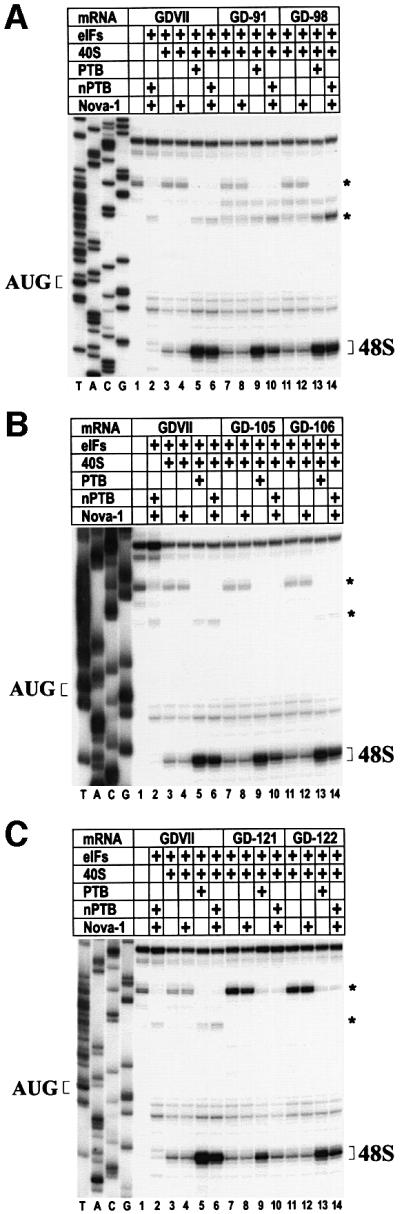
Fig. 6. Toeprint analysis of 48S complexes assembled on wt GDVII and mutant RNAs in the presence of PTB, nPTB and Nova-1. Reactions were assembled with translation components as indicated in the tables above the gels. For more details, see the legend to Figure 1.
Nova-1 by itself had no effect on 48S complex formation on any RNA (Figure 6), although the addition of Nova-1 to nPTB showed differences compared with nPTB alone in the case of some of the mutants, e.g. the efficiency of 48S complex formation with nPTB in GD-91 was slightly less than with PTB and even less with nPTB in the presence of Nova-1 (data not shown).
Discussion
Picornaviruses have a distinctive strategy of translation, which includes a major cis-acting control element for the internal ribosome entry and initiation of viral RNA translation. The regulation of picornaviral RNA translation depends on canonical and host cell-specific factors that are known to bind to the IRES (reviewed in Jackson and Kaminski, 1995; Pestova et al., 2001). There is some evidence that the binding of these factors affects translation and plays a role in picornavirus-induced disease. In the case of Sabin poliovirus vaccine strains, a point mutation in the IRES attenuates the virus (Evans et al., 1985) and decreases translation efficiency in neural cells with little effect on virus growth and translation in HeLa cells (La Monica and Racaniello, 1989). Previously, we demonstrated that both PTB and ITAF45 (Mpp1), a cell-specific proliferation-associated factor, are critical for efficient translation initiation mediated by the IRES of FMDV (Pilipenko et al., 2000). Replacement of the GDVII IRES by the corresponding region of FMDV resulted in virus attenuation, suggesting that a failure of growth in neurons was due to the absence of ITAF45 and a resultant defect in translation in these non-proliferating cells. Thus, the presence of a cell-specific translation factor can determine viral tropism, virulence and disease phenotype.
Here, we examined the contribution of tissue-specific translation control to the disease phenotype in the case of GDVII. PTB is known to be critical for efficient 48S complex formation on GDVII IRES (Pilipenko et al., 2000). Although the CNS has limited expression of PTB, it is enriched in nPTB (Markovtsov et al., 2000; Lillevali et al., 2001). Our study was carried out with both Nova-1 and nPTB since these are physically associated and because Nova-1 is present in motor neurons, a major host cell for GDVII. Our results demonstrate that nPTB binds to the IRES and regulates the efficiency of translation initiation in a cell-specific manner.
Footprints demonstrated that PTB and nPTB bind similarly to specific sites in the GDVII IRES, some of which have previously been identified as PTB-binding sites in EMCV (Kolupaeva et al., 1996); however, sites 1ss, 2ss and 2ds (see Figure 2 for details) were not identified as PTB-binding sites in the case of EMCV since the fragment of the EMCV RNA that was probed in these studies did not include this region of the IRES. In addition to single-stranded regions, we noted protection of several double-stranded regions. The protection of double-stranded sites could be due to direct interaction of PTB with the base-paired regions or related to PTB binding to one or more of the adjacent single-stranded sites. An identical 4 bp segment, C-G/A-U/C-G/A-U, is present in 2ds, 4ds and 6ds, suggesting that there may be a specific double-stranded motif for PTB binding. The double-stranded binding sites are probably functionally important since mutation of 4ds in GD-91 affected interactions of nPTB with the IRES more than PTB, led to a significant decrease in 48S complex formation in the presence of nPTB–Nova and attenuated the neurovirulence of the mutant virus.
We found that PTB and nPTB in an equal molar amount with the GDVII IRES protected six single-stranded and five double-stranded sites. The fact that a PTB- or an nPTB-dimer contains an array of eight RRMs suggests that some RRMs of PTB or nPTB interact with composite sites which are comprised of adjacent single-stranded and double-stranded RNA sequences, similarly to the U1A spliceosomal protein (Oubridge et al., 1994). The composite nature of some PTB- and nPTB-binding sites is supported by the apparently interdependent protection of single-stranded and the adjacent double-stranded sites by PTB, e.g. mutation of 6ss abrogated protection of 6ds in GD-105 and GD-106. Interestingly, some binding sites resemble others: the long interdomain polypyrimidine stretch in 1ss resembles a region in 7ss, and the putative single-stranded/double-stranded composite sites 2ss/2ds and 3ss/3ds resemble 6ss/6ds and 5ss/5ds, respectively (see Figure 2 for details). These similarities raise the possibility that certain pairs of RRMs in a PTB- or an nPTB-dimer selectively interact with particular pairs of PTB (nPTB)-binding sites.
PTB and nPTB binding affects the overall IRES structure, exemplified by enhanced cleavage of RNA segments distant from the binding sites (Figure 2) and by the prominent changes seen in the pattern of RT stops in toeprints following addition of PTB and nPTB (Figure 1). These observations are consistent with their proposed function as RNA chaperones, fostering a functionally competent folding of the IRES needed for efficient assembly of 48S complex. Their functional role as RNA chaperones is also supported by the decreased efficiency of 48S complex formation on mutant RNA, which has a destroyed PTB (nPTB)-binding site(s).
PTB and nPTB binding to an array of putative binding sites can occur if the free energy loss due to PTB or nPTB interactions with these sites exceeds the free energy gain that results from changes in the IRES conformation (following the binding of PTB or nPTB to this array of binding sites). Out of a number of thermodynamically permissive combinations of PTB and nPTB interactions with binding sites, an array of interactions with the lowest free energy is realized; the particular array that is selected is not predictable without a more detailed knowledge of the thermodynamic parameters. We found that mutation of a PTB- or an nPTB-binding site in the GDVII IRES interfered with the protein’s interactions with some non-mutated sites that were distantly located (e.g. mutation of 6ss in GD-105 affected the protein’s interaction with 4ss, 4ds and 5ds). These observations suggest that following mutation of a PTB- or an nPTB-binding site, the protein’s binding to a few of the non-mutated sites may be more thermodynamically favorable than binding to all of the remaining (non-mutated) sites. Our studies also demonstrated that mutation of a PTB- or nPTB-binding site led to a more severe decrease in binding of nPTB than PTB at one or more non-mutated sites [e.g. the binding of nPTB, but not PTB, was disturbed at site 7ss in the cases of GD-105 (Figure 4C) and GD-91, and at sites 2ds, 3ss/3ds, 4ds in the case of GD-121 (Figure 5B)]. The more prominent loss in nPTB binding correlated with the more significant decrease in 48S complex formation following addition of nPTB rather than PTB. These results provide an explanation for the better growth of these mutant attenuated viruses in BHK-21 cells (which express PTB) than in neurons (which express nPTB, but not PTB) and their loss of neurovirulence. Interestingly, a second-site mutation at nt 649 in revertant viruses generated a new PTB (nPTB)-binding site by changing the sequence to a consensus PTB-binding motif that is present in aphthoviruses and all other cardioviruses, including other TMEV strains. The new binding site stabilized PTB and nPTB interactions with non-mutated sites, stimulated 48S complex formation and restored the neurovirulent phenotype.
The fact that some disturbances in PTB interactions with the IRES following mutation of a PTB-binding site did not dramatically affect the ability of PTB to stimulate 48S complex formation (and the ability of a mutant virus to grow in BHK-21 cells) demonstrates a remarkable plasticity of functional IRES conformations. This plasticity is also supported by our finding that the addition of a new binding site, 5ss, led to a restoration of efficient 48S complex formation of the mutant RNA in the presence of nPTB. These findings show that the multiple PTB- and nPTB-binding sites in the IRES provide an opportunity for different arrays of PTB and nPTB interactions that can lead to a competent IRES conformation. A PTB or an nPTB interaction with a particular binding site may or may not be essential for the IRES function, depending on specific conditions. For example, disruption of PTB or nPTB interaction with site 7ss had only a small effect on IRES function in the case of GD-121–PTB, GD-122–PTB and GD-122–nPTB complexes (most likely because PTB and nPTB interactions with the other binding sites were not perturbed in the above complexes), but led to the loss of the IRES function in the case of GD121–nPTB, GD-91–nPTB and GD-105–nPTB complexes (most likely because nPTB interactions with other binding sites were perturbed). The multiple PTB- and nPTB-binding sites in the GDVII IRES were presumably selected in order to ensure proper IRES function in cells containing different forms of PTB, including ones that predominantly express nPTB (e.g. neurons).
Our data show a correlation between nPTB binding, stimulation of 48S complex formation and neurovirulence. In addition, our findings suggest that a mutation of a PTB (nPTB)-binding site is more likely to destabilize nPTB binding more than PTB binding to non-mutated sites. We suspect that nPTB–Nova-1 binds to the RNA of other neurotropic viruses to determine virus-induced CNS disease. Of note, critically important mutations responsible for the attenuation of Sabin poliovirus vaccine strains reside in the IRES and impair IRES function in a cell-dependent manner (La Monica and Racaniello, 1989; Haller et al., 1996). PTB and PCBP have been shown to bind to and stimulate translation of poliovirus RNA (Hellen et al., 1994; Blyn et al., 1996, 1997; Hunt et al., 1999). PCBP is ubiquitously expressed, while, as demonstrated in the present study, nPTB substitutes for the function of PTB in neurons. The attenuation mutation in poliovirus type 3 reduced cross-linking of a fragment of the poliovirus 5′ UTR to PTB in neuroblastoma cells but not in HeLa cells (Gutierrez et al., 1997). It may be that PTB binds to the IRES of Sabin strains more stably than nPTB, thereby allowing for better translation and growth in non-neural cells compared with those in neurons. In this way, cell- and IRES-specific RNA chaperone proteins such as PTB, nPTB–Nova-1 and ITAF45 can regulate cell-specific translation of viral RNA, determining virus tropism, virulence and disease phenotype. In an analogous way, these and other host cell proteins may control cell-specific expression of cellular IRES-containing mRNAs.
PTB is known as a negative regulator of splicing that binds several intronic pyrimidine-rich sequences and represses exon inclusion into mature RNA (reviewed in Valcarcel and Gebauer, 1997). The alternative splicing of several pre-mRNAs in neurons was attributed, at least in part, to the reciprocal expression and somewhat different function of PTB and nPTB (Ashiya and Grabowski, 1997; Markovtsov et al., 2000; for a review, see Grabowski, 1998). We demonstrated that PTB and nPTB can bind different arrays of recognizable motifs present in a particular RNA. It may be that this difference in RNA-binding properties contributes to the alternative pre-mRNA splicing.
Sequestration of RNA-binding proteins and disturbances in RNA transcription and processing have been proposed as mechanisms underlying cell death in a number of neurodegenerative diseases, including ones involving the motor neuron (reviewed in Dredge et al., 2001; McCampbell and Fischbeck, 2001). Our studies raise the possibility that the binding of nPTB–Nova-1 to the GDVII IRES leads to a decrease in the availability of these or similar factors, which are required for neuronal specific splicing of certain mRNAs and are critical for motor neuronal function and viability. In this way, the dysfunction or death of motor neurons (or other cell types) in virus-induced diseases may be related to the sequestration of essential cellular proteins.
Materials and methods
Plasmids
In order to clone and express nPTB, total RNA was extracted from the mouse CNS using ‘Ultraspec RNA’ according to the manufacturer’s protocol (Biotecx Laboratories, Houston, TX), amplified by RT–PCR using primers flanking the nPTB coding region (DDBJ/EMBL/GenBank accession No. AF095718; 5′-TGTTGTGCGGCCGCTTAGATTGTTGACTTGGAGAAAGACACTCT-3′ and 5′-TGGTCGAGCTCAATGGACGGAATTGTCACTGAGGTTGC-3′), digested with SacI and NotI, and inserted into pET-28b (Novagen, Madison, WI). In order to clone and express Nova-1, whole Marathon-Ready human brain cDNA (Clontech Laboratories, Palo Alto, CA) was amplified by PCR using primers flanking the Nova-1 coding region (DDBJ/EMBL/GenBank accession No. U04840; 5′-CACACTCGAGTCAACCCACTTTCTGAGGATTGGC-3′ and 5′-AGAGAGAGCTCCATGATGGCGGCAGCTCCCA-3′), digested with SacI and XhoI, and inserted into pET-28b. Plasmids pET(His6-eIF4A), pET(His6-eIF4B) (Pestova et al., 1996) and pE15 (Hellen et al., 1993) have been described.
Mutations were introduced by two-step PCR amplification of the full-length pGDVII infectious clone with primers containing the desired nucleotide changes. Full-length plasmids pGD-91 and pGD-98 were assembled following ligation of fragments BglII–KpnI (nt 395–935 containing the engineered mutations), KpnI–ApaI (nt 936–13) and ApaI–BglII (nt 14–394). pGD-105 and pGD-121 were assembled from fragments KpnI–AatII (nt 936–1206 containing the engineered mutations), AatII–AatII (nt 1207–3916) and AatII–KpnI (nt 3917–935). pGD-106 and pGD-122 were assembled from fragments MluI–KpnI (nt 638–935 with the engineered nt 649 mutation) and KpnI–MluI (nt 936–637) derived from pGD-105 and pGD-121, respectively.
Viruses
Viruses were recovered from transfection of BHK-21 cells with RNA derived from in vitro transcription of linearized plasmids and characterized as described previously (Pilipenko et al., 1994, 1995). A region of the viral RNA (between nt 550 and 1120) was amplified by RT–PCR and sequenced (Pilipenko et al., 1995).
Animal studies
In order to determine the PD50 and LD50 values, 3-week-old SJL/J mice (The Jackson Laboratory, Bar Harbor, ME) were intracerebrally inoculated with 10-fold virus dilutions using at least five animals per dilution. 129S-Rag1™1Mom (The Jackson Laboratory) were inoculated with 102 and 105 PFU of GD-121 using four animals per dose. Viral genome in CNS homogenates of paralyzed or dead animals was detected by RT–PCR and the appropriate region of the viral RNA was sequenced as described above.
Purification of factors and 40S ribosomal subunits
Recombinant proteins were expressed in Escherichia coli BL21(DE3; Novagen). Recombinant nPTB was purified by Ni2+-NTA–agarose (Qiagen, Valencia, CA) and poly(U)–Sepharose (Amersham Pharmacia Biotech, Piscataway, NJ) column chromatography. Recombinant Nova-1 was purified by Ni2+-NTA–agarose (Qiagen) and monoQ HiTrap (Amersham Pharmacia Biotech) column chromatography. Ribosomal subunits eIF2, eIF3 and eIF4F were purified from rabbit reticulocyte lysates, and recombinant eIF4A, eIF4B and PTB were purified as described (Pestova et al., 1996).
Assembly and analysis of ribosomal complexes
To form 48S complexes, an RNA transcript (nt 1–1333) was incubated with purified translation components for 6 min at 37°C essentially as previously described (Pestova et al., 1996; Pilipenko et al., 2000). A primer complementary to GDVII nt 1194–1213 (5′-CCAGAAGAC GTCATCGTCCA-3′) was added and cDNA generated with avian myeloblastosis virus RT (Promega, Madison, WI) as described (Pestova et al., 1996; Pilipenko et al., 2000). 48S complexes assembled on the GDVII mRNA inhibited the primer extension 17–19 nt downstream of the A of the initiator AUG (nt 1067–1069), resulting in shorter cDNA products seen after gel electrophoresis. If not stated otherwise, PTB, nPTB and Nova-1 dimers were added at an equal molar amount with mRNA. Experiments were repeated at least twice with reproducible results.
Footprinting
RNA transcripts corresponding to GDVII nt 1–1333 were incubated for 10 min at 37°C with an equal molar amount of PTB, nPTB and Nova-1 to form complexes and then probed with RNase ONE (which cleaves single-stranded RNA without base specificity), CMCT (which reacts with unpaired uracil and to a lesser extent guanine residues), DMS (which reacts with unpaired adenine and to a lesser extent cytosine residues) and RNase V1 (which cleaves base-paired regions) essentially as described (Pilipenko et al., 1989; Kolupaeva et al., 1996). After treatment, RNA was deproteinized, precipitated, dissolved in water and divided into aliquots for subsequent primer extension from different primers. Cleaved or modified sites were identified by the primer extension as described (Pilipenko et al., 1989) using primers complementary to nt 1107–1121 (5′-CGTCAACGGCTGTGC-3′) and nt 740–758 (5′-CTTTGTGTGGTC GCTACAG-3′). In addition, primers complementary to nt 650–667, 864–881 and 977–995 were used for wt GDVII RNA footprinting. Experiments were repeated at least two times with reproducible results.
Acknowledgments
Acknowledgements
This study was supported in part by NIH 1RO1 NS37958-01 to R.P.R and grants from the National Multiple Sclerosis Society to E.V.P, R.P.R. and V.I.A.
References
- Ashiya M. and Grabowski,P.J. (1997) A neuron-specific splicing switch mediated by an array of pre-mRNA repressor sites: evidence of a regulatory role for the polypyrimidine tract binding protein and a brain-specific PTB counterpart. RNA, 3, 996–1015. [PMC free article] [PubMed] [Google Scholar]
- Blyn L.B., Swiderek,K.M., Richards,O., Stahl,D.C., Semler,B.L. and Ehrenfeld,E. (1996) Poly(rC) binding protein 2 binds to stem–loop IV of the poliovirus RNA 5′ non-coding region: identification by automated liquid chromatography-tandem mass spectrometry. Proc. Natl Acad. Sci. USA, 93, 11115–11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyn L.B., Towner,J.S., Semler,B.L. and Ehrenfeld,E. (1997) Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J. Virol., 71, 6243–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell R.B. (1996) Onconeural antigens and the paraneoplastic neurologic disorders: at the intersection of cancer, immunity and the brain. Proc. Natl Acad. Sci. USA, 93, 4529–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dredge B.K., Polydorides,A.D. and Darnell,R.B. (2001) The splice of life: alternative splicing and neurological disease. Nature Rev. Neurosci., 2, 43–50. [DOI] [PubMed] [Google Scholar]
- Duke G.M., Hoffman,M.A. and Palmenberg,A.C. (1992) Sequence and structural elements that contribute to efficient encephalomyocarditis virus RNA translation. J. Virol., 66, 1602–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D.M., Dunn,G., Minor,P.D., Schild,G.C., Cann,A.J., Stanway,G., Almond,J.W., Currey,K. and Maizel,J.V.,Jr (1985) Increased neuro-virulence associated with a single nucleotide change in a noncoding region of the Sabin type 3 poliovaccine genome. Nature, 314, 548–550. [DOI] [PubMed] [Google Scholar]
- Ghetti A., Pinol-Roma,S., Michael,W.M., Morandi,C. and Dreyfuss,G. (1992) hnRNP I, the polypyrimidine tract-binding protein: distinct nuclear localization and association with hnRNAs. Nucleic Acids Res., 20, 3671–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski P.J. (1998) Splicing regulation in neurons: tinkering with cell-specific control. Cell, 92, 709–712. [DOI] [PubMed] [Google Scholar]
- Gromeier M., Alexander,L. and Wimmer,E. (1996) Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc. Natl Acad. Sci. USA, 93, 2370–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A.L., Denova-Ocampo,M., Racaniello,V.R. and del Angel,R.M. (1997) Attenuating mutations in the poliovirus 5′ untranslated region alter its interaction with polypyrimidine tract-binding protein. J. Virol., 71, 3826–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller A.A., Stewart,S.R. and Semler,B.L. (1996) Attenuation stem–loop lesions in the 5′ noncoding region of poliovirus RNA: neuronal cell-specific translation defects. J. Virol., 70, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen C.U., Witherell,G.W., Schmid,M., Shin,S.H., Pestova,T.V., Gil,A. and Wimmer,E. (1993) A cytoplasmic 57-kDa protein that is required for translation of picornavirus RNA by internal ribosomal entry is identical to the nuclear pyrimidine tract-binding protein. Proc. Natl Acad. Sci. USA, 90, 7642–7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen C.U., Pestova,T.V., Litterst,M. and Wimmer,E. (1994) The cellular polypeptide p57 (pyrimidine tract-binding protein) binds to multiple sites in the poliovirus 5′ nontranslated region. J. Virol., 68, 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt S.L., Hsuan,J.J., Totty,N. and Jackson,R.J. (1999) unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev., 13, 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R.J. and Kaminski,A. (1995) Internal initiation of translation in eukaryotes: the picornavirus paradigm and beyond. RNA, 1, 985–1000. [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T., Ichikawa,M., Arai,J., Tateiwa,H., Fu,L., Higuchi,K. and Yoshimura,N. (2000) Molecular cloning and characterization of a new neuron-specific homologue of rat polypyrimidine tract binding protein. J. Biochem., 128, 811–821. [DOI] [PubMed] [Google Scholar]
- Kolupaeva V.G., Hellen,C.U. and Shatsky,I.N. (1996) Structural analysis of the interaction of the pyrimidine tract-binding protein with the internal ribosomal entry site of encephalomyocarditis virus and foot-and-mouth disease virus RNAs. RNA, 2, 1199–1212. [PMC free article] [PubMed] [Google Scholar]
- La Monica N. and Racaniello,V.R. (1989) Differences in replication of attenuated and neurovirulent polioviruses in human neuroblastoma cell line SH-SY5Y. J. Virol., 63, 2357–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillevali K., Kulla,A. and Ord,T. (2001) Comparative expression analysis of the genes encoding polypyrimidine tract binding protein (PTB) and its neural homologue (brPTB) in pre-natal and post-natal mouse brain. Mech. Dev., 101, 217–220. [DOI] [PubMed] [Google Scholar]
- Markovtsov V., Nikolic,J.M., Goldman,J.A., Turck,C.W., Chou,M.Y. and Black,D.L. (2000) Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol. Cell. Biol., 20, 7463–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCampbell A. and Fischbeck,K.H. (2001) Polyglutamine and CBP: fatal attraction? Nature Med., 7, 528–530. [DOI] [PubMed] [Google Scholar]
- Meerovitch K., Svitkin,Y.V., Lee,H.S., Lejbkowicz,F., Kenan,D.J., Chan,E.K., Agol,V.I., Keene,J.D. and Sonenberg,N. (1993) La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J. Virol., 67, 3798–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oubridge C., Ito,N., Evans,P.R., Teo,C.H. and Nagai,K. (1994) Crystal structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature, 372, 432–438. [DOI] [PubMed] [Google Scholar]
- Perez I., Lin,C.H., McAfee,J.G. and Patton,J.G. (1997a) Mutation of PTB binding sites causes misregulation of alternative 3′ splice site selection in vivo. RNA, 3, 764–778. [PMC free article] [PubMed] [Google Scholar]
- Perez I., McAfee,J.G. and Patton,J.G. (1997b) Multiple RRMs contribute to RNA binding specificity and affinity for polypyrimidine tract binding protein. Biochemistry, 36, 11881–11890. [DOI] [PubMed] [Google Scholar]
- Pestova T.V., Hellen,C.U. and Shatsky,I.N. (1996) Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol., 16, 6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova T.V., Kolupaeva,V.G., Lomakin,I.B., Pilipenko,E.V., Shatsky,I.N., Agol,V.I. and Hellen,C.U. (2001) Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl Acad. Sci. USA, 98, 7029–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilipenko E.V., Blinov,V.M., Chernov,B.K., Dmitrieva,T.M. and Agol,V.I. (1989) Conservation of the secondary structure elements of the 5′-untranslated region of cardio- and aphthovirus RNAs. Nucleic Acids Res., 17, 5701–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilipenko E.V., Gmyl,A.P., Maslova,S.V., Belov,G.A., Sinyakov,A.N., Huang,M., Brown,T.D. and Agol,V.I. (1994) Starting window, a distinct element in the cap-independent internal initiation of translation on picornaviral RNA. J. Mol. Biol., 241, 398–414. [DOI] [PubMed] [Google Scholar]
- Pilipenko E.V., Gmyl,A.P., Maslova,S.V., Khitrina,E.V. and Agol,V.I. (1995) Attenuation of Theiler’s murine encephalomyelitis virus by modifications of the oligopyrimidine/AUG tandem, a host-dependent translational cis element. J. Virol., 69, 864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilipenko E.V., Viktorova,E.G., Khitrina,E.V., Maslova,S.V., Jarousse,N., Brahic,M. and Agol,V.I. (1999) Distinct attenuation phenotypes caused by mutations in the translational starting window of Theiler’s murine encephalomyelitis virus. J. Virol., 73, 3190–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilipenko E.V., Pestova,T.V., Kolupaeva,V.G., Khitrina,E.V., Poperechnaya,A.N., Agol,V.I. and Hellen,C.U. (2000) A cell cycle-dependent protein serves as a template-specific translation initiation factor. Genes Dev., 14, 2028–2045. [PMC free article] [PubMed] [Google Scholar]
- Polydorides A.D., Okano,H.J., Yang,Y.Y., Stefani,G. and Darnell,R.B. (2000) A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proc. Natl Acad. Sci. USA, 97, 6350–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin Y.V., Maslova,S.V. and Agol,V.I. (1985) The genomes of attenuated and virulent poliovirus strains differ in their in vitro translation efficiencies. Virology, 147, 243–252. [DOI] [PubMed] [Google Scholar]
- Svitkin Y.V., Pestova,T.V., Maslova,S.V. and Agol,V.I. (1988) Point mutations modify the response of poliovirus RNA to a translation initiation factor: a comparison of neurovirulent and attenuated strains. Virology, 166, 394–404. [DOI] [PubMed] [Google Scholar]
- Svitkin Y.V., Meerovitch,K., Lee,H.S., Dholakia,J.N., Kenan,D.J., Agol,V.I. and Sonenberg,N. (1994) Internal translation initiation on poliovirus RNA: further characterization of La function in poliovirus translation in vitro. J. Virol., 68, 1544–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcarcel J. and Gebauer,F. (1997) Post-transcriptional regulation: the dawn of PTB. Curr. Biol., 7, R705–R708. [DOI] [PubMed] [Google Scholar]