

Abstract
Ligation of mycolic acids to structural components of the mycobacterial cell wall generates a hydrophobic, impermeable barrier that provides resistance to toxic compounds such as antibiotics. Secreted proteins FbpA, FbpB, and FbpC attach mycolic acids to arabinogalactan, generating mycolic acid methyl esters (MAME) or trehalose, generating α,α′-trehalose dimycolate (TDM; also called cord factor). Our studies of Mycobacterium smegmatis showed that disruption of fbpA did not affect MAME levels but resulted in a 45% reduction of TDM. The fbpA mutant displayed increased sensitivity to both front-line tuberculosis-targeted drugs as well as other broad-spectrum antibiotics widely used for antibacterial chemotherapy. The irregular, hydrophobic surface of wild-type M. smegmatis colonies became hydrophilic and smooth in the mutant. While expression of M. smegmatis fbpA restored defects of the mutant, heterologous expression of the Mycobacterium tuberculosis fbpA gene was less effective. A single mutation in the M. smegmatis FbpA esterase domain inactivated its ability to provide antibiotic resistance. These data show that production of TDM by FbpA is essential for the intrinsic antibiotic resistance and normal colonial morphology of some mycobacteria and support the concept that FbpA-specific inhibitors, alone or in combination with other antibiotics, could provide an effective treatment to tuberculosis and other mycobacterial diseases.
Mycobacteria are notorious for their extremely high levels of intrinsic drug resistance, traditionally attributed to their impermeable, hydrophobic cell envelope. The principle components of the envelope have been identified chemically and rationalized in a structural model originally proposed by Minnikin in 1982 (24). Since then, many biochemical, biophysical, and electron microscopic analyses have supported and extended this model (3, 8, 12, 22). Mycobacterial plasma membrane and peptidoglycan layers have features that are similar to those of other gram-positive bacteria. The complex outer layers of the cell wall are only found in certain related genera within the Actinomycetales taxon, including Corynebacterium, Nocardia, Gordona, Rhodococcus, and Mycobacterium (3, 8). In Mycobacterium, peptidoglycan is covalently linked to a surrounding network of arabinogalactans, which is itself covered by a layer of mycolates (24). Passage of low-molecular-weight nutrients and waste products is apparently facilitated by porins that are similar to those found in the outer membranes of gram-negative bacteria (27). The mycolic acid layer forms an effective barrier against the passage of both hydrophilic and hydrophobic compounds (8) and is thought to render mycobacteria resistant to antibiotics even though they have corresponding drug-sensitive targets.
Mycolic acids are synthesized as two alkyl chains by enzymes similar to those that assemble fatty acids. These chains are fused and subject to numerous modifications (3, 8, 12, 22) to generate a complex family of lipids that are incorporated into the cell envelope. Interestingly, a Mycobacterium smegmatis mutant defective for mycolate biosynthesis (37) displayed increased rates of uptake and sensitivity to erythromycin (23). Similarly, a transposon mutation in a mycolic acid biosynthetic gene of Mycobacterium tuberculosis (kasB) made it more sensitive to some antibiotics as well as mammalian antimicrobial proteins lysozyme and defensin (13).
Mycolic acids in the cell wall of mycobacteria are esterified either to trehalose in forms that can be extracted in organic solvents (α,α′-trehalose dimycolate, also called TDM or cord factor, and α,α′-trehalose monomycolate [TMM]) or covalently linked to the arabinogalactan in the cell wall (mycolic acid methyl esters [MAMEs]). TDM catalyzes formation of elongated mycobacterial aggregates (“cord structures”) in liquid medium or on the surface of colonies. Esterifications of mycolic acids within the cell wall are carried out by a family of at least three homologous enzymes that provide partially overlapping activities. These exported proteins were initially characterized as major antigens of M. tuberculosis, “the antigen 85 complex” (38). Later they were found to be fibronectin-binding proteins (Fbp), a property originally thought to be involved in phagocytosis into macrophages (29). Most recently, these proteins were shown to catalyze a mycolyltransferase reaction that is essential for cell wall biogenesis (5). Analysis of an M. tuberculosis fbpC mutant demonstrated that this gene was dispensable for growth but needed for the construction of cell walls containing normal amounts of MAMEs. Furthermore, both chenodeoxycholate, a hydrophobic compound, and glycerol, a hydrophilic compound, diffused faster through the cell envelope of the fbpC mutants (17). Curiously, resistance to the limited spectrum of antibiotics tested was unaffected. Genetic analysis showed that all three M. tuberculosis fbp genes could be disrupted individually and that they played partially redundant roles in cell wall biosynthesis (30). The fact that a synthetic analog of a Fbp substrate was able to inhibit growth and cell wall biosynthesis demonstrated that these proteins, or others having similar activities, were essential and thus attractive targets for new antimycobacterial drugs (5). In this paper, we show that the M. smegmatis fbpA gene provides a nonredundant function in cell wall biosynthesis that is needed for intrinsic antibiotic resistance, hydrophobicity of the cell wall, and colonial structure.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
All strains and plasmids used in this study are listed in Table 1. Wild-type M. smegmatis strain MC2155 (35) and its transposon-derived mutants were grown in 7H9 liquid and 7H10 (Difco) or LB agar medium supplemented with 0.5% Tween 80. Kanamycin was used at a final concentration of 50 μg ml−1. Hygromycin was used at 100 μg ml−1 and 75 μg ml−1 for Escherichia coli and mycobacteria, respectively. Genomic DNA from M. smegmatis was isolated using the DNAzol kit (MRC). Transformation was carried out as described elsewhere (7).
TABLE 1.
Strains, plasmids, and primers used in this studya
| Strain, plasmid, or primer | Relevant features | References |
|---|---|---|
| Strains | ||
| MC2155 | Mycobacterium smegmatis wild type, high transformation efficiency | 35 |
| MAR1 | MC2155-derived pMycoMar transposon multidrug-sensitive mutant | This study |
| Plasmids | ||
| pMycoMar | Himar1 transposon carrying vector, Ts mycobacterial replicon | 33 |
| pMV361 | E. coli-Mycobacterium shuttle integrative vector, Kanr, built-in heat shock promotor for translational fusion | 36 |
| pMycVec2 | E. coli-Mycobacterium shuttle replicative vector, Hygr | 18 |
| Primers | ||
| MS_FbpA.EB | 5′-ggatccgaattcatgaagttcgttgggagaatgc-3′ | |
| MS_FbpA.HXb | 5′-tctagaaagctttcaactcgatcaggcggtc-3′ | |
| S171fbpAMS-up | 5′-gttgtcggtctgGcgatggcc-3′ | |
| S171fbpAMS-down | 5′-ggccatcgCcagaccgacaac-3′ | |
| MS_FbpD.EB | 5′-ggatccgaattcatgctgaccgtggtgctcgcc-3′ | |
| MS_FbpD.PHXb | 5′-tctagaaagcttttaattaactacttgatggtggcgaccagct-3′ | |
| TB_FbpA.EB | 5′-ggatccgaattcatgcagcttgttgacagggttcgtg-3′ | |
| TB_FbpA.C1XbE | 5′-gaattctctagaatcgatcgctagatgttgtgtctgttcggag-3′ | |
| Mar-Ext1 | 5′-gggaatcatttgaaggttggt-3′ | |
| Mar-Ext2 | 5′-gtcaattcgagctcgggta-3′ | |
| Mar-Int1 | 5′-tagcgacgccatctatgtgtc-3′ | |
| Mar-Int2 | 5′-cttgaagggaactatgttg-3′ | |
| ARB1 | 5′-ggccacgcgtcgactagtacnnnnnnnnnngatat-3′ | |
| ARB6 | 5′-ggccacgcgtcgactagtacnnnnnnnnnnacgcc-3′ | |
| ARB2 | 5′-ggccacgcgtcgactagtac-3′ |
Italic letters in primer sequences indicate engineered restriction sites. Capital letters indicate mutated nucleotides.
Mariner transposon-based library screen of antibiotic-sensitive mutants.
The pMycoMar vector that carries a Himar1 transposon was used to make the mutation library (33). Wild-type M. smegmatis MC2155 was transformed with pMycoMar. Transformed bacteria were cultivated at 28°C overnight to recover and amplify the library before plating on LB agar plates containing 50 μg ml−1 kanamycin. After incubation for 3 to 5 days at 40°C, single colonies were picked and spotted in arrays on kanamycin-containing plates. These plates were used as “master plates” to replicate to NE plates (26) containing different antibiotics. Colonies which grew on kanamycin NE plates but failed to grow on selected antibiotic plates were subjected to antibiotic disk tests to confirm their sensitivity profile.
Arbitrary PCR identification of drug-sensitive transposants.
The identification of transposon mutants by using arbitrary PCR was carried out as described previously (28). A first round of PCR was done using the Roche Expand long-template PCR system with the random annealing primers ARB1/ARB6 and the pMycoMar-specific primers MarExt1 and pMarExt2 (Table 1). Cells from colonies grown on kanamycin plates were directly used as template for the PCR. Annealing temperature was set at 45°C. Products of the first-round PCR were used as template for the second-round PCR, which used Taq polymerase (Roche) and the primers ARB2 and MarInt1/MarInt2. PCR products from the second round were cleaned up using a QIAGEN PCR purification kit and sequenced. PCR using primers flanking the identified open reading frame or Southern blot were used to identify the precise insertion site.
Cloning of fbp genes and complementation.
The GC-rich PCR system (Roche) was used to clone genes from M. smegmatis genomic DNA. The fbpAMS gene was PCR amplified using the primers MS_FbpA.EB and MS_FbpA.HXb. fbpDMS was amplified using the primers MS_FbpD.EB and MS_FbpD.PHXb (Table 1). PCR products were ligated to the pGEM-T Easy vector (Promega) and sequenced. DNA fragments were then removed by digestion with EcoRI and HindIII and cloned into pMV361 (36) cut with the same enzymes to fuse them with the heat shock promoter hsp60. Genes fused with the hsp60 promoter were then excised from pMV361 using NheI/XbaI and ligated into pMycVec2 (18) digested with SpeI. These plasmids were electroporated into the MAR1 mutant, and transformants were selected on hygromycin-containing LB agar plates. The M. tuberculosis fbpAMTB gene was PCR amplified using the primers TB_FbpA.EB and TB_FbpA.ClXbE and cloned into pMV361 using EcoRI/ClaI. The fused hsp60-fbpAMTB was cloned into pMycVec2 as described above. To construct the vector expressing fbpAMS and fbpDMS, the NheI/XbaI DNA fragment carrying hsp60-fbpAMS was filled in with Klenow polymerase I and blunt-end ligated into the EcoRV site of pMycVec2-hsp60-fbpDMS.
The mutated allele fbpAMS(S171A) was made using the primers S171fbpAMS-up and S171fbpAMS-down in combination with MS_FbpA.EB and MS_FbpA.HXb in a two-round PCR protocol. This changed the TCG (serine) codon into GCG (alanine), corresponding to amino acid residue 171.
Drug resistance measurements.
Antibiotic susceptibilities of strains were measured on NE medium (26) using a diffusion assay with antibiotic disks (Pasteur Diagnostics). MICs were determined using E-test antibiotic strips (AB Biodisk) following instructions of the manufacturer. Exponential cultures grown in Middlebrook 7H9 broth supplemented with 0.5% Tween 80 were harvested after 20 h. Cultures were normalized to an optical density at 600 nm of 1.5, and 100 μl was seeded in top agar on NE medium. E-test strips were applied, and MICs were scored after 60 h at 37°C. Values were read from the scale (in μg ml−1) at the point of intersection between the inhibition ellipse edge and the E-test strip.
Extraction and analyses of MAMEs.
MAMEs were extracted as described previously (6). Briefly, 2 ml of 15% tetrabutylammonium hydroxide was added to 50 mg of either defatted cells or whole cells of M. smegmatis in an 8.5-ml tube and heated at 100°C, overnight. After cooling, the reaction mixture was diluted with 2 ml water followed by addition of 1 ml dichloromethane and 250 μl iodomethane and stirred for 30 min. The upper layer was then removed and the lower organic layer was washed with 3 ml of 1 M hydrochloric acid, followed by 3 ml of water. The samples were dried under a stream of nitrogen using a sand bath at 37°C. The crude MAMEs thus obtained were dissolved in 0.5 ml of dichloromethane, transferred to a polypropylene microcentrifuge tube, and evaporated to dryness under nitrogen. The residue was dissolved in a mixture of 0.2 ml toluene and 0.1 ml acetonitrile and a further addition of 0.2 ml acetonitrile. Samples were incubated for 1 h at 4°C. The precipitated MAMEs were removed from the supernatant by centrifugation and resuspended in dichloromethane prior to thin-layer chromatography (TLC) analysis. These MAMEs were analyzed by TLC using a solvent system containing petroleum ether-diethyl ether (95:5) (6). Individual MAMEs were revealed by charring at 110°C for 15 min using 5% ethanolic molybdophosphoric acid.
Extraction and analyses of TDM.
Extraction of TDM was performed as described elsewhere (16). Bacteria were vortexed vigorously in petroleum ether for 2 min, followed by 5 min of incubation at room temperature. The culture was centrifuged at 500 × g for 10 min. The supernatant was removed, and the extraction process was repeated twice. The petroleum ether extracts of mycobacteria contain primarily TDM (>95% of the total extract), with relatively small quantities of free mycolic acid glycerides, menaquinones, and hydrocarbons present. The resulting TDM was analyzed by TLC (Merck 5554-silica gel 60F254; 6.6 cm by 6.6 cm) using chloroform-methanol (9:1) as the solvent system. The spots were visualized by spraying with 5% sulfuric acid containing 10% α-naphthol followed by charring for 10 min.
The TDM of M. smegmatis strains were quantified by radioactive labeling. Briefly, 5 Ci [14C]acetate (2.22 GBq mmol−1; Amersham) was added to 100 ml of exponential-phase bacterial cultures, which were incubated for 3 to 5 h. Labeling was stopped by centrifugation, and the cell pellets were extracted with petroleum ether as described above. The crude TDM (>95% pure) thus obtained was purified by TLC and quantified by scintillation counting. A minimum of three sequential determinations was performed from separate preparations. Infrared (IR) and matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) analyses were carried out to verify the identity of the TDM (see below).
Infrared spectroscopic analysis of TDM.
The identity of the extracted TDM of M. smegmatis was confirmed by IR spectroscopy. IR spectra of samples were recorded using a Perkin-Elmer (Spectrum One) FT-IR spectrometer. Mycobacterium tuberculosis TDM (Sigma) was used as the standard. The purified TDM was dissolved in chloroform to obtain the IR spectra.
MALDI-TOF mass spectrometry analysis of TDM and MAMEs.
MALDI-TOF mass spectra (in the positive mode) were acquired on a Voyager-DE STR mass spectrometer (PerSeptive Biosystems) equipped with a pulsed nitrogen laser emitting at 337 nm. Samples were analyzed in the reflectron mode using an extraction delay time of 100 ns and an accelerating voltage operating in positive ion mode of 20 kV (21). To improve the signal-to-noise ratio, 150 single shots were averaged for each mass spectrum and, typically, four individual spectra were accumulated to generate a summed spectrum.
RESULTS
Isolation of a multidrug-sensitive mutant of M. smegmatis from a mariner transposon library.
We used the Himar1 transposon system (20) to mutagenize wild-type M. smegmatis MC2155. This library of 15,000 isolated insertion mutants was replica plated and screened for increased sensitivity to various antibiotics, including erythromycin, spectinomycin, spiramycin, and chloramphenicol (see Materials and Methods).
A mutant (MAR1), found in a screen for erythromycin sensitivity (MIC less than 8 μg ml−1), displayed additional sensitivities to many other antibiotics. Disk diffusion assays showed that MAR1 was more sensitive to many clinically important antibiotics with diverse chemical structures and functions. These included hydrophilic and hydrophobic antibiotics targeted to the ribosome (macrolides, lincosamides, streptogramins, spectinomycin, and chloramphenicol), cell wall assembly enzymes (vancomycin, imipenem, and daptomycin), and RNA polymerase (rifampin). MICs for the four representative antibiotics were quantified using E-test diffusion strips; MAR1 was 24-, 85-, ≫8-, and 12-fold more susceptible to erythromycin, imipenem, rifampin, and vancomycin, respectively (Fig. 1).
FIG. 1.

Effect of fbpA allele expression on antibiotic susceptibility of Mycobacterium smegmatis. Antibiotic sensitivities of the wild-type MC2155, the MAR1 mutant, and the MAR1 mutants expressing fbpAMS, fbpAMS(S171A), fbpAMTB, fbpDMS, or both fbpAMS and fbpDMS were assayed using E-test strips containing antibiotic gradients. A. Representative E-test assay with rifampin (left) and erythromycin (right) strips. B. MICs (in μg ml−1) of M. smegmatis strains to vancomycin (VA), rifampin (RI), erythromycin (EM), and imipenem (IP) as determined using this assay.
MAR1-a fbpA mutant.
Arbitrary PCR first localized the transposon mutation in MAR1 to a gene encoding a protein of the “antigen 85 complex,” also known as “fibronectin binding proteins” (Fbps). The amino acid sequence of the mutated gene (fbpAMS) had highest similarities to M. tuberculosis fbpA (fbpAMTB) and homologs in other mycobacteria; the locus displayed synteny with the M. tuberculosis and Mycobacterium leprae genomes (Fig. 2A). All three genomes encode a fbpA-like gene upstream of a fbpD-like gene (Fig. 2A). Interestingly, the M. smegmatis locus has an additional fbp-like gene upstream of fbpAMS (fbpEMS) (Fig. 2A), which was less homologous to fbpA.
FIG. 2.

Identification of transposon mutation of MAR1 and the orthologous loci in Mycobacterium smegmatis, Mycobacterium tuberculosis, and Mycobacterium leprae genomes. A. Synteny of the orthologous loci in three mycobacterial genomes. MAR1 had a transposon mutation in a gene homologous to fbpAMTB. Sequencing mapped the insertion to a TA dinucleotide that introduced a stop codon after the Leu79 residue. B. PCR amplification using primers flanking the fbpAMS open reading frame of wild-type M. smegmatis MC2155. The transposon insertion in MAR1 resulted in a corresponding increase in the size of the PCR product. C. Western blot assay results using monoclonal antibody against the M. tuberculosis antigen 85 complex. The higher-molecular-weight signal (arrow) was missing in the MAR1 protein filtrate sample, and it reappeared when MAR1 was complemented with fbpAMS. D. Alignment of the carboxyesterase domain of Fbp proteins from M. smegmatis (MS), M. tuberculosis (MTB), and the human carboxyesterase D (CarbestD). The carboxylesterase consensus sequence is underlined. and the conserved serine 171 is marked in bold. Amino acid shading indicates the degree of conservation.
Alignment of amino acid sequences of these proteins with other Fbp proteins revealed that FbpEMS and FbpAMS contained a carboxyesterase domain with a conserved serine 171 (Fig. 2D) that is essential for in vitro mycolyltransferase activity (5). Phylogenic analyses also supported the homology of FbpA and FbpD proteins in the three genomes (not shown). FbpEMS was equally similar to M. tuberculosis FbpA, FbpB, and FbpC.
The disruption was further confirmed by recovering the mutant locus by PCR using primers flanking the putative open reading frame. The mutant gene generated a larger fragment corresponding to the inserted transposon. Insertion of the transposon resulted in a decreased mobility of the PCR product on an agarose gel (Fig. 2B). Sequencing of the junction region from these PCR products identified the insertion site at a dinucleotide TA (Fig. 2A), thus introducing a stop codon after the triplet encoding the Leu79 residue. To confirm loss of the fbpAMS gene product, Western blot assays were done using an antibody against the M. tuberculosis antigen 85 complex HYT27 (Antibodyshop A/S, Gentofte, Denmark). There were two protein bands that cross-reacted with HYT27 in MC2155 culture filtrate; the higher-molecular-weight, weaker band was missing in the MAR1 filtrate (Fig. 2C).
Complementation of MAR1 with the fbpAMS gene transcribed from the heat shock promoter hsp60 (see Materials and Methods) only partially restored resistance to antibiotics (Fig. 1). Expression of the M. tuberculosis fbpA gene in MAR1 provided a lower level of antibiotic resistance (Fig. 1).
The fbpAMS mutation caused major alterations in colonial morphology and hydrophobicity.
The MAR1 mutant had significant alterations in colonial morphology. The edges of wild-type colonies were irregular, while those of MAR1 were smooth and rounded (Fig. 3A). On the surface, colonies of the wild type formed wrinkled and acne-like structures after a few days of cultivation, while MAR1 colonies were shiny and smooth (Fig. 3A). In addition, while wild-type colonies were dry and fragile, MAR1 colonies were wetter and stickier. This phenotype was partially suppressed when culture media were supplemented with Tween 80 detergent. These observations first suggested that the mutant surface topology might be due to alterations in its surface tension. This was verified by a simple experiment in which droplets of water or oil were applied to wild-type or mutant lawns (Fig. 3B). On a lawn of a wild-type M. smegmatis culture, a drop of water formed a bead while a drop of oil readily spread over the surface. This assay provided a simple visualization of the characteristic hydrophobicity of mycobacteria. In contrast, water spread on a MAR1 lawn while oil formed a bead. This phenotype was suppressed when MAR1 was complemented with fbpAMS. Colonial morphology and hydrophobicity of MAR1 were partially rescued by expression of fbpAMTB (not shown). These observations implied that the characteristic hydrophobicity of the mycobacterial cell wall was lost in the MAR1 mutant and was due to fbpA activity.
FIG. 3.
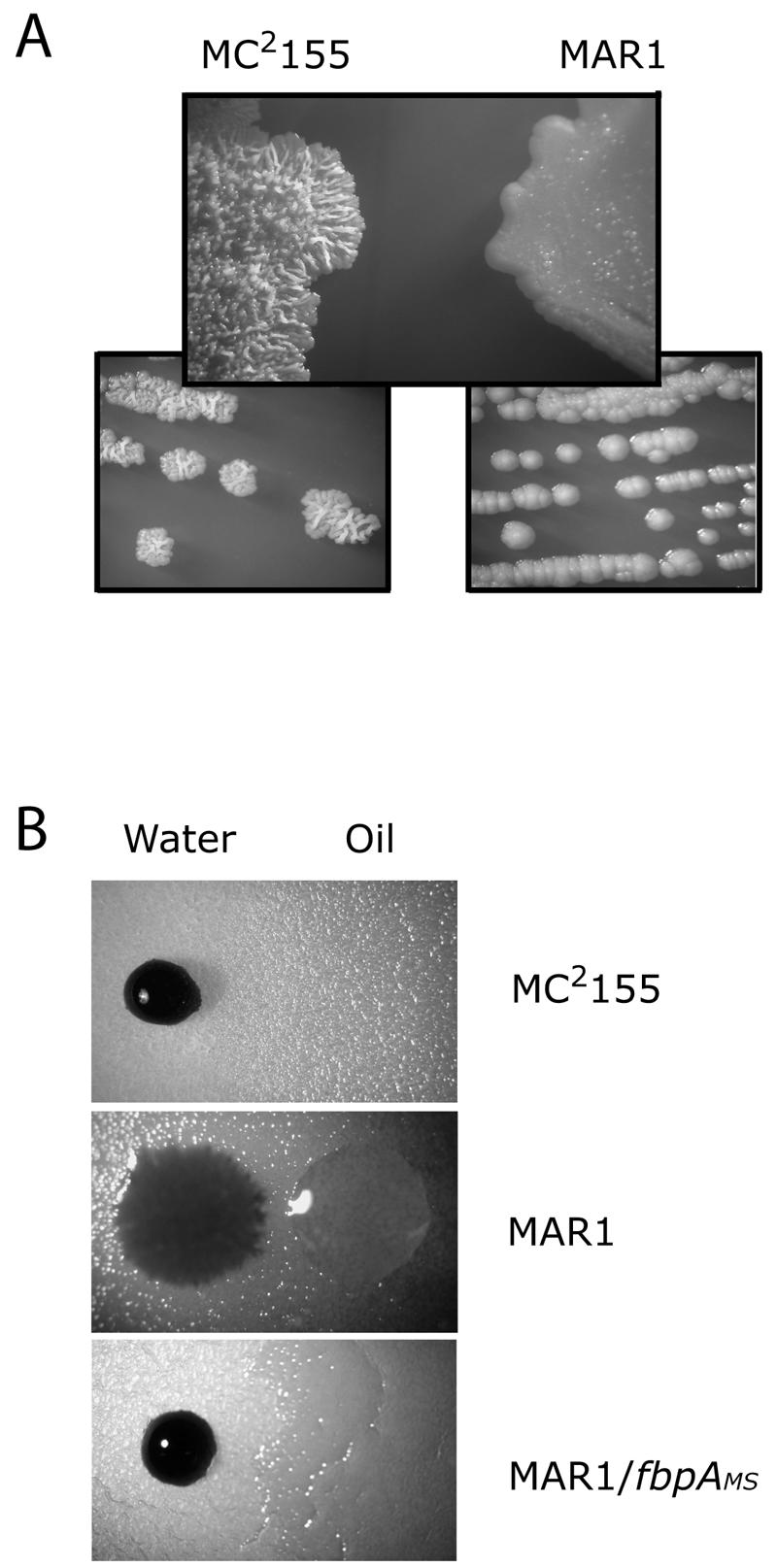
Altered morphologies and hydrophobicities of MAR1 colonies. A. The MAR1 colonial morphology was smooth and shiny compared to the rough and irregular surface and edge of wild-type M. smegmatis MC2155. B. The MAR1 surface was more hydrophilic compared to wild-type M. smegmatis MC2155. Droplets of oil or water containing trypan blue were applied to the surface of mutant and wild-type colonies. In the upper and lower panels, the oil spread into a thin film over the surface.
FbpDMS expression does not play a role in the phenotypes of MAR1.
To test if fbpDMS, the gene downstream of fbpAMS, was involved in the observed phenotypes of MAR1, cloned fbpDMS was expressed from the heat shock promoter in MAR1 (as described above for fbpAMS). fbpDMS expression could not rescue the antibiotic resistance (Fig. 1) or morphological defects (not shown) of MAR1. However, when this fbpDMS expression plasmid was supplemented with another DNA fragment containing hsp60-fbpAMS, both the defects were partially rescued (antibiotic resistance results are shown in Fig. 1). This suggested that the MAR1 phenotypes were FbpAMS specific and did not involve FbpDMS.
FbpAMS activity requires the serine esterase domain.
The reduced antibiotic resistance in the fbpAMS disruption mutant was likely a consequence of a loss of mycolyltransferase activity. Fbp enzymatic activity is dependent on a serine esterase domain (5), which forms a trehalose-binding pocket (32). Mutagenesis studies revealed that the conserved serine 171 in this domain was essential for in vitro mycolyltransferase activity, ligating mycolate to trehalose (5). In order to test whether the FbpAMS-dependent antibiotic resistance was due to this activity, we constructed a vector expressing a mutated allele of FbpAMS in which serine 171 was changed to alanine (S171A). This allele, fbpAMS(S171A), failed to rescue the defects of MAR1 in antibiotic resistance and colonial morphology, suggesting that the MAR1 phenotypes were due to a loss of mycolyltransferase activity (Fig. 1).
The MAR1 cell envelope had a decreased trehalose-dimycolate content.
The two principle mycolate complexes (TDM and MAMEs) were analyzed in the wild type, MAR1, and the complemented strain. Thin-layer chromatography of the products suggested a significant decrease in the TDM content of MAR1 (Fig. 4A). To confirm this result, total lipids of M. smegmatis cultures were labeled by growth in medium supplemented with 14C-radiolabeled acetate. TDM from equal amounts of labeled cells, as measured by weight, was extracted and purified in organic solvents; its identity was confirmed by IR spectroscopy (Fig. 4C) and MALDI-TOF (not shown). Scintillation counting revealed a 45% reduction of TDM in MAR1 compared to the wild type (Fig. 4B).
FIG.4.
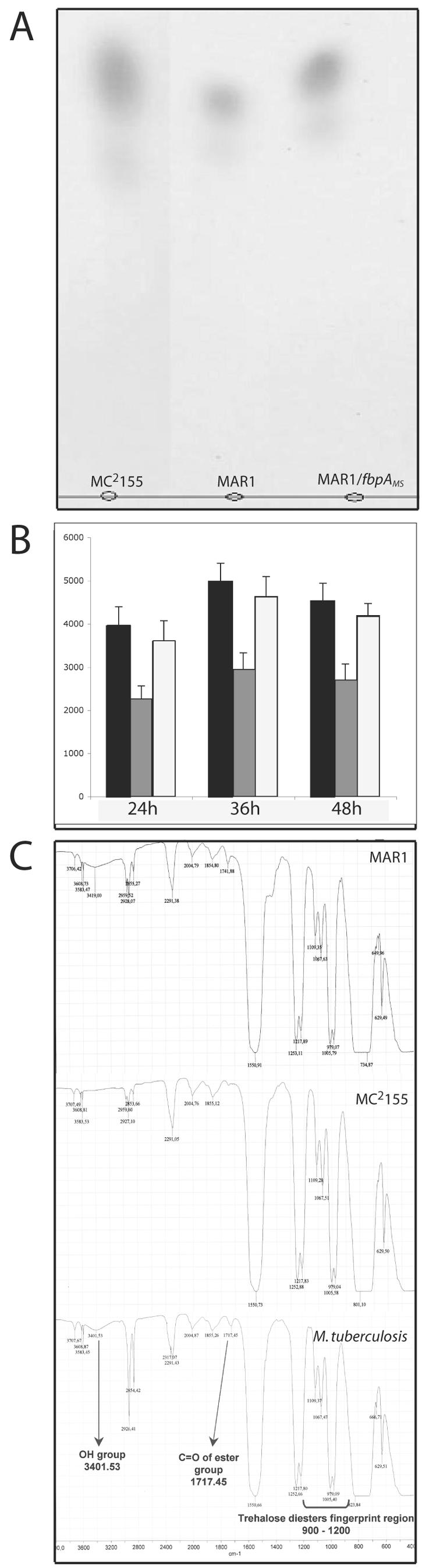
α,α′-trehalose dimycolate from Mycobacterium smegmatis MC2155, MAR1, and the fbpAMS-complemented strain. A. Thin-layer chromatography of TDM extracts from equal amount of cells of mycobacterial strains. B. Quantitative analysis of TDM extracts from [14C]acetate-labeled mycobacterial cultures of wild-type M. smegmatis MC2155 (dark bars), MAR1 (shaded bars), and the fbpAMS complemented strain (empty bars) during growth, by scintillation counting of TLC-purified TDM. C. Identities of TDM extracts from mycobacterial cultures were compared to purified Mycobacterium tuberculosis TDM by IR spectroscopy.
MAMEs derived from the mycolates covalently linked to arabinogalactan were, however, unchanged in MAR1. Thin-layer chromatography of radiolabeled extracts showed the content of α-MAME, α′-MAME, and epoxy-MAME were similar in MC2155 and MAR1 (Fig. 5). Detailed MALDI-TOF analysis of MAMEs isolated from TLC plates also showed no difference in their masses (not shown).
FIG. 5.

MAMEs of Mycobacterium smegmatis. The mycolates covalently linked to arabinogalactans of M. smegmatis MC2155, MAR1, and the fbpAMS-complemented strain were extracted and revealed by thin-layer chromatography, including α-MAME, α′-MAME, and epoxy-MAME. Radioisotope labeling experiments (as described for Fig. 4) indicated that the three strains incorporated equal levels of [14C]acetate into each of the three MAMEs.
DISCUSSION
Without direct evidence, mycobacterial cell wall structure and antibiotic resistance is believed to depend on mycolyltransferase activities that catalyze the assembly of mycolic acids into an impermeable envelope. In vitro and in vivo experiments established that this is carried out by the proteins of the “antigen 85 complex” (38), which includes FbpA, FbpB, and FbpC (5, 17, 19); however, the antibiotic sensitivities or altered colonial morphologies of these fbp mutants were not reported.
The amphiphilic properties of TDM, rather than its chemical specificity, may determine many of its important biological activities (4, 31). Using hydrophobic beads as a model system for TDM in the outer cell wall of mycobacteria, Retzinger et al. (31) demonstrated that at low concentrations TDM formed a monolayer, with trehalose head groups exposed to the aqueous surface within a matrix of hydrophobic tails extended along the hydrophobic surface of the bead. At higher concentrations, TDM formed extended cord-like structures resembling the irregular cording of mycobacterial rods on the colony surface. Similarly, decreased surface TDM concentration may correlate with the change from an irregular hydrophobic colony surface that grows upward in the wild type to the flattened smooth hydrophilic surface of the MAR1 mutant. In addition, the ability of TDM to readily entrap peptides and proteins in such amphiphilic monolayers may allow it to neutralize antibiotics or antimicrobial compounds of the immune system (2, 11, 16).
A more conventional model would explain the antibiotic sensitivity of MAR1 by changes in permeability rather than entrapment. Sensitivity to hydrophilic antibiotics such as vancomycin and β-lactam-based antibiotics might be explained by a structural defect in the hydrophobic mycolate layer that normally provides a protective permeability barrier, thus allowing antibiotics access to targets in interior peptidoglycan layer (15). Decreased mycolic acid content can also increase the sensitivity of M. smegmatis to hydrophobic antibiotics that have cytoplasmic targets associated with increases in the fluidity of the envelope (23, 37).
Antibiotic resistance and colonial morphology of MAR1 were only partially restored by expression of fbpAMS from the heat shock promoter hsp60 (Fig. 1). This promoter is much less active than the native fbpAMTB promoter (1) and presumably not synchronized with expression of other cell envelope biosynthesis enzymes. Heterologous expression of fbpAMTB in M. smegmatis MAR1 had even less activity, suggesting variations in specificity and enzymatic activities among Fbp proteins from different mycobacterial species.
M. tuberculosis, Mycobacterium bovis, and M. leprae genomes encode another fbp-like gene adjacent to and potentially cotranscribed with fbpA (annotated as fbpD or fbpC1) (9, 10, 14). However, these FbpD paralogs do not contain the carboxyesterase domain needed for mycolyltransferase activity (Fig. 2D); furthermore, structural determination showed that FbpDMTB lacked catalytic elements required for mycolyltransferase activity as well as α,α′-trehalose monomycolate-binding sites (39, 40). Mycolyltransferase activity of FbpDMTB was not detected in vitro (19). Nevertheless, retention of the gene, especially in the downsized M. leprae genome, suggests its utility, perhaps providing an altered catalytic activity or a nonenzymatic function (39, 40). To examine whether a polar effect of the transposon on fbpDMS expression was involved in MAR1 phenotypes, the gene was placed under the control of the hsp60 promoter. These studies showed that fbpDMS could not rescue either the antibiotic resistance or morphological defects of MAR1.
Mutation of the conserved serine in the esterase domain to alanine had been shown to inactivate trehalose mycolyltransferase activity of Fbp proteins in vitro (5, 32). Our in vivo data showed that this mutation inactivated the ability of fbpAMS to rescue the defects of MAR1. Thus, the antibiotic sensitivity and abnormal morphology of MAR1 were the consequences of a loss in a rather specific mycolyltransferase activity provided by FbpAMS.
Consistent with previous analyses of an M. tuberculosis fbpA mutant (30), our analyses of MAR1 showed a difference in TDM but not MAMEs. Thin-layer chromatography and mass spectrometry analyses showed no alteration in quantity or structure of MAME components. However, the TDM content of MAR1 was 45% lower than wild type. In principle, other M. smegmatis fbp genes or enzymes having similar catalytic functions may account for the remaining TDM synthesis. Belisle et al. (5) reported that trehalose mycolyltransferase activity purified from M. smegmatis was composed of two different proteins which have identical isoelectric points (∼5.1) and N-terminal amino acid sequences (RPGLPVEY) but slightly different molecular masses (31 and 34 kDa) (5, 34). This sequence corresponded to the N-terminal sequence of secreted FbpAMS but not FbpEMS and FbpDMS. A BLAST search of the unfinished M. smegmatis genome sequence database revealed a fourth fbp gene homolog that also has this N-terminal sequence. This suggested that FbpAMS was one of the trehalose mycolyltransferases purified by Belisle et al. (5) and that the other Fbp homolog may provide the lower levels of TDM retained in MAR1. It will be interesting to see whether disruption of this fbp gene completely eliminates TDM in the cell wall of MAR1 and leads to even higher sensitivity to antibiotics.
Our in vivo results, in addition to previous reports (5, 17, 19, 30), suggested that FbpA (and probably also FbpB) mycolyltransferase activity preferred trehalose as a substrate, while FbpC prefers arabinogalactan. These preferences may help explain the apparent redundancy of Fbp paralogs. However, it is still unclear why all three proteins (FbpAMTB, FbpBMTB, and FbpCMTB) have trehalose mycolyltransferase activities in vitro (5, 19) and why overexpression of fbpAMTB and fbpBMTB was able to rescue the ability of the fbpCMTB mutant to synthesize MAMEs (30). It is possible that the partial specificities of these enzymes are due to their preferred spatial cellular localizations, despite their comparable biochemical activities in vitro. Studies on mycobacterial cell biology in our laboratory suggest that the peptidoglycan layer of the mycobacterial cell wall is remodeled at the cell poles during growth (unpublished data). Proteins such as FbpC, involved in the coordinated assembly of the arabinogalactan mycolate layer of the cell wall matrix, may be colocalized at the pole, while enzymes such as FbpA that synthesize TDM and other more mobile mycolates may not have the same spatial constraints.
Our results suggested that FbpA-mediated biosynthesis of trehalose dimycolate is an intrinsic determinant of antibiotic resistance in mycobacteria. Recently, we have also discovered a regulatory protein (WhiB7) that provides resistance to diverse antibiotics that have entered the cytoplasm (25). These proteins may serve as attractive targets for the identification of inhibitors that render M. tuberculosis more sensitive to numerous antibiotics that are presently available. Such inhibitors could open up a powerful repertoire of currently redundant clinical antibiotics and minimize the problems associated with prolonged and limited chemotherapy.
Acknowledgments
We thank Eric J. Rubin, Nicholas Judson, John Mekalanos, William R. Jacobs, Jr., and Michael Niederweis for providing mycobacterial plasmids and strains used in this work, Richard W. Stokes for critical reading of the manuscript, and Paul Jenö for MALDI-TOF MS analyses. Preliminary sequence data of M. smegmatis were obtained from The Institute for Genomic Research through their website at http://www.tigr.org.
This work was supported by the Swiss National Science Foundation (NRP 4049-69384).
REFERENCES
- 1.Al-Zarouni, M., and J. W. Dale. 2002. Expression of foreign genes in Mycobacterium bovis BCG strains using different promoters reveals instability of the hsp60 promoter for expression of foreign genes in Mycobacterium bovis BCG strains. Tuberculosis (Edinburgh) 82:283-291. [DOI] [PubMed] [Google Scholar]
- 2.Armitige, L. Y., C. Jagannath, A. R. Wanger, and S. J. Norris. 2000. Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infect. Immun. 68:767-778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barry, C. E., III, R. E. Lee, K. Mdluli, A. E. Sampson, B. G. Schroeder, R. A. Slayden, and Y. Yuan. 1998. Mycolic acids: structure, biosynthesis and physiological functions. Prog. Lipid Res. 37:143-179. [DOI] [PubMed] [Google Scholar]
- 4.Behling, C. A., B. Bennett, K. Takayama, and R. L. Hunter. 1993. Development of a trehalose 6,6′-dimycolate model which explains cord formation by Mycobacterium tuberculosis. Infect. Immun. 61:2296-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belisle, J. T., V. D. Vissa, T. Sievert, K. Takayama, P. J. Brennan, and G. S. Besra. 1997. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 276:1420-1422. [DOI] [PubMed] [Google Scholar]
- 6.Besra, G. 1998. Preparation of cell-wall fractions from Mycobacteria, p. 91107. In T. Parish and N. G. Stoker (ed.), Mycobacteria protocols. Humana Press, Totowana, New Jersey. [DOI] [PubMed]
- 7.Braunstein, M., S. S. Bardarov, and W. R. Jacobs, Jr. 2002. Genetic methods for deciphering virulence determinants of Mycobacterium tuberculosis. Methods Enzymol 358:67-99. [DOI] [PubMed] [Google Scholar]
- 8.Brennan, P. J., and H. Nikaido. 1995. The envelope of mycobacteria. Annu. Rev. Biochem. 64:29-63. [DOI] [PubMed] [Google Scholar]
- 9.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, B. G. Barrell, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 10.Cole, S. T., K. Eiglmeier, J. Parkhill, K. D. James, N. R. Thomson, P. R. Wheeler, N. Honore, T. Garnier, C. Churcher, D. Harris, K. Mungall, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. M. Davies, K. Devlin, S. Duthoy, T. Feltwell, A. Fraser, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, C. Lacroix, J. Maclean, S. Moule, L. Murphy, K. Oliver, M. A. Quail, M. A. Rajandream, K. M. Rutherford, S. Rutter, K. Seeger, S. Simon, M. Simmonds, J. Skelton, R. Squares, S. Squares, K. Stevens, K. Taylor, S. Whitehead, J. R. Woodward, and B. G. Barrell. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007-1011. [DOI] [PubMed] [Google Scholar]
- 11.Copenhaver, R. H., E. Sepulveda, L. Y. Armitige, J. K. Actor, A. Wanger, S. J. Norris, R. L. Hunter, and C. Jagannath. 2004. A mutant of Mycobacterium tuberculosis H37Rv that lacks expression of antigen 85A is attenuated in mice but retains vaccinogenic potential. Infect. Immun. 72:7084-7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daffe, M., and P. Draper. 1998. The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39:131-203. [DOI] [PubMed] [Google Scholar]
- 13.Gao, L. Y., F. Laval, E. H. Lawson, R. K. Groger, A. Woodruff, J. H. Morisaki, J. S. Cox, M. Daffe, and E. J. Brown. 2003. Requirement for kasB in Mycobacterium mycolic acid biosynthesis, cell wall impermeability and intracellular survival: implications for therapy. Mol. Microbiol. 49:1547-1563. [DOI] [PubMed] [Google Scholar]
- 14.Garnier, T., K. Eiglmeier, J. C. Camus, N. Medina, H. Mansoor, M. Pryor, S. Duthoy, S. Grondin, C. Lacroix, C. Monsempe, S. Simon, B. Harris, R. Atkin, J. Doggett, R. Mayes, L. Keating, P. R. Wheeler, J. Parkhill, B. G. Barrell, S. T. Cole, S. V. Gordon, and R. G. Hewinson. 2003. The complete genome sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. USA 100:7877-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goffin, C., and J. M. Ghuysen. 2002. Biochemistry and comparative genomics of SxxK superfamily acyltransferases offer a clue to the mycobacterial paradox: presence of penicillin-susceptible target proteins versus lack of efficiency of penicillin as therapeutic agent. Microbiol. Mol. Biol. Rev. 66:702-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Indrigo, J., R. L. Hunter, Jr., and J. K. Actor. 2002. Influence of trehalose 6,6′-dimycolate (TDM) during mycobacterial infection of bone marrow macrophages. Microbiology 148:1991-1998. [DOI] [PubMed] [Google Scholar]
- 17.Jackson, M., C. Raynaud, M. A. Laneelle, C. Guilhot, C. Laurent-Winter, D. Ensergueix, B. Gicquel, and M. Daffe. 1999. Inactivation of the antigen 85C gene profoundly affects the mycolate content and alters the permeability of the Mycobacterium tuberculosis cell envelope. Mol. Microbiol. 31:1573-1587. [DOI] [PubMed] [Google Scholar]
- 18.Kaps, I., S. Ehrt, S. Seeber, D. Schnappinger, C. Martin, L. W. Riley, and M. Niederweis. 2001. Energy transfer between fluorescent proteins using a co-expression system in Mycobacterium smegmatis. Gene 278:115-124. [DOI] [PubMed] [Google Scholar]
- 19.Kremer, L., W. N. Maughan, R. A. Wilson, L. G. Dover, and G. S. Besra. 2002. The M. tuberculosis antigen 85 complex and mycolyltransferase activity. Lett. Appl. Microbiol. 34:233-237. [DOI] [PubMed] [Google Scholar]
- 20.Lampe, D. J., M. E. Churchill, and H. M. Robertson. 1996. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 15:5470-5479. [PMC free article] [PubMed] [Google Scholar]
- 21.Laval, F., M. A. Laneelle, C. Deon, B. Monsarrat, and M. Daffe. 2001. Accurate molecular mass determination of mycolic acids by MALDI-TOF mass spectrometry. Anal. Chem. 73:4537-4544. [DOI] [PubMed] [Google Scholar]
- 22.Liu, J., C. E. Barry III, and H. Nikaido. 1999. Cell wall: physical structure and permeability, p. 220-239. In J. Dale (ed.), Mycobacteria—molecular biology and virulence. Blackwell Sciences Ltd., Oxford, England.
- 23.Liu, J., and H. Nikaido. 1999. A mutant of Mycobacterium smegmatis defective in the biosynthesis of mycolic acids accumulates meromycolates. Proc. Natl. Acad. Sci. USA 96:4011-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minnikin, D. 1982. Complex lipids: their chemistry, biosynthesis, and roles, p. 94-184. In C. Ratledge and J. Standford (ed.), The biology of mycobacteria, vol. 1. Academic Press, London, England. [Google Scholar]
- 25.Morris, R. P., L. Nguyen, J. Gatfield, K. Visconti, K. T. Nguyen, D. Schnappinger, S. Ehrt, Y. Liu, L. Heifets, J. Pieters, G. Schoolnik, and C. J. Thompson. 2005. Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 102:12200-12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murakami, T., H. Anzai, S. Imai, A. Satoh, K. Nagaoka, and C. J. Thompson. 1986. The bialaphos biosynthetic genes of Streptomyces hygroscopicus: molecular cloning of the gene cluster. Mol. Gen. Genet. 205:42-50. [Google Scholar]
- 27.Niederweis, M. 2003. Mycobacterial porins—new channel proteins in unique outer membranes. Mol. Microbiol. 49:1167-1177. [DOI] [PubMed] [Google Scholar]
- 28.O'Toole, G. A., L. A. Pratt, P. I. Watnick, D. K. Newman, V. B. Weaver, and R. Kolter. 1999. Genetic approaches to study of biofilms. Methods Enzymol. 310:91-109. [DOI] [PubMed] [Google Scholar]
- 29.Peake, P., A. Gooley, and W. J. Britton. 1993. Mechanism of interaction of the 85B secreted protein of Mycobacterium bovis with fibronectin. Infect. Immun. 61:4828-4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puech, V., C. Guilhot, E. Perez, M. Tropis, L. Y. Armitige, B. Gicquel, and M. Daffe. 2002. Evidence for a partial redundancy of the fibronectin-binding proteins for the transfer of mycoloyl residues onto the cell wall arabinogalactan termini of Mycobacterium tuberculosis. Mol. Microbiol. 44:1109-1122. [DOI] [PubMed] [Google Scholar]
- 31.Retzinger, G. S., S. C. Meredith, K. Takayama, R. L. Hunter, and F. J. Kezdy. 1981. The role of surface in the biological activities of trehalose 6,6′-dimycolate. Surface properties and development of a model system. J. Biol. Chem. 256:8208-8216. [PubMed] [Google Scholar]
- 32.Ronning, D. R., T. Klabunde, G. S. Besra, V. D. Vissa, J. T. Belisle, and J. C. Sacchettini. 2000. Crystal structure of the secreted form of antigen 85C reveals potential targets for mycobacterial drugs and vaccines. Nat. Struct. Biol 7:141-146. [DOI] [PubMed] [Google Scholar]
- 33.Rubin, E. J., B. J. Akerley, V. N. Novik, D. J. Lampe, R. N. Husson, and J. J. Mekalanos. 1999. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. USA 96:1645-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sathyamoorthy, N., and K. Takayama. 1987. Purification and characterization of a novel mycolic acid exchange enzyme from Mycobacterium smegmatis. J. Biol. Chem. 262:13417-13423. [PubMed] [Google Scholar]
- 35.Snapper, S. B., R. E. Melton, S. Mustafa, T. Kieser, and W. R. Jacobs, Jr. 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4:1911-1919. [DOI] [PubMed] [Google Scholar]
- 36.Stover, C. K., V. F. de la Cruz, T. R. Fuerst, J. E. Burlein, L. A. Benson, L. T. Bennett, G. P. Bansal, J. F. Young, M. H. Lee, G. F. Hatfull, et al. 1991. New use of BCG for recombinant vaccines. Nature 351:456-460. [DOI] [PubMed] [Google Scholar]
- 37.Wang, L., R. A. Slayden, C. E. Barry III, and J. Liu. 2000. Cell wall structure of a mutant of Mycobacterium smegmatis defective in the biosynthesis of mycolic acids. J. Biol. Chem. 275:7224-7229. [DOI] [PubMed] [Google Scholar]
- 38.Wiker, H. G., and M. Harboe. 1992. The antigen 85 complex: a major secretion product of Mycobacterium tuberculosis. Microbiol. Rev. 56:648-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson, R. A., W. N. Maughan, L. Kremer, G. S. Besra, and K. Futterer. 2004. The structure of Mycobacterium tuberculosis MPT51 (FbpC1) defines a new family of non-catalytic alpha/beta hydrolases. J. Mol. Biol. 335:519-530. [DOI] [PubMed] [Google Scholar]
- 40.Wilson, R. A., S. Rai, W. N. Maughan, L. Kremer, B. M. Kariuki, K. D. Harris, T. Wagner, G. S. Besra, and K. Futterer. 2003. Crystallization and preliminary X-ray diffraction data of Mycobacterium tuberculosis FbpC1 (Rv3803c). Acta Crystallogr. D 59:2303-2305. [DOI] [PubMed] [Google Scholar]