

Abstract
Evidence from in vivo studies suggests that some imputs to cardiac hypertrophy are opposed by the actions of estrogen. However, the mechanisms of E2 action in this respect are mainly unknown. An important pathway that is utilized by multiple hypertrophic stimuli involves the activation of the tyrosine phosphatase, calcineurin (PP2B). Here we show that 17β -estradiol (E2) significantly prevents angiotensin II (AngII)- or endothelin-1 (ET-1)-induced new protein synthesis, skeletal muscle actin expression, and increased surface area in cultured rat cardiomyocytes. ET-1 stimulated calcineurin phosphatase activity, resulting in new protein synthesis, and both were prevented by E2. E2 induced the MCIP1 gene, an inhibitor of calcineurin activity, via phosphatidylinositol 3-kinase, transcriptional, and mRNA stability mechanisms. Small interfering RNA for MCIP1 significantly reversed both the E2 restraint of protein synthesis and the inhibition of AngII-induced calcineurin activity. AngII-induced the translocation of the hypertrophic transcription factor, NF-AT, to the nucleus of the cardiomyocyte and stimulated NF-AT transcriptional activity. Both were prevented by E2. AngII also stimulated the activation of ERK and protein kinase C, contributing to cardiac hypertrophy. E2 inhibited these pathways, related to the stimulation of atrial natriuretic peptide production and secretion. Thus, restraint of calcineurin and kinase signaling to the hypertrophic program underlie these important effects of E2.
Cardiac hypertrophy develops in response to hypertension and is consequent to 80% of all myocardial infarctions. Hypertrophy is an independent risk factor for the development of ischemia, arrhythmia, and sudden death (1, 2). The most important vascular hormone that contributes to the development of hypertrophy is angiotensin II (AngII)1 (3).
Myocardial hypertrophy frequently develops in older humans. Women have a lower overall incidence of left ventricular hypertrophy than men, but left ventricular hypertrophy in postmenopausal women exceeds the incidence in age-matched males (4). The latter can be reversed in postmenopausal women by hormone replacement therapy (5).
Animal studies support a possible anti-hypertrophic action of estrogen in the heart. In ovariectomized mice, estrogen supplementation causes a 30% reduction in pressure overload-induced hypertrophy (6). Although the basis is not known, one mechanism could be related to intracellular calcium. In this regard, the FKBP12.6 gene encodes a protein that modulates the intracellular ryanodine receptor and calcium store release. Disruption of this gene results in severe cardiac hypertrophy only in male mice. However, when female FKBP12.6 null mice are treated with tamoxifen, a specific estrogen receptor (ER) antagonist, hypertrophic changes similar to those in the male mice are observed (7). Perhaps estrogen dampens either the unregulated intracellular calcium sparking or resulting events that cause cardiac hypertrophy. The responsible mechanism for this important steroid action is obscure.
In general, the actions of estrogen are mediated through nuclear ER by transcribing genes that encode proteins that lead to the biological effects. Additional actions of estrogen are mediated by plasma membrane ER-initiated steroid signaling. Such signaling impacts both transcriptional (8) and nontranscriptional effects of the sex steroid. We postulated that genes up-regulated by membrane ER signaling could oppose the development of cardiac hypertrophy and impact intracellular calcium signaling to this disorder.
Here we report that estradiol (E2) limits vascular hormone-induced cardiomyocyte hypertrophy. In isolated cardiomyocytes, E2 inhibits the important hypertrophic pathway involving the calcium-sensitive protein phosphatase, calcineurin, as induced by the vascular peptides AngII or endothelin-1. We also found that AngII stimulates additional hypertrophic signaling involving ERK and PKC. E2 inhibits this signaling, dependent upon the ability of the sex steroid to up-regulate atrial natriuretic peptide (ANP) production and secretion. Thus, E2 modulates multiple signal inputs to prevent cardiomyocyte hypertrophy in vitro.
MATERIALS AND METHODS
Isolation and Culture of Rat Neonatal Cardiomyocytes
Myocytes were isolated from the hearts of 1-to-3-day-old rats or pregnant female rats (Charles River Laboratories) using a cardiomyocyte isolation kit (Worthington), according to the manufacturer’s instructions. The cells were incubated in Dulbecco’s modified Eagle’s medium/F-12 supplemented with 10% fetal bovine serum, 1× ITS (insulin/transferrin/selenium) (Sigma) antibiotic and antimycotic, and 10 μ g/ml fibronectin (to aid adherence).
Measurement of Protein Synthesis, Protein Secretion, and Cell Area
After 24 h in media without serum, the cells were treated with either 100 nmol/liter AngII or 10 nmol/liter ET-1 or no treatment, in the presence or absence of 10 nmol/liter E2. New protein synthesis, a marker of hypertrophy, was determined using [3H]leucine uptake. The cells were cultured as above with the addition of 1 μCi/ml [3H]leucine for 48 h. After 48 h, the cells were rinsed in Dulbecco’s modified Eagle’s medium/F-12 and lysed with Trizol. The protein and RNA were extracted per the manufacturer’s instructions. The protein fraction was counted in a scintillation counter, and the mean ± S.E. of the counts was plotted. Differences between treatments were compared for combined experimental data, and the results were analyzed by analysis of variance plus Schefe’s test (p < 0.05 was significant). ANP protein secretion into the cell incubation medium was determined after 24 h of incubation with AngII ± E2 or E2 alone. The medium was concentrated, and Western blot using ANP antisera (Phoenix Pharmaceutical) was performed. Cell surface area was determined under various conditions after culturing on coverslips and imaging by confocal microscopy. This was done at 40–50% confluency of the cells. Surface area was quantified by imaging to the complete boundary of 30 individual cells/condition using ImageJ software (National Institutes of Health). From this, a mean ± S.E. value was calculated for the surface area in each condition. The planar aspect and deviations of individual cells in this respect (z axis) were determined and adjusted.
RT-PCR
Analysis of gene expression in the cardiomyocytes was investigated using RT-PCR. PCR was run with primer sets for hypertrophic genes, common markers of cardiac hypertrophy in vivo and in vitro, or MCIP1. GAPDH was used as a standard. The primer sequences were as follows: skeletal muscle actin forward primer, AAGGACCTGTATGCCAACAA, and reverse primer, GAGAGGATTCGTCGTCCTGA; MCIP1, GACTGGAGCTTCATTGACTGCGAGA and AAGGAACCTACAGCCTCTTGGAAAG; GAPDH, AGCCACATCGCTCAGAACAC and GAGGCATTGCTGATGATCTTG; ANP, CGTGCCCCGACCCACGCCAGCATGGGCTCC and GGCTCCGAGGGCCAGCGAGCAGAGCCCTCA; and BNP, GGGCGCTCCTGCTCCTGCTCTTC and ACACCTGTGGGACGGGGGCTCTC.
Calcineurin Activity and NF-AT Transactivation
Phosphatase activity was determined using a BioMol kit per the manufacturer’s instructions. Cells were treated as above for 24 h and lysed, and the proteins were isolated. Calcineurin activity was determined by running duplicates of the samples in the presence of EGTA and subtracting this value from the total phosphatase activity. All samples were split into 6 wells (3 for total phosphatase and 3 for relative calcineurin), and the experiments were carried out three times.
Activity of the NF-AT transcription factor was determined by transfecting cardiomyocytes with a luciferase reporter linked to a promoter with three NF-AT sites (a gift from Dr. Chris Hughes, University of California, Irvine), using Lipofectamine Plus (Invitrogen). A control vector, TK-luc, contained the promoter sequence without NF-AT sites. Transfection efficiency was determined by co-transfecting a GFP construct. Cells were lysed, added to (200 μl) detection buffer, and immediately read on a luminometer. The luciferase values indicated are the mean of six determinations/condition.
NF-AT translocation from the cytoplasm to the nucleus was determined over time by Western blot. Cell fractions were obtained by centrifugation as described previously (9).
Nuclear Run-on and mRNA Stability
Nuclear run-on studies were performed as follows. Nuclei were isolated, followed by sucrose gradient centrifugation. The nuclei were transferred to 2× reaction buffer (10 mol/liter Tris-Cl, 5 mmol/liter MgCl2, 0.3 mol/liter KCl) with 0.5 mmol/liter nucleotides, and 0.3 μmol/liter radiolabeled [32P]UTP (>3000 μCi/mmol/liter). The radiolabeled RNA was isolated to probe a nylon blot with equal amounts of cDNA for GAPDH and MCIP1. Hybridizations were performed in Ultrahyb (Ambion) at 42 °C overnight and then washed twice in 2× SSC at 65 °C.
Stability assays were performed on MCIP1 mRNA by incubating the cardiomyocytes with/without E2 for 6 h and then blocking transcription after 6 h with 50 nmol/liter 5,6-dichlorobenzimidazole (DRB). Total RNA was isolated over 8 h after the addition of the DRB. RT-PCR using primers specific to MCIP1 was used to determine the degree of mRNA stability. GAPDH primers were used as an internal control. The intensities of the MCIP1 bands were normalized to GAPDH and plotted for each of the time points.
RNA Interference
siRNA molecules were designed using Qiagen’s website and transfected into cardiomyocytes with Oligofectamine. A fluorescein isothiocyanate-labeled nonsense siRNA molecule controlled specificity. Transfected cells were recovered for 24 h and treated with hypertrophic agents with/without estrogen as before. Knockdown in gene expression was identified after isolating the RNA and performing RT-PCR (MCIP1) or protein blots (AKT) 48 h after expression of the siRNAs. Preliminary time courses of protein knockdown were carried out, leading to selection of 48 h of post-transfection for experiments (data not shown).
Kinase Activity and Nitric Oxide Assays
ERK and AKT activities were determined at the times shown by methods published previously (8, 9). PKC activity was determined by immunoprecipitating PKC protein with antibody H-300, 24 h after incubation of the cardiomyocytes with AngII ± E2. This antibody against the C terminus of the kinase protein (Santa Cruz Biotechnology) recognized most PKC family members. Immunoprecipitates were used for an in vitro tube assay, utilizing histone-H1 as substrate for PKC activity. Kinase proteins were shown below the activities, as controls. Nitric oxide (NO) was measured as the conversion of [3H]arginine to citrulline by the cardiomyocyte, as we described previously (10).
Presence of ER in Cardiomyocytes
Isoforms of ER were determined in whole cells by confocal microscopy, using techniques we published previously (9). The presence of ER isoforms at the plasma membrane of neonatal cardiomyocytes was determined by Western blot, after separating membrane proteins by native gel electrophoresis, as we described previously (11). Antibodies for ERα and ERβ, respectively, were from Santa Cruz Biotechnology and Zymed Laboratories Inc., and the cardiac myosin heavy chain antibody was from Abcam.
RESULTS
Estrogen Inhibits AngII or ET-1-induced Cardiomyocyte Hypertrophy
Treatment of cardiomyocytes with the hypertrophic peptides AngII or ET-1 stimulated incorporation of leucine into newly formed protein (Fig. 1A, left). Co-incubation with 10 nm E2 caused as much as an 80% loss of the stimulated protein synthesis. The inhibition by sex steroid occurred in a dose-related fashion, at a concentration as low as 1 nm E2, and was substantially reversed by ICI182780, an ER antagonist (Fig. 1A, right). We also determined the expression of a classical hypertrophic marker, skeletal muscle actin (Fig. 1B). RT-PCR showed a clear increase in actin mRNA levels in cells treated with AngII or ET-1. However, in cells treated with either peptide plus E2, the increases in mRNA expression were significantly smaller.
Fig. 1. Estradiol inhibits vascular peptide-induced cardiomyocyte hypertrophy.
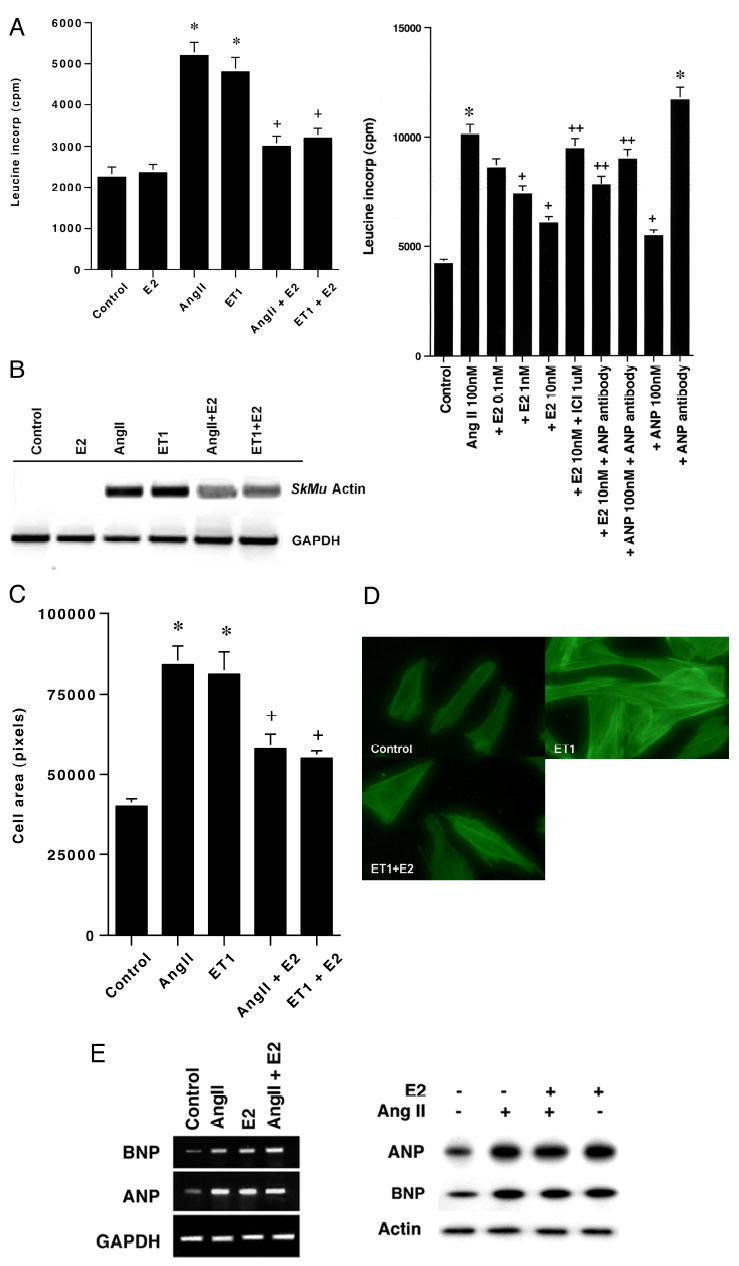
A, left, new protein synthesis in rat cardiomyocytes is stimulated by AngII or ET-1 and inhibited by E2. Three experiments were combined for the bar graph (mean ± S.E. of response), and the data were analyzed by analysis of variance plus Schefe’s test. *, p < 0.05 for control or E2 alone versus AngII or ET-1; +, p < 0.05 for AngII or ET-1 versus same + E2. Right, concentration-related E2 inhibition of protein synthesis is reversed by ICI182780 and is partially dependent on ANP secretion/action. The bar graph represents three experiments combined. *, p < 0.05 for control versus condition;+, p < 0.05 for AngII versus AngII plus either E2 or ANP, 100 nm; ++-, p < 0.05 for 10 nm E2 versus E2 + ICI, E2 + ANP antibody, or ANP + ANP antibody. B, skeletal muscle actin gene expression is stimulated by hypertrophic peptides and inhibited by E2. Cells were incubated with peptides ± steroid for 48 h. Extracted RNA from each experimental condition was reverse-transcribed and used for the PCR determination. A single representative RT-PCR experiment of three is shown, and GAPDH serves as control. C, cell surface area in myocytes incubated with AngII, ET-1, or each with E2. Cardiomyocytes were cultured on coverslips for 24 h, and surface area was quantified. The bar graph represents three experiments combined, 30 cells counted per condition in each experiment. E2 itself had no effect on surface area (data not shown). Statistical analysis is the same as A. D, microscopic images of cells incubated with ET-1 or ET-1 plus E2. E2 reduced the surface area and actin (phalloidin staining) of ET-1/E2- exposed cells, compared with ET-1 alone. E, ANP and BNP are stimulated in response to AngII or E2. The cells were incubated with AngII ± E2 or E2 alone for 24 h. Left, gene expression was determined by RT-PCR. Right, secretion of ANP and BNP into the incubation medium was assessed by Western blot. A representative study of two is shown.
Cardiomyocyte cell size defines hypertrophy in vitro. We found that the mean cell area was substantially stimulated by the hypertrophic peptides and was reduced ~65% in E2-treated cells (Fig. 1C). ET-1 induced an increase in cell size and actin staining compared with control, and both parameters were reversed by concomitant E2 exposure (Fig. 1D). E2 alone had no effect on these parameters (data not shown). Because similar results were observed regardless of the hypertrophic stimulus (either AngII or ET-1), and because AngII induces hypertrophy in part through ET-1 (12), many subsequent studies were performed with one of the two stimuli.
Estrogen Stimulates ANP and BNP Expression
Several investigators have reported that E2 stimulates ANP gene expression, correlating to sex steroid prevention of hypertrophy (13, 14). It is also well known that ANP and BNP expression in the heart is up-regulated by a variety of hypertrophic factors, and the natriuretic peptides are anti-hypertrophic (15). Thus, natriuretic peptide stimulation may be a compensatory response that limits the extent of cardiac enlargement, because severe hypertrophy is ultimately maladaptive. We therefore asked whether E2 stimulates ANP and BNP, and what role this may have on the effects of E2.
We first determined the effects of AngII, E2, or both on ANP and BNP gene expression at 24 h. We found that individually, AngII and E2 each stimulated the gene expression of ANP and BNP (Fig. 1E, left). We also found that AngII and E2 each stimulated ANP and BNP secretion into the incubation medium (Fig. 1E, right). We then asked whether the ability of E2 to stimulate ANP production impacted the anti-hypertrophy effects of the sex steroid. We determined leucine incorporation in the setting of AngII, E2, or both, with or without ANP antibody added to the culture medium.
As seen in Fig. 1A, right, E2 inhibited AngII-induced protein synthesis. This was partially prevented by ANP anti- body, at a concentration of antisera that fully blocked the anti-hypertrophic effect of the exogenous added ANP, 100 nm (Fig. 1A, right). ANP antibody also slightly augmented AngII-induced leucine incorporation.
Estrogen Prevents Calcineurin Activation
Cardiomyocyte hypertrophy often involves calcineurin, inducing NF-AT translocation to the nucleus (16). We found that the calcineurin inhibitor, FK506, caused a significant decrease in ET-1-stimulated leucine incorporation (Fig. 2A). These results suggest that hypertrophic effects of ET-1 are mediated in part through calcineurin.
Fig. 2. Modulation and function of calcineurin activity in cardiomyocytes.
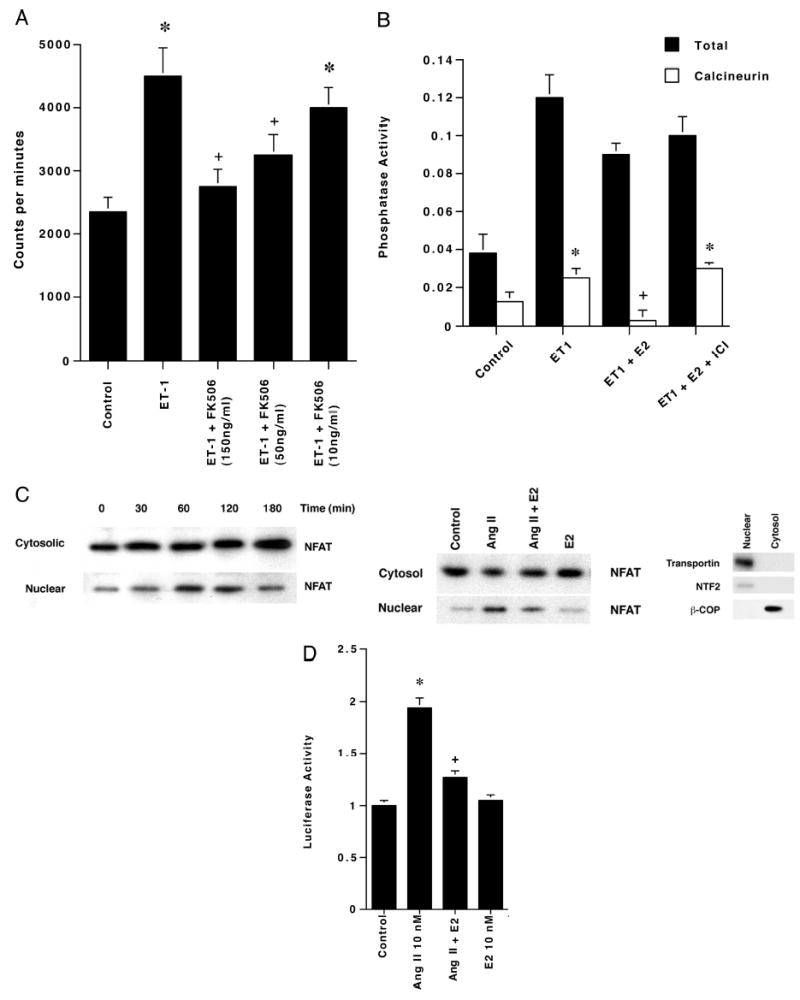
A, leucine incorporation is stimulated by ET-1 and is prevented by the calcineurin inhibitor FK506. Leucine incorporation studies were carried out in the presence of ET-1 ± several concentrations of the calcineurin inhibitor FK506. The bar graph is three experiments combined. *, p < 0.05 for control versus ET-1 or ET-1 + 10 ng/ml FK506; +, p < 0.05 for ET-1 versus ET-1 plus 100 or 150 ng/ml FK506. B, E2 inhibits ET-1-stimulated calcineurin activity. Calcineurin activity was determined at 24 h of incubation using a Biomol kit. Data are from three experiments. *, p < 0.05 for control versus ET-1 or ET-1 + E2 + ICI; +, p < 0.05 for ET-1 versus ET-1 + E2. C, left, AngII stimulates the translocation of NF-ATc3 from cytosol to nucleus. Western blot of NF-ATc3 from both cytosolic and nuclear compartments was determined in cardiomyocytes treated with AngII for 0–180 min. Right, estrogen inhibits AngII-induced NF-ATc3 translocation to the nucleus. Cardiomyocytes were incubated with AngII ± E2 for 60 min, and Western blots were carried out. The studies were repeated twice. A representative Western blot of nuclear (NTF2 and transportin) and cytosolic (β-COP) proteins is also shown to establish fraction purity. D, E2 inhibits AngII-activated NF-AT-induced transcription. Cardiomyocytes were transfected with a 3xNF-AT-binding site-luciferase reporter plasmid and then incubated with AngII ± E2 for 6 h. Luciferase activity was determined as under “Materials and Methods.” Three experiments comprise the bar graph data. *, p < 0.05 for control versus AngII;+, p < 0.05 for AngII versus AngII plus E2.
To investigate a possible interaction between calcineurin and E2, we performed assays to determine overall phosphatase activity and calcineurin activity specifically. Cardiomyocytes treated with ET-1 had large increases in both total phosphatase and calcineurin activity (Fig. 2B). In cells treated with ET-1 and E2, there was a small reduction in overall phosphatase activity and a large reduction in calcineurin activity. This suggests that the E2 effect is relatively specific to this phosphatase and not to overall activity stimulated by ET-1. The ER antagonist, ICI182780, inhibited the effect of E2 (Fig. 2B) but had no action by itself (the latter data is not shown). This precludes a non-ER action of E2.
Downstream events in the calcineurin pathway were analyzed by investigating the localization and activity of the transcription factors, NF-ATc3 and NF-ATc4. We first determined that NF-ATc3 translocated to the nucleus maximally after 60 min of exposure to AngII, and the effect persisted for an additional hour (Fig. 2C, left). The cytoplasmic pool of this transcription factor was much more abundant and did not appreciably diminish. This suggests that a small but important subpopulation of NF-AT participates in the hypertrophic response. We also determined that E2 significantly blocks the AngII-induced increase in NF-ATc3 translocation to the nucleus (Fig. 2C, right). Similar results were found for NF-ATc4 (data not shown).
These results suggested that E2 might block AngII-induced NFAT transcriptional activity. To determine this, cardiomyocytes were transfected with a 3xNF-AT-luc plasmid (which has three NF-AT-binding sites linked to the luciferase gene) or the promoter-less vector and treated with AngII and/or E2. AngII-treated cells demonstrated increased luciferase activity, compared with cells expressing the promoter-less vector or to cells transfected with the NF-AT reporter, but were not exposed to angiotensin (control) (Fig. 2D). No significant change in activity was observed when cells were treated with E2 alone. In cells treated with both AngII and E2, ~75% of the increased luciferase activity observed in the AngII alone cells was lost. Thus, E2 prevents the stimulation of NF-AT transcriptional activity, related in part to preventing nuclear translocation of the transcription factor.
MCIP1 Is Induced by Estrogen to Down-regulate Calcineurin Activity
How might E2 oppose the calcineurin-regulated hypertrophic pathway? Perhaps this involves the calcineurin inhibitor MCIP1 (17, 18). To determine this, we carried out RT-PCR. Cardiomyocytes treated with AngII or ET-1 showed low basal levels of MCIP1 expression, insignificantly above base line (Fig. 3A). However, in E2-treated cells, abundant MCIP1 levels were detected, and AngII or ET-1 did not affect this. The PI3K inhibitor LY294002 abrogated the increased MCIP1 expression in cardiomyocytes because of estrogen. These results extend our previous findings in endothelial cells (8) and suggest that this estrogen action in cardiomyocytes occurs in part through PI3K.
Fig. 3. MCIP expression in rat neonatal cardiomyocytes.
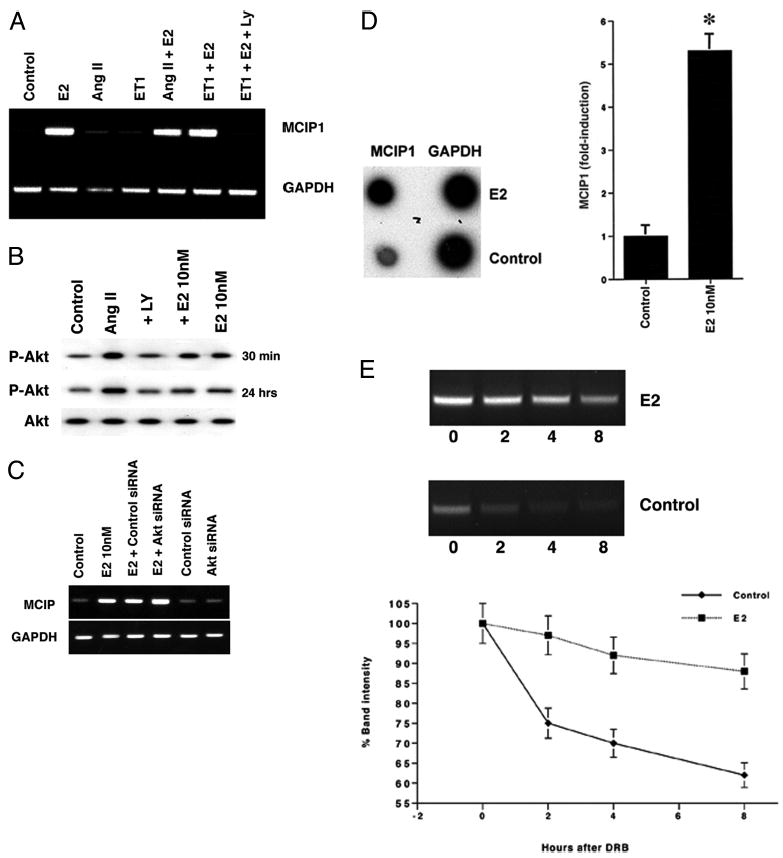
A, RT-PCR of MCIP1. Cells were incubated for 24 h with 10 nmol/liter ET-1 or 100 nmol/liter AngII with/without 10 nmol/liter E2. RNA was extracted, and RT-PCR was performed using primers for MCIP1 or GAPDH (loading and amplification control). This representative experiment was repeated. B, E2 and AngII-induced AKT activity. AKT activation was determined after 30 min and 24 h of cell incubation by Western blot, using a phospho-specific antibody to Ser-473. The data are representative of two separate experiments. LY, LY294002. C, silencing of AKT does not influence E2-induced MCIP1 gene expression. siRNA to GFP (control) or AKT was singly introduced into cardiomyocytes, and the cells were then recovered and incubated with E2. PCR for MCIP1 expression was then carried out. D, E2 induces the transcription of MCIP1. Cardiomyocytes were/were not incubated with 10 nmol/liter E2 for 6 h, and nuclear run-on was accomplished as described under “Materials and Methods.” A representative study is shown, and the bar graph represents three experiments combined. *, p < 0.05 for control versus E2. E, E2 stabilizes MCIP1 mRNA. mRNA levels were determined over 8 h in response to 10 nmol/liter E2 (or no E2) after adding the transcription inhibitor, DRB. Three studies, using triplicate determinations at each point, are combined.
Downstream of PI3K, AKT activation may play a role in estrogen induction of MCIP1. To investigate this we first determined the ability of AngII, E2, or both to modulate AKT activation (reflected by Ser-473 phosphorylation). We found that at 30 min of incubation, either E2 or AngII stimulated AKT activation, and there was no significant effect from combining both (Fig. 3B). However, at 24 h the AngII effect continued; E2 alone caused an insignificant activation of AKT, and the steroid significantly inhibited AngII-induced activation. The effects of E2 on MCIP1 and many of the hypertrophic parameters were determined at 24 h, indicating it is unlikely that E2 is acting through AKT. To test this further, we examined the effects of silencing AKT (via siRNA). In this setting, we also did not find a significant effect on E2-stimulated MCIP1 (Fig. 3C). These results indicate that E2-induced PI3K signaling to MCIP up-regulation does not occur through AKT.
We then investigated whether E2 stimulates MCIP1 gene expression by transcriptional transactivation or by increasing the stability of the message. To determine this, we performed nuclear run-on and mRNA stability assays. Nuclei from cardiomyocytes treated with or without estrogen were isolated, and the new RNA was used to probe a blot of cDNA for GAPDH (control) or MCIP1. Blots hybridized with RNA from cardiomyocytes treated with E2 had significantly increased expression of MCIP1, compared with control cells (Fig. 3D). GAPDH levels were similar from both pools of transcripts. These results clearly indicate that E2 stimulates MCIP1 gene transcription.
To investigate stabilization of the transcript, the cells were treated with E2 as before, but transcription was stopped at 6 h by blocking with 50 μg/ml DRB. At various times after addition of the transcriptional inhibitor, mRNA was isolated and prepared for RT-PCR (Fig. 3E). At the time of addition of the DRB, MCIP1 levels were higher in the estrogen-treated cells compared with the nontreated controls. With the passage of time, estrogen-treated cells maintained relatively high levels of MCIP1, whereas mRNA isolated from the nontreated cells had little detectable MCIP1 by 4 h from the point of addition of the DRB. Together, estrogen stimulates both the transcription of MCIP and the stability of the message.
MCIP1 Underlies E2 Inhibition of Cardiac Hypertrophy
It is important to determine whether the anti-hypertrophic effects of E2 were significantly related to stimulating MCIP1. To address this, we designed siRNA molecules and transfected these into cardiomyocytes. Antibodies to MCIP were not available to determine protein knockdown, and RT-PCR was performed on cells treated with control or MCIP1 siRNA (Fig. 4A). Compared with nontreated cells (Fig. 3) or cells transfected with a control GFP siRNA (Fig. 4A), MCIP1 siRNA caused a strong reduction in RNA levels. The MCIP1 siRNA had no significant effect on GAPDH transcripts.
Fig. 4. MCIP contributes to E2-inhibition of cardiac hypertrophy.
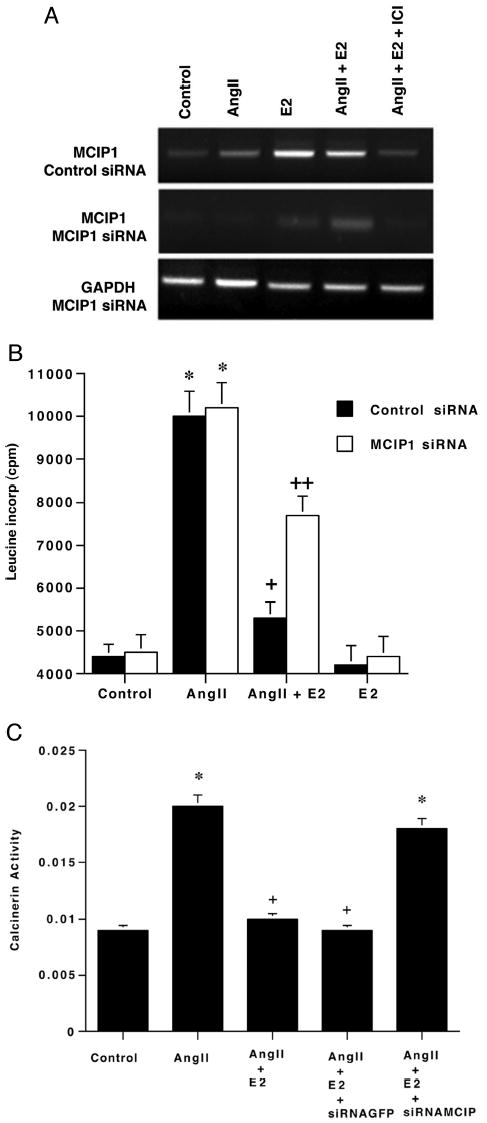
A, RT-PCR of MCIP after siRNA treatment. E2 induces MCIP1 gene expression following transfection with a control siRNA and is reversed by ICI182780, the ER antagonist. MCIP1 is barely evident after MCIP1 siRNA expression. The representative study here was carried out three times. B, E2 inhibits leucine incorporation and is reversed by MCIP siRNA. AngII-stimulated leucine incorporation in the presence or absence of E2 was determined after first transfecting control or MCIP1 siRNAs into the cardiomyocytes. The cells were recovered for 24 h, and leucine incorporation studies were carried out. Data are three experiments combined. *, p < 0.05 for control versus AngII or AngII + control or MCIP siRNA; +, p < 0.05 for AngII versus AngII + E2 + control siRNA, + +, p < 0.05 for AngII + E2 + control siRNA versus AngII + E2 + the MCIP siRNA. C, calcineurin activity as a function of AngII ± E2 in the setting of siRNA. Calcineurin activity was determined as described above from cells transfected to express the control or MCIP siRNA. Three studies were combined. *, p < 0.05 for control versus AngII or AngII + E2 MCIP siRNA; +, p < 0.05 for AngII versus AngII+E2 or same + GFP siRNA.
We then evaluated interfering RNA for MCIP1 on cardiomyocyte protein synthesis. Leucine incorporation stimulated by AngII was no different in the setting of either control or MCIP siRNAs (Fig. 4B). E2 significantly reduced leucine incorporation in cells incubated with both the steroid and AngII in the presence of control siRNA. However, MCIP siRNA prevented this anti-hypertrophic effect of E2 by 50%, limited by our transfection efficiency of 60%. These results indicate that MCIP induction/function is important in the inhibition of hypertrophy by E2.
To understand further the role of MCIP1, we treated cardiomyocytes with siRNA and analyzed them for calcineurin activity. In control siRNA-treated cells, E2 substantially reduced AngII-stimulated calcineurin activity. However, in cells treated with siRNA molecules against MCIP1, the decrease in calcineurin activity because of E2 was reversed. This indicates an important role of MCIP1 to mediate E2-down-regulation of calcineurin activity (Fig. 4C) and related hypertrophy.
Regulation of Hypertrophic Signaling through ERK and PKC by E2
It has been shown previously that AngII and ET-1 enact hypertrophic signaling in part through ERK and PKC activation (19, 20). Both the AngII and ET-1 receptors are G protein-coupled, and Gq-dependent signaling triggers the activation of ERK and PKC, resulting in cardiac hypertrophy (21–23). It is possible that E2 down-regulates AngII-induced ERK and PKC activation in the cardiomyocytes.
We first asked whether ERK and PKC activity might be relevant to AngII-induced hypertrophy. As seen in Fig. 5A, the ability of AngII to stimulate leucine incorporation was significantly blocked by E2, calphostin C (PKC inhibitor), or by PD98059 (ERK inhibitor). Thus, multiple signaling pathways contribute to the overall hypertrophic effects of AngII.
Fig. 5. E2 prevents AngII-induced signaling through ERK and PKC to hypertrophy.
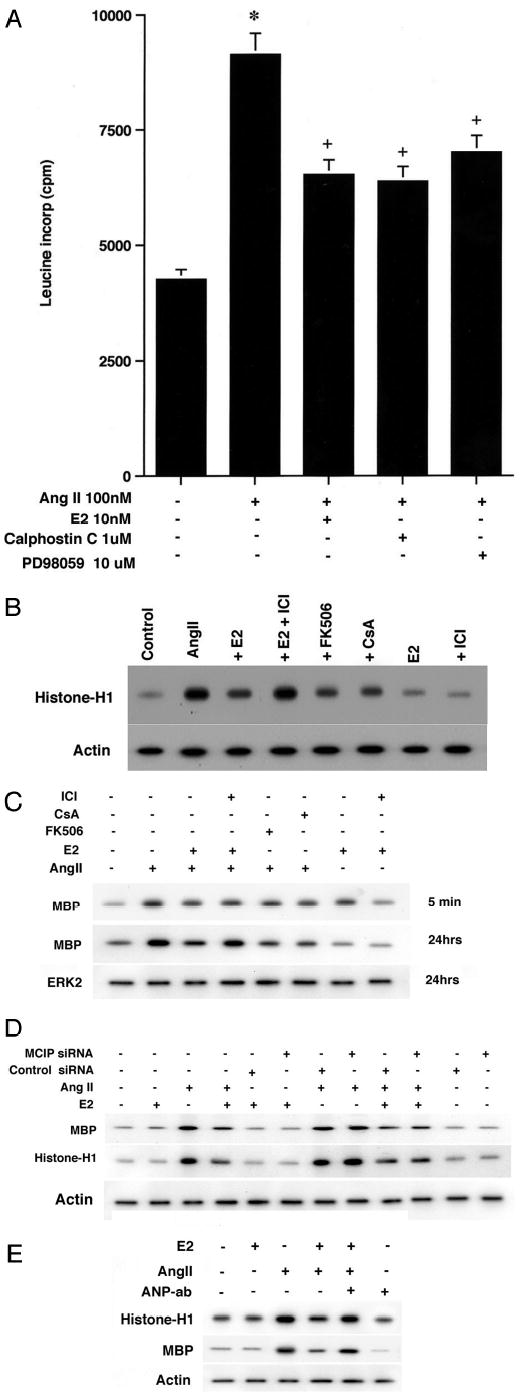
A, AngII-stimulated protein synthesis is partially prevented by PD98059 (ERK mitogen-activated protein kinase inhibitor) or calphostin C (PKC inhibitor). Three studies were combined for the bar graph. *, p < 0.05 for control versus AngII;+, p < 0.05 for AngII versus AngII plus steroid or AngII plus kinase inhibitors calphostin C (PKC) or PD98059 (ERK mitogen-activated protein kinase). B, PKC activity is stimulated by AngII, dependent upon calcineurin activity, and is inhibited by E2. Cardiomyocytes were exposed to AngII for 24 h, in the presence or absence of E2 (± ICI182780) or in the presence of the calcineurin inhibitors FK506 or cyclosporin A (5 μm). A representative study is shown and was repeated. Protein loading is assessed by actin. C, ERK activity. The cells were similarly incubated for the times shown, and ERK activity was determined against myelin basic protein (MBP) as substrate. ERK2 protein from each condition serves as control. A representative study shown was repeated. D, MCIP silencing does not affect ERK or PI3K activity. Cells were transfected with control or MCIP siRNA, recovered, and then incubated with AngII ± E2 or E2 alone. Kinase activity was determined as described. The study shown was repeated once. E, ANP mediates E2 inhibition of PKC and ERK activity. Cells were incubated with AngII ± E2 in the absence or presence of ANP antibody. PKC and ERK activities were determined. Actin protein is included as a loading control. A representative study of two is shown.
We then asked whether E2 modulates AngII-induced kinase activity. We found that AngII significantly stimulated PKC activity, directed against histone H1 as substrate (Fig. 5B). This was prevented by E2 and in turn reversed by ICI182780. We further found that FK506 and cyclosporin A each prevented AngII-induced PKC activity. This suggests a cross-talk from calcineurin to PKC activation. Similar results were found for ERK activity (Fig. 5C). Most interestingly, the ability of E2 to inhibit AngII-induced ERK was only significant at 24 h, when hypertrophic parameters were determined in many of our studies. After only 5 min, both AngII and E2 individually stimulated ERK kinase activity. Thus, the duration of E2 exposure is critical to modulate kinase activity in a fashion consistent with its anti-hypertrophic effects.
Does the ability of E2 to stimulate MCIP1 impact the regulation of PKC and ERK? To answer this, we introduced an siRNA to MCIP1 or GFP (control) into the cardiomyocytes, recovered the cells over 24 h, and then incubated the cells with AngII ± E2 for 24 h. As shown in Fig. 5D, silencing MCIP1 did not significantly reverse the ability of AngII to stimulate or E2 to inhibit either kinase activity. This suggests that E2 acts independently of MCIP1 to down-regulate ERK and PKC activity.
It has been published previously that ANP inhibits ET-1-and AngII-induced ERK and PKC activation (24, 25). Therefore, we asked whether E2-induced ANP secretion may underlie the inhibition of the two kinases. We found that ANP antibody significantly blocked the ability of E2 to inhibit AngII-induced PKC or ERK activity (Fig. 5E). Thus, this aspect of the anti-hypertrophic actions of the sex steroid specifically depends on ANP production/secretion.
Finally, it has been shown that NO functions as an anti-hypertrophy factor (26, 27). Here, we demonstrate that NO production is stimulated by E2, and generation is reduced by AngII (the latter probably mediated through the AT1 receptor). However, the ability of E2 to stimulate NO is not mediated through MCIP, as shown using RNA interference to this protein (Table I).
Table I. Effects of E2 and AngII on nitric-oxide synthase activity.
Data are the mean ± S.E. from three determinations per experiment, repeated twice.
| Conditions | [3H]Citrulline |
|---|---|
| cpm | |
| Control | 3965 ± 256 |
| E2 10 nm | 7472 ± 562a |
| AngII 1 μm | 2369 ± 305 |
| E2 + AngII | 5574 ± 685b |
| E2 + control siRNA | 7145 ± 685 |
| AngII + control siRNA | 2143 ± 268 |
| E2 + MCIP siRNA | 7012 ± 874a |
| AngII + MCIP siRNA | 2568 ± 321 |
| E2 + AngII + control siRNA | 5632 ± 598 |
| E2 + AngII + MCIP siRNA | 6455 ± 578 |
| Control siRNA | 3714 ± 417 |
| MCIP siRNA | 4002 ± 396 |
p < 0.05 for control versus condition.
p < 0.05 for E2 versus E2 + AngII by analysis of variance plus Schefe’s test.
Estrogen Receptors in Neonatal and Adult Cardiomyocytes
Our results indicate that E2 acts through the ER to mediate its anti-hypertrophic actions, indicated specifically using the ER antagonist ICI182780. Furthermore, if our results in neonatal cardiomyocytes are applicable to adult heart, then ER should be present as well. To demonstrate this, we carried out confocal microscopy studies of cultured neonatal and adult cardiomyocytes (Fig. 6A). In both types of cells, we saw the presence of each ER isoform. The distribution was somewhat different in that ERα was predominantly localized in the nucleus, with some cytoplasmic/membrane presence. In contrast, ERβ was predominantly localized to the cytoplasmic/perinuclear areas of the cell, with evidence of membrane localization as well. Few ERβ receptors could be seen in the nucleus of the cardiomyocytes.
Fig. 6. Presence of ERαand ERβin neonatal and adult rat cardiomyocytes.
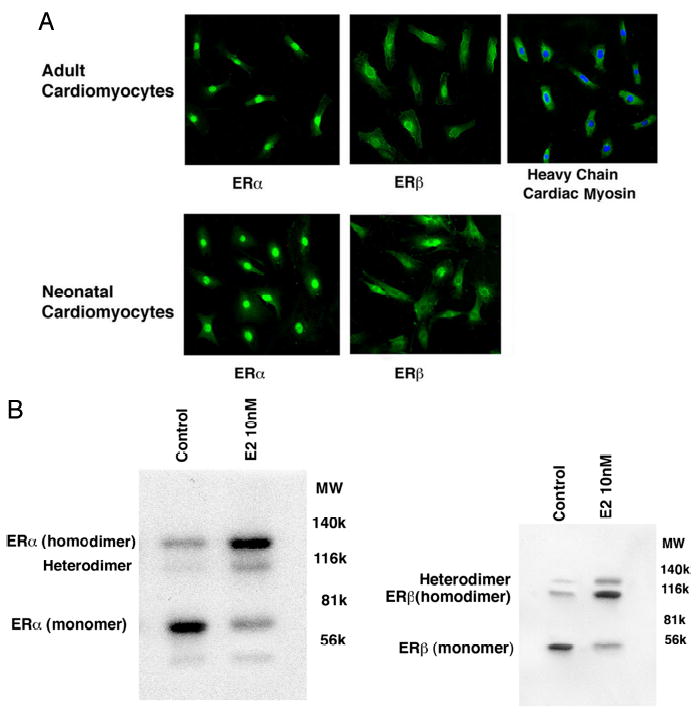
A, confocal microscopy identification of ER in cardiomyocytes from neonatal or pregnant female rats. Live cells were first stained with Hoechst 33342 dye (nucleus), then fixed with 4% paraformaldehyde, and incubated with antibodies to ERα, ERβ, or myosin heavy chain. Secondary antibodies conjugated with fluorescein were used for subsequent microscopy. B, Western blots showing ERα and ERβ in membrane fractions from cardiomyocytes. Membranes were isolated by sucrose gradient centrifugation and separated by native gel electrophoresis under nonreducing conditions, as described previously (9, 11). The proteins on the gels were transferred to nitrocellulose and subsequently blotted with ERα and ERβ antibodies.
Because we showed that E2 up-regulation of MCIP occurs through PI3K-dependent signaling, this implicates signaling from plasma membrane-localized ER (8). We therefore evaluated the presence of the membrane ER in the cardiomyocytes. ERα and ERβ homodimers were the predominant forms of the receptors in the plasma membrane, upon incubation of the cells with E2 (Fig. 6B). In the absence of E2, the monomer for each isoform predominated, and the presence of the ERα/ERβ heterodimer was also seen, augmented by the steroid ligand. Our results are very similar to the presence and forms of ER in membranes from endothelial cells (11). These data support the ability of E2 to act through ER to oppose cardiac hypertrophy, in part mediated through membrane receptors.
DISCUSSION
Estrogen has recently been proposed to prevent cardiac hypertrophy. In ovariectomized mice, E2 administration prevents ventricular remodeling after myocardial infarction (28). For postmenopausal women, sex hormone replacement therapy (HRT) results in a 20% decrease in left ventricular mass, compared with women not taking HRT (29). Similarly, left ventricular mass index is significantly reduced in hypertensive women taking HRT (5, 30). More interestingly, blood pressure was not significantly affected in these studies, suggesting a direct action of sex steroids on the heart. Heart failure and death from post-myocardial infarction occur less frequently in women taking HRT (31). In all these studies, the mechanism of estrogen action was not determined.
In contrast, results from the Women’s Health Initiative suggest that estrogen does not provide protection against the development of arteriosclerotic coronary artery disease and resulting myocardial infarction (32). The conclusions of this study have recently been challenged (33). It is certainly possible that E2 has specific beneficial effects on some related aspects of cardiovascular disease (hypertension and cardiac hypertrophy) although possibly not modulating the atherosclerotic process.
In the studies here, we show that E2 strongly inhibits the cardiomyocyte hypertrophic response to AngII or ET-1. The response to hypertrophic peptides was dependent on calcineurin, and E2 prevented the up-regulation of the activity of this phosphatase.
Much data support a central role for the protein phosphatase, calcineurin, in the development of cardiac hypertrophy (16, 18, 34, 35). Hypertrophic agents AngII, ET-1, and phenylephrine up-regulate the activity of this enzyme in a calcium-dependent fashion. Mice that are null for the catalytic subunit of calcineurin demonstrate an impaired hypertrophic response to AngII, aortic constriction, or isoproterenol (34). When calcineurin is induced, it dephosphorylates and promotes the translocation of the transcription factor NF-ATc3 to the nucleus. In the nucleus, NF-AT cooperates with the transcription factor GATA-4 to up-regulate hypertrophic genes (36, 37). We found that hypertrophic peptide-induced new protein synthesis depends on calcineurin activity, and estrogen signaling dampens this phosphatase activity, preventing cardiomyocyte hypertrophy.
The ability of E2 to reduce calcineurin activity depended on stimulation of the potent calcineurin inhibitor MCIP1. This protein binds directly to the catalytic unit of the calcineurin holoenzyme (35). Functionally, the overexpression of MCIP1 reduces cardiac hypertrophy in vivo following aortic banding (38). We found that siRNA directed to this gene substantially reversed the inhibitory effects of E2 on calcineurin activity and on new protein synthesis. This novel mechanism accounted for the downstream modulation of NF-AT translocation to the nucleus and activity that we demonstrated here, up-regulated by AngII and inhibited by E2.
Previous work proposed that E2 up-regulates the ANP gene, leading to a decrease in phenylephrine-induced cardiomyocyte hypertrophy (13). Genetic deletion of the guanylate cyclase A protein, the functional receptor for ANP action, leads to pronounced hypertrophy as induced by several stimuli (15). Thus, ANP stimulates cGMP to defend against cardiomyocyte hypertrophy. Here we show that ANP and BNP production and secretion are stimulated by E2. Furthermore, antibodies to ANP partially reversed the ability of E2 to block AngII-induced protein synthesis in the cardiomyocytes. There is likely to be specificity to E2 mechanisms, depending upon the hypertrophic stimulus. However, the ability of E2 overall to oppose the hypertrophic effects of multiple relevant inputs supports the importance of this function. We have preliminarily found that estrogen inhibits angiotensin II-induced hypertrophy in mice, mediated through both ERα and ERβ receptors.2
A potential mechanism for E2 action can be postulated from our investigations and by studies of genetically altered mice. Deletion of the FKBP12.6 gene in the in vivo cardiomyocyte leads to disordered calcium sparking through the ryanodine receptor (7). In these mice, only the post-natal males subsequently developed cardiac hypertrophy. However, female mice developed severe hypertrophy when administered the ER antagonist tamoxifen. It is unknown from that study how estrogen modulates the dysregulation of intracellular calcium and downstream events that produce the hypertrophic phenotype. We speculate that deleted FKBP12.6 might enhance calcium-dependent calcineurin activity and subsequent cardiac hypertrophy (7). Estrogen might mitigate the loss of the FKBP12.6 gene by increasing MCIP1 to down-regulate calcium-induced calcineurin activity and prevent hypertrophy. Mutation and functional loss of this calcium-stabilizing protein (FKBP12.6) also predisposes mice or humans to fatal cardiac arrhythmias (39), but it is unknown whether a sexual dichotomy exists for this predisposition.
The ability of E2 to signal through PI3K has been implicated to protect rats from an ischemia-reperfusion injury in muscle (40). Kinase signaling results from E2 binding to membrane-localized ER (9, 12) and impacts both the transcriptional and post-transcriptional actions of the sex steroid. Thus, an integrative action occurs where membrane ER/E2 induces PI3K activation to up-regulate MCIP1 transcription, and this opposes hypertrophy. The MCIP1 gene utilizes four different promoters, in a cell context-specific fashion, and so a detailed analysis is beyond the scope of the current study. Most interestingly, the regulation of MCIP by E2 was independent of AKT, shown after silencing this kinase that is sometimes functionally downstream of PI3K.
AngII also induced ERK and PKC, pathways that have been implicated previously in hypertrophic signaling (19–23). We report here that AngII-induced kinase activation contributes to its overall hypertrophic action, and that E2 inhibits this signaling. We further found that kinase activation by AngII stems from cross-talk by calcineurin to ERK and PKC activation, because activation is prevented by cyclosporin A or FK506. Thus, the ability of E2 to block calcineurin activity impacts hypertrophic signaling through ERK and PKC. Related to this, ERK and calcineurin-NFAT signaling have recently been shown to cooperate in the induction of cardiomyocyte hypertrophic genes via several distinct mechanisms (41). We did not find, however, that the E2 inhibition of ERK and PKC occurred through MCIP induction. Rather, we found that E2-induced ANP secretion mediated the inhibition of AngII-stimulated ERK and PKC. A schematic of the overall E2 modulation of hypertrophic signaling is seen in Fig. 7.
Fig. 7. E2 inhibits hypertrophic signaling induced by AngII.
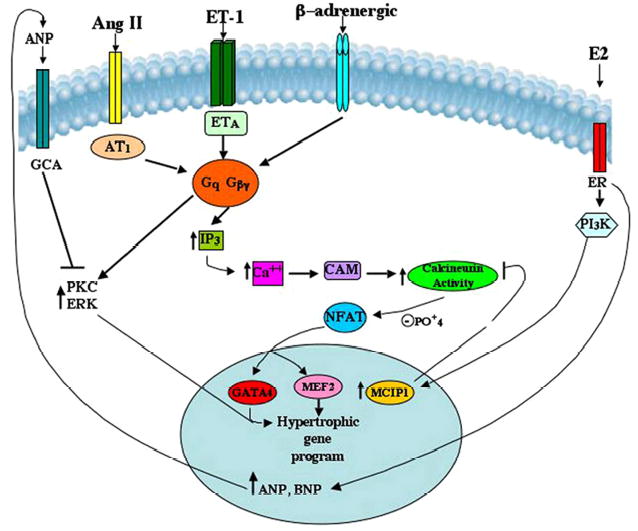
ETA is the endothelin A receptor; AT1 is the angiotensin 1 receptor; GCA is the guanylate cyclase A receptor; CAM is calmodulin; IP3 is inositol 1,4,5-trisphosphate.
Implicating ANP in E2 modulation of hypertrophic signaling is consistent with previous reports (24, 25) that ANP inhibits ET-1 and AngII signaling through ERK and PKC. A possible link from ANP to calcineurin may be relevant. It was reported recently (42) that the combined cardiac GC-A receptor KO/endothelial nitric-oxide synthase null mouse had marked cardiac activation of calcineurin and subsequent hypertrophy. This implies that loss of ANP action at its receptor leads to calcineurin up-regulation. ANP inhibition of this phosphatase could underlie the ability of E2 (and ANP) to inhibit PKC and ERK activation in the hypertrophic setting.
Several caveats need to be appreciated regarding our findings. First, our studies, as well as many others in the cardiac hypertrophy area, utilize neonatal cardiomyocytes. It is not known whether this model fully recapitulates the situation in the adult myocyte. Germane to this issue, there is some controversy whether adult cardiomyocytes express ER, with studies supporting (43, 44) or not supporting (45) the presence of ER in this cell. We show here that both neonatal and adult rat cardiomyocytes express abundant ER. Most interestingly, the receptor distribution between the two isoforms, ERα and ERβ, are not identical, with ERβ present predominantly in the cytoplasm. Future studies will define the specific contributions of each receptor isoform to the actions of the sex steroid. In addition, the anti-hypertrophic effects of E2 on the myocardium (7) could be, in part, indirectly mediated through ER expressed on myocyte-adjacent cells or even distant cells.
In summary, E2 mitigates important mechanisms of cardiomyocyte hypertrophy utilized by hypertrophic peptides. The effects of E2 may provide a protection against the progression of heart disease in women, a concept that could be evaluated in women currently taking hormone replacement after the menopause.
Footnotes
This work was supported by grants from the Research Service of the Department of Veterans Affairs and by National Institutes of Health Grants HL-59890 and CA-100366 (to E. R. L.).
The abbreviations used are: AngII, angiotensin II; E2, 17-β estradiol; ET-1, endothelin-1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; siRNA, small interfering RNA; ET-1, endothelin-1; PI3K, phosphatidylinositol 3-kinase; ERK, extracellular signal-regulated kinase; PKC, protein kinase C; DRB, 5,6-dichlorobenzimidazole; RT, reverse transcription; BNP, brain natriuretic peptide; ANP, atrial natriuretic peptide; GFP, green fluorescent protein; ER, estrogen receptor; HRT, hormone replacement therapy;
A. Pedram, M. Razandi, M. Aitkenhead, and E. R Levin, unpublished observations.
References
- 1.Dunn FG, Pfeffer MA. N Engl J Med. 1999;340:1279–1280. doi: 10.1056/NEJM199904223401610. [DOI] [PubMed] [Google Scholar]
- 2.Saxon LA, De Marco T. Card Electrophysiol Rev. 2002;6:18–25. doi: 10.1023/a:1017914517113. [DOI] [PubMed] [Google Scholar]
- 3.Wagenaar LJ, Voors AA, Buikema H, van Gilst WH. Can J Cardiol. 2002;18:1331–1339. [PubMed] [Google Scholar]
- 4.Agabiti-Rosei E, Muiesan ML. J Hypertens. 2002;20:S34–S38. [PubMed] [Google Scholar]
- 5.Miya Y, Sumino H, Ichikawa S, Nakamura T, Kanda T, Kumakura H, Takayama Y, Mizunuma H, Sakamaki T, Kurabayashi M. Hypertens Res. 2002;25:153–159. doi: 10.1291/hypres.25.153. [DOI] [PubMed] [Google Scholar]
- 6.van Eickels M, Grohe C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA. Circulation. 2001;104:1419–1423. doi: 10.1161/hc3601.095577. [DOI] [PubMed] [Google Scholar]
- 7.Xin HB, Senbonmatsu T, Cheng DS, Wang YX, Copello JA, Ji GJ, Collier ML, Deng KY, Jeyakumar LH, Magnuson MA, Inagami T, Kotlikoff MI, Fleischer S. Nature. 2002;416:334–338. doi: 10.1038/416334a. [DOI] [PubMed] [Google Scholar]
- 8.Pedram A, Razandi M, Aitkenhead M, Hughes CCW, Levin ER. J Biol Chem. 2002;277:50768–50775. doi: 10.1074/jbc.M210106200. [DOI] [PubMed] [Google Scholar]
- 9.Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER. Mol Endocrinol. 2002;16:100–115. doi: 10.1210/mend.16.1.0757. [DOI] [PubMed] [Google Scholar]
- 10.Razandi M, Pedram A, Rubin T, Levin ER. Am J Physiol. 1996;270:H1342–H1349. doi: 10.1152/ajpheart.1996.270.4.H1342. [DOI] [PubMed] [Google Scholar]
- 11.Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER. Mol Endocrinol. 2004;18:2854–2865. doi: 10.1210/me.2004-0115. [DOI] [PubMed] [Google Scholar]
- 12.Xia Y, Karmazyn M. J Pharmacol Exp Ther. 2004;310:43–51. doi: 10.1124/jpet.104.065185. [DOI] [PubMed] [Google Scholar]
- 13.Babiker FA, De Windt LJ, van Eickels M, Thijssen V, Bronsaer RJ, Grohe C, van Bilsen M, Doevendans PA. Circulation. 2004;109:269–276. doi: 10.1161/01.CIR.0000105682.85732.BD. [DOI] [PubMed] [Google Scholar]
- 14.Jankowski M, Rachelska G, Donghao W, McCann SM, Gutkowska J. Proc Natl Acad Sci U S A. 2001;98:11765–11770. doi: 10.1073/pnas.201394198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klinger JR, Warburton RR, Pietras L, Oliver P, Fox J, Smithies O, Hill NS. Am J Physiol. 2002;284:H58–H65. doi: 10.1152/ajpheart.2002.282.1.H58. [DOI] [PubMed] [Google Scholar]
- 16.Vega RB, Bassel-Duby R, Olson EN. J Biol Chem. 2003;278:36981–36984. doi: 10.1074/jbc.R300023200. [DOI] [PubMed] [Google Scholar]
- 17.Vega RB, Rothermel BA, Weinheimer CJ, Kovacs A, Naseem RH, Bassel-Duby R, Williams RS, Olson EN. Proc Natl Acad Sci U S A. 2003;100:669–674. doi: 10.1073/pnas.0237225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vega RB, Yang J, Rothermel BA, Bassel-Duby R, Williams RS. J Biol Chem. 2002;277:30401–30407. doi: 10.1074/jbc.M200123200. [DOI] [PubMed] [Google Scholar]
- 19.Aoki H, Richmond M, Izumo S, Sadoshima J. Biochem J. 2000;347:275–284. [PMC free article] [PubMed] [Google Scholar]
- 20.Yue TL, Gu JL, Wang C, Reith AD, Lee JC, Mirabile RC, Kreutz R, Wang Y, Maleeff B, Parsons AA, Ohlstein EH. J Biol Chem. 2000;275:37895–37901. doi: 10.1074/jbc.M007037200. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Proud CG. Circ Res. 2002;91:821–829. doi: 10.1161/01.res.0000041029.97988.e9. [DOI] [PubMed] [Google Scholar]
- 22.Eskildsen-Helmond YE, Bezstarosti K, Dekkers DH, van Heugten HA, Lamers JM. J Mol Cell Cardiol. 1997;29:2545–2559. doi: 10.1006/jmcc.1997.0491. [DOI] [PubMed] [Google Scholar]
- 23.Bayer AL, Heidkamp MC, Howes AL, Heller Brown J, Byron KL, Samarel AM. J Mol Cell Cardiol. 2003;35:1121–1133. doi: 10.1016/s0022-2828(03)00228-1. [DOI] [PubMed] [Google Scholar]
- 24.Kumar R, von Geldern TW, Calle RA, Pandey KN. Biochim Biophys Acta. 1997;1356:221–228. doi: 10.1016/s0167-4889(96)00168-1. [DOI] [PubMed] [Google Scholar]
- 25.Murohara T, Kugiyama K, Ota Y, Doi H, Ogata N, Ohgushi M, Yasue H. J Cardiovasc Pharmacol. 1999;34:870–878. doi: 10.1097/00005344-199912000-00015. [DOI] [PubMed] [Google Scholar]
- 26.Brede M, Roell W, Ritter O, Wiesmann F, Jahns R, Haase A, Fleischmann BK, Hein L. Hypertension. 2003;42:1177–1182. doi: 10.1161/01.HYP.0000100445.80029.8E. [DOI] [PubMed] [Google Scholar]
- 27.Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, Matsuura H, Chayama K, Teranishi Y, Iba O, Amano K, Matsubara H. Hypertension. 2003;41:99–107. doi: 10.1161/01.hyp.0000050101.90932.14. [DOI] [PubMed] [Google Scholar]
- 28.Cavasin MA, Sankey SS, Yu AL, Menon S, Yang XP. Am J Physiol. 2003;284:H1560–H1569. doi: 10.1152/ajpheart.01087.2002. [DOI] [PubMed] [Google Scholar]
- 29.Lim WK, Wren B, Jepson N, Roy S, Caplan G. Am J Cardiol. 1999;83:1132–1134. doi: 10.1016/s0002-9149(99)00029-6. [DOI] [PubMed] [Google Scholar]
- 30.Light KC, Hinderliter AL, West SG, Grewen KM, Steege JF, Sherwood A, Girdler SS. J Hypertens. 2001;19:269–278. doi: 10.1097/00004872-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 31.Shlipak MG, Angeja BG, Go AS, Frederick PD, Canto JG, Grady D. Circulation. 2001;104:2300–2304. doi: 10.1161/hc4401.98414. [DOI] [PubMed] [Google Scholar]
- 32.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J, Writing Group for the Women’s Health Initiative Investigators. J Am Med Assoc. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 33.Turgeon JL, McDonnell DP, Martin KA, Wise PM. Science. 2004;304:1269–1273. doi: 10.1126/science.1096725. [DOI] [PubMed] [Google Scholar]
- 34.Bueno OF, Wilkins BJ, Tymitz KM, Glascock BJ, Kimball TF, Lorenz JN, Molkentin JD. Proc Natl Acad Sci U S A. 2002;99:4586–4591. doi: 10.1073/pnas.072647999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams RS. Circulation. 2002;105:2242–2243. doi: 10.1161/01.cir.0000017141.01845.fa. [DOI] [PubMed] [Google Scholar]
- 36.Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Mol Cell Biol. 2002;22:7603–7613. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuentes JJ, Genesca L, Kingsbury TJ, Cunningham KW, Perez-Riba M, Estivill X, de la Luna S. Hum Mol Genet. 2000;9:1681–1690. doi: 10.1093/hmg/9.11.1681. [DOI] [PubMed] [Google Scholar]
- 38.Rothermel BA, McKinsey TA, Vega RB, Nicol RL, Mammen P, Yang J, Antos CL, Shelton JM, Bassel-Duby R, Olson EN, Williams RS. Proc Natl Acad Sci U S A. 2001;98:3328–3333. doi: 10.1073/pnas.041614798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Science. 2004;304:292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 40.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanna B, Bueno OF, Dai YS, Wilkins BJ, Molkentin JD. Mol Cell Biol. 2005;25:865–878. doi: 10.1128/MCB.25.3.865-878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bubikat A, De Windt LJ, Zetsche B, Fabritz L, Sickler H, Eckardt D, Goedecke A, Baba HA, Kuhn M. J Biol Chem. 2005;280:21594–21599. doi: 10.1074/jbc.M501103200. [DOI] [PubMed] [Google Scholar]
- 43.Nuedling S, Kahlert S, Loebbert K, Doevendans PA, Meyer R, Vetter H, Grohe C. Cardiovasc Res. 1999;43:666–674. doi: 10.1016/s0008-6363(99)00093-0. [DOI] [PubMed] [Google Scholar]
- 44.Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM, Jr, Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW. Proc Natl Acad Sci U S A. 2004;101:4130–4135. doi: 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forster C, Kietz S, Hultenby K, Warner M, Gustafsson JA. Proc Natl Acad Sci U S A. 2004;101:14234–14239. doi: 10.1073/pnas.0405571101. [DOI] [PMC free article] [PubMed] [Google Scholar]