

Abstract
Enterotoxigenic Escherichia coli (ETEC) is an enteric pathogen that causes cholera-like diarrhea in humans and animals. ETEC secretes a heat-labile enterotoxin (LT), which resembles cholera toxin, but the actual mechanism of LT secretion is presently unknown. We have identified a previously unrecognized type II protein secretion pathway in the prototypic human ETEC strain, H10407 (serotype O78:H11). The genes for this pathway are absent from E. coli K-12, although examination of the K-12 genome suggests that it probably once possessed them. The secretory pathway bears significant homology at the amino acid level to the type II protein secretory pathway required by Vibrio cholerae for the secretion of cholera toxin. With this in mind, we determined whether the homologous pathway of E. coli H10407 played a role in the secretion of LT. To this end, we inactivated the pathway by inserting a kanamycin-resistance gene into one of the genes (gspD) of the type II secretion pathway by homologous recombination. LT secretion by E. coli H10407 and the gspD mutant was assayed by enzyme immunoassay, and its biological activity was assessed by using Y-1 adrenal cells. This investigation showed that the protein secretory pathway is functional and necessary for the secretion of LT by ETEC. Our findings have revealed the mechanism for the secretion of LT by ETEC, which previously was unknown, and provide further evidence of close biological similarities of the LT and cholera toxin.
Enterotoxigenic Escherichia coli (ETEC) is an enteric pathogen that causes watery cholera-like diarrhea in animals and humans (1). Infections with ETEC pose a major health problem in developing countries, accounting for more than 200 million cases of diarrhea and approximately 380,000 deaths annually among children under 5 years of age (2). ETEC is also the most common cause of diarrhea among travelers from industrialized to less developed countries, including military troops on deployment (2).
ETEC secrete at least one of two types of enterotoxins, known as heat-labile (LT) and heat-stable enterotoxin, respectively (1, 3). LT is an 84-kDa multimeric protein comprised of a single A subunit and a pentamer of identical B subunits. The pentameric B subunit mediates binding to GM1 ganglioside on intestinal epithelial cells, after which the toxin is internalized and processed. The free A subunit then catalyses the ADP-ribosylation of Gsα, a GTP-binding regulatory protein, leading to activation of adenylate cyclase, production of excessive amounts of cAMP, disruption of electrolyte transport across the intestinal lumen, and diarrhea (4). Prostaglandins and neurotransmitters of the enteric nervous system also play a role in the induction of fluid secretion by LT (5, 6). Although a great deal is known about the structure and biological activity of LT, little is known about the mechanism of its secretion by ETEC.
LT is structurally and biologically related to cholera toxin (CT), the major virulence determinant of Vibrio cholerae, with over 77% nucleotide (4) and protein homology (1). Both CT and plasmid-encoded LT are secreted by V. cholerae (7–9), with secretion taking place in two membrane translocation steps (10). Initially, the A and B subunits are produced with N-terminal signal peptides that are cleaved during Sec-dependent translocation across the cytoplasmic membrane via the general secretory pathway into the periplasmic space (7, 10, 11). Here they undergo folding and assembly to form the mature holotoxin, which is then transported across the outer membrane via a type II protein secretion pathway (10) known as the main terminal branch of the general secretory pathway (12). In contrast to V. cholerae, when the nonpathogenic laboratory strain of E. coli K-12 is transformed with either an LT- or a CT-expressing plasmid, the toxins are not efficiently secreted but are retained mainly within the periplasm, indicating that E. coli K-12 does not possess or express the pathway used by V. cholerae to secrete these toxins (11, 13, 14). Although E. coli K-12 does possess the genes for a type II secretion pathway (15), which appears to play a role in the secretion of endogenous endochitinase (16), this pathway is evidently unable to transport CT or LT. This inability to transport CT or LT may be because the level of expression of the genes encoding this pathway is extremely low under standard growth conditions (17). Nevertheless, when human isolates of ETEC are grown in culture, LT is detectable in the supernatant (18, 19), suggesting that ETEC possesses a functional pathway for the secretion of LT. This suggestion is supported by other work that shows LT does not remain in the periplasm of these bacteria (20).
In this paper, we report the identification of a type II protein secretion pathway in ETEC that is absent from E. coli K-12. This secretion pathway is highly homologous to the pathway responsible for the secretion of CT and LT by V. cholerae and is required for the secretion of LT by ETEC.
Materials and Methods
Bacterial Strains, Plasmids, and Growth Conditions.
The bacterial strains and plasmids used in this study are listed in Table 1 (21, 22). Strains were routinely grown at 37°C in Luria broth (1% tryptone/0.5% yeast extract/171 mM NaCl) (23) or CAYE medium [2% Casamino acids/0.6% yeast extract/43 mM NaCl/38 mM K2HPO4/0.25% glucose/0.1% (vol/vol) trace salts solution consisting of 203 mM MgSO4/25 mM MnCl2/18 mM FeCl3] (24) supplemented with 100 μg/ml of ampicillin (CSL, Parkville, Victoria, Australia) or 50 μg/ml of kanamycin (Sigma–Aldrich) when necessary. Y1 mouse adrenal cells (CSL) were maintained in DMEM (23) supplemented with 10% FBS (CSL) and 100 μg/ml of gentamicin (Sigma–Aldrich) and incubated at 37°C in an atmosphere containing 5% CO2.
Table 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Relevant characteristics | Ref. or source |
|---|---|---|
| E. coli | ||
| H10407 | ETEC serotype O78:H11, LT+ ST+ | 21 |
| MT13 | H10407gspD∷KmR | This study |
| XL1-Blue | F′∷Tn10 proA+B+lacIq Δ(lacZ)M15/recA1 endA1 gyrA96 (NalR) thi hsdR17(r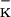 m m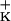 )supE44 relA1 lac )supE44 relA1 lac
|
Stratagene |
| Plasmids | ||
| pST76-A | Temperature-sensitive suicide vector derived from pSC101, ApR | 22 |
| pUC4-KIXX | pUC4K derivative containing KmR and BmR genes from Tn5 | Pharmacia |
| pGEM-T Easy | High copy number vector, ApR | Promega |
| pMT37 | pGEM-T Easy containing 1.9-kb PCR fragment encompassing gspD from H10407, ApR | This study |
| pMT39 | pMT37 containing 1.4-kb SmaI fragment encompassing the KmR gene from pUC4-KIXX cloned into SnaBI site, ApR KmR | This study |
| pMT42 | pST76-A containing 3.3-kb EcoRI fragment of pMT39 bearing gspD with an inserted KmR gene cloned into the EcoRI site, ApR KmR | This study |
| pMT6 | pGEM-T Easy containing 5.5-kb PCR fragment encompassing gspD, gspE, and gspF from H10407, ApR | This study |
| pMT44 | Deletion derivative of pMT6 containing 3.8-kb fragment encompassing gspD from H10407, ApR | This study |
ApR, ampicillin resistance; KmR, kanamycin resistance; BmR, bleomycin resistance.
Growth and Viability Studies.
Bacterial growth was assayed by measuring the optical density at OD600 of cultures every hour by using an Ultrospec III spectrophotometer (Pharmacia). To determine the extent of cell death (lysis), the Molecular Probes Baclight Live/Dead kit was used (Molecular Probes). Both live and dead bacteria were labeled simultaneously as instructed by the manufacturer, after which they were visualized by using a Leica DM-LB HC microscope with a I3 fluorescein filter (excitation 450–490 nm). The percentage of live and dead bacteria was ascertained by counting at least 100 cells in at least five separate random fields.
Preparation and Manipulation of DNA.
Plasmid DNA was isolated by using the Wizard Plus SV DNA Purification System (Promega). Standard restriction digestion and cloning procedures using DNA-modifying enzymes supplied by Promega or New England Biolabs were used (23). Electrocompetent E. coli XL1-Blue, H10407, and MT13 cells were obtained by growing the bacteria to midlogarithmic phase (OD600 0.5–0.8) at 37°C. Cells were then washed three times with sterile cold 10% glycerol in distilled water and resuspended in 1/75 the original culture volume. Transformation was achieved by using a Bio-Rad Gene Pulser and electroporation conditions 1.80 kV, 200 Ω, and 25 μF.
DNA Amplification, Sequencing, and Analysis.
PCR was used to amplify the genes for the putative type II secretion pathway and to confirm insertional inactivation of gspD by allelic exchange. Genomic DNA of E. coli H10407 was prepared by the boiling lysis method (23). PCR amplifications were performed with Vent DNA polymerase (New England Biolabs), which has proofreading activity, or with AmpliTaq polymerase (Applied Biosystems) in a reaction volume of 25–50 μl in a Gene Amp PCR System 9700 thermal cycler (Applied Biosystems). The PCR conditions involved denaturation for 5 min at 94°C, followed by 35 cycles of 30 sec at 94°C, 30 sec at 55°C, and 1–4 min at 72°C, with a final extension for 5 min at 72°C.
Nucleotide sequencing was performed by using an abi prism Big Dye Terminators, Ver. 3.0, Cycle Sequencing Kit (Applied Biosystems). Reactions were analyzed on an Applied Biosystems ABI PRISM 377 DNA sequencer. Nucleotide sequence data were edited and assembled into contiguous sequence with the sequencher program (Gene Codes, Ann Arbor, MI).
Nucleotide sequence and amino acid similarity searches with sequences in the public databases were performed by using the blastn, blastp, and blastx programs (25) available from the National Center for Biotechnology Information web site (www.ncbi.nlm.nih.gov).
Construction of a Functionally Nonpolar gspD Mutant.
A 1.9-kb fragment containing 1.6 kb of gspD was amplified by PCR from E. coli H10407 genomic DNA by using oligonucleotide primers P35 (5′-ATTGTCGGTTATGCAGCGAAGC) and P109 (5′-TCCACCTTCGAGACTTCC). The PCR product was ligated into pGEM-T Easy and electroporated into XL1-Blue to generate pMT37. A SmaI flanked kanamycin-resistance gene was excised from pUC4-KIXX and inserted into the unique SnaBI site of pMT37 generating plasmid, pMT39. A 3.3-kb EcoRI fragment of pMT39, which contained the kanamycin-resistance gene and flanking gspD sequences, was ligated into the EcoRI site of the temperature sensitive suicide vector pST76-A (22). The resulting construct, pMT42, was transformed into E. coli H10407 by electroporation. Bacteria were plated on LB agar containing kanamycin and incubated at 30°C overnight. Transformants of E. coli H10407 were then incubated at 42°C for 24 h to obtain cells cured of the free plasmid. Bacterial colonies were replica plated on LB agar containing ampicillin or kanamycin to identify ampicillin-sensitive kanamycin-resistant transformants. One of these was E. coli MT13, in which inactivation of gspD by insertion of the kanamycin-resistant gene was confirmed by using PCR analysis.
Complementation of the gspD Mutant.
A 5.5-kb fragment containing gspD, gspE, and gspF was amplified by PCR from E. coli H10407 genomic DNA by using Vent DNA polymerase and oligonucleotide primers P35 (5′-ATTGTCGGTTATGCAGCGAAGC) and P36 (5′-ACTCCTGCAAATTCCAGTTACC). The PCR product was A-tailed with AmpliTaq polymerase, ligated into pGEM-T Easy behind the lac promoter, and electroporated into XL1-Blue to generate pMT6. The correct orientation of the insert was verified by restriction analysis, and its sequence was determined to ensure that fidelity had been maintained during amplification. pMT6 was digested with NcoI to generate a deletion derivative, pMT44, in which only gspD was retained on a 3.8-kb fragment. pMT44 was then transformed into E. coli MT13 by electroporation.
Preparation of Culture Supernatants and Periplasmic Extracts.
E. coli H10407, MT13, and MT13(pMT44) were cultured overnight in 5 ml of LB broth at 37°C in a rotary shaker set at 180 rpm. Five milliliters of prewarmed CAYE medium in 50-ml Erlenmeyer flasks was then inoculated with 125 μl of the overnight bacterial cultures and incubated at 37°C with vigorous aeration (220 rpm in a rotary shaker). To isolate culture supernatants, 4-, 8-, and 24-h cultures were clarified of cells by centrifugation at 3,000 × g for 20 min at 4°C and by passage through a filter with a 0.20-μm-diameter pore size (Sartorius).
To prepare periplasmic extracts, bacterial pellets from cultures used to prepare the supernatants were washed in 1 ml of PBS, pH 7.2 and resuspended in an equal volume of PBS containing 12,000 units/ml polymyxin B (Sigma–Aldrich). After incubation with gentle shaking at 37°C for 30 min, the periplasmic extracts were separated from cell debris by centrifugation at 16,000 × g for 3 min. They were then filtered as described above, after which the volume of the extract was adjusted to match that of the culture supernatant. Supernatants and periplasmic extracts were assayed immediately or stored at 4°C and assayed within 48 h.
The integrity of the supernatant and periplasmic fractions was determined by assaying these fractions for the periplasmic enzyme alkaline phosphatase by using the Sigma 104 alkaline phosphatase kit as follows: 0.05 ml of 221 phosphate buffer, 0.05 ml of p-nitrophenyl phosphate (PNPP), and 0.01 ml of either periplasmic or supernatant extracts were incubated at room temperature for 30 min, after which the reactions were stopped by the addition of 0.1 ml of 5.0 M NaOH. Hydrolysis of PNPP was detected at OD420.
Assays for LT.
LT was assayed for receptor binding by enzyme immunoassay (EIA) (26) and for biological activity by using Y-1 adrenal cells (27). The EIA was modified from a previously described method (26), with all washes performed three times by using PBS containing 0.5% (vol/vol) Tween-20 (PBS-T), and all samples, standards, and antibodies diluted in PBS-T containing 1% (vol/vol) FBS. In addition, all incubations were at 37°C. Briefly, microtiter plates (Nalge Nunc) were coated with 0.1 μg of GM1 ganglioside (Sigma–Aldrich) in 0.06 M sodium carbonate–bicarbonate buffer (pH 9.6) and incubated overnight at 4°C. Plates were washed, blocked with 5% (vol/vol) FBS in PBS for 1 h, and washed again. After initial dilution (culture supernatants 1 in 2 and periplasmic extracts 1 in 5), samples were serially diluted 5-fold and added to the wells. Standard curves were generated in each assay plate by using 2-fold serial dilutions of purified LT (Sigma–Aldrich), at a starting concentration of 400 ng/ml. Plates were incubated for 2 h, washed, and incubated for 1 h with rabbit anti-CT antiserum (Sigma–Aldrich) diluted 1 in 5,000. After washing, horseradish peroxidase-conjugated sheep anti-rabbit IgG (Sigma–Aldrich), diluted 1 in 1,000, was added and incubated for 1 h. After further washing, bound antibody was detected by using the chromogenic substrate, TMB (Kirkegaard & Perry Laboratories). After 3 min at room temperature, the reaction was stopped by the addition of 2 M H2SO4 (30 μl/well), and the OD450 was measured in an ELX800 Universal microplate reader (Bio-Tek, Winooski, VT). Each assay was performed in triplicate on at least three separate occasions. Regression analysis (R2 > 0.96) was used to generate a standard curve for determination of LT concentrations in the test samples.
To measure biological activity of the toxin preparations, Y1 mouse adrenal cells were seeded in 96-well microtiter plates at a concentration of 2 × 104 cells per well. After 48 h, the growth medium was replaced with DMEM supplemented with 1% FBS and gentamicin (100 μl), containing duplicate samples of bacterial culture supernatant and periplasmic extracts, serially diluted 2-fold after an initial dilution of 1 in 10. The cells were incubated for 18 h and then examined for typical rounding by a blinded operator by using phase-contrast microscopy at a magnification of ×200. The end point of the assay was defined as the highest dilution that showed more than 50% cell rounding. Each assay was performed in duplicate on at least two separate occasions. Two-fold dilutions of purified LT at a starting concentration of 10 ng/ml were assayed in the same plates and the result used to determine the sensitivity of the assay.
Prevalence of the Type II Secretion Pathway in ETEC Strains.
To determine the prevalence of the novel type II secretion pathway in ETEC strains of clinical origin, a variety of ETEC isolates of different serotypes and toxin profiles were examined by PCR for a 1.0-kb fragment of gspD by using primers P78 (5′-TTCGGAAATCGCCCGCGTGC) and P109 (5′-TCCACCTTCGAGACTTCC), and for a 1.2-kb fragment of gspK by using primers P5 (5′-GCAGCAGGTGACTAACGGC) and P12 (5′-CAGGGCTTAACCACGGGTC).
Statistical Analysis.
All analyses were performed by using Student's t test. A value of P < 0.05 was taken to indicate statistical significance.
Nucleotide Sequence Accession Number.
The E. coli H10407 type II secretion locus has been assigned GenBank accession no. AY056599.
Results
Identification, Sequencing, and Characterization of the Type II Secretion Genes.
In our work, to characterize pathogenicity islands in rabbit-specific strains of enteropathogenic E. coli (28), we identified and sequenced a novel type II protein secretion pathway from one particular strain (GenBank accession no. AF426313). Using primers generated from this sequence, we found that several other pathogenic strains of E. coli possessed this pathway, including the prototypic human ETEC strain H10407. We then determined the sequence of the H10407 chromosome encompassing the putative genes for this pathway by amplifying and sequencing overlapping fragments (GenBank accession no. AY056599). Computer analysis of 11,765 bp of this sequence revealed 13 ORFs, 11 of which were organized in a single operon, with several of the ORFs overlapping. Because the genetic organization of the type II gene cluster is relatively well conserved (29), we designated the newly identified genes gspC-M to comply with current nomenclature (Fig. 1A). Interestingly, a search of the public databases for homologous sequences revealed that the 5′ and 3′ ends of the ETEC type II secretion pathway are also present in the E. coli K-12 strain, MG1655 (15) (Fig. 1B).
Figure 1.

(A) Schematic diagram of the genetic organization of the chromosomal region containing the genes (solid arrows) for the putative type II secretion pathway in ETEC H10407. *, Insertion site of the K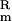 gene in E. coli MT13. (B) Chromosomal region common to ETEC strain, H10407 and E. coli K-12 strain, MG1655, with the percentage identity/similarity of homologous proteins shown in parentheses. †, Percentage identity/similarity over 286 amino acid residues; ‡, percentage identity/similarity over 276 amino acid residues. (C) Arrangement of the genes encoding the type II pathway for the secretion of CT by V. cholerae [accession nos. AF109904 (30), AF055371 (31), and L33796 (10)], and, in parentheses, the percentage identity/similarity of homologous proteins of ETEC H10407 and V. cholerae, TRH7000. §, In V. cholerae TRH7000, the prepilin peptidase gene, pilD/vcpD, is not linked to the type II secretion gene cluster. ¶, There is no homologue of epsN in the ETEC H10407 type II secretion gene cluster. (D) Arrangement of the genes encoding the type II secretion pathway in E. coli K-12, strain MG1655 [accession no. AE000409 (15)], and, in parentheses, the percentage identity/similarity of homologous proteins of ETEC H10407 and E. coli K-12, strain MG1655. ∥, NSH, no significant homology.
gene in E. coli MT13. (B) Chromosomal region common to ETEC strain, H10407 and E. coli K-12 strain, MG1655, with the percentage identity/similarity of homologous proteins shown in parentheses. †, Percentage identity/similarity over 286 amino acid residues; ‡, percentage identity/similarity over 276 amino acid residues. (C) Arrangement of the genes encoding the type II pathway for the secretion of CT by V. cholerae [accession nos. AF109904 (30), AF055371 (31), and L33796 (10)], and, in parentheses, the percentage identity/similarity of homologous proteins of ETEC H10407 and V. cholerae, TRH7000. §, In V. cholerae TRH7000, the prepilin peptidase gene, pilD/vcpD, is not linked to the type II secretion gene cluster. ¶, There is no homologue of epsN in the ETEC H10407 type II secretion gene cluster. (D) Arrangement of the genes encoding the type II secretion pathway in E. coli K-12, strain MG1655 [accession no. AE000409 (15)], and, in parentheses, the percentage identity/similarity of homologous proteins of ETEC H10407 and E. coli K-12, strain MG1655. ∥, NSH, no significant homology.
The first complete ORF of the ETEC type II secretion gene cluster is pppA; PppA is 96% identical to its homologue in MG1655. The next closest homology (46% identity, 63% similarity) is to VcpD (PilD), the prepilin peptidase required for the secretion of CT by V. cholerae (30, 31). Downstream of pppA is a small ORF, yghG, which encodes a putative outer membrane lipoprotein, showing 24% identity and 49% similarity to a conserved hypothetical protein of unknown function from V. cholerae, but which is not colocated with either vcpD (pilD) or the type II gene cluster. The proteins encoded by the genes gspC-M, which evidently comprise a single operon, show the closest homology to the type II secretion proteins of V. cholerae (10), with sequence identities ranging from 76 to 24% and similarities ranging from 86 to 47% (Fig. 1C).
To determine the prevalence of the type II secretion pathway in clinical isolates of ETEC, we performed PCR analysis for gspD and gspK on a variety of ETEC strains of serotypes O6:H16, O8:H9, O20:H-, O25:H-, O25:H42, O75:H4, O78:H11, O114:H21, O128:H21, O148:H28, O149:H10, O27:H20, O159:H34, and O-nontypeable:H9. The test bacteria included 11 that produced LT only, 9 that produced both LT and heat-stable enterotoxin, and 4 that produced heat-stable enterotoxin only. All 24 of these strains tested positive for both gspD and gspK.
Determination of the Role of the Type II Pathway in the Secretion of LT by ETEC Strain H10407.
Because V. cholerae requires a type II protein secretory pathway to secrete CT, we investigated whether the homologous pathway of E. coli H10407 played a role in the secretion of LT. To this end, we inactivated the pathway by inserting a kanamycin resistance gene into gspD by homologous recombination (Fig. 1A). We chose this gene for inactivation because in Pseudomonas aeruginosa, the homologue of GspD forms multimeric complexes, is localized within the outer membrane, and is the putative pore of the type II secretion apparatus (32, 33). Insertional inactivation of gspD in ETEC H10407 yielded the mutant strain, MT13.
To determine whether MT13 was able to secrete LT, we used an EIA (26) to compare LT secretion by MT13 with that by E. coli H10407. Comparison of the growth curves of E. coli H10407 and MT13 showed no difference between them and indicated that suitable time points to assay the culture medium for LT were 4 h (representing midlogarithmic phase), 8 h (early-stationary phase), and 24 h (late-stationary phase) (Fig. 2A). The detection limit of the EIA using purified LT was determined to be 0.4 ng/ml, with a standard curve that was linear between 2.6 and 24.0 ng/ml (data not shown). This portion of the curve was used to estimate the amount of LT in the test samples. The results of these assays (Fig. 2B) showed that by 4 h, H10407 had secreted 58.5 ± 3.7 (mean ± SD) ng/ml of LT into the supernatant, but the amount of LT in the supernatant of the gspD mutant MT13 was below the level of detection (<0.4 ng/ml; P = 0.001). At the same time point, however, the amount of LT in the periplasm of the mutant (31.3 ± 2.6 ng/ml) was approximately 10-fold greater than that of the wild type (3.4 ± 0.1 ng/ml; P < 0.001) (Fig. 2C). These results indicate that MT13 could synthesize LT but could not secrete it. By 8 h the amount of LT secreted into the medium by H10407 had increased to 90.3 ± 7.6 ng/ml, but the amount of LT in the supernatant of MT13 was still below the level of detection (P = 0.002). At the same time point the amount of toxin retained in the periplasm of the mutant had fallen to 1.5 ± 0.1 ng/ml, but was still significantly greater than that found in the wild type, which was below detectable levels (P = 0.002). The reduction in the amount of LT in the periplasm of the mutant could be due to proteolytic degradation/turnover (14), as the A subunit is susceptible to a variety of proteases (11). By 24 h, E. coli H10407 had secreted 193.6 ± 1.2 ng/ml of LT into the medium. At this time point, 29.1 ± 1.1 ng/ml of LT was detectable in the supernatant for MT13 (P < 0.001), but the amount of toxin retained in the periplasm of MT13 was still significantly greater than the wild type (P < 0.005). The integrity of the cell fractions used for these studies was confirmed by assays for the periplasmic enzyme alkaline phosphatase. These assays showed no alkaline phosphatase activity in the supernatants from E. coli H10407 or MT13 at 4 and 8 h and comparable amounts of enzyme activity in supernatants at 24 h and in the periplasmic extracts from both strains at all time points.
Figure 2.

(A) Growth curves of E. coli H10407 (interrupted line) and MT13 (solid line) in CAYE broth. (B) Concentration of LT (detected by *, EIA, and †, Y1 adrenal cell assay) in supernatants of CAYE broth, 4, 8, and 24 h after inoculation with E. coli H10407 or MT13. (C) Concentration of LT (detected by *, EIA, and †, Y1 adrenal cell assay) in centrifuged bacteria pellets obtained from the broth cultures depicted in B. Error bars indicate SD.
Complementation of MT13 with a wild-type copy of gspD in trans on plasmid pMT44 restored LT secretion into culture supernatants to 74% of wild-type levels at 4 h, 76% at 8 h, and 89% at 24 h. These findings are similar to those reported when an epsD (gspD homologue) mutant of V. cholerae, strain N16961, was trans-complemented by a plasmid clone of wild-type epsD (34). Together, these data show that the type II secretion pathway is required for the secretion of LT in E. coli H10407.
The results of the bacterial viability/cell lysis assays showed no difference between E. coli H10407 and MT13. At 4 and 8 h, there was less than 1% cell lysis, and at 24 h there was 34% cell lysis, which could account for the presence of LT in the supernatant of MT13.
Biological Activity of LT.
To determine whether the LT produced by the mutant strain MT13 was biologically active, we used the Y1 adrenal cell bioassay. This assay is based on the observation that LT causes Y1 adrenal cells to change from their usual elongated spindle morphology to a more rounded shape. These morphological changes are indistinguishable from those induced by CT (18) and are concentration dependent. Y1 cells seeded in 96-well microtiter plates were incubated with cell-free supernatants and periplasmic extracts from E. coli H10407 and MT13. The morphological changes induced in the Y1 cells by these preparations were identical, indicating that the LT produced by strain MT13 was biologically active (Fig. 3).
Figure 3.

Phase-contrast photomicrographs of cultured Y1 adrenal cells 18 h after addition of a 1 in 40 dilution of cell free CAYE culture medium from 4-h cultures of (A) E. coli H 10407 or (B) E. coli MT13. C shows the effect of a 1 in 40 dilution of a periplasmic extract from a 4-h culture of E. coli MT13.
A semiquantitative assay (27) using Y1 adrenal cells and 2-fold dilutions of the cell-free supernatants and periplasmic extracts was used to determine the amount of LT in these fractions. Using purified LT, we determined the sensitivity of the assay (amount of toxin that induced rounding in 50% of the cells) to be 49 pg/ml. This value was used to estimate the amount of LT in the test samples. The result of this assay confirmed those of the EIA, namely that the gspD mutant was capable of LT synthesis but was markedly defective in LT secretion (Figs. 2 and 3).
Discussion
Heat-labile enterotoxin is a major virulence determinant of ETEC, which must be released from the bacteria before it can act on host cells. The failure to identify a protein secretory pathway in ETEC fostered the view that ETEC release LT only when the bacteria undergo lysis (35). Contradicting this view are the data, confirmed in this study, that the kinetics of LT secretion by ETEC resemble those exhibited by V. cholerae when secreting CT, with an increase in toxin concentration that increases with cell density and less than 10% of toxin retained within the periplasm (8). Moreover, Horstman and Kuehn (20) demonstrated that in ETEC strain, ETEC 2 (ATCC 43886), LT does not remain in the periplasm but is found associated with the cell exterior. They propose that in the absence of an identified LT secretory mechanism, such as a type II secretion pathway, vesicles may play a role in the secretion of the toxin. Fleckenstein et al. (36) reported a gene, leoA, which is required for the secretion of LT by E. coli strain H10407. However, LeoA bears no homology to any known protein involved in the secretion of CT (36), hence its discovery does not shed any light on the mechanism of LT secretion by ETEC.
In this study, we have shown that the prototypic ETEC strain, H10407, possesses a protein secretory pathway highly homologous to that used by V. cholerae to secrete CT, and that genes that encode components of this pathway are present in a wide variety of ETEC strains, suggesting that this protein secretory pathway is highly conserved among ETEC strains. Inactivation of this pathway by mutagenesis of the gspD gene completely ablated LT secretion, yet LT secretion was restored when the gspD mutant was complemented in trans with a copy of the wild-type gspD gene. We also showed that the gspD mutant retained the ability to synthesize biologically active LT, and that the kinetics of toxin release by this strain closely resembled those in LT-producing derivatives of E. coli K-12 (37), which also lack this secretory pathway. Taken together, these data demonstrate that ETEC possess a type II secretion pathway that is responsible for the secretion of LT.
Although E. coli K-12 does contain the genes for a complete type II secretion pathway at minute 74.5 of the genome (15), these genes are not expressed under standard laboratory conditions (16). The percentage identity between the predicted proteins of these genes and their homologues present in the type II secretion pathway of ETEC H10407 ranges from no significant homology to 65% (Fig. 1D). Interestingly, the E. coli K-12 type II secretion pathway is absent from ETEC H10407 (data not shown). If it were present and functional, it may have complemented the gspD mutant, as occurred with a strain of P. aeruginosa that retained the capacity for extracellular secretion despite carrying a mutation in gspD (xcpQ), because it contained a second gspD homologue, xqhA (38).
An interesting feature of this recently identified ETEC type II secretion pathway is that remnants of it are present in the E. coli K-12 strain, MG1655, at minute 67 on the genetic map (15). This finding suggests that K-12 strains may have originally possessed this pathway and that it was lost during the course of evolution. Francetic et al. have cloned the pppA gene from E. coli K-12 (39), which encodes prepilin peptidase (PppA). They determined that it is functional and present in two LT-producing ETEC strains. The putative prepilin peptidase encoded in the type II secretion locus of ETEC H10407 is 96% identical to PppA from E. coli K-12 and presumably acts by processing individual components of the pathway before assembly.
Both LT and CT are encoded by mobile genetic elements. They share over 77% homology at the nucleotide and amino acid levels for their A and B subunits (7), and the plasmid-borne genes encoding LT are presumed to have originated from the ancestral genes for CT (40), which are encoded by a prophage (41). Bacteria with a type II secretory pathway that acquired toxin-encoding genes by interbacterial transfer would secrete the toxins relatively efficiently and consequently would be expected to have a survival advantage over those that do not, because their enhanced virulence would lead to greater numbers of bacteria being excreted in feces with improved prospects of their transmission to new hosts.
Acknowledgments
We are indebted to Dr. M. M. Levine, University of Maryland School of Medicine, and to Dr. H. R. Smith Public Health Laboratory Service, London, U.K., for the gift of bacterial strains that were used in this study; and to R. Good for advice and assistance. This study was supported in part by a grant from the Australian National Health and Medical Research Council. M.T. was supported by an Australian Postgraduate Research Award.
Abbreviations
- CT
cholera toxin
- EIA
enzyme immunoassay
- ETEC
enterotoxigenic E. coli
- LT
heat-labile enterotoxin
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AY056599).
References
- 1.Nataro J P, Kaper J B. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. Wkly Epidemiol Rec. 1999;74:98–101. [Google Scholar]
- 3.Gyles C L. Can J Microbiol. 1992;38:734–746. doi: 10.1139/m92-120. [DOI] [PubMed] [Google Scholar]
- 4.Spangler B. Microbiol Rev. 1992;56:622–647. doi: 10.1128/mr.56.4.622-647.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beubler E, Schuligoi R. Ann NY Acad Sci. 2000;915:339–346. doi: 10.1111/j.1749-6632.2000.tb05262.x. [DOI] [PubMed] [Google Scholar]
- 6.Mourad F H, Nassar C F. Gut. 2000;47:382–386. doi: 10.1136/gut.47.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connell T D, Metzger D J, Wang M, Jobling M G, Holmes R K. Infect Immun. 1995;63:4091–4098. doi: 10.1128/iai.63.10.4091-4098.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirst T R, Sanchez J, Kaper J B, Hardy S J, Holmgren J. Proc Natl Acad Sci USA. 1984;81:7752–7756. doi: 10.1073/pnas.81.24.7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neill R J, Ivins B E, Holmes R K. Science. 1983;221:289–291. doi: 10.1126/science.6857285. [DOI] [PubMed] [Google Scholar]
- 10.Sandkvist M, Michel L O, Hough L P, Morales V M, Bagdasarian M, Koomey M, DiRita V J, Bagdasarian M. J Bacteriol. 1997;179:6994–7003. doi: 10.1128/jb.179.22.6994-7003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirst T R, Holmgren J. J Bacteriol. 1987;169:1037–1045. doi: 10.1128/jb.169.3.1037-1045.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pugsley A P. Microbiol Rev. 1993;57:50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michel L O, Sandkvist M, Bagdasarian M. Gene. 1995;152:41–45. doi: 10.1016/0378-1119(94)00691-k. [DOI] [PubMed] [Google Scholar]
- 14.Pearson G D, Mekalanos J J. Proc Natl Acad Sci USA. 1982;79:2976–2980. doi: 10.1073/pnas.79.9.2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blattner F R, Plunkett G, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, et al. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 16.Francetic O, Belin D, Badaut C, Pugsley A P. EMBO J. 2000;19:6697–6703. doi: 10.1093/emboj/19.24.6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Francetic O, Pugsley A P. J Bacteriol. 1996;178:3544–3549. doi: 10.1128/jb.178.12.3544-3549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donta S T, Moon H W, Whipp S C. Science. 1974;183:334–336. doi: 10.1126/science.183.4122.334. [DOI] [PubMed] [Google Scholar]
- 19.Kunkel S L, Robertson D C. Infect Immun. 1979;23:652–659. doi: 10.1128/iai.23.3.652-659.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horstman A L, Kuehn M J. J Biol Chem. 2000;275:12489–12496. doi: 10.1074/jbc.275.17.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans D G, Evans D J, Jr, Pierce N F. Infect Immun. 1973;7:873–880. doi: 10.1128/iai.7.6.873-880.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Posfai G, Koob M D, Kirkpatrick H A, Blattner F R. J Bacteriol. 1997;179:4426–4428. doi: 10.1128/jb.179.13.4426-4428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A. Current Protocols in Molecular Biology. New York: Wiley; 1991. [Google Scholar]
- 24.Mundell D H, Anselmo C R, Wishnow R M. Infect Immun. 1976;14:383–388. doi: 10.1128/iai.14.2.383-388.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 26.Ristaino P A, Levine M M, Young C R. J Clin Microbiol. 1983;18:808–815. doi: 10.1128/jcm.18.4.808-815.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sack D A, Sack R B. Infect Immun. 1975;11:334–336. doi: 10.1128/iai.11.2.334-336.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tauschek, M., Strugnell, R. A. & Robins-Browne, R. M. (2002) Mol. Microbiol., in press. [DOI] [PubMed]
- 29.Sandkvist M. Infect Immun. 2001;69:3523–3535. doi: 10.1128/IAI.69.6.3523-3535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fullner K J, Mekalanos J J. Infect Immun. 1999;67:1393–1404. doi: 10.1128/iai.67.3.1393-1404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marsh J W, Taylor R K. Mol Microbiol. 1998;29:1481–1492. doi: 10.1046/j.1365-2958.1998.01031.x. [DOI] [PubMed] [Google Scholar]
- 32.Bitter W, Koster M, Latijnhouwers M, de Cock H, Tommassen J. Mol Microbiol. 1998;27:209–219. doi: 10.1046/j.1365-2958.1998.00677.x. [DOI] [PubMed] [Google Scholar]
- 33.Hardie K R, Lory S, Pugsley A P. EMBO J. 1996;15:978–988. [PMC free article] [PubMed] [Google Scholar]
- 34.Ali A, Johnson J A, Franco A A, Metzger D J, Connell T D, Morris J G, Jr, Sozhamannan S. Infect Immun. 2000;68:1967–1974. doi: 10.1128/iai.68.4.1967-1974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wai S N, Takade A, Amako K. Microbiol Immunol. 1995;39:451–456. doi: 10.1111/j.1348-0421.1995.tb02228.x. [DOI] [PubMed] [Google Scholar]
- 36.Fleckenstein J M, Lindler L E, Elsinghorst E A, Dale J B. Infect Immun. 2000;68:2766–2774. doi: 10.1128/iai.68.5.2766-2774.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirst T R, Randall L L, Hardy S J. J Bacteriol. 1984;157:637–642. doi: 10.1128/jb.157.2.637-642.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez A, Ostrovsky P, Nunn D N. Mol Microbiol. 1998;28:1235–1246. doi: 10.1046/j.1365-2958.1998.00888.x. [DOI] [PubMed] [Google Scholar]
- 39.Francetic O, Lory S, Pugsley A P. Mol Microbiol. 1998;27:763–775. doi: 10.1046/j.1365-2958.1998.00723.x. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto T, Gojobori T, Yokota T. J Bacteriol. 1987;169:1352–1357. doi: 10.1128/jb.169.3.1352-1357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waldor M K, Mekalanos J J. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]