

Abstract
This article presents a Bayesian method for model-based clustering of gene expression dynamics. The method represents gene-expression dynamics as autoregressive equations and uses an agglomerative procedure to search for the most probable set of clusters given the available data. The main contributions of this approach are the ability to take into account the dynamic nature of gene expression time series during clustering and a principled way to identify the number of distinct clusters. As the number of possible clustering models grows exponentially with the number of observed time series, we have devised a distance-based heuristic search procedure able to render the search process feasible. In this way, the method retains the important visualization capability of traditional distance-based clustering and acquires an independent, principled measure to decide when two series are different enough to belong to different clusters. The reliance of this method on an explicit statistical representation of gene expression dynamics makes it possible to use standard statistical techniques to assess the goodness of fit of the resulting model and validate the underlying assumptions. A set of gene-expression time series, collected to study the response of human fibroblasts to serum, is used to identify the properties of the method.
Both cDNA (1) and synthetic oligonucleotide (2) microarrays enable investigators to simultaneously measure the expression of thousands of genes and hold the promise to cast new light onto the regulatory mechanisms of the genome. Different unsupervised methods have been used to analyze these data to characterize gene functional behaviors. Among others (3–5), correlation-based hierarchical clustering (6) is today one of the most popular analytical methods to characterize gene-expression profiles. Given a set of expression values measured for a set of genes under different experimental conditions, this approach recursively clusters genes according to the correlation of their measurements under the same experimental conditions. The intuition behind this approach is that correlated genes are acting together because they belong to similar, or at least related, functional categories. The clustering process returns a sorted representation of expression profiles that allows the investigator to identify sets of coregulated genes. The sorted set of gene expression profiles is used to support the operation of partitioning the profiles in separated clusters, which is left to the visual inspection of the investigator.
This clustering approach has become widely popular and it has been successfully applied to the genomewide discovery and characterization of the regulatory mechanisms of several processes and organisms (7–10). Several of these applications of genomewide clustering methods focus on the temporal profiling of gene expression patterns. Temporal profiling offers the possibility of observing the cellular mechanisms in action and tries to break down the genome into sets of genes involved in the same, or at least related, processes. However, correlation-based clustering methods rest on the assumption that the set of observations for each gene are independent and identically distributed (iid). While this assumption holds when expression measures are taken from independent biological samples, it is known to be no longer valid when the observations are actually realizations of a time series, where each observation may depend on prior ones (e.g., refs. 11 and 12). Pairwise similarity measures currently used for clustering gene expression data, such as correlation or Euclidean distance, are invariant with respect to the order of observations: if the temporal order of a pair of series is permuted, their correlation or Euclidean distance will not change. Biomedical informatics investigators over the past decade have demonstrated the risks incurred by disregarding the dependency among observations in the analysis of time series (13, 14). Not surprisingly, the functional genomic literature is becoming increasingly aware of the specificity of temporal profiles of gene expression data, as well as of their fundamental importance in unraveling the functional relationships between genes (15–17).
We introduce here a Bayesian model-based clustering method to profile gene expression time series, which explicitly takes into account the dynamic nature of temporal gene expression profiles; that is, that time series data are not iid observations. This method is a specialized version of a more general class of methods called Bayesian clustering by dynamics (BCD) (18), which have been applied to a variety of time series data, ranging from cognitive robotics (19) to official statistics (20). The main novelty of BCD is the concept of similarity: two time series are similar when they are generated by the same stochastic process. With this concept of similarity, the Bayesian approach to the task of clustering a set of time series consists of searching the most probable set of processes generating the observed time series. The method presented here models temporal gene-expression profiles by autoregressive equations (11) and groups together the profiles with the highest posterior probability of being generated by the same process.
Besides its ability to account for the dynamic nature of temporal gene expression profiles, this method automatically identifies the number of clusters and partitions the gene expression time series in different groups on the basis of the principled measure of the posterior probability of the clustering model. In this way, it allows the investigator to assess whether the experimental data convey enough evidence to support the conclusion that the behavior of a set of genes is significantly different from the behavior of another set of genes. This feature is particularly important as decades of cognitive science research have shown that the human eye tends to overfit observations by selectively discount variance and “seeing” patterns in randomness (e.g., refs. 21–23). In our case, we therefore expect that visual inspection will find more clusters than those supported by the available evidence. By contrast, a recognized advantage of a Bayesian approach to model selection, like the one adopted in this article, is the ability to automatically constrain model complexity (24, 25) and to provide appropriate measures of uncertainty.
Since the number of possible clustering models grows exponentially with the number of time series, our method uses a bottom-up distance-based heuristic to make the search process amenable. The result of this clustering process can be represented by a set of trees, graphically displaying the model in an intuitive form. In this way, the method retains the important visualization capability of distance-based clustering but acquires an independent, principled measure to decide whether two series are similar enough to belong to the same cluster. Furthermore, the use of the posterior probability of the model as an independent clustering metrics allows the comparison of different similarity measures, which are typically chosen in a somewhat arbitrary way. In the analysis presented here, for instance, we will use several similarity measures—Euclidean distance, correlation, delayed correlation, and Kullback–Leiber distance—and select the one returning the most probable clustering model. Another important character of the method here presented is its reliance on an explicit statistical model of gene expression dynamics. This reliance makes it possible to use standard statistical techniques to assess the goodness of fit of the resulting model and validate the underlying assumptions. A set of gene expression time series, collected to study the response of human fibroblasts to serum (8), is used to study the properties of the method. Our results confirm the advantages of taking into account the dynamic nature of temporal data and return a different picture from the one offered by the original correlation-based cluster analysis. Statistical diagnostics and biological insights support our results.
Methods
We regard a set of temporally oriented gene expression observations as a set of time series S = {S1, S2, … , Sm}, generated by an unknown number of stochastic processes. The task here is to iteratively merge time series into clusters, so that each cluster groups the time series generated by the same process. Our clustering method has two components: a stochastic description of a set of clusters, from which we derive a probabilistic scoring metric, and a heuristic search procedure. The derivation of the scoring metric assumes that the processes generating the data can be approximated by autoregressive models.
Autoregressive Models.
Let S = {yj1, … yjt, … yjn} be a stationary time series of continuous values. The series follows an autoregressive model of order p, say ar(p), if the value of the series at time t > p is a linear function of the values observed in the previous p steps. More formally, we can describe the model in matrix form as
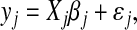 |
1 |
where yj is the vector
(yj(p+1), … , yjn)T,
Xj is the (n − p) × q
regression matrix whose tth row is (1,
yj(t−1), … , yj(t−p)), for
t > p, and q = p + 1. The
elements of the vector βj =
{βj0, βj1, … ,
βjp} are the autoregressive coefficients, and
ɛj = (ɛj(p+1), … ,
ɛjn)T is a vector of uncorrelated errors
that we assume normally distributed, with expected value
E(ɛjt) = 0 and variance
V(ɛjt) = σ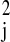 , for any
t. The value p is the autoregressive order and
specifies that, at each time point t, yjt is
independent of the past history given the previous p steps.
The time series is stationary if it is invariant by temporal
translations. Formally, stationarity requires that the coefficients
βj are such that the roots of the polynomial
f(u) = 1 −
∑
, for any
t. The value p is the autoregressive order and
specifies that, at each time point t, yjt is
independent of the past history given the previous p steps.
The time series is stationary if it is invariant by temporal
translations. Formally, stationarity requires that the coefficients
βj are such that the roots of the polynomial
f(u) = 1 −
∑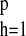 βjhuh have
moduli greater than unity. The model in Eq. 1 represents the
evolution of the process around its mean μ, which is related to the
βj coefficients by the equation
μ = βj0/(1 −
∑
βjhuh have
moduli greater than unity. The model in Eq. 1 represents the
evolution of the process around its mean μ, which is related to the
βj coefficients by the equation
μ = βj0/(1 −
∑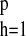 βjh). In particular, μ is
well defined as long as
∑
βjh). In particular, μ is
well defined as long as
∑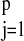 βj ≠ 1. When
the autoregressive order p = 0, the series S
becomes a sample of independent observations from a normal distribution
with mean μ = βj0 and variance
σ
βj ≠ 1. When
the autoregressive order p = 0, the series S
becomes a sample of independent observations from a normal distribution
with mean μ = βj0 and variance
σ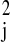 . Note that the model in Eq.
1 is a special case of a state-space model (12).
. Note that the model in Eq.
1 is a special case of a state-space model (12).
Probabilistic Scoring Metric.
We describe a set of c clusters of time series as a
statistical model Mc, consisting of c
autoregressive models with coefficients βk and
variance σ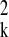 . Each cluster
Ck groups mk time series
that are jointly modeled as
. Each cluster
Ck groups mk time series
that are jointly modeled as
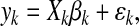 |
where the vector yk and the matrix Xk are defined by stacking the mk vectors ykj and regression matrices Xkj, one for each time series, as follows
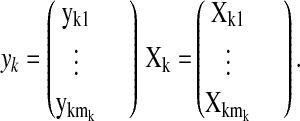 |
Note that we now label the vectors yj
assigned to the same cluster Ck with the double
subscript kj, and k denotes the cluster
membership. The vector ɛk is the vector of
uncorrelated errors with zero expected value and constant variance
σ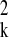 . Given a set of possible clustering
models, the task is to rank them according to their posterior
probability. The posterior probability of each clustering model
Mc is
. Given a set of possible clustering
models, the task is to rank them according to their posterior
probability. The posterior probability of each clustering model
Mc is
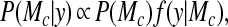 |
where P(Mc) is the prior probability of Mc, y consists of the data {yk}, and the quantity f(y|Mc) is the marginal likelihood. The marginal likelihood f(y|Mc) is the solution of the integral
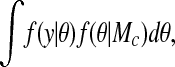 |
where θ is the vector of parameters specifying the clustering model Mc, and f(θ|Mc) is its prior density. Assuming uniform prior distributions on the model parameters and independence of the time series conditional on the cluster membership, f(y|Mc) can be computed as
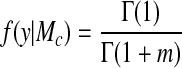 |
2 |
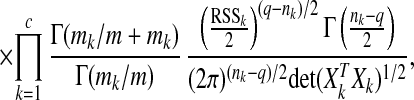 |
where nk is the dimension of the vector
yk, and rssk= y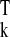 (In −
Xk(X
(In −
Xk(X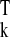 Xk)−1X
Xk)−1X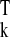 )yk
is the residual sum of squares in cluster Ck.
When all clustering models are a priori equally likely, the
posterior probability P(Mc|y) is proportional
to the marginal likelihood f(y|Mc), which
becomes our probabilistic scoring metric.
)yk
is the residual sum of squares in cluster Ck.
When all clustering models are a priori equally likely, the
posterior probability P(Mc|y) is proportional
to the marginal likelihood f(y|Mc), which
becomes our probabilistic scoring metric.
Agglomerative Bayesian Clustering.
The Bayesian approach to the clustering task is to choose the model Mc with maximum posterior probability. As the number of clustering models grows exponentially with the number of time series, we use an agglomerative, finite-horizon search strategy that iteratively merges time series into clusters. The procedure starts by assuming that each of the m observed time series is generated by a different process. Thus, the initial model Mm consists of m clusters, one for each time series, with score f(y|Mm). The next step is the computation of the marginal likelihood of the m(m − 1) models in which two of the m series are merged into one cluster. The model Mm−1 with maximal marginal likelihood is chosen and, if f(y|Mm) ≥ f(y|Mm−1), no merging is accepted and the procedure stops. If f(y|Mm) < f(y|Mm−1), the merging is accepted, a cluster Ck merging the two time series is created, and the procedure is repeated on the new set of m − 1 clusters, consisting of the remaining m − 2 time series and the cluster Ck.
Heuristic Search.
Although the agglomerative strategy makes the search process feasible, the computational effort can still be extremely demanding when the number m of time series is large. To reduce this effort further, we use a heuristic strategy based on a measure of similarity between time series. The intuition behind this strategy is that the merging of two similar time series has better chances of increasing the marginal likelihood of the model. The heuristic search starts by computing the m(m − 1) pairwise similarity measures of the m time series and selects the model Mm−1 in which the two closest time series are merged into one cluster. If f(y|Mm−1) > f(y|Mm), the merging is accepted, the two time series are merged into a single cluster. The average profile of this cluster is computed by averaging the two observed time series, and the procedure is repeated on the new set of m − 1time series, containing the new cluster profile. If this merging is rejected, the procedure is repeated on pairs of time series with decreasing degree of similarity until an acceptable merging is found. If no acceptable merging is found, the procedure stops. Note that the decision of merging two clusters is actually made on the basis of the posterior probability of the model and that the similarity measure is used only to improve efficiency and limit the risk of falling into local maxima.
Several measures can be used to assess the similarity of two time series, both model-free, such as Euclidean distance, correlation and lag-correlation, and model-based, such as Kullback–Leiber distance. Model-free distances are calculated on the raw data. Because the method uses these similarity measures as heuristic tools rather than scoring metrics, we can actually assess the efficiency of each of these measures to drive the search process toward the model with maximum posterior probability. In this respect, the Euclidean distance of two time series Si = {yi1, … , y1n} and Sj = {yj1, … , yjn}, computed as
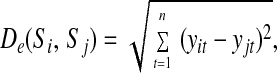 |
performs best on the short time series of our data set. This finding is consistent with the results of ref. 9, claiming a better overall performance of Euclidean distance in standard hierarchical clustering of gene expression profiles.
Validation.
Standard statistical diagnostics are used as independent assessment
measures of the cluster model found by the heuristic search. Once the
procedure terminates, the coefficients βk of
the ar(p) model associated with each cluster
Ck are estimated as
β̂k =
(X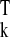 Xk)−1X
Xk)−1X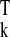 yk,
while σ̂
yk,
while σ̂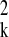 =rssk/(nk −
q) is the estimate of the within-cluster variance
σ
=rssk/(nk −
q) is the estimate of the within-cluster variance
σ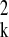 . The parameter estimates can be used
to compute the fitted values for the series in each cluster as
ŷkj =
Xkjβ̂k, from which we compute the
standardized residuals rkj =
(ykj −
ŷkj)/σ̂k. If
ar(p) models provide an accurate approximation
of the processes generating the time series, the standardized residuals
should behave like a random sample from a standard normal distribution.
A normal probability plot or the residuals histogram per cluster are
used to assess normality. Departures from normality cast doubt on the
autoregressive assumption, so that some data transformation, such as a
logarithmic transformation, may be needed. Plots of fitted vs. observed
values and of fitted values vs. standardized residuals in each cluster
provide further diagnostics. To choose the best autoregressive order,
we repeat the clustering for p = 0, 1, … ,
w, for some preset w—by using the same
p for every clustering model—and compute a goodness of fit
score defined as
. The parameter estimates can be used
to compute the fitted values for the series in each cluster as
ŷkj =
Xkjβ̂k, from which we compute the
standardized residuals rkj =
(ykj −
ŷkj)/σ̂k. If
ar(p) models provide an accurate approximation
of the processes generating the time series, the standardized residuals
should behave like a random sample from a standard normal distribution.
A normal probability plot or the residuals histogram per cluster are
used to assess normality. Departures from normality cast doubt on the
autoregressive assumption, so that some data transformation, such as a
logarithmic transformation, may be needed. Plots of fitted vs. observed
values and of fitted values vs. standardized residuals in each cluster
provide further diagnostics. To choose the best autoregressive order,
we repeat the clustering for p = 0, 1, … ,
w, for some preset w—by using the same
p for every clustering model—and compute a goodness of fit
score defined as
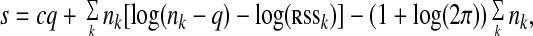 |
where c is the number of clusters, nk is the size of the vector yk in Ck, q = p + 1, where p is the autoregressive order, and rssk is the residual sum of squares of cluster Ck. This score is derived by averaging the log-scores cumulated by the series assigned to each clusters. The derivation of this score is detailed in the technical report available from the Supporting Appendixes, which are published as supporting information on the PNAS web site, www.pnas.org. The resulting score trades off model complexity—measured by the quantity cq + ∑knklog(nk − q)—with lack of fit—measured by the quantity ∑nklog(rssk), and it generalizes the well known Akaike information criterion goodness of fit criterion of ref. 26 to a set of autoregressive models. We then choose the clustering model with the autoregressive order p that maximizes this goodness of fit score.
Display.
As in ref. 6, a colored map is created by displaying the rows of the original data table according to the pairwise merging of the heuristic search. In our case, a set of binary trees (dendrogram), one for each cluster, is appended to the colored map. Each branching node is labeled with the ratio between the marginal likelihood of the merging accepted at that node and the marginal likelihood of the model without this merging. This ratio measures how many times the model accepting the merging is more likely than the model refusing it.
Materials
Iyer et al. (8) report the results of a study of the temporal deployment of the transcriptional program underlying the response of human fibroblasts to serum. The study uses two-dye cDNA microarrays to measure the changes of expression levels of 8,613 human genes over 24 h. The actual data described in the study comprise a selection of 517 genes whose expression level changed in response to serum stimulation. At the time of their original publication, 238 genes were unknown expressed sequence tags (ESTs). We relabeled the data set with the most recent UniGene database (http://www.ncbi.nlm.nih.gov/UniGene), and 45 genes were left unknown. The UniGene classification was used to identify repeated genes in the data set. We found that 20 genes appear at least twice in the data set and were not known to be part of the same UniGene cluster at the time of the original report.
Results
Our clustering method was applied to the 517 gene expression time series of length 13, described in Materials. As in the original analysis, expression values were log-transformed. We ran the clustering algorithm with four autoregressive orders p = 0, 1, 2, 3 and several similarity measures including Euclidean distance, correlation, and lag-correlation to account for the temporal dependency of the data.
Statistical Analysis.
Euclidean distance gave the best results: it systematically returned
clustering models with higher posterior probability than
correlation-based distances. The number of clusters found for
p = 0, 1, 2, 3 varied between 4 (p = 0,
1) and 3 (p = 2, 3). To choose a clustering model
among these four, we used the goodness of fit score described in
Methods. The scores for the four models were, for increasing
p, 10130.78, 13187.15, 11980.38, and 11031.12, and the model
with autoregressive order p = 1 was therefore selected.
This model merges the 517 gene time series into four clusters of 3,
216, 293, and 5 time series, with estimates of the autoregressive
coefficients and within-cluster variance
β̂10 = 0.518;
β̂11 = 0.708; σ̂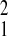 =
0.606 in cluster 1, β̂20 = 0.136;
β̂21 = 0.776; σ̂
=
0.606 in cluster 1, β̂20 = 0.136;
β̂21 = 0.776; σ̂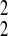 =
0.166 in cluster 2, β̂30 = −0.132;
β̂31 = 0.722; σ̂
=
0.166 in cluster 2, β̂30 = −0.132;
β̂31 = 0.722; σ̂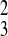 =
0.091 in cluster 3; and β̂40 =
−0.661; β̂41 = 0.328;
σ̂
=
0.091 in cluster 3; and β̂40 =
−0.661; β̂41 = 0.328;
σ̂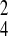 = 0.207 in cluster 4. In this model,
merging any of these clusters decreases the posterior probability of
the clustering model of at least 10.05 times, a strong
evidence in favor of their separation (27). Colored maps and dendrogram
of the four clusters are displayed in Fig. 2.
= 0.207 in cluster 4. In this model,
merging any of these clusters decreases the posterior probability of
the clustering model of at least 10.05 times, a strong
evidence in favor of their separation (27). Colored maps and dendrogram
of the four clusters are displayed in Fig. 2.
Figure 2.

Binary tree (dendrogram) and labeled gene expression display showing the clustering model obtained by our method on the data reported in Iyer et al. (8). The numbers on the branch points of the tree represent how many times the merging of two series renders the model more probable. The model identifies four distinct clusters containing 3 (Cluster 1), 216 (Cluster 2), 293 (Cluster 3), and 5 (Cluster 4) time series.
The symmetry of the standardized residuals in Fig. 1, together with the lack of any significant patterns in the scatter plot of the fitted values vs. the standardized residuals and the closeness of fitted and observed values suggests that ar(1) models provide a good approximation of the processes generating these time series. This impression is reinforced further by the averages of the fitted time series in each cluster, shown in Fig. 1, which follow closely their respective cluster average profiles.
Figure 1.

Diagnostic plots for the clustering model identified by the method when the autoregressive order is p = 1. The first row reports histogram of standardized residuals. The second row reports the scatter plot of fitted values vs. observed values. The third row shows the scatter plot of fitted values vs. standardized residuals. The fourth row displays, in black, the four cluster average profiles—computed as averages of the observed time series in each cluster—and, in blue, the averages of the fitted time series in each cluster. In these plots, the x axis reports time in hours.
Comparison with Correlation-Based Clustering.
The most evident difference between the model in Fig. 2 and the model obtained by the original analysis is the number of clusters: our method detects four distinct clusters, characterized by the autoregressive models described above, while hierarchical clustering merges all 517 genes into a single cluster. Across the four clusters, both average profiles and averages of the fitted time series appear to capture different dynamics. Iyer et al. (8) identify, by visual inspection, eight subgroups of genes—labeled A, B, C, … , I, J—from eight large contiguous patches of color. With the exception of a few genes, our cluster 2 merges the subgroups of time series labeled as D, E, F, G, H, I, and J, and cluster 3 merges the majority of time series assigned to subgroups A, B, and C. Interestingly enough, the members of subgroups A, B, and C differ, on average, by one single value. Similarly, groups D and G differ by a single value, as well as F, H, J, and I.
Our cluster 1 collects the temporal patterns of three genes—IL-8, prostaglandin-endoperoxide synthase 2, and IL-6 (IFN-β2). These time series were assigned by ref. 8 to the subgroups F, I, and J, respectively. Cluster 4 collects the time series of five genes—receptor tyrosine kinase-like orphan receptor, TRABID protein, death-associated protein kinase, DKFZP586G1122 protein, and transcription termination factor-like protein. Three of these time series were assigned by ref. 8 to the A and B subgroups. These two smaller clusters—clusters 1 and 4—are particularly noteworthy because they illustrate how our method automatically identifies islands of particular expression profiles. The first of these two clusters merges cytokines involved in the processes of the inflammatory response and chemotaxis and the signal transduction and cell–cell signaling underlying these processes. The cluster includes IL-8, IL-6, and prostaglandin-endoperoxide synthase 2, which catalyzes the rate-limiting step in the formation of inflammatory prostaglandins. The second small cluster includes genes that are known to be involved in the cell-death/apoptosis processes. This cluster includes kinases and several transcription factors reported to be involved in these processes. The cluster includes receptor tyrosine kinase-like orphan receptor 2, TRAF-binding protein domain, and Death-associated protein kinase. The cluster also includes the transcription termination factor-like protein, which plays a central role in the control of rRNA and mRNA synthesis in mammalian mitochondria (28), and DKFZP586G1122 protein, which has unknown function but has strong homology with murine zinc finger protein Hzf expressed in hematopoiesis.
The number of clusters found by our algorithm is directly inferred from the data, which also provide evidence in favor of a temporal dependency of the observations: the goodness of fit score of the ar(0) clustering model, where the observations are assumed to be marginally independent, is lower than the goodness of fit score of the ar(1) clustering model, which assumes that each observation depends on its immediate predecessor. The allocation of the 20 repeated genes in the data set seems to support our claim that identifying subgroups of genes by visual inspection may overfit the data: with the exception of the two repeats of the DKFZP566O1646 protein, our model assigns each group of repeated genes to the same cluster, whereas four of the repeated genes are assigned to different subgroups in ref. 8. Details are shown in Table 1. The risks of overfitting by visual inspection can be easily appreciated by looking at the color patterns in Fig. 2. As the dendrogram is built, genes with highly similar temporal profiles are merged first, thus producing subtrees with similar patterns of colors. However, according to our analysis, the data do not provide enough evidence to conclude that such subtrees contain time series generated by different processes and they are therefore merged into a single cluster.
Table 1.
Assignment of gene repeats to subgroups by Iyer et al. (8) (column 2) and by our method (column 3)
| Gene name | Group membership | Cluster membership |
|---|---|---|
| Serum/glucocorticoid regulated kinase | J, J | 2, 2 |
| Pre-B-cell colony-enhancing factor | J, NA | 2, 2 |
| Myeloid cell leukemia sequence 1 | J, J | 2, 2 |
| Serine proteinase inhibitor | I, I | 2, 2 |
| Stromal cell-derived factor 1 | NA, H | 2, 2 |
| Neurotrimin | H, H | 2, 2 |
| Dual specificity phosphatase 6 | F, F | 2, 2 |
| V-ets avian erythroblastosis virus E26 | F, F | 2, 2 |
| Expressed sequence tags | H, H | 2, 2 |
| DKFZP566O1646 protein | B, A | 2, 3 |
| Stearoyl-CoA desaturase | C, C, C | 3, 3, 3 |
| Pregnancy-associated plasma protein A | C, C | 3, 3 |
| DEAD/H box polypeptide 17 | B, B | 3, 3 |
| KIAA0923 protein | B, B, B, B | 3, 3, 3, 3 |
| WW Domain-containing gene | B, B | 3, 3 |
| Bardet–Biedl syndrome 2 | B, B | 3, 3 |
| Calcium/calmodulin-dependent protein kinase | B, B | 3, 3 |
| Tax1 (human T cell leukemia virus type I) | A, B | 3, 3 |
| AD036 protein | A, A | 3, 3 |
| DKFZp586I1823 | A, A | 3, 3 |
The first column reports the UniGene name of the repeated genes. Subgroups in column 2 are identified by A–J letters, with NA denoting a placement outside the eight clusters identified by the authors.
An example of this phenomenon is shown in detail by Fig. 3, which enlarges part of the dendrogram in Fig. 2. The subtree on the top half of the figure merges 29 time series that appear to be more homogenous to visual inspection, and the large Bayes factors, in log scale, which shows that at each step of the iterative procedure, merging the time series determines a model which is more likely than the model determined by not merging them. Similarly, the bottom half subtree merges 16 time series that appear to be more similar. The Bayes factors attached to the terminal node of the picture are exp(33), meaning that the model in which the two subtrees are merged together is exp(33) times more likely than the model in which these subtrees are taken as two separate clusters.
Figure 3.

A zoom of the dendrogram in Fig. 2, with details of the probability of merging.
Discussion
The analysis of gene expression data collected along time is at the basis of critical applications of microarray technology. This contribution addresses a fundamental property of temporal data—their directed dependency along time—in the context of cluster analysis. We have introduced the application to microarray data of a clustering algorithm able to account for dependency of temporal observations and to automatically identify the number and the members of the clusters.
We have represented the dependency of temporal observations as autoregressive equations and we have taken a Bayesian approach to the problem of selecting the number and members of clusters. To explore the exponential number of possible clustering models, we have devised a heuristic search procedure based on pairwise distances to guide the search process. In this way, our method retains the important visualization capability of traditional distance-based clustering and acquires a principled measure to decide when two time series are different enough to belong to different clusters. It is worth noting that the measure here adopted, the posterior probability of the clustering model, takes into account all the available data, and such a global measure also offers a principled way to decide whether the available evidence is sufficient to support an empirical claim. Our analysis shows that sometimes the available evidence is not sufficient to support the claim that two time series are generated by two distinct processes. Fig. 2 shows contiguous patches of colors, but the posterior probability of the model does not support the claim that these subgroups are sufficiently distinct to be viewed as distinct processes. This finding has interesting implications for experiment design and sample size determination, because it allows the analyst to assess whether the available information is sufficient to support significant differentiations among gene profiles and, if necessary, collect more data. A third feature of the method presented here is the reliance of the clustering process on an explicit statistical model. Contrary to other approaches (16), our method builds the clustering model by using the parametric content of the statistical model rather than providing statistical content to an established clustering model. This stochastic content allows us to use standard statistical techniques to validate the goodness of fit of the clustering model, as illustrated at the end of Results. While the biological validation of microarray experiments plays a critical role in the development of modern functional genomics, practical considerations often limit this validation to few genes, while the claims and the scope of a microarray experiment involve thousands. A proper use of available statistical diagnostics provides analytical tools to independently assess the global validity of a clustering model.
Autoregressive equations are very simple representations of process dynamics and they rely on the assumption that the modeled time series are stationary. Our reason to choose this representation is its simplicity: since the time series of gene expression experiments are typically very short, more sophisticated representations could be prone to overfitting. Stationarity conditions can be checked with the method described at the end of Methods but, both in the data analyzed here and in our general experience, the clustering process seems to be largely unaffected by the presence of nonstationary time series. In principle, however, more sophisticated representations can be integrated within the Bayesian framework described in this article. The method here described is implemented in a computer program called caged (Cluster Analysis of Gene Expression Dynamics), available from http://genomethods.org/caged.
Supplementary Material
Acknowledgments
We thank Stefano Monti (Whitehead Institute) and Alberto Riva (Harvard Medical School) for their insightful comments on an early draft of this article. This research was supported in part by the National Science Foundation (Bioengineering and Environmental Systems Division/Biotechnology) under Contract ECS-0120309.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Schena M, Shalon D, Davis R W, Brown P O. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 2.Lockhart D J, Dong H, Byrne M C, Follettie M T, Gallo M V, Chee M S, Mittmann M, Wang C, Kobayashi M, Horton H, Brown E L. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 3.Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E S, Golub T R. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butte A J, Tamayo P, Slonim D, Golub T R, Kohane I S. Proc Natl Acad Sci USA. 2000;97:12182–12186. doi: 10.1073/pnas.220392197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alter O, Brown P O, Botstein D. Proc Natl Acad Sci USA. 2000;97:10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisen M, Spellman P, Brown P, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen X, Fuhrman S, Michaels G S, Carr D B, Smith S, Barker J L, Somogyi R. Proc Natl Acad Sci USA. 1998;95:334–339. doi: 10.1073/pnas.95.1.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iyer V R, Eisen M B, Ross D T, Schuler G, Moore T, Lee J C, Trent J M, Staudt L M, Hudson J, Jr, Boguski M S, Lashkari D, et al. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- 9.Golub T R, Slonim D K, Tamayo P, Huard C, Gaasenbeek M, Mesirov J P, Coller H, Loh M L, Downing J R, Caligiuri M A, et al. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 10.Lossos I S, Alizadeh A A, Eisen M B, Chan W C, Brown P O, Botstein D, Staudt L M, Levy R. Proc Natl Acad Sci USA. 2000;97:10209–10213. doi: 10.1073/pnas.180316097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Box G E P, Jenkins G M. Time Series Analysis: Forecasting and Control. 2nd Ed. San Francisco: Holden–Day; 1976. [Google Scholar]
- 12.West M, Harrison J. Bayesian Forecasting and Dynamic Models. 2nd Ed. New York: Springer; 1997. [Google Scholar]
- 13.Shahar Y, Tu S, Musen M. Knowl Acquis. 1992;1:217–236. [Google Scholar]
- 14.Haimowitz I J, Le P P, Kohane I S. Artif Intell Med. 1995;7:471–472. doi: 10.1016/0933-3657(95)00023-6. [DOI] [PubMed] [Google Scholar]
- 15.Reis B Y, Butte A S, Kohane I S. J Biomed Inform. 2001;34:15–27. doi: 10.1006/jbin.2001.1005. [DOI] [PubMed] [Google Scholar]
- 16.Holter N S, Maritan A, Cieplak M, Fedoroff N V, Banavar J R. Proc Natl Acad Sci USA. 2001;98:1693–1698. doi: 10.1073/pnas.98.4.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aach J, Church G M. Bioinformatics. 2001;17:495–508. doi: 10.1093/bioinformatics/17.6.495. [DOI] [PubMed] [Google Scholar]
- 18.Ramoni M, Sebastiani P, Cohen P R. Mach Learn. 2002;47:91–121. [Google Scholar]
- 19.Ramoni M, Sebastiani P, Cohen P R. Proceedings of the 2000 National Conference on Artificial Intelligence (AAAI-2000) San Francisco: Morgan Kaufmann; 2000. pp. 633–638. [Google Scholar]
- 20.Sebastiani P, Ramoni M. Res Off Stat. 2001;4:169–183. [Google Scholar]
- 21.Tversky A, Kahneman D. Science. 1974;185:1124–1131. doi: 10.1126/science.185.4157.1124. [DOI] [PubMed] [Google Scholar]
- 22.Kahneman D, Slovic P, Tversky A. Judgment Under Uncertainty: Hueristic and Biases. New York: Cambridge Univ. Press; 1982. [Google Scholar]
- 23.Gilovich T, Vallone R, Tversky A. Cognit Psychol. 1985;17:295–314. [Google Scholar]
- 24.MacKay D J C. Neural Comput. 1992;4:415–447. [Google Scholar]
- 25.Tenenbaum J B, Griffiths T L. Behav Brain Sci. 2001;24:629–640. doi: 10.1017/s0140525x01000061. [DOI] [PubMed] [Google Scholar]
- 26.Akaike H. 2nd International Symposium on Information Theory. Budapest: Akademiai Kiado; 1973. pp. 267–281. [Google Scholar]
- 27.Kass R E, Raftery A. J Am Stat Assoc. 1995;90:773–795. [Google Scholar]
- 28.Fernandez-Silva P, Martinez-Azorin F, Micol V, Attardi G. Embo J. 1997;16:1066–1079. doi: 10.1093/emboj/16.5.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.