

Abstract
Human cytomegalovirus (HCMV) encodes several proteins that can modulate components of the cell cycle machinery. The UL123 gene product, IE1-72, binds the Rb-related, p107 protein and relieves its repression of E2F-responsive promoters; however, it is unable to induce quiescent cells to enter S phase in wild-type (p53+/+) cells. IE1-72 also induces p53 accumulation through an unknown mechanism. We present here evidence suggesting that IE1-72 may activate the p53 pathway by increasing the levels of p19Arf and by inducing the phosphorylation of p53 at Ser15. Phosphorylation of this residue by IE1-72 expression alone or HCMV infection is found to be dependent on the ataxia-telangiectasia mutated kinase. IE2-86 expression leads to p53 phosphorylation and may contribute to this phenotype in HCMV-infected cells. We also found that IE1-72 promotes p53 nuclear accumulation by abrogating p53 nuclear shuttling. These events result in the stimulation of p53 activity, leading to a p53- and p21-dependent inhibition of cell cycle progression from G1 to S phase in cells transiently expressing IE1-72. Thus, like many of the small DNA tumor viruses, the first protein expressed upon HCMV infection activates a p53 response by the host cell.
The p53 tumor suppressor is a transcription factor with sequence-specific DNA-binding activity that plays an important role in regulating cell cycle progression and cellular growth (41). Upon DNA damage or in response to certain cellular stresses, the induction of p53 leads to growth arrest. This arrest requires p53-mediated transactivation of the gene encoding the cyclin-dependent kinase inhibitor, p21 (22, 27). In some circumstances, apoptosis can also occur as a result of p53 activation (8).
Under most conditions, p53 exists at low levels in normal cells due to its short half-life. The rapid turnover of p53 is primarily attributed to Mdm2, an E3-ubiquitin ligase that facilitates the degradation of p53 by the 26S proteosome (28, 30, 40). A feedback loop exists wherein p53 positively regulates Mdm2 levels by activating Mdm2 transcription (6, 78) and Mdm2 negatively regulates p53 by promoting its degradation. Upstream of Mdm2 is p19Arf, a nucleolar protein that binds and inhibits Mdm2 activity (6, 31, 37, 52, 71, 78). Deregulated expression of numerous cellular oncoproteins such as Ras, Myc, and E2F can modulate p53 levels by inducing p19Arf expression (7, 50, 57, 82). In addition, viral factors, such as the polyomavirus middle T antigen, have also been shown to increase p19Arf expression thereby leading to the stabilization of p53 (42).
In response to various cellular stress signals, p53 is stabilized by covalent modifications that prevent p53 degradation. The phosphorylation of p53 at specific N-terminal serine residues significantly enhances p53 stability by disrupting the Mdm2/p53 interaction, thereby activating p53 (for a review, see reference 54). In addition, the phosphorylation of Ser15 has been shown to promote p53 nuclear accumulation by inhibiting nuclear export (80).
Most of the information known about the pathways leading to p53 phosphorylation stem from studies of cellular responses to DNA damage or hypoxia. After exposure to UV or ionizing radiation, the activation of a number of cellular kinases leads to the phosphorylation of p53 at numerous N- and C-terminal serine and threonine residues (54). Among the kinases activated in response to DNA damage are the product of the ataxia telangiectasia mutated gene (ATM), the ATM-Rad3-related protein (ATR), DNA protein kinase (DNA-PK), and the checkpoint kinase proteins, CHK1 and CHK2, which can each phosphorylate p53 at key N-terminal residues. Therefore, it appears that multiple proteins regulate p53 stability and function and different stimuli can activate pathways that modulate p53 activity.
Human cytomegalovirus (HCMV) has divergent effects on the cell cycle (for reviews, see references 10 and 35). Early reports suggest that in human foreskin fibroblasts, HCMV infection causes cells to arrest in either G1 or G2/M (9, 20, 32, 43). Although these studies show that HCMV induces fibroblasts to undergo what has been described as a “G1 arrest,” biochemically these cells exhibit hallmarks of S phase, including pRb hyperphosphorylation, cyclin E and cyclin A kinase activation, and expression of many S-phase genes such as DHFR, DNA polymerase α, PCNA, and topoisomerase II. In addition, infection of a differentiated embryonic carcinoma cell line with HCMV causes entry into S phase (66). These observations illustrate the variety and seemingly contradictory effects HCMV has on the cell cycle.
The apparent capacity of HCMV infection to deregulate aspects of the cell cycle may be attributed to the ability of certain viral proteins to modulate key cell cycle regulatory protein activities. In addition to altering the levels of phosphorylated pRb protein, HCMV infection also leads to increases in p53 levels in both human fibroblasts and smooth muscle cells (32, 47, 69). In addition, p21 levels transiently accumulate in HCMV-infected cells during immediate-early (IE) times of infection. Although it is not clear how HCMV increases p53 or p21 levels, it has been suggested that the HCMV IE gene products may be the viral factors responsible for modulating p53 expression (46, 47, 69).
HCMV encodes a number of proteins that mediate effects on the cell cycle, including immediate-early (IE) and virion-associated factors (for reviews, see references 10 and 35). Both HCMV UL122 and UL123 encode nuclear proteins, designated IE2-86 (also referred to as IE86 or IE2) and IE1-72 (also referred to as IE72 or IE1), respectively, that transactivate viral and cellular promoters (10). These IE proteins also directly modulate components of the cell cycle machinery. IE2-86 can interact with pRb and is capable of alleviating pRb repression of E2F-responsive promoters (23, 26, 67). IE2-86 also binds p53 and inhibits its transactivation activity (69, 72). As a result, IE2-86 expression induces quiescent cells into S phase in human and rodent fibroblasts (11, 48, 75). The IE1-72 protein interacts with p107, another member of the RB protein family, thereby derepressing E2F transcriptional activity (51, 81). Although IE1-72 inactivates p107 and induces E2F activity, IE1-72 is unable to induce entry into S phase in wild-type fibroblasts. However, IE1-72 can induce entry into S phase in quiescent rodent fibroblasts that are null for p53 (11). IE1-72 expression also leads to an increase in p53 levels (11), which may contribute to the inability of IE1-72 to promote proliferation in the presence of p53.
Given that IE1-72 does not transactivate a p53 promoter-reporter construct nor does it appear to interact with p53 (46), the mechanism by which IE1-72 elevates the levels of and presumably activates p53 is unclear. Therefore, we investigated the mechanisms contributing to p53 protein accumulation and cell cycle arrest after IE1-72 expression. We found that IE1-72 may modulate p53 levels by two mechanisms, including increasing the levels of p19Arf and activating an ATM-dependent phosphorylation of p53, resulting in p53 nuclear accumulation and activation. This increase in p53 levels leads to p21 induction and a p21-dependent cell cycle arrest in synchronized cells transiently expressing IE1-72. These findings raise the possibility that the expression of IE1-72 in HCMV-infected cells elevates p53 levels at IE times and that the previously reported HCMV-induced cell cycle arrest may be attributed in part to these effects of IE1-72 expression.
MATERIALS AND METHODS
Cell culture.
REF52 cells were cultured as described previously (11). Early-passage wild-type (WT), as well as genetically matched p53-deficient (p53−/−) mouse embryo fibroblasts (MEFs), Mdm2−/−/p53−/− MEFs, and SAOS-2 cells were generous gifts from Stephen Jones (University of Massachusetts Medical School, Worcester, MA). The p19Arf−/− MEFs were provided by Charles Sherr (St. Jude's Medical Center, Memphis, TN). Early passage p21+/+ and p21−/− MEFs were provided by Tyler Jacks (Massachusetts Institute of Technology, Boston, MA). MEFs were cultured as described previously (11, 57). Human embryonic lung fibroblasts (HELs) were a generous gift from Eng-Shang Huang (University of North Carolina, Chapel Hill, NC). The SAOS-2 cells and HEL fibroblasts were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Human dermal fibroblasts from an ataxia-telangiectasia patient (GM03395C; AT), as well as age-matched, normal dermal fibroblasts (GM00316B; WT), were obtained from the Coriell Institute for Medical Research (Camden, NJ) and cultured according to conditions recommended by the provider.
To induce growth arrest, cells were first seeded at a plating density between 2 × 103 to 3 × 103 cells/cm2. The WT and p21−/− MEFs were then serum starved by a wash with phosphate-buffered saline (PBS) and then culture in DMEM containing 0.25% serum for a minimum of 48 h. The conditions used were based on empirical studies that optimized serum concentration and culturing times to induce growth arrest in over 80% of each cell population (unpublished observations).
HCMV infections.
HEL fibroblasts were infected with HCMV AD169 or CR208 strain at a multiplicity of infection (MOI) of 5. AD169 was obtained from the American Type Culture Collection, CR208, an AD169-derived virus that is deleted for UL123 (25), is used with permission from Ed Mocarski (Stanford). Viral infections were performed in serum-free medium for 1 h at 37°C and 5% CO2. The viral inoculum was then removed and replaced with DMEM containing 10% FBS and cultured as described above. Cells were harvested at 24 h postinfection (hpi), and lysates were generated as described previously (11).
Adenovirus vectors and infection.
The recombinant adenovirus encoding the HCMV IE gene product IE1-72 (AdIE1-72) and IE2-86 (AdIE2-86) have been described (77) and characterized previously (3, 11). An empty vector virus (38) or a recombinant adenovirus expressing β-galactosidase was used as a control (labeled as AdCon) for virus infection in the experiments (56, 57). An E2F1-encoding adenovirus, AdE2F1 (38), and a p53-encoding adenovirus, Adp53 (39), have been described previously. Viruses were grown and titers were determined in a human embryonic kidney cell line (i.e., cell line 293) and subsequently purified on cesium-chloride gradients (49). Virus titers were determined by immunohistochemical staining of the adenovirus type 2 hexon (11, 57). Infections were done as described previously (11, 58). Cells were infected with either AdIE1-72, AdIE2-86, or a control adenovirus at an MOI of 250 unless otherwise noted.
Immunoblot analysis.
Whole-cell extracts were prepared as described previously (11) with the following exception. To assess p21 protein levels, washed cells were scraped into 1 ml of radioimmunoprecipitation assay buffer (PBS, 0.1% NP-40, 1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, sodium vanadate, phenylmethylsulfonyl fluoride, and aprotinin) and incubated on ice for 1 h. Samples were lightly sonicated for 15 s, and soluble proteins were collected by centrifugation for 10 min at 13,000 rpm in a microcentrifuge. Aliquots of cell extracts were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the resolved proteins were transferred to nitrocellulose membranes by electroblotting. Detection of p53, phospho-Ser15 p53, phospho-Ser20 p53, p21, p19Arf, and HCMV IE1-72 and IE2-86 proteins was performed with antibodies specific for total p53 (Ab-6 or Ab-7; Oncogene Research Products), phospho-Ser15 and phospho-Ser20 p53 (Cell Signaling Technology), p21 (C-19; Santa Cruz Biotechnology), p19Arf (NB 200-106; Novus Biologics), and IE1-72 and IE2-86 (MAB8130 and MAB810, respectively; Chemicon International). ATM and phospho-Ser1981 ATM were detected with antibodies from Rockland Immunochemicals (Gilbertsville, PA). Blots were stripped and probed for actin levels as a control for sample loading.
p53 nuclear shuttling.
Heterokaryons were generated as described previously (79). Essentially, subconfluent cultures of SAOS-2 cells were coinfected with Adp53 and either AdIE1-72 or AdCon. After infection, cells were cultured in normal media at 37°C for 1.5 h. The SAOS-2 cells were replated with an equal number of Mdm2−/−/p53−/− MEFs onto glass coverslips in 35-mm plates and incubated overnight at 37°C. Cells were then washed twice with PBS and incubated in medium containing cycloheximide (50 μg/ml) for 20 min at 37°C. Cells were subsequently washed with PBS and then incubated for 2 min in 50% polyethylene glycol (MW 3350; Sigma) dissolved in DMEM to facilitate cell fusion. Cells were washed with PBS and incubated in medium containing cycloheximide for 1 h at 37°C. Cells were then fixed with 3.7% formaldehyde in PBS for 10 min at room temperature and permeabilized with cold 0.2% Triton X-100 in PBS for 10 min at 4°C. To detect heterokaryons and p53 localization, coverslips were blocked with 10% FBS in PBS for 15 min at room temperature prior to incubation with primary antibodies specific for p53 (Ab-1; Oncogene Research Products) and human Ku-86 (Santa Cruz Biotechnology). After incubation with the primary antibodies, the coverslips were washed three times with PBS then incubated with isotype-specific fluorescein isothiocyanate-conjugated and rhodamine red-X-conjugated antibodies to detect p53 and human Ku-86, respectively. Cells were washed three times with PBS and once with distilled water. Glass coverslips containing the cells were mounted onto glass slides with Vectashield containing DAPI (4′,6′-diamidino-2-phenylindole; Vector Laboratories). p53 nuclear shuttling was scored by assessing the localization of p53 in human-murine heterokaryons with a Zeiss immunofluorescence microscope. The results were presented as the percentage of heterokaryons in which p53 shuttled from human to murine nuclei.
Luciferase assay.
HEL fibroblasts were plated at subconfluent densities onto six-well plates and grown overnight at 37°C. Cells were cotransfected with a p21-specific reporter construct, WAF-1-Luc (generously provided by Karen Vousden, National Cancer Institute, Frederick, MD) (22) (1 μg/well) and either a plasmid encoding IE1-72 cDNA (pcDNA3-IE1-72) or a control plasmid (pcDNA3) (2 μg/well), along with a Renilla luciferase-expressing plasmid, pRL-TK (kindly provided by Zdenka Matijasevic, University of Massachusetts Medical School, Worcester, MA) (0.25 μg/well) as an internal control to normalize for transfection efficiency. Transfection reactions were done with Lipofectamine Plus (Invitrogen) and were incubated for 5 h in 37°C. Afterward, transfection medium was replaced with DMEM containing 10% FBS, and cells were incubated at 37°C for 24 h. The cells were then lysed, and the luciferase activity was determined by using a Dual-Luciferase reporter assay system (Promega). The results are presented as the fold induction of p21 promoter activity relative to the control plasmid-transfected cells.
Immunohistochemical staining for p21 protein accumulation.
Subconfluent cultures of REF52 cells were infected with the appropriate recombinant adenovirus constructs and immunohistochemical staining for p21 protein was performed. At 24 hpi, the cells were fixed for 5 min in formaldehyde (0.37% final concentration), followed by 5 min in methanol. Cells were blocked with 1% bovine serum albumin (BSA) in PBS-0.5% Tween 20 (PBS/Tw) and incubated with an anti-p21 MAb (Santa Cruz Biotechnology) diluted in 1% BSA in PBS/Tw for 1 h at room temperature. The cells were washed with PBS/Tw, and bound antibody was detected by using a Vectastain 3,3′-diaminobenzidine (DAB) substrate kit (Vector Laboratories) as described by the manufacturer.
Cell cycle analysis by flow cytometry.
Cells were infected with the appropriate recombinant adenoviruses and processed for flow cytometry as described previously (11). Flow cytometry data was processed by using the FlowJo FACS analysis program (Treestar). S-phase cells were defined as the population of cells with more than 2 N but less than 4 N DNA content.
RESULTS
HCMV IE1-72 expression induces p53 and p19ARF protein.
We previously reported that IE1-72 expression induces quiescent cells to enter S phase in the absence of p53 (11). Because increased p53 levels typically correlate with function, we examined p53 levels in cells expressing IE1-72. We used REF-52 cells and MEFs for this experiment for comparison with the earlier study. As shown in Fig. 1A, the IE1-72-expressing cells exhibited a two- to threefold increase in the levels of p53 compared to that observed in the control virus-infected cells, confirming our earlier finding (11). Given that IE1-72 can function as a transcriptional coactivator, we looked for changes in p53 mRNA levels in cells transduced with an IE1-72-encoding recombinant adenovirus. We found that IE1-72 expression did not affect p53 mRNA levels, suggesting that IE1-72 does not transactivate p53 expression through its endogenous promoter (data not shown).
FIG. 1.

Expression of HCMV IE1-72 results in p53 and p19Arf accumulation. (A to C) Immunoblot detection of p53 and p19Arf. (A) Immunoblot analyses were performed with an anti-p53 polyclonal antibody on whole-cell extracts from WT MEFs infected with either AdIE1-72 or control virus. Cells were harvested at the indicated times postinfection. An extract from p53−/− MEFs was included as a negative control for p53 detection. (B) Whole-cell extracts from REF52 cells infected with either AdIE1-72 or control virus were probed for endogenous p19Arf by immunoblotting with an anti-p19Arf polyclonal antibody. An extract from p19Arf−/− MEFs was included as a negative control for p19Arf detection. (C) Immunoblot analysis for p53 was performed on whole-cell extracts from p19Arf−/− MEFs infected with either AdIE1-72 or a control virus. Cells were harvested at the indicated times postinfection. An extract from uninfected p53−/− MEFs was included as a negative control for p53 detection. An extract from UV-treated (60J/m2) p19Arf−/− MEFs was included as a positive control for p53 accumulation.
Another way that p53 levels are regulated is by the p19Arf/Mdm2 pathway in which increased levels of p19Arf stabilizes p53 through its ability to inactivate the E3 ubiquitin ligase activity of Mdm2 (30). We determined whether IE1-72 might alter p53 levels by inducing p19Arf accumulation. As shown in Fig. 1B, approximately a threefold increase in the levels of p19Arf was observed in cells expressing IE1-72. Given that increased levels of p19Arf is sufficient to stabilize p53, it would appear that IE1-72 modulates p53 levels by increasing the levels of p19Arf in cells.
We next measured p53 levels in p19Arf−/− MEFs to determine whether p19Arf is necessary for p53 accumulation following IE1-72 expression. As shown in Fig. 1C, low levels of p53 were detected in extracts from control virus-infected p19Arf−/− MEFs. Reduced basal levels of p53 in early passage p19Arf−/− MEFs have been observed previously (55). Unexpectedly, increased levels of p53 were observed in the IE1-72-expressing cells compared to the control virus-infected cells. These findings suggest that p19Arf is not responsible for the most of the change in p53 levels associated with IE1-72 expression and raise the possibility that IE1-72 can act through an additional mechanism(s) to alter p53 levels.
IE1-72 and HCMV increase the levels of phospho-Ser15/18 p53.
In addition to the p19Arf/Mdm2 pathway, modulation of p53 levels can occur through other mechanisms, including the covalent modification of p53. A well-studied model of this process is the upregulation of p53 after UV or ionizing radiation. DNA damage induces p53 phosphorylation at several residues, resulting in protein stabilization (54). Phosphorylation at the Ser18 residue on mouse p53 (Ser15 on human p53) is a common event in response to DNA damage and other stresses and leads to enhanced p53 stability by disrupting the binding of Mdm2 to p53 so that p53 is not ubiquitinated and degraded (64, 65). To examine the consequences of IE1-72 expression on the levels of phospho-Ser18 p53, we expressed IE1-72 in WT MEFs and measured p53 levels with a phospho-specific antibody. As shown in Fig. 2A, higher levels of phospho-Ser18 p53 were observed in WT MEFs expressing IE1-72 compared to extracts from the control cells. Increased levels of phospho-Ser18 p53 were also observed in the p19Arf−/− MEFs after IE1-72 expression, suggesting that the p19Arf/Mdm2 pathway is not required for this modification to p53 (Fig. 2B). Infection of HEL fibroblasts with HCMV or AdIE1-72 also resulted in an accumulation of total p53 and phospho-Ser15 p53 (Fig. 2C). IE1-72 expression alone had a greater influence on p53 and phospho-Ser15 p53 than HCMV infection, which was likely due to the higher levels of IE1-72 protein expressed from the adenovirus vector (Fig. 2C). Another serine residue on p53 that is often phosphorylated after DNA damage and other chromatin stresses is Ser20 (13, 14, 29, 63). We found that levels of phospho-Ser20 p53 are not affected by IE1-72 expression alone but are increased in HCMV-infected cells, indicating that additional p53 phosphorylation pathways are activated during HCMV replication (Fig. 2C). Thus, HCMV infection and IE1-72 expression alter p53 by promoting its phosphorylation.
FIG. 2.
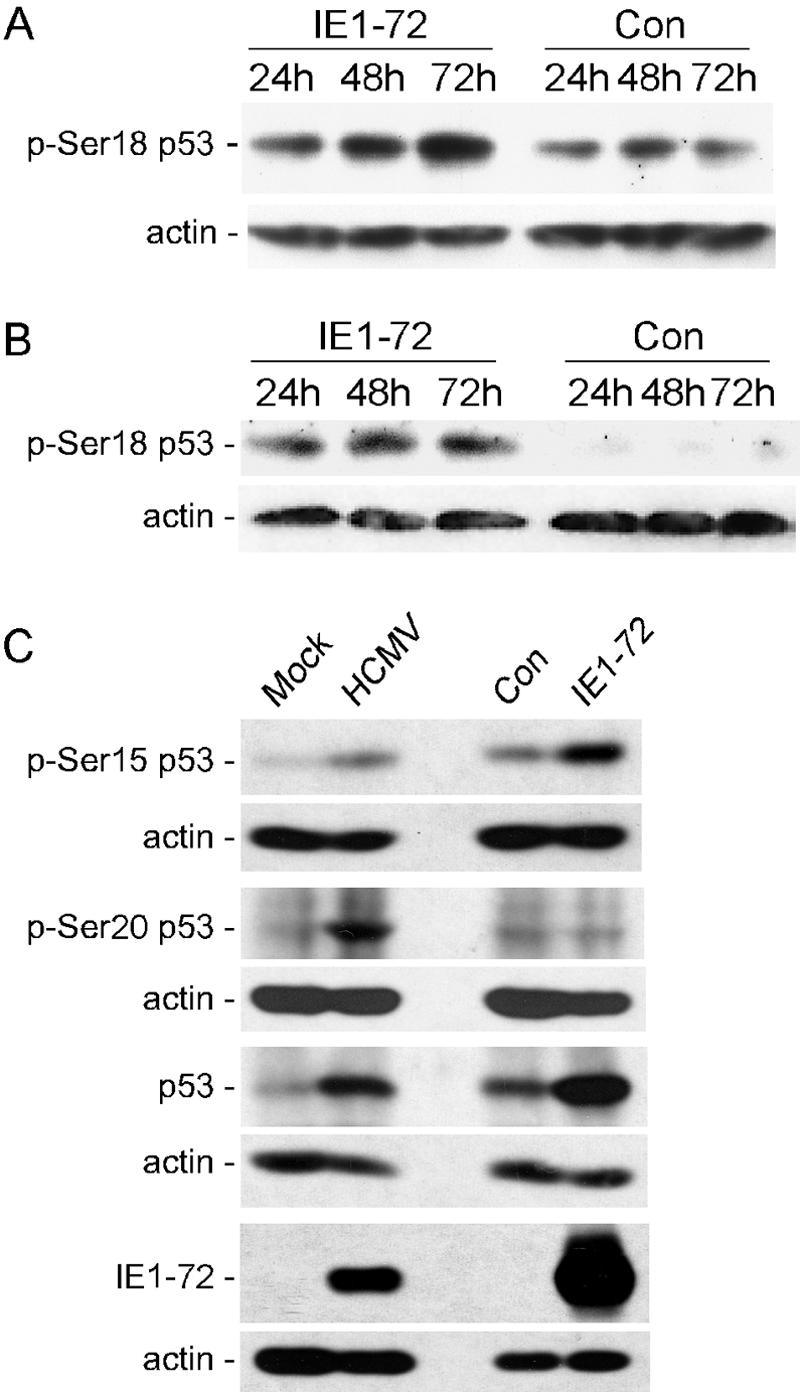
IE1-72 expression and HCMV infection increase the levels of phosphorylated p53. Immunoblot analyses for the phospho-Ser18 form of p53 were performed on whole-cell extracts from WT MEFs (A) and p19Arf−/− MEFs (B) infected with either AdIE1-72 or a control virus. (C) Detection of total and modified forms of p53 in HCMV or AdIE1-72-infected cells. HEL fibroblasts were infected with HCMV, AdIE1-72, or a control virus and whole-cell extracts derived 24 hpi. Phosphorylated forms of p53 at Ser15 or Ser20 were detected by using antibodies specific for each modification. IE1-72 levels were detected with a monoclonal antibody.
Phosphorylation of p53 at Ser15 after IE1-72 expression requires ATM.
Several kinases have been shown to phosphorylate p53 at Ser15/18 after DNA damage, including ATM, ATR, and DNA-PK. These cellular kinases can be inhibited by caffeine (60). Therefore, we determined whether the increase in phospho-Ser15 p53 induced by IE1-72 expression was sensitive to caffeine in HEL fibroblasts. In the absence of caffeine, IE1-72-expressing cells displayed a higher level of phospho-Ser15 p53 relative to that seen in the control virus-infected cells (Fig. 3A). In the presence of caffeine, dose-dependent decreases in the levels of total and phospho-Ser15 p53 were observed in IE1-72-expressing cells.
FIG. 3.
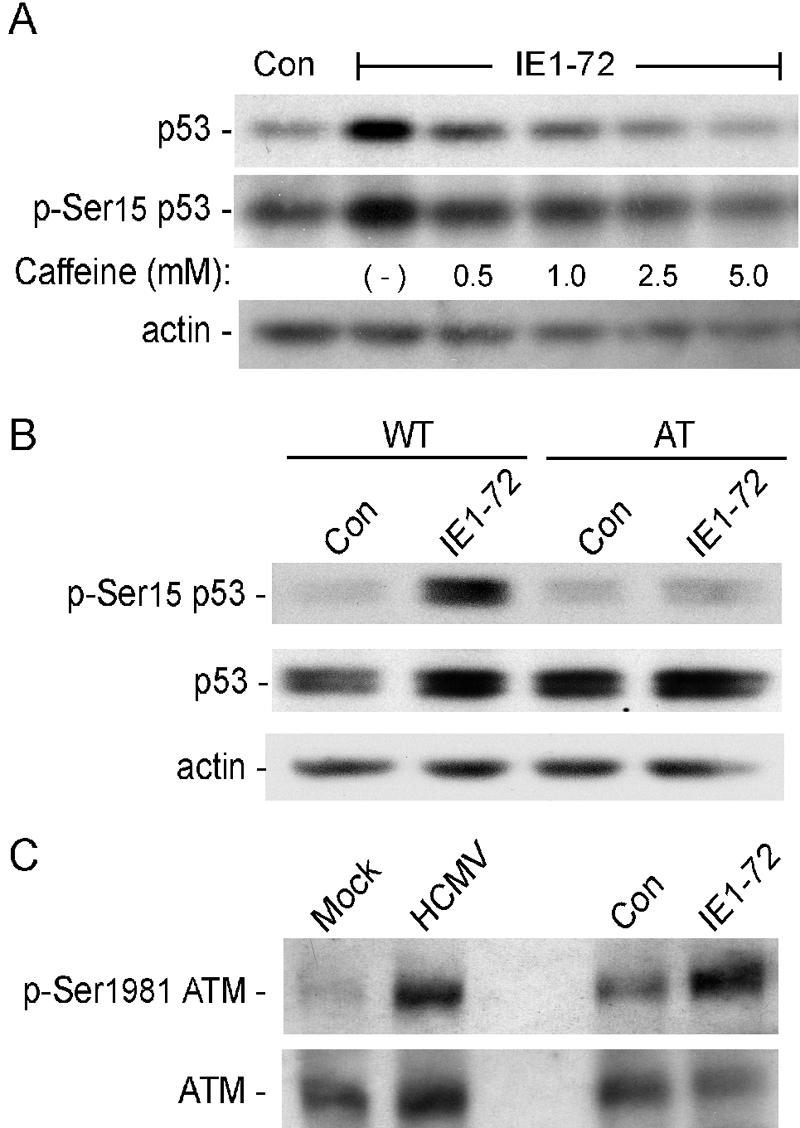
Phosphorylation of p53 at Ser15 after IE1-72 expression requires activated ATM. (A) Immunoblot analysis for total and phospho-Ser15 form of p53 in response to caffeine treatment. HEL fibroblasts were infected with either AdIE1-72 or a control virus and cultured in the presence of increasing doses of caffeine for the 6 h prior to harvesting at 24 hpi. (B) Immunoblot analyses for total and phospho-Ser15 p53 in human cells. Cultures of dermal fibroblasts isolated from an AT patient or an age- and gender-matched normal donor (WT) were infected with either AdIE1-72 or a control virus, and whole-cell extracts were harvested at 24 hpi. (C) Immunoblot analyses for total and phospho-Ser1981 ATM. Whole-cell extracts from HCMV, AdIE1-72 or control virus-infected HEL fibroblasts were generated at 24 hpi.
We used human dermal fibroblasts from ataxia telangiectasia (AT) patients to determine whether ATM is required for p53 phosphorylation after IE1-72 expression. AT patients have an inactivated atm gene and, as a result, fail to fully execute many of the cellular responses to chromatin stress, including the ATM-dependent phosphorylation of p53 at Ser15. The levels of p53 phosphorylation observed after IE1-72 expression in AT fibroblasts were compared to the levels seen in age and gender-matched normal human dermal fibroblasts (WT) expressing IE1-72. IE1-72 expression in the WT fibroblasts resulted in nearly a fivefold increase in the levels of phospho-Ser15 p53 compared to the level observed in the control virus-infected WT fibroblasts, whereas the levels of total p53 were only slightly elevated (Fig. 3B). In contrast, the levels of phospho-Ser15 p53 detected in the AT fibroblasts expressing IE1-72 were similar to that detected in control virus-infected AT and WT fibroblasts. IE1-72 expression also had little effect on the levels of total p53 in AT fibroblasts, suggesting a role for phosphorylation in p53 stabilization.
ATM is autophosphorylated on Ser1981 when stimulated by DNA damage or chromatin changes (5). To confirm that ATM kinase was activated by IE1-72, we examined its phosphorylation status. As shown in Fig. 3C, we observed increased levels of phospho-Ser1981 ATM in cells expressing IE1-72, whereas the levels of total ATM did not increase and may even be reduced slightly. We also found that HCMV-infected cells displayed enhanced levels of phospho-Ser1981 ATM, as well as total ATM (Fig. 3C). Therefore, ATM is activated by IE1-72 expression alone and HCMV infection. Taken together, these findings demonstrate that IE1-72 induces an ATM-dependent phosphorylation of p53.
Contribution of IE1-72 and possibly IE2-86 proteins to p53 phosphorylation in HCMV-infected cells.
We have shown that deregulation of E2F leads to ATM autophosphorylation and p53 activation (56, 57, 58; F. M. Frame, et al., submitted for publication). Given that both IE1-72 and IE2-86 deregulate E2F activity through inactivation of RB family members, we sought to determine whether IE2-86 could also stimulate p53 phosphorylation. Expressing IE2-86 in HEL fibroblasts resulted in an increase in the phospho-Ser15 form of p53, whereas total p53 levels were unchanged (Fig. 4A). The absence of a change in total p53 was unexpected given that an earlier study has suggested that IE2-86 binds to and stabilizes p53 (69). However, results similar to ours have recently been published (68), although Song and Stinski analyzed exogenously expressed p53. Regardless of p53 accumulation, IE2-86, like IE1-72, expression leads to p53 phosphorylation.
FIG. 4.

IE protein expression leads to ATM activation and p53 phosphorylation. (A) Immunoblot analysis for p53 in human cells. HEL fibroblasts were infected with AdIE2-86 or control virus and whole-cell extracts derived at 24 hpi. (B) Immunoblot detection of ATM and p53 forms in HEL fibroblasts. Cells were mock-infected or infected with HCMV or CR208 and harvested at 24 hpi.
We next compared HCMV to a mutant (CR208) virus devoid of IE1-72 expression. Both HCMV and CR208 virus infections resulted in an increase in total p53 levels, whereas CR208-infected cells had reduced levels of phospho-Ser15 p53 and phospho-Ser1981 ATM relative to HCMV-infected cells (Fig. 4B). On the surface, these results would suggest that IE1-72 is solely responsible for these changes to ATM and p53. However, at the MOI used in these experiments, UL122 expression (IE2-86) is apparently still dependent on transactivation by IE1-72 because IE2-86 levels are not detected in the CR208-infected cells (Fig. 4B).
IE1-72 expression results in nuclear retention of p53.
We previously noted that p53 accumulates in the nuclei of IE1-72-expressing cells (11). One possible explanation for our observation is that IE1-72 inhibits the ability of p53 to shuttle in and out of the nucleus from the cytoplasm. The p53 protein contains three nuclear localization sequences (NLS) and one nuclear export sequence (NES). The NLS are sufficient to mediate the shuttling of p53 from the cytoplasm into the nucleus (70, 74), and the NES can mediate the export of p53 from the nucleus in a Crm-dependent manner (70). Mdm2 can promote the export of p53 out of the nucleus (59, 73), whereas p53 phosphorylation abrogates the ability of p53 to shuttle from the nucleus to the cytoplasm (80). We generated heterokaryons containing human and mouse nuclei to determine whether IE1-72 expression also interferes with p53 shuttling. p53-deficient SAOS-2 cells were infected with a recombinant adenovirus encoding a human p53 cDNA (Adp53) and, in some instances, coinfected with AdIE1-72 prior to being fused with mdm2−/−/p53−/− MEFs. After fusion, the resulting heterokaryons were identified via immunofluorescence staining for human Ku-86 and the location of p53 was assessed (Fig. 5A). Nuclear shuttling of p53 between human and mouse nuclei was observed in 55% of the control heterokaryons, whereas p53 shuttled between nuclei in only 25% of the AdIE1-72-infected heterokaryons (MOI = 100) (Fig. 5B). p53 nuclear shuttling occurred in only 5% of heterokaryons infected with a higher dose of AdIE1-72 (MOI = 250) (Fig. 5B). Coinfection with a control adenovirus vector did not impede the ability of p53 to shuttle from human nuclei to mouse nuclei in heterokaryons, demonstrating that the increased vector load did not affect shuttling (compare Fig. 5B and C). Therefore, the nuclear accumulation of p53 after IE1-72 expression appears to be due, at least in part, to the ability of IE1-72 to abrogate p53 nuclear shuttling.
FIG. 5.

IE1-72 expression induces p53 nuclear retention. (A) Immunofluorescent detection of p53 shuttling. SAOS-2 cells were coinfected with Adp53 and either AdIE1-72 or AdCon. At 2 h after infection, cells were replated with Mdm2−/−/p53−/− MEFs and cultured overnight at 37°C. The cells were then fused together by treatment with polyethylene glycol and de novo protein synthesis was inhibited with cycloheximide. Cells were fixed 1 h after fusing. Immunofluorescent detection of human Ku-86 (red) and p53 (green) was performed to identify the human-mouse heterokaryons and p53 localization, respectively. (B and C) Quantitation of p53 shuttling. Human-mouse heterokaryons (n = 20) were identified in each experiment, and the detection of p53 in the mouse (Ku-86-negative) nuclei was scored as positive for p53 shuttling. The averaged results from two experiments are shown in panel B. The averaged results from three experiments that include infections of AdCon and the calculated standard errors are depicted in panel C. Numbers in parentheses indicate the MOIs of the recombinant adenovirus used.
IE1-72 expression increases p21 levels in a p53-dependent manner.
Given that p53 phosphorylation and nuclear accumulation should result in increased p53 activity, we next analyzed the consequences of IE1-72 expression on p21, a transcriptional target of p53 (22, 27). Initially, we determined whether IE1-72 expression activates the p21 promoter in reporter assays. As shown in Fig. 6A, IE1-72 expression increased p21 promoter-reporter activity 17-fold over the control plasmid-transfected fibroblasts. IE1-72 expression also resulted in a more modest increase in p21 levels relative to that observed in control virus-infected cells (Fig. 6B). To quantify the accumulation of p21 on a per-cell basis, we immunohistochemically stained cells infected with AdIE1-72 or control virus for p21 and scored the number of p21-positive cells in each population. In both the mock-infected and the control virus-infected cells, 17 to 29% of the population stained positive for p21 during a 48-h time course (Fig. 6C). Cells infected with either a low (MOI = 100) or a high (MOI = 250) dose of AdIE1-72 exhibited a higher percentage (56 to 73%) of p21-positive cells during this time course. The percentage of p21-positive cells observed after IE1-72 expression was similar to the percentage of p21-positive cells after p53 overexpression, which served as a positive control for p21 induction in this experiment. The IE1-72-mediated increase in the number of p21-positive cells required p53, since IE1-72 expression did not alter the percentage of p21-positive cells in p53−/− MEFs, whereas transduction of p53 into p53−/− MEFs did increase the number of p21-positive cells (Fig. 6D). Therefore, IE1-72 expression leads to a p53-dependent increase in p21 levels.
FIG. 6.

IE1-72 expression induces an accumulation of p21 levels in a p53-dependent manner. (A) Analysis of transcriptional activation of a p21 promoter-reporter. HEL fibroblasts were cotransfected with a p21-promoter luciferase construct, WAF-1-Luc, and either an IE1-72 cDNA expressing plasmid, pcDNA3-IE1-72, or a control plasmid, pcDNA3. A Renilla luciferase plasmid was also cotransfected and served as a normalization control for transfection efficiency in our reporter assays. Samples were harvested at 24 h posttransfection, and the luciferase activity was analyzed as described in the text. The results are depicted as fold induction of p21 promoter activity relative to the control plasmid transfected cells. Error bars indicate the standard error of the mean. (B) Immunoblot analysis for p21. Whole-cell extracts were harvested at 24 hpi from WT MEFs infected with either AdIE1-72 or a control virus. (C and D) Detection of p21 expression by immunohistochemistry. (C) REF52 cells were infected with either AdIE1-72 or a control virus and immunohistochemically stained for p21 protein at 24 and 48 hpi. Cells infected with Adp53 (MOI = 20) served as a positive control for p21 induction. A minimum of 300 cells was scored for each condition. The results are shown as the percentage of cells within the population and represent the average number of p21-positive cells from three separate analyses. Error bars indicate the standard error of the mean. (D) Number of p21-positive cells within populations of p53−/− MEFs infected with AdIE1-72, a control virus, or Adp53. The result from a representative experiment is shown.
IE1-72 expression induces quiescent cells to enter S-phase in the absence of p21.
Since induction of p21 expression by p53 is required for p53-mediated growth arrest, we determined whether p21 was responsible for the inability of IE1-72 to induce S phase in quiescent cells. Both WT and p21−/− MEFs were rendered quiescent by serum withdrawal prior to infection with either AdIE1-72 or control virus. As shown in Fig. 7, IE1-72 expression did not increase the percentage of cells in S phase relative to the control virus-infected population of serum-starved, WT MEFs (3.1% versus 3.2%, respectively). However, IE1-72 expression in quiescent p21−/− MEFs resulted in an increased percentage of S-phase cells over control infections (20.8% versus 6.7%, respectively). We included E2F1 as a positive control for S-phase induction (16, 17, 33, 38) and, as expected, E2F1 expression in MEFs caused an increase in S-phase cells in both WT and p21−/− MEFs. The percentage of p21−/− MEFs in S phase after IE1-72 expression was similar to that observed in E2F1-expressing cells. These results suggest that the inability of IE1-72 to induce quiescent cells into S phase is due to the activation of the p53/p21 growth arrest pathway.
FIG. 7.
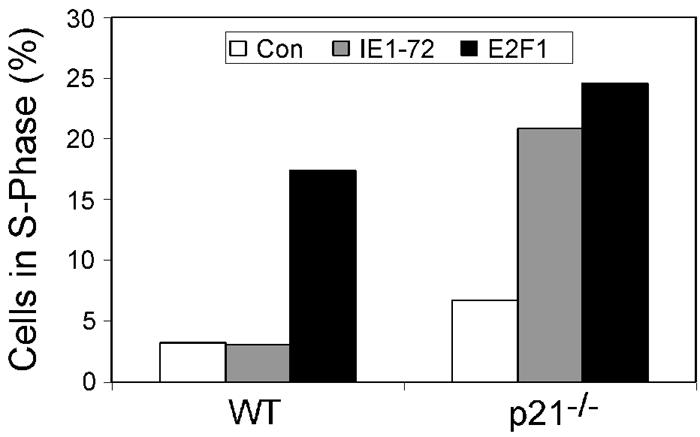
IE1-72 expression induces quiescent cells to enter S phase in the absence of p21. Serum-starved, early-passage WT and p21−/− MEFs were infected with AdIE1-72 or a control virus and maintained under low-serum conditions. At 24 hpi, MEFs were harvested, and DNA was stained with propidium iodide. Flow cytometry analysis was performed, and the percentage of cells in S phase was scored. The averaged results from two separate experiments are represented as histograms for each cell type.
DISCUSSION
We previously reported that IE1-72 promotes the nuclear accumulation of p53 and suggested this accumulation likely contributes to the inability of IE1-72 to induce reentry into the cell cycle in p53-expressing cells. Consistent with this hypothesis, IE1-72 expression results in p53 accumulation and is only able to induce quiescent cells to enter S phase in the absence of p53 (11). However, the mechanism by which p53 accumulates and inhibits the S-phase-promoting ability of IE1-72 remained unclear. We present evidence here that IE1-72 may activate p53 through two mechanisms: (i) perhaps by inducing the accumulation p19Arf protein and, more prominently, (ii) by activating an ATM-dependent p53 phosphorylation on Ser15. In addition, we find that IE1-72 promotes p53 nuclear accumulation by abrogating p53 nuclear shuttling. The consequences of IE1-72-mediated p53 accumulation and modification are a p53-dependent induction of p21 expression and a p21-dependent growth arrest.
Small DNA tumor viruses modulate the host cell cycle by encoding viral oncoproteins that inactivate p53 and members of the RB family. By overriding the functions of the RB proteins and abrogating p53 activity, the small DNA tumor viruses stimulate E2F transcriptional activity, induce S phase, and permit viral DNA replication to occur. Analogous to the small DNA tumor viruses, HCMV encodes several proteins that modulate cell cycle regulating proteins. Expression of the pp71 tegument protein induces quiescent cells to enter S phase by targeting the RB proteins for degradation by the proteosome (34, 36). Two IE proteins, IE1-72 and IE2-86, interact with RB family members p107 and pRb, respectively, and alleviate the repression of E2F-responsive promoters by binding with their respective RB family members (23, 26, 51, 67, 81). Although IE2-86 expression induces S-phase entry presumably by blocking pRb and p53 function (11, 48, 75), IE1-72 can mediate a similar effect in cells lacking p53 by relieving p107-mediated repression of E2F-responsive promoters (11, 51, 81). IE1-72 can also alleviate p107 inhibition of cyclin E/Cdk2 activity by displacing its binding to p107 (81). Thus, it appears that targeting of the RB proteins by these HCMV proteins is crucial for their ability to induce S phase in cells.
Oncoproteins such as E2F can stabilize p53 protein levels by increasing p19Arf levels through transactivation of the p19Arf promoter (7, 18, 57). Because IE1-72 can relieve p107-mediated repression of E2F-responsive promoters (51), it is possible that IE1-72 induces p19Arf through E2F. However, unlike the adenovirus E1A protein and polyomavirus middle T antigen, which require p19Arf to induce p53 accumulation (18, 42), IE1-72 can still modulate p53 levels in cells lacking p19Arf. Phosphorylation of Ser15/18 likely contributes to the overall increase in p53 levels since this modification has been shown to enhance p53 stability (12). The importance of this particular modification to p53 is demonstrated by the fact that polyomavirus infection also results in the phosphorylation of p53 on Ser18 in mouse cells, leading to elevated levels of p53 and increased activity (19). Likewise, we find that HPV-E7 expression in diploid human cells leads to Ser15 phosphorylation on p53 (58). In both cases, pRb targeting is required to induce this phosphorylation on p53. Moreover, it has been suggested that phosphorylation of p53 at Ser15 prevents p53 nuclear shuttling (80). Thus, our finding that IE1-72 expression promotes the phosphorylation and nuclear retention of p53 likely contribute to its activation.
It has been proposed that IE1-72 exhibits kinase activity, thereby raising the possibility that IE1-72 might directly phosphorylate p53. However, p53 has not been shown to be a direct substrate (45). Rather, we find that ATM is autophosphorylated and required for Ser15 modification on p53. ATM is a primary signal transducer activated in response to different stresses, including DNA damage (for a review, see reference 21) and deregulation of E2F activity (53, 56-58), and as part of G1/S-, G2/M-, and S-phase checkpoints (1). IE1-72 expression and HCMV replication might trigger ATM activity by modulating one of these checkpoints and/or by inactivating RB family members and inducing E2F activity. Presumably, then, IE1-72 contributes to ATM activation during HCMV infection. However, demonstrating this causality is not feasible because every viral factor that stimulates E2F activity should also induce ATM activity and p53 phosphorylation. Indeed, IE2-86 expression led to Ser15 phosphorylation on p53 as well (Fig. 4) (68). Our attempt to discern the contributions of each IE protein to p53 phosphorylation during HCMV infection were not fruitful because the UL123 (IE1-72) mutant used did not express either IE protein. Also, given the precedence of IE proteins activating E2F and stimulating p53 phosphorylation, we predict that pp71, which induces E2F activity (34), will lead to p53 phosphorylation. Finally, IE1-72 has also been described as being potentially mutagenic (44, 61). Thus, IE1-72 (and perhaps IE2-86) expression might somehow lead to DNA damage and activate components of the DNA damage pathway, including ATM.
Activation of p53 transcriptional activity results in increased expression of numerous target genes including p21, which is responsible for the growth arrest mediated by p53. IE1-72 induces p21 expression in a p53-dependent manner. The induction of p53 and p21 negatively impacts Cdk2 activity and delays S-phase progression. Therefore, the observed increase in p21 levels most likely accounts for the inability of IE1-72 to induce S phase in quiescent cells in our study. A very transient increase in p21 observed during immediate-early times of productive HCMV infections in laboratory experiments (15) raises the possibility that IE1-72 is responsible for (or contributes to) this induction in HCMV-infected fibroblasts. The levels of p21 then rapidly decline after this initial increase during HCMV infection, perhaps to counter the response of p53-p21 signaling. Intriguingly, elevated levels of p53 and p21 are also observed in a subset of HCMV-positive cells observed in tissues biopsies. Infected cells that are positive for p53 and p21 are inclusion body negative, which, according to the authors, are indicative of early stages of HCMV infection and may represent an intrinsic cellular defense to virus replication (24).
In addition to IE1-72 activating ATM, we and others (62) have found that HCMV infection leads to ATM autophosphorylation and Ser15 phosphorylation on p53. A question left unanswered is why HCMV does not block this host response to infection. At least in endothelial cells, stimulation of ATM and p53 activity appear to contribute to the death of cells at late times of HCMV infections, which may facilitate the dissemination of virus (62). Alternately, Wilkinson and Weller show that DNA damage recognition and repair proteins are recruited to ND10 nuclear domains in herpes simplex virus-infected cells (76). Some of these DNA damage recognition and signaling proteins, including NBS1 and p53, are substrates of ATM kinase activity (76). The periphery of ND10 domains (or PODs) are also sites of viral DNA replication during HCMV infection (2, 4). Thus, it seems reasonable to speculate that ATM kinase activity might facilitate viral DNA replication during an HCMV infection.
Acknowledgments
We thank Rachel Gerstein, Steve Jones, and members of our laboratory for comments on the manuscript. We thank Alonzo Ross for assistance with fluorescence microscopy. We thank Yanping Zhang and Yue Xiong for technical assistance with the heterokaryon assays.
An NIH National Research Service Award (HL 10334-01 to J.P.C.) and grants from the American Heart Association (9830085N to T.F.K. and 0365207B to A.D.Y.) and the NIH (CA86038 to T.F.K. and AI56077 to A.D.Y.) supported this work. The Flow Cytometry Core Facility is supported by an NIH Center grant (DK32520).
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
REFERENCES
- 1.Abraham, R. T. 2001. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15:2177-2196. [DOI] [PubMed] [Google Scholar]
- 2.Ahn, J. H., and G. S. Hayward. 2000. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 274:39-55. [DOI] [PubMed] [Google Scholar]
- 3.Ahn, J. H., and G. S. Hayward. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 71:4599-4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahn, J. H., W. J. Jang, and G. S. Hayward. 1999. The human cytomegalovirus IE2 and UL112-113 proteins accumulate in viral DNA replication compartments that initiate from the periphery of promyelocytic leukemia protein-associated nuclear bodies (PODs or ND10). J. Virol. 73:10458-10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bakkenist, C. J., and M. B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499-506. [DOI] [PubMed] [Google Scholar]
- 6.Barak, Y., T. Juven, R. Haffner, and M. Oren. 1993. mdm2 expression is induced by wild type p53 activity. EMBO J. 12:461-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bates, S., A. C. Phillips, P. A. Clark, F. Stott, G. Peters, R. L. Ludwig, and K. H. Vousden. 1998. p14ARF links the tumour suppressors RB and p53. Nature 395:124-125. [DOI] [PubMed] [Google Scholar]
- 8.Bates, S., and K. H. Vousden. 1996. p53 in signaling checkpoint arrest or apoptosis. Curr. Opin. Genet. Dev. 6:12-18. [DOI] [PubMed] [Google Scholar]
- 9.Bresnahan, W. A., I. Boldogh, E. A. Thompson, and T. Albrecht. 1996. Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology 224:150-160. [DOI] [PubMed] [Google Scholar]
- 10.Castillo, J. P., and T. F. Kowalik. 2002. Human cytomegalovirus immediate-early proteins and cell growth control. Gene 290:19-34. [DOI] [PubMed] [Google Scholar]
- 11.Castillo, J. P., A. D. Yurochko, and T. F. Kowalik. 2000. Role of human cytomegalovirus immediate-early proteins in cell growth control. J. Virol. 74:8028-8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao, C., S. Saito, C. W. Anderson, E. Appella, and Y. Xu. 2000. Phosphorylation of murine p53 at ser-18 regulates the p53 responses to DNA damage. Proc. Natl. Acad. Sci. USA 97:11936-11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chehab, N. H., A. Malikzay, M. Appel, and T. D. Halazonetis. 2000. Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 14:278-288. [PMC free article] [PubMed] [Google Scholar]
- 14.Chehab, N. H., A. Malikzay, E. S. Stavridi, and T. D. Halazonetis. 1999. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl. Acad. Sci. USA 96:13777-13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen, Z., E. Knutson, A. Kurosky, and T. Albrecht. 2001. Degradation of p21cip1 in cells productively infected with human cytomegalovirus. J. Virol. 75:3613-3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeGregori, J., T. F. Kowalik, and J. R. Nevins. 1995. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis and G1/S regulatory genes. Mol. Cell. Biol. 15:4215-4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeGregori, J., G. Leone, A. Miron, L. Jakol, and J. R. Nevins. 1997. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. USA 94:7245-7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Stanchina, E., M. E. McCurrach, F. Zindy, S. Y. Shieh, G. Ferbeyre, A. V. Samuelson, C. Prives, M. F. Roussel, C. J. Sherr, and S. W. Lowe. 1998. E1A signaling to p53 involves the p19ARF tumor suppressor. Genes Dev. 12:2434-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dey, D., J. Dahl, S. Cho, and T. L. Benjamin. 2002. Induction and bypass of p53 during productive infection by polyomavirus. J. Virol. 76:9526-9532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dittmer, D., and E. S. Mocarski. 1997. Human cytomegalovirus infection inhibits G1/S transition. J. Virol. 71:1629-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durocher, D., and S. P. Jackson. 2001. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr. Opin. Cell Biol. 13:225-231. [DOI] [PubMed] [Google Scholar]
- 22.el-Deiry, W. S., T. Tokino, V. E. Velculescu, D. B. Levy, R. Parsons, J. M. Trent, D. Lin, W. E. Mercer, K. W. Kinzler, and B. Vogelstein. 1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75:817-825. [DOI] [PubMed] [Google Scholar]
- 23.Fortunato, E. A., M. H. Sommer, K. Yoder, and D. H. Spector. 1997. Identification of domains within the human cytomegalovirus major immediate-early 86-kilodalton protein and the retinoblastoma protein required for physical and functional interaction with each other. J. Virol. 71:8176-8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia, J. F., M. A. Piris, E. Lloret, J. L. Orradre, P. G. Murillo, and J. C. Martinez. 1997. p53 expression in CMV-infected cells: association with the alternative expression of the p53 transactivated genes p21/WAF1 and MDM2. Histopathology 30:120-125. [DOI] [PubMed] [Google Scholar]
- 25.Greaves, R. F., and E. S. Mocarski. 1998. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus ie1 mutant. J. Virol. 72:366-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagemeier, C., R. Caswell, G. Hayhurst, J. Sinclair, and T. Kouzarides. 1994. Functional interaction between the HCMV IE2 transactivator and the retinoblastoma protein. EMBO J. 13:2897-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harper, J. W., G. R. Adami, N. Wei, K. Keyomarsi, and S. J. Elledge. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805-816. [DOI] [PubMed] [Google Scholar]
- 28.Haupt, Y., R. Maya, A. Kazaz, and M. Oren. 1997. Mdm2 promotes the rapid degradation of p53. Nature 387:296-299. [DOI] [PubMed] [Google Scholar]
- 29.Hirao, A., A. Cheung, G. Duncan, P. M. Girard, A. J. Elia, A. Wakeham, H. Okada, T. Sarkissian, J. A. Wong, T. Sakai, E. De Stanchina, R. G. Bristow, T. Suda, S. W. Lowe, P. A. Jeggo, S. J. Elledge, and T. W. Mak. 2002. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol. Cell. Biol. 22:6521-6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Honda, R., H. Tanaka, and H. Yasuda. 1997. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 420:25-27. [DOI] [PubMed] [Google Scholar]
- 31.Honda, R., and H. Yasuda. 1999. Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 18:22-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jault, F. M., J. M. Jault, F. Ruchti, E. A. Fortunato, C. Clark, J. Corbeil, D. D. Richman, and D. H. Spector. 1995. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J. Virol. 69:6697-6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson, D., J. K. Schwarz, W. D. Cress, and J. R. Nevins. 1993. Expression of transcription factor E2F1 induces quiescent cells to enter S-phase. Nature 378:206-208. [DOI] [PubMed] [Google Scholar]
- 34.Kalejta, R. F., J. T. Bechtel, and T. Shenk. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23:1885-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalejta, R. F., and T. Shenk. 2002. Manipulation of the cell cycle by human cytomegalovirus. Front. Biosci. 7:d295-306. [DOI] [PubMed] [Google Scholar]
- 36.Kalejta, R. F., and T. Shenk. 2003. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc. Natl. Acad. Sci. USA 100:3263-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamijo, T., J. D. Weber, G. Zambetti, F. Zindy, M. F. Roussel, and C. J. Sherr. 1998. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc. Natl. Acad. Sci. USA 95:8292-8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kowalik, T., J. DeGregori, J. K. Schwarz, and I. R. Nevins. 1995. E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J. Virol. 69:2491-2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kowalik, T. F., J. DeGregori, G. Leone, L. Jakoi, and J. R. Nevins. 1998. E2F1-specific induction of apoptosis and p53 accumulation, which is blocked by Mdm2. Cell Growth Differ. 9:113-118. [PubMed] [Google Scholar]
- 40.Kubbutat, M. H., S. N. Jones, and K. H. Vousden. 1997. Regulation of p53 stability by Mdm2. Nature 387:299-303. [DOI] [PubMed] [Google Scholar]
- 41.Levine, A. J. 1997. p53, the cellular gatekeeper for growth and division. Cell 88:323-331. [DOI] [PubMed] [Google Scholar]
- 42.Lomax, M., and M. Fried. 2001. Polyomavirus disrupts ARF signaling to p53. Oncogene 20:4951-4960. [DOI] [PubMed] [Google Scholar]
- 43.Lu, M., and T. Shenk. 1996. Human cytomegalovirus infection inhibits cell cycle progression at multiple points, including the transition from G1 to S. J. Virol. 70:8850-8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukac, D. M., and J. C. Alwine. 1999. Effects of human cytomegalovirus major immediate-early proteins in controlling the cell cycle and inhibiting apoptosis: studies with ts13 cells. J. Virol. 73:2825-2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Margolis, M. J., S. Pajovic, E. L. Wong, M. Wade, R. Jupp, J. A. Nelson, and J. C. Azizkhan. 1995. Interaction of the 72-kilodalton human cytomegalovirus IE1 gene product with E2F1 coincides with E2F-dependent activation of dihydrofolate reductase transcription. J. Virol. 69:7759-7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muganda, P., R. Carrasco, and Q. Qian. 1998. The human cytomegalovirus IE2 86-kDa protein elevates p53 levels and transactivates the p53 promoter in human fibroblasts. Cell Mol. Biol. 44:321-331. [PubMed] [Google Scholar]
- 47.Muganda, P., O. Mendoza, J. Hernandez, and Q. Qian. 1994. Human cytomegalovirus elevates levels of the cellular protein p53 in infected fibroblasts. J. Virol. 68:8028-8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murphy, E. A., D. N. Streblow, J. A. Nelson, and M. F. Stinski. 2000. The human cytomegalovirus IE86 protein can block cell cycle progression after inducing transition into the S phase of permissive cells. J. Virol. 74:7108-7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nevins, J. R., J. DeGregori, L. Jakoi, and G. Leone. 1997. Functional analysis of E2F transcription factor. Methods Enzymol. 283:205-219. [DOI] [PubMed] [Google Scholar]
- 50.Palmero, I., C. Pantoja, and M. Serrano. 1998. p19ARF links the tumour suppressor p53 to Ras. Nature 395:125-126. [DOI] [PubMed] [Google Scholar]
- 51.Poma, E. E., T. F. Kowalik, L. Zhu, J. H. Sinclair, and E. S. Huang. 1996. The human cytomegalovirus IE1-72 protein interacts with the cellular p107 protein and relieves p107-mediated transcriptional repression of an E2F-responsive promoter. J. Virol. 70:7867-7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pomerantz, J., N. Schreiber-Agus, N. J. Liegeois, A. Silverman, L. Alland, L. Chin, J. Potes, K. Chen, I. Orlow, H. W. Lee, C. Cordon-Cardo, and R. A. DePinho. 1998. The Ink4a tumor suppressor gene product, p19ARF, interacts with MDM-2 and neutralizes MDM-2's inhibition of p53. Cell 92:713-723. [DOI] [PubMed] [Google Scholar]
- 53.Powers, J. T., S. Hong, C. N. Mayhew, P. M. Rogers, E. S. Knudsen, and D. G. Johnson. 2004. E2F1 uses the ATM signaling pathway to induce p53 and Chk2 phosphorylation and apoptosis. Mol. Cancer Res. 2:203-214. [PubMed] [Google Scholar]
- 54.Prives, C., and P. A. Hall. 1999. The p53 pathway. J. Pathol. 187:112-126. [DOI] [PubMed] [Google Scholar]
- 55.Rao, L., M. Debbas, P. Sabbatini, D. Hockenbery, S. Korsmeyer, and E. White. 1992. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 89:7742-7746. Erratum, 89:9974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rogoff, H. A., and T. F. Kowalik. 2004. Life, death, and E2F: linking proliferation control and DNA damage signaling via E2F1. Cell Cycle 3:845-846. [DOI] [PubMed] [Google Scholar]
- 57.Rogoff, H. A., M. T. Pickering, M. E. Debatis, S. Jones, and T. F. Kowalik. 2002. E2F1 induces phosphorylation of p53 that is coincident with p53 accumulation and apoptosis. Mol. Cell. Biol. 22:5308-5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rogoff, H. A., M. T. Pickering, F. M. Frame, M. E. Debatis, Y. Sanchez, S. Jones, and T. F. Kowalik. 2004. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol. Cell. Biol. 24:2968-2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roth, J., M. Dobbelstein, D. A. Freedman, T. Shenk, and A. J. Levine. 1998. Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 17:554-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sarkaria, J. N., E. C. Busby, R. S. Tibbetts, P. Roos, Y. Taya, L. M. Karnitz, and R. T. Abraham. 1999. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 59:4375-4382. [PubMed] [Google Scholar]
- 61.Shen, Y., H. Zhu, and T. Shenk. 1997. Human cytomegalovirus IE1 and IE2 proteins are mutagenic and mediate “hit-and-run” oncogenic transformation in cooperation with the adenovirus E1A proteins. Proc. Natl. Acad. Sci. USA 94:3341-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen, Y. H., B. Utama, J. Wang, M. Raveendran, D. Senthil, W. J. Waldman, J. D. Belcher, G. Vercellotti, D. Martin, B. M. Mitchelle, and X. L. Wang. 2004. Human cytomegalovirus causes endothelial injury through the ataxia telangiectasia mutant and p53 DNA damage signaling pathways. Circ. Res. 94:1310-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shieh, S. Y., J. Ahn, K. Tamai, Y. Taya, and C. Prives. 2000. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 14:289-300. [PMC free article] [PubMed] [Google Scholar]
- 64.Shieh, S. Y., M. Ikeda, Y. Taya, and C. Prives. 1997. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91:325-334. [DOI] [PubMed] [Google Scholar]
- 65.Siliciano, J. D., C. E. Canman, Y. Taya, K. Sakaguchi, E. Appella, and M. B. Kastan. 1997. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 11:3471-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sinclair, J., J. Baillie, L. Bryant, and R. Caswell. 2000. Human cytomegalovirus mediates cell cycle progression through G1 into early S phase in terminally differentiated cells. J. Gen. Virol. 81(Pt. 6):1553-1565. [DOI] [PubMed] [Google Scholar]
- 67.Sommer, M. H., A. L. Scully, and D. H. Spector. 1994. Transactivation by the human cytomegalovirus IE2 86-kilodalton protein requires a domain that binds to both the TATA box-binding protein and the retinoblastoma protein. J. Virol. 68:6223-6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Song, Y. J., and M. F. Stinski. 2005. Inhibition of cell division by the human cytomegalovirus IE86 protein: role of the p53 pathway or cyclin-dependent kinase 1/cyclin B1. J. Virol. 79:2597-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Speir, E., R. Modali, E. S. Huang, M. B. Leon, F. Shawl, T. Finkel, and S. E. Epstein. 1994. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science 265:391-394. [DOI] [PubMed] [Google Scholar]
- 70.Stommel, J. M., N. D. Marchenko, G. S. Jimenez, U. M. Moll, T. J. Hope, and G. M. Wahl. 1999. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J. 18:1660-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stott, F. J., S. Bates, M. C. James, B. B. McConnell, M. Starborg, S. Brookes, I. Palmero, K. Ryan, E. Hara, K. H. Vousden, and G. Peters. 1998. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 17:5001-5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tanaka, K., J. P. Zou, K. Takeda, V. J. Ferrans, G. R. Sandford, T. M. Johnson, T. Finkel, and S. E. Epstein. 1999. Effects of human cytomegalovirus immediate-early proteins on p53-mediated apoptosis in coronary artery smooth muscle cells. Circulation 99:1656-1659. [DOI] [PubMed] [Google Scholar]
- 73.Tao, W., and A. J. Levine. 1999. Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc. Natl. Acad. Sci. USA 96:3077-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vousden, K. H., and G. F. Woude. 2000. The ins and outs of p53. Nat. Cell Biol. 2:E178-E180. [DOI] [PubMed] [Google Scholar]
- 75.Wiebusch, L., and C. Hagemeier. 2001. The human cytomegalovirus immediate-early 2 protein dissociates cellular DNA synthesis from cyclin-dependent kinase activation. EMBO J. 20:1086-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilkinson, D. E., and S. K. Weller. 2003. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life 55:451-458. [DOI] [PubMed] [Google Scholar]
- 77.Wilkinson, G. W., and A. Akrigg. 1992. Constitutive and enhanced expression from the CMV major IE promoter in a defective adenovirus vector. Nucleic Acids Res. 20:2233-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu, X., J. H. Bayle, D. Olson, and A. J. Levine. 1993. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7:1126-1132. [DOI] [PubMed] [Google Scholar]
- 79.Zhang, Y., and Y. Xiong. 1999. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol. Cell 3:579-591. [DOI] [PubMed] [Google Scholar]
- 80.Zhang, Y., and Y. Xiong. 2001. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science 292:1910-1915. [DOI] [PubMed] [Google Scholar]
- 81.Zhang, Z., S. M. Huong, X. Wang, D. Y. Huang, and E. S. Huang. 2003. Interactions between human cytomegalovirus IE1-72 and cellular p107: functional domains and mechanisms of up-regulation of cyclin E/cdk2 kinase activity. J. Virol. 77:12660-12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zindy, F., C. M. Eischen, D. H. Randle, T. Kamijo, J. L. Cleveland, C. J. Sherr, and M. F. Roussel. 1998. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 12:2424-2433. [DOI] [PMC free article] [PubMed] [Google Scholar]