

Abstract
The distinct lifestyle of obligately intracellular bacteria can alter fundamental forces that drive and constrain genome change. In this study, sequencing the 792-kb genome of Blochmannia pennsylvanicus, an obligate endosymbiont of Camponotus pennsylvanicus, enabled us to trace evolutionary changes that occurred in the context of a bacterial–ant association. Comparison to the genome of Blochmannia floridanus reveals differential loss of genes involved in cofactor biosynthesis, the composition and structure of the cell wall and membrane, gene regulation, and DNA replication. However, the two Blochmannia species show complete conservation in the order and strand orientation of shared genes. This finding of extreme stasis in genome architecture, also reported previously for the aphid endosymbiont Buchnera, suggests that genome stability characterizes long-term bacterial mutualists of insects and constrains their evolutionary potential. Genome-wide analyses of protein divergences reveal 10- to 50-fold faster amino acid substitution rates in Blochmannia compared to related bacteria. Despite these varying features of genome evolution, a striking correlation in the relative divergences of proteins indicates parallel functional constraints on gene functions across ecologically distinct bacterial groups. Furthermore, the increased rates of amino acid substitution and gene loss in Blochmannia have occurred in a lineage-specific fashion, which may reflect life history differences of their ant hosts.
Genome sequencing provides a rich data set to predict the metabolic capabilities of organisms, and comparative analyses among closely related species offer a powerful approach to examine mechanisms of genome flux. Since 2000, genomics has shed light on the metabolism and evolution of obligately intracellular, mutualistic bacteria that have coevolved with various insect groups for tens to hundreds of millions of years (Baumann et al. 2000). Fully sequenced genomes of Buchnera associated with aphids (Shigenobu et al. 2000; Tamas et al. 2002; van Ham et al. 2003), Wigglesworthia of tsetse flies (Akman et al. 2002), and Blochmannia associated with Camponotus (Gil et al. 2003) are extremely streamlined yet retain basic cellular processes and specific biosynthetic abilities required by the insect host. Genome comparisons within Buchnera have shown stability, with no gene acquisition, inversions, or translocations throughout 50–70 million years (Myr) of evolution within aphids (Tamas et al. 2002) and near-perfect synteny since the establishment of this association 150–200 million years ago (Mya) (van Ham et al. 2003). This exceptional stasis of genome architecture contrasts with lability of free-living and parasitic bacterial genomes. Genome stability may reflect the dearth of molecular tools for gene exchange (e.g., phage, certain rec genes, and repeated DNA sequences) in this mutualist and limited ecological opportunities to recombine with genetically distinct bacteria (Tamas et al. 2002; van Ham et al. 2003; Moran and Plague 2004). Such constraints on genome changes in stable mutualists may profoundly affect the evolutionary potential of these bacteria and their hosts. However, owing to the lack of multiple sequenced genomes within endosymbiont groups, genome stability in other long-term endosymbionts has remained untested.
In order to contribute to a more comprehensive model of genome evolution in ancient endosymbiotic associations, we have evaluated genome dynamics in Blochmannia, a bacterial genus that is closely related to Buchnera and has cospeciated with ants for ∼30 Myr. The wide range of interactions between ants and other species, including plants, fungi, trophobionts, other insects, and diverse bacteria (Dasch et al. 1984; Currie 2001; Zientz et al. 2001), may explain the huge ecological success of ants, which play dominant roles in nutrient turnover in terrestrial ecosystems and include more than twice as many species as mammals (Hölldobler and Wilson 1990). Blochmannia is the most evolutionarily stable ant associate and lives exclusively within cells of the closely related genera Polyrhachis, Colobopsis, and Camponotus (Dasch et al. 1984; Schröder et al. 1996; Sameshima et al. 1999; Sauer et al. 2000; Degnan et al. 2004). Blochmannia has been studied most extensively in Camponotus, the second largest ant genus, with ∼1000 species (Bolton 1995) ranging from omnivores to specialists on plant secretions and homopteran exudates (Dasch 1975; Hölldobler and Wilson 1990; Bolton 1995; Davidson 1997, 1998) and with nesting habitats including wood, soil beneath rocks, and the rainforest canopy (Bolton 1995).
The 706-kb sequence of B. floridanus (Blochmannia of host Camponotus floridanus) indicated this ant endosymbiont retains numerous metabolic pathways that may be involved in host nutrition, including nitrogen recycling and assimilation, biosynthesis of amino acids and fatty acids, and sulfate reduction (Gil et al. 2003). Blochmannia is thought to be important during host development (Sauer et al. 2002; Wolschin et al. 2004), but its specific roles in host physiology and ecology and its functional variation across ant species remain unclear. We have sequenced the 792-kb genome of Blochmannia pennsylvanicus (Blochmannia associated with Camponotus pennsylvanicus) to examine genome changes since this lineage and B. floridanus diverged from a common ancestor ∼16–20 Mya (Degnan et al. 2004). Comparing genome inventories and architectures of the two Blochmannia strains allowed us to trace functional and structural changes that occurred in the context of this bacterium–ant interaction and to contrast genome dynamics and protein evolution in two mutualist groups (Blochmannia and Buchnera).
Results
Genome features
The B. pennsylvanicus genome consists of a 791,654-bp circular chromosome that we sequenced to 12× coverage (Table 1; Fig. 1; Supplemental Fig. S1; GenBank entry CP000016). This AT-rich genome has a relatively low percentage of coding DNA (76.7%) and shows a GC skew that distinguishes the leading and the lagging strands of replication [(G–C)/(G+C) averaged across 2.5-kb sliding windows]. We identified predicted ribosomal binding sites (RBSs) for 479 of 610 open reading frames (ORFs), but only 42 had the canonical AGGAG motif. Surprisingly, two pseudogenes (rpmD and uvrD) had detectable RBSs.
Table 1.
Comparison of general genome features among obligate insect mutualists
| B. pennsylvanicus | B. floridanus | W. glossinidia | Buchnera-BP | Buchnera-SG | Buchnera-APS | E. coli K12 | |
|---|---|---|---|---|---|---|---|
| Chromosome, bp | 791,654 | 705,557 | 697,724 | 615,980 | 641,454 | 640,681 | 4,639,221 |
| Plasmids | — | — | 1 | 1 | 2 | 2 | — |
| G+C content total, % | 29.6 | 27.38 | 22 | 25.3 | 26.3 | 26.2 | 50.8 |
| Total gene number | 658 | 636 | 677 | 553 | 629 | 621 | 4550 |
| ORFs | 610 | 590 | 621 (6) | 507 (3) | 554 (9) | 571 (9) | 4284 |
| rRNAs | 3 | 3 | 6 | 3 | 3 | 3 | 22 |
| tRNAs | 39 | 37 | 34 | 32 | 32 | 32 | 86 |
| RNAs | 2 | 2 | 2 | 2 | 2 | 2 | 8 |
| Pseudogenes | 4 | 4 | 14 | 9 | 38 | 13 | 150 |
| Chromosomal protein-coding regions, % | 76.7 | 83.8 | 86.9 | 80.9 | 83.1 | 86.7 | 87.8 |
| Average length CDS, bp | 995 | 1,002 | 983 | 996 | 979 | 984 | 950 |
Total open reading frames (ORFs) include predicted genes located on plasmids (noted in parentheses). The repeated trpEG genes on Buchnera-APS and -SG plasmids were counted once. Minor discrepancies in gene numbers (ORFs and pseudogenes) from the original publications reflect our efforts to present the most current annotation data from the literature, available at GenBank (http://www.ncbi.nlm.nih.gov) and the Comprehensive Microbial Resource (CMR) database (http://www.tigr.org/tigr-scripts/CMR2/CMRHomePage.spl). Fusion of yidCD in B. pennsylvanicus counted as a single ORF.
Figure 1.

Circular map of the B. pennsylvanicus genome and genome features. The origin of replication was putatively set upstream of gidA, in accordance with the shift of GC skew. The concentric rings denote the following features: (1) numbered base pair coordinates beginning with base one of the gidA open reading frame (ORF); (2) GC skew as calculated by (G–C)/(G+C) using a 2.5-kb sliding window; (3) ORFS present on the leading strand (+); (4) ORFS present on the lagging strand (–); (5) pseudogenes of B. pennsylvanicus that are present in (orange) or absent from (black) B. floridanus; (6) ORFS of B. pennsylvanicus that are shared with (red), pseudogenes in (yellow), or absent in (dark blue) the genome of B. floridanus; (7) transfer RNAs (tRNAs) that are shared with (gray) or absent in (pink) B. floridanus; (8) ribosomal RNAs (rRNAs) 5S, 16S, and 23S (purple). Circular plot and GC skew analysis were generated by the JENA Prokaryotic Genome Viewer–Export Version v3.06.04.
Differential gene loss, yet complete stability of genome architecture within Blochmannia
The 86-kb size difference between the Blochmannia genomes (B. pennsylvanicus, 792 kb; B. floridanus, 706 kb) largely reflects differential gene loss between the two lineages, with a greater extent of loss in B. floridanus. Both genomes possess several intact genes that are missing or pseudogenes in the other genome (Fig. 2). Although lateral gene transfer could, in principle, account for genes distinct to one Blochmannia genome, all genes grouped closely with enterobacterial homologs and showed amino acid and codon usage patterns typical of their resident genome. Notably, the order and strand orientation (i.e., genome architecture) is completely conserved for their 635 shared genes, including cases in which a functional gene in one genome is a pseudogene in the second.
Figure 2.

Comparison of B. pennsylvanicus and B. floridanus gene contents. Genes in the outer section of each circle are unique to one genome, while those listed in the intersection of the two circles are shared. Note that seven shared genes are apparently functional in one genome, but pseudogenes in the other. The truncation of B. floridanus dnaX (see text) is not included as a pseudogene here. The fusion of yidCD in B. pennsylvanicus is counted as two intact genes. (*) The 628 shared intact genes include genes that have a single frameshift within long poly(A) tracts but are otherwise intact (see text). These loci include B. pennsylvanicus hisH, ytfM, ubiF, and ybiS and B. floridanus ytfM, ybiS, and gmhB.
Assuming that the common ancestor of the two Blochmannia strains encoded at least their combined set of 615 ORFs, then gene loss or inactivation in the lineage leading to B. floridanus occurred at an approximate rate of one ORF per 0.64–0.8 Myr [(25 ORFs lost)/(16 Myr) to (25 ORFs lost)/(20 Myr)]. Gene loss in the B. pennsylvanicus lineage is apparently ∼6.5 times slower, with loss or inactivation of one ORF per 4.0–5.0 Myr [(4 ORFs lost)/(16 Myr) to (4 ORFs lost)/(20 Myr)]. Estimated rates of gene loss in B. floridanus exceed those in Buchnera, in which one ORF was lost or inactivated per 2.70–3.60 Myr between Buchnera–B. pistaciae and Buchnera–Acyrthosiphon pisum [(111 ORFs lost)/(150 Myr × 2) to (111 ORFs lost)/(200 Myr × 2)] and per 1.70–2.38 Myr between Buchnera–A. pisum and Buchnera–Schizaphis graminum [(59 ORFs lost)/(50 Myr × 2) to (59 ORFs lost)/(70 Myr × 2)]. Rates of loss may be underestimated if the same genes were deleted independently along lineages. Mechanisms that influence rates of gene loss may include changes in the strength and/or efficacy of selection to maintain genes, as well as differences in underlying rates of gene knockouts and deletion (see Discussion).
Truncations and frameshifts in otherwise conserved Blochmannia genes
In all, 13 Blochmannia genes show a significant (20%–40%) truncation compared to Escherichia coli orthologs but apparently encode functional proteins. These genes include aceF, aroK, aroQ, ftsK, ftsY, hfq, mreC, pheA, rpoZ, thrS, trpD, yfcB, and yqeI. In all but one case (hfq, see below), truncations are shared by B. pennsylvanicus and B. floridanus and thus likely occurred before the divergence of these lineages. Certain truncations are also shared between Blochmannia and Wigglesworthia (yfcB, rpoZ, ftsK), Buchnera (aroQ), or among all three mutualist groups (aceF, ftsY).
Although a >20% length reduction is often interpreted as evidence for loss of gene function (e.g., Lerat and Ochman 2004), the sequence conservation of truncated Blochmannia genes suggests they encode functional proteins. First, nonsynonymous divergence (dN) between B. pennsylvanicus and B. floridanus is relatively low at the 12 truncated genes they share (average of 0.3012 ± 0.14), whereas synonymous divergence (dS) exceeds 2 for each gene. Although dN/dS is difficult to calculate because of saturation of synonymous sites, this ratio is below 0.13 for each gene. Moreover, apart from RpoZ, protein divergences at truncated genes are generally comparable to other ORFs (average protein divergence of 0.569 ± 0.281, compared to the genome-wide average of 0.565 ± 0.309). RpoZ is quite divergent (2.16), but the fact that dN/dS ≪ 1 suggests the truncation did not eliminate gene function. The one truncated gene (hfq) in B. pennsylvanicus that lacks an ortholog in B. floridanus has a relatively low protein divergence with the closest outgroup, Wigglesworthia (0.576). Thus, in the absence of detailed biochemical and structural information for each of these genes, a 20% truncation appears too conservative a criterion for inferring loss of function. In Blochmannia, truncated genes clearly differ from annotated pseudogenes, in which frameshift and nonsense mutations introduce multiple stop codons throughout the gene.
We also detected a single, short frameshift in each of B. pennsylvanicus ytfM, ybiS, hisH, and ubiF. The first two genes are unclassified proteins, while the latter are required for histidine and ubiquinone biosynthesis, respectively (see Fig. 2 legend). Each indel occurred within a 9–11-bp string of consecutive As (Fig. 3). Reanalysis of B. floridanus pseudogenes revealed a similar pattern at ytfM, ybiS, and gmhB, in which single frameshifts occur within poly(A) or poly(T) tracts. Poly(A) tracts are very common within Blochmannia ORFs (e.g., B. pennsylvanicus has 133 poly(A) or (T) tracts >9 bp long) and likely reflect AT mutational bias and reduced selective pressures. The indels in ytfM, ybiS, hisH, and ubiF each show high coverage in the B. pennsylvanicus shotgun assembly (5–15 independent clones). In the case of hisH, the frameshift occurs across independent PCR products sequenced directly with varying types of sequencing chemistry.
Figure 3.

Sequence context of homopolymeric frameshift mutations within five Blochmannia genes. The single frameshifts in Blochmannia genes (A) hisH, (B) ubiF, (C) ybiS, (D) ytfM, and (E) gmhB were “corrected” manually, based on comparisons with sequences that lack the frameshift. Genes were then aligned by their inferred amino acid sequence against E. coli. Underlined nucleotides indicate poly(A) and poly(T) tracts in which frameshifts occur. Regions highlighted in gray indicate the putative sites of transcriptional or translational slippage that may restore the proper reading frame. (•) Amino acid identical to the top sequence (B. pennsylvanicus).
Despite their consistency, we hesitate to interpret these frameshifts as firm evidence of pseudogenes. Notably, apart from a single frameshift, these genes would otherwise encode intact proteins that are relatively conserved between the two Blochmannia genomes, with an average protein divergence (0.67 ± 0.16) that is only slightly above the average for other ORFs (0.565 ± 0.309). The occurrence of frameshifts within homopolymeric tracks is consistent with slippage during transcription (Wagner et al. 1990; Baranov et al. 2005) or during translation (Baranov et al. 2002; Gurvich et al. 2003) that could restore the full-length protein (see Discussion). If the frameshifts have, indeed, disrupted protein functions, these mutations must have occurred very recently since rapid mutation in Blochmannia (Degnan et al. 2004) is expected to erode pseudogenes quickly.
Metabolic similarities of Blochmannia spp.
Analysis of B. pennsylvanicus and reanalysis of other insect mutualist genomes using MultiFun (Serres and Riley 2000) allowed more comprehensive metabolic comparisons across groups (see Fig. 2 and Supplemental Tables S1 and S2). The two Blochmannia genomes share nearly all metabolic pathways thought to contribute to host nutrition, including most biosynthetic functions, sulfate assimilation and metabolism, and hydrolysis of urea (see Gil et al. 2003).
Metabolic differences between Blochmannia spp.
The 30 ORFs that distinguish the two Blochmannia genomes have a range of predicted cellular functions that may alter host–symbiont metabolic exchanges (Figs. 2 and 4). B. pennsylvanicus retains coaADE and dfp for the biosynthesis of coenzyme A, an essential cofactor and substrate for the TCA cycle (Fig. 4A). In contrast, the deletion of these genes from B. floridanus implies that this endosymbiont does not require coenzyme A, uses other enzymes for its synthesis, or imports this cofactor from the host.
Figure 4.

Distinct biosynthetic capabilities of B. pennsylvanicus and B. floridanus. The two mutualist genomes differ in genes encoded for the biosynthesis of (A) coenzyme A, and (B) isoprenoids. Gene names in gray are missing or pseudogenes (noted by ψ).
Several differences between the Blochmannia genomes involve the biosynthesis, transport, and mediation of cellular wall and membrane components. B. pennsylvanicus retains six distinct ORFs that contribute to the de novo synthesis of peptidoglycan (murein), the major constituent of Gram-negative bacterial cell walls (Park 1996). In addition to MurI, it retains the complete pathway for the biosynthesis of isoprenoids (Fig. 4B), essential substrates for synthesis of peptidoglycan (El Ghachi et al. 2004), and several organic compounds including carotinoids, glycosyl carrier lipids, and the side chains of ubiquinone (Ogura et al. 1997; Kainou et al. 2001). In B. floridanus, the absence of murI and the interruption of isoprenoid biosynthesis implies this bacterium may import both D-glutamate and trans, trans-farnesyl diphosphate (isoprenoid precursor) from the host, might synthesize these components using other enzymes, or produce a cell wall lacking these structural elements (i.e., isoprenoids).
B. pennsylvanicus is the first fully sequenced insect mutualist that retains the entire sec-dependent secretory pathway, including the chaperonin SecB. This general pathway mediates the export and translocation of numerous proteins to the periplasm or inner membrane (Pugsley 1993). The other insect mutualist genomes lack certain components (typically secBDF) of this pathway (Shigenobu et al. 2000; Akman et al. 2002; Tamas et al. 2002; Gil et al. 2003; van Ham et al. 2003), although these losses are not expected to eliminate function (Mushegian and Koonin 1996). B. pennsylvanicus also retains the inner-membrane-bound heat-induced protease HtpX and the periplasmic chaperonins LolB and DsbA. Together, these results suggest that B. pennsylvanicus is better able to respond to cellular stress and ensure proper transport localization, conformation, and decomposition of gene products. Distinct membrane features in B. pennsylvanicus also include its retention of the inner membrane proteins YciC, putatively involved in transport, and BacA, which confers bacitracin resistance.
While insect mutualists have lost many regulatory genes, B. pennsylvanicus encodes three that are missing from B. floridanus (hns, hfq, and mntR). Like B. floridanus and Wigglesworthia, B. pennsylvanicus lacks DnaA, considered important in the initiation of DNA replication (Akman et al. 2002; Gil et al. 2003). In B. floridanus, the HU-like nucleoprotein HlpA might be involved in starting the nucleosome (Gil et al. 2003), and in B. pennsylvanicus, the nucleoprotein Hns might also be recruited to the replication origin for this purpose. Neither Blochmannia genome encodes the θ subunit of the holoenzyme DNA polymerase III, but B. pennsylvanicus retains the τ and γ subunits of DnaX. The τ subunit acts as a molecular tether that couples DnaB (DNA helicase) to the core of DNA polymerase III (α and ε) as the replication fork progresses from the origin (Walker et al. 2000; Gao and McHenry 2001). Thus, the inability of B. floridanus, Buchnera, and Wigglesworthia to transcribe the τ subunit is expected to decrease the efficiency, accuracy, and processivity of the holoenzyme (van Ham et al. 2003). These distinct features of the B. pennsylvanicus replication machinery might contribute to the apparent 29-kb shift observed in B. floridanus GC skew relative to B. pennsylvanicus (see Supplemental Fig. S1).
Long intergenic spacers in B. pennsylvanicus
Intergenic spacers in B. pennsylvanicus are significantly longer (average 291 bp) than homologous spacers in B. floridanus (average 180 bp; Wilcoxon Rank sum test, p < 0.0001), are longer than spacers in most other bacteria (Bacillus subtilis, average 121.9 bp; Vibrio cholerae, average 156.4 bp; Escherichia coli K12 (http://ecocyc.org), average 128.8 bp) (Mira et al. 2001) and contribute to its larger genome size compared to B. floridanus (77% vs. 84% coding regions) (Table 1). While spacers of the two Blochmannia genomes are too divergent to align, the strict conservation of gene order allowed us to predict homologous spacers based on their position in the chromosome. A detectable relationship between lengths of spacers in the two genomes suggests a certain degree of conservation of spacer length (Fig. 5A) (Spearman's ρ = 0.7895; p < 0.0001), although some short and medium-length spacers in B. floridanus are quite long (often >1 kb) in B. pennsylvanicus. Multiple gene-finding tools did not detect ORFs or recognizable pseudogenes in any spacers, which have lower GC contents (average of 20.0%) compared to ORFs (average of 32.1%) (Fig. 5B) and may represent eroded pseudogenes and/or regions involved in gene regulation.
Figure 5.

Comparison of intergenic spacers (IGSs) in B. pennsylvanicus and B. floridanus. (A) A significant relationship in the lengths of homologous spacers implies a certain conservation of spacer length (Spearman's ρ = 0.7895; p < 0.0001), but B. pennsylvanicus IGSs are generally longer (average 290 bp) than those of B. floridanus (average 180 bp) (Wilcoxon Rank sum test, p < 0.0001). Homologous spacers were identified based on their positions in the genomes. IGSs that lacked a homolog were excluded. (B) B. pennsylvanicus IGSs (open circles) generally have lower %GC and shorter lengths than ORFs (filled squares). Horizontal lines mark the average %GC for ORFS (32.1%; solid line) and IGSs (20.0%; dashed line). Additionally the base composition of all homologous, nonzero IGSs were compared and show no strong correlation, yet cluster around ∼20% GC in both genomes (unpublished data).
Polymorphism within B. pennsylvanicus
Polymorphisms in the pooled symbiont population used for library construction were detectable as well-supported (Phred scores >40) discrepancies in the genome assembly that were represented by at least two independent clones. Nearly all (445/497) polymorphisms are single nucleotide polymorphisms (SNPs). Those located within ORFs occur primarily at third codon positions (Table 2). Although the majority of SNPs are located within ORFs, a disproportionate number of SNPs (35%) and indels (96% of insertions and/or deletions) occur within the intergenic regions (which comprise 23% of the B. pennsylvanicus genome). The two polymorphic indels within ORFs produce amino acid insertions/deletions in sucA and aroE.
Table 2.
Polymorphisms detected in the assembled B. pennsylvanicus genome
| Polymorphisms | 497 |
| Indels | 52 |
| Average size, bp | 1.58 |
| Intergenic regions | 50 |
| Pseudogenes | 0 |
| RNAs | 0 |
| ORFs | 2 |
| SNPs | 445 |
| Transitions | 331 |
| Transversions | 114 |
| Ratio | 2.9 |
| Intergenic Spacers | 156 |
| Pseudogenes | 5 |
| RNAs | 1 |
| ORFs | 283 |
| Synonymous | 176 |
| 1st | 7 |
| 2nd | 1 |
| 3rd | 168 |
| Nonsynonymous | 107 |
| 1st | 66 |
| 2nd | 37 |
| 3rd | 4 |
Nucleotide polymorphisms were recorded for all sites in the genome where high quality (>40 PHRED score) disagreements occurred, and where both variants were represented by multiple clones (at least two clones, and typically many more). The two indels within ORFs did not disrupt the reading frame, but rather resulted in amino acid insertions or deletions, that is, insertion/deletion of a leucine and lysine in sucA (sites 348-349 of the protein sequence) and the insertion/deletion of an alanine in aroE (between sites 126 and 127 of the protein sequence).
Comparison of protein divergences
Wide variation in protein divergence across loci indicates variable functional constraint across Blochmannia proteins (Fig. 6; Supplemental Tables S3 and S5). Amino acid biosynthetic genes, ribosomal proteins, and particular chaperonins are exceptionally conserved (a mean protein divergence of 0.448, with a 99% confidence interval of 0.400–0.504), while surface structures [mean of 0.725 (0.533–0.991)] and hypothetical or unclassified proteins [mean of 0.775 (0.667–0.898)] are quite divergent (Supplemental Table S5). The membrane proteins tolA and tonB are particularly divergent (divergences of 1.74 and 2.64, respectively) (Supplemental Table S3).
Figure 6.

Correlated protein divergences suggest parallel selective constraints in endosymbionts and free-living bacteria. Protein divergence in Blochmannia shows a strong correlation with divergence at orthologous genes of (A) Buchnera (–A. pisum versus –S. graminum) and (B) E. coli versus P. luminescens. Lines that best fit the data and intercept zero are shown, but significance was tested using the nonparametric test of association (JMP 4.0; SAS Institute Inc.). For all bacterial pairs, the most conserved genes include certain chaperonins and translation functions (mostly ribosomal proteins), while the most divergent genes include surface structures. Substitution rates above 2 are prone to saturation but are included here for comparison. Supplemental Table S3 lists all of the pairwise divergences.
Previous studies have demonstrated a negative relationship between GC content of endosymbiont genes and their level of divergence from free-living relatives (Herbeck et al. 2003; Banerjee et al. 2004), suggesting that amino acid changes in proteins under strong functional constraint are less severely affected by AT mutational bias. A strong negative association between GC content and protein divergence in Blochmannia (Supplemental Fig. S3) indicates this relationship also holds when protein divergences are estimated within a mutualist group (rather than to a more distant free-living relative). Protein divergences were, on average, ∼1.88 times faster in B. floridanus lineage compared to B. pennsylvanicus lineage. Genes with particularly elevated rates in B. floridanus include secG, rpsJ, rpsR, and rnpA, each of which evolves more than 10-fold faster in B. floridanus compared to B. pennsylvanicus (Fig. 7; Supplemental Table S4). A 14-fold rate acceleration at ispE may reflect the loss or disruption of other genes involved in isoprenoid biosynthesis in the B. floridanus genome and relaxed selection at this remaining isp gene.
Figure 7.

Accelerated rates of evolution in the lineage leading to B. floridanus compared to B. pennsylvanicus, since their divergence from a common ancestor. Rates of protein divergence were compared using a relative rates test with E. coli as an outgroup (see Methods). The analysis included 516 proteins for which RSD identified orthologs in the two genomes, and for which divergences between the two Blochmannia or between either endosymbiont and E. coli did not exceed 2.0. On average, proteins evolved 1.88 times faster in B. floridanus compared to in B. pennsylvanicus. Only 50 of the proteins tested evolved more slowly in B. pennsylvanicus than in B. floridanus or at the same rate, with values ≤1on the histogram, while 467 genes evolved faster in B. floridanus. Supplemental Table S4 lists proteins with particularly accelerated evolutionary rates in B. floridanus.
The B. pennsylvanicus genome offered the first opportunity to compare genome-wide patterns of protein evolution in the context of distinct endosymbiotic associations. A previous study showed accelerated evolution at 16S rDNA and at synonymous positions of select Blochmannia genes compared to enteric bacteria and even compared to the rapidly evolving Buchnera (Degnan et al. 2004). Here, we tested whether the Blochmannia genome also undergoes exceptionally fast rates of protein evolution. Strong correlations in divergences at homologous genes indicate parallel functional constraints in Blochmannia compared to Buchnera and E. coli–Photorhabdus luminescens (Fig. 6). However, notably higher protein divergences between Blochmannia spp., despite their relatively recent split, reflects a several-fold acceleration in absolute rates of protein evolution. Among the 302 genes for which RSD detected orthologs across Blochmannia and Buchnera and estimated divergences within each mutualist pair, the average substitution rate in Blochmannia was 0.0132 to 0.0166 amino acid substitutions/site per million years (based on a 20- to 16-Myr divergence, respectively). This is ∼10 times faster than rates in Buchnera for the same gene set (0.00258–0.00362 amino acid substitutions/site per million years, based on a 70–50-Myr divergence). Previous studies have shown that nonsynonymous sites in Buchnera evolve an average of twofold (Canbäck et al. 2004) and up to 10-fold (Clark et al. 1999) faster than in the enterics. Likewise, we found an average ∼50-fold rate acceleration in Blochmannia compared to E. coli–Salmonella typhimurium across the 538 genes for which RSD detected orthologs (0.0142–0.0177 in Blochmannia, and 0.000245–0.000367 for E. coli–S. typhimurium assuming a divergence time of 150–100 Myr). Deviations from this general trend highlight genes that may experience different levels of functional constraint among genomes. For example, relative to other genes in the genome, amino acid biosynthetic genes evolve slightly slower in Blochmannia than in the enterics, while genes involved in translation (comprised largely of ribosomal proteins) evolve slightly faster than expected (Fig. 6).
Comparisons of average protein divergences within and between major functional categories confirmed many of these observations above (Supplemental Table S5). Blochmannia, Buchnera, and E. coli–P. luminescens show similar relative divergences across most functional categories, such as relatively high divergences of unclassified and hypothetical genes, loci for surface structures, cell membrane components; moderate divergences of genes encoding cofactor biosynthesis, information transfer, cell processes, metabolism, information transfer; and relatively low divergences of genes for nucleotide biosynthesis and amino acid biosynthesis. Striking differences among pairs include relatively high divergence of chaperonins and fatty acid biosynthetic genes in Buchnera, the relative conservation of regulation genes in both endosymbionts, and the slightly higher divergence of translation genes in Blochmannia, as noted above.
Discussion
In contrast to many pathogenic and free-living bacterial species in which lateral gene transfer, chromosomal inversions, and translocations drive changes in gene order and content, we found complete conservation in gene order and orientation between two genomes of Blochmannia that diverged 16–20 Mya. This exceptional genome stability, first demonstrated in Buchnera (Tamas et al. 2002; van Ham et al. 2003), suggests that Blochmannia also lacks genetic machinery for gene inversions or translocation. Notably, like Buchnera, both Blochmannia strains lack RecA, numerous other recombination functions (RadA and Rec-FOR), phage, and have relatively low levels of repeated DNA.
The gene content of B. pennsylvanicus differs from B. floridanus only by 3.6% (24/659); however, genes specifically retained in B. pennsylvanicus span varied functional categories that may affect its metabolic capabilities and host interaction. The retention of genes for cell wall integrity, chaperonins, gene regulation, and DNA replication in B. pennsylvanicus may reflect different bacterial requirements for the maintenance of cell processes and structures within C. pennsylvanicus bacteriocytes. Furthermore, the ability to synthesize both coenzyme A and isoprenoids likely benefits B. pennsylvanicus and their ant hosts. Genome stasis implies gene losses in either Blochmannia lineage are irreversible, such that deletions of metabolic functions may constrain the evolutionary potential of this association. Such constraints have been proposed in Buchnera (Tamas et al. 2002), where the loss of genes for sulfur reduction and cysteine biosynthesis in Buchnera–SG may constrain the S. graminum host to its relatively cysteine-rich grass diet.
Certain aspects of Blochmannia metabolism remain unclear, owing to the uncertainty of whether or not single frameshifts in particular genes eliminate function. For example, if single indels within poly(A) tracts of hisH and ubiF are subject to correction of some type, the encoded proteins might be functional in B. pennsylvanicus. One possible mechanism for correction may be instability of such frameshifts during DNA replication, such that populations include heterogeneous genomes with different numbers of adenines in the homopolymeric repeats (e.g., Parkhill et al. 2000). However, among the many C. pennsylvanicus colonies used for symbiont library construction, we found no evidence for variation in lengths of these or any homopolymeric tracts. Alternatively, transcriptional slippage, or “stuttering” within repeat mononucleotides is well-documented in E. coli (Chamberlin and Berg 1962), where it often occurs within poly(A) or poly(T) tracts (Wagner et al. 1990). In a survey of published bacterial genomes, Baranov et al. (2005) identified several “pseudo pseudogenes,” for which transcriptional slippage could correct a frameshift within poly(A) or poly(T) tracts and restore uninterrupted ORFs. These authors describe a mechanism in which the RNA chain dissociates from the DNA template and reassociates in a new location. Third, functional proteins may be restored by frame-shifting during translation, or “recoding,” a phenomenon that occurs in yeast (Hansen et al. 2003), archea (Cobucci-Ponzano et al. 2005), and E. coli (Gurvich et al. 2003) and can also follow poly(A) tracts, especially in the form of A_AAA_AAG (Baranov et al. 2002, 2003). Such slippage has been proposed to explain aberrant indels in animal mitochondrial genes (Beckenbach et al. 2005), and in principle, might operate in endosymbionts. Given the multiple levels at which frameshifts within homopolymeric sequences may be corrected, we argue that these mutations should be interpreted cautiously, and with consideration of whether the gene otherwise encodes a full-length ORF. Although it is possible that such genes represent very recent loss of function, we take the approach of Baranov et al. (2005) in questioning whether such genes should be annotated as pseudogenes.
Conservation of genome architecture and overall similarity in gene content within Blochmannia contrasts with the exceptionally fast rates of sequence evolution observed in this group. Namely, protein divergences are higher for the two ant mutualists than within much older bacterial pairs. A previous study showed that amino acid changes were influenced by AT mutational pressure to a greater extent in basal lineages in Buchnera, suggesting endosymbiont proteins were more tolerant of these presumably deleterious changes early in the association (Clark et al. 1999). By the same token, rapid rates of protein evolution in Blochmannia might reflect the younger age of this association (∼30 Myr, compared to ∼150–200 Myr for Buchnera). Despite an overall rate acceleration in Blochmannia, the relative levels of divergence among proteins were strikingly similar to those of Buchnera and the enterobacteria. Although different types of selection shape free-living and endosymbiotic bacteria, this observed correlation suggests parallel functional constraints across many shared proteins.
A deviation from this pattern occurs in Blochmannia, where genes involved in translation are not the most conserved functional category relative to the enterics (Supplemental Table S5). We compared the ribosomal proteins that deviated from the best fit line in Figure 6 and those that showed a significant acceleration in the B. floridanus lineage (Fig. 7; Supplemental Table S4) to the detailed crystal structures of the 30S and 50S ribosomal subunits (Wimberly et al. 2000; Harms et al. 2001). These subunits assemble from 52 ribosomal proteins that form scaffolds around the 5S, 16S, and 23S ribosomal RNAs. Notably, many of the genes that show elevated substitution rates are positioned at the periphery of the ribosomal subunits, well away from the active regions at their interface. Furthermore, those proteins specifically involved the initiation and aggregation of the 30S and 50S ribosomes (RplL, RpsG) and those that have regulatory functions (RplADKT, RpsH), while exhibiting elevated rates were not among the genes with the highest divergences. These fast rates of substitution at ribosomal proteins might be influenced by several factors, including the effects of genome-wide patterns of nucleotide substitution; changes in regulatory interactions; compensatory changes due to the AT bias and consequently the secondary structure of the 16S, 23S, and 5S rRNAs; and the inactivation of the rpmD large ribosomal subunit locus. These amino acid substitutions in the ribosomal subunits might reduce the stability of the ribosome and thus decrease translation efficiency.
Within Blochmannia, faster divergence in the B. floridanus lineage at nearly all (∼90%) proteins may reflect elevated mutation rates, reduced selective coefficients, or smaller effective population size of this symbiont and/or its host with associated increased genetic drift. Although data to distinguish these alternatives are limited, an analysis of four gene regions (groEL, rpsB, atpB, and gidA, all of which evolve faster in the B. floridanus than B. pennsylvanicus lineage) showed no consistent increase in dN/dS in B. floridanus or its close relatives (Fry and Wernegreen 2005) that would be predicted under relaxed selection or drift hypotheses. Evidence supporting the mutation hypothesis includes a lower genomic GC content for B. floridanus than B. pennsylvanicus (Table 1). Because the two Blochmannia genomes have the same set of DNA repair genes, there is no a priori reason to propose that B. floridanus has a faster rate of mutations per replication. However, the year-round activity of C. floridanus and its relatives in the subgenus Myrmothrix contrasts with the winter dormancy of C. pennsylvanicus and related temperate species in the subgenus Camponotus. This activity may increase the number of host and bacterial generations per year and, consequently, the rate of mutations per unit time. Likewise, a combination of elevated mutation, relaxed selection, and/or increased genetic drift in the B. floridanus lineage may account for faster rates of gene loss compared to the B. pennsylvanicus lineage (see Lawrence and Roth [1999] for theoretical framework). However, given that current data cannot distinguish points along the B. floridanus lineage at which evolutionary rates accelerated or at which particular genes were lost, at this time we cannot link these changes to specific aspects of host ecology. More intensive sampling of Blochmannia across ecologically diverse hosts should allow such connections to be made.
Given the wide variation in Camponotus nutritional ecology, ranging from plant-specialists to omnivorous species, it seems unlikely that a single nutrient is lacking in the diet of all species that house Blochmannia. Rather than supplementing specific dietary deficiencies, nutritional functions of Blochmannia may play critical roles during two “starvation” phases of the host when metabolic demands exceed the available food supply—metamorphosis and colony founding (Wheeler and Martinez 1995). Recent work has shown that Blochmannia proliferate during pupation (Wolschin et al. 2004), a stage of metamorphosis when the host must construct all components of the adult body plan with no food intake (Wheeler and Martinez 1995). Genome sequence data provide a starting point for experimental analyses to clarify the functional significance of this mutualism, to explore the implications of genome variability on the physiology and ecology of both symbiotic partners, and to clarify the levels and timing of selection that shape this long-term bacterium–ant association.
Methods
Blochmannia genome sequencing and assembly
B. pennsylvanicus genomic DNA (gDNA) was prepared from worker and larvae C. pennsylvanicus collected from five colonies at two sites in Falmouth, Massachusetts, USA. The gDNA was either extracted directly from the agarose plugs containing the purified bacterial cells (Charles and Ishikawa 1999) or gelpurified from a chromosomal fragment resolved through Pulsed Field Gel Electrophoresis (PFGE) (Wernegreen et al. 2002). Short (1.5–2.5 kb) insert libraries were generated from hydrosheared DNA using a double adaptor kit (SeqWright Inc.) (Andersson et al. 1996). Plasmid clones were purified and bidirectionally sequenced using BigDye v3.0 chemistry on either an ABI3700 or an ABI3730xl (Applied Biosystems). Detailed methods for library construction and sequencing are provided in the Supplemental material.
Raw sequence data were analyzed by PHRED (Ewing and Green 1998; Ewing et al. 1998) (http://www.phrap.org/phredphrapconsed.html) and screened using BLASTN/X (Altschul et al. 1990) for ant host contamination. Sequence reads that were putatively identified as γ-Proteobacterial (E ≤ 1–10) were assembled using ARACHNE 2 (Jaffe et al. 2003) (http://www.genome.wi.mit.edu/wga). The resulting contigs were analyzed by hand in CONSED (Gordon et al. 1998) and using BAMBUS (Pop et al. 2004). The B. pennsylvanicus assembly was aligned using LAGAN (Brudno et al. 2003) (http://lagan.stanford.edu/lagan_web/index.shtml) to the published B. floridanus genome (NC005061), which facilitated primer design for gap closure by PCR.
Annotation and metabolic reconstruction
Open reading frames (ORFs) were identified iteratively using GLIMMER v2.10a (Delcher et al. 1999) (http://www.tigr.org/software/glimmer) and gene orthology predictions based on BLASTP sequence similarity to the NR, SWISS-PROT, and ECOLI databases; HMMR searches against Pfam_ls (Bateman et al. 2004); and identification of E. coli orthologs using the Reciprocal Sequence Distance (RSD) program (Wall et al. 2003) (details of the RSD method are noted below under “Comparison of protein divergences”). The few discrepancies among methods were limited to cases of differential loss of one gene from a pair of paralogs (ilvBG, tufAB, argFI), the presence of gene fusions (yidCD) or split genes (trpDG), or failure of RSD to identify a given ortholog because of high sequence divergence.
Three pseudogenes (uvrD, yqiC, rpmD) were detected as regions with similarity to functional ORFs in other genomes, but with multiple indels and missense mutations resulting in stop codons throughout each gene. The two most degraded pseudogenes (rpmD and yqiC) were undetectable by BLASTX and were only identified because of the conservation of gene order between the two Blochmannia genomes. Among the three pseudogenes, uvrD retains the longest (107 amino acids) intact reading frame with similarity to functional orthologs, but even this region is just 15% of the length of UvrD in E. coli and other outgroups. In this sense, annotated pseudogenes clearly differ from truncated B. pennsylvanicus ORFs, which retain at least 60% of the length of orthologous proteins. The three B. pennsylvanicus pseudogenes also differ from genes with single frameshifts within homopolymeric regions (hisH, ubiF, ytfM, ybiS), since the latter would encode intact, relatively conserved proteins if the frame-shift were corrected by slippage during transcription or translation (see Fig. 3, Results, and Discussion).
ORFS and RNAs were manually curated using a Generic Model Organism Database (GMOD) Web browser. ORFs that lacked sequence similarity to any entry in GenBank or the Comprehensive Microbial Resource (Peterson et al. 2001) and lacked any predicted protein domains in Pfam_ls were excluded from the annotation. Functional and pseudo-transfer RNAs (tRNAs) were identified using tRNAscan-SE (http://selab.wustl.edu/cgibin/selab.pl?mode=software), and ribosomal RNAs and structural RNAs were identified by BLASTN searches of the intergenic regions. Blochmannia gene functions and interactions were inferred from orthologs of E. coli K12 MG1655 described in GenProtEC (Serres et al. 2004) (http://genprotec.mbl.edu) and characterized by MultiFun (Serres and Riley 2000), two resources that represent functions of ∼80% of the 4401 genes in E. coli K12. Metabolic pathways were evaluated using the reference pathways available for E. coli at EcoCyc (Karp et al. 2004) and KEGG (Kanehisa and Goto 2000). Genomes of other insect mutualists were reanalyzed in the same manner for a consistent metabolic comparison.
Comparison of protein divergences
The RSD algorithm (Wall et al. 2003) was used to identify the reciprocal best BLAST hits (rbh) between translated ORFs of select bacterial genomes. The program used BLAST to identify potential matches of a given translated gene, aligned all potential matches using CLUSTALW (Thompson et al. 1994), and calculated a maximum likelihood estimation of amino acid substitutions between proteins using PAML (Yang 1997). Protein divergences were based on an empirical amino acid substitution rate matrix (Jones et al. 1992) and accounted for variation in evolutionary rates among protein sites using a γ distribution with shape parameter α = 1.53 (as recommended by Nei et al. 2001). The protein with the lowest divergence was then BLASTed against the first genome, followed by the alignment and divergence calculations. If the protein match with the lowest divergence was the same as the original query sequence, the pair was considered orthologous and the divergence was retained in the output.
Such comparisons were performed within endosymbiont groups: B. pennsylvanicus versus B. floridanus; Buchnera–A. pisum versus Buchnera–S. graminum; and Buchnera–A. pisum versus Buchnera–Baizongia pistaciae. Divergences in endosymbionts were compared to the enterobacterial pairs E. coli versus S. typhimurium, and E. coli versus P. luminescens. Genomes were downloaded from NCBI in June 2004. All genomes were compared to E. coli using RSD for ortholog detection and MultiFun-based functional assignments (detailed in Supplemental material). Rates of protein divergence along the lineages leading to B. pennsylvanicus and B. floridanus were compared using E. coli as an outgroup. Relative rates were calculated as follows, with “B0” representing the common ancestor of the two Blochmannia lineages:
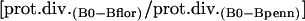 |
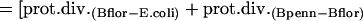 |
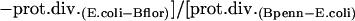 |
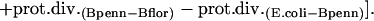 |
Pairwise dN and dS values reported for certain genes [e.g., truncated genes or ORFs with apparent frameshifts within poly(A) tracts] were calculated using PAML (runmode –2) (Yang 1997). In order to account for the extreme base composition of endosymbiont sequences, we implemented a maximum likelihood model in which codon frequencies were calculated from the average nucleotide frequencies at the three codon positions (CodonFreq = 2). In these cases, translated Blochmannia genes were aligned to each other and to appropriate outgroups using CLUSTALX (Thompson et al. 1997) and back-translated to nucleotide sequences using the program RevTrans (Wernersson and Pedersen 2003).
Acknowledgments
We thank Andrew McArthur, Margrethe Serres, Seth Bordenstein, Seth Kauppinen, and four anonymous reviewers for comments on the manuscript, and are grateful to Daniel Hahn, Diana Wheeler, Stefan Cover, and Diana Davidson for helpful discussion of Camponotus biology. Amy McCurley and Josh Larson contributed to the library construction, and Jeremy Brozek confirmed the B. pennsylvanicus hisH sequence. We also thank S. Kauppinen for assistance in aligning and analyzing pseudogene sequences. Andrew McArthur, Margrethe Serres, Michael Cipriano, Sulip Goswami, Hilary Morrison, and Matt Beverly provided intellectual and technical support with genome assembly and annotation. Alessandro Romualdi provided software and support for the JENA Prokaryotic Genome Viewer, Dennis P. Wall provided access to RSD, and D. Wall and Richard Fox offered assistance implementing this program. We thank Brian Dale for permission to collect C. pennsylvanicus for this study. This research was supported by grants to J.J.W. from NIH (R01 GM62626-01) and the NSF (DEB 0089455). Additional support was provided by the NASA Astrobiology Institute (NCC2-1054 and NNA04CC04A). Template preparation and DNA sequencing was performed in the W.M. Keck Facility for Ecological Genomics at the Marine Biological Laboratory.
[Supplemental material is available online at www.genome.org. The complete, annotated genome sequence has been submitted to GenBank under accession no. CP000016.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3771305.
References
- Akman, L., Yamashita, A., Watanabe, H., Oshima, K., Shiba, T., Hattori, M., and Aksoy, S. 2002. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat. Genet. 32: 402–407. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, W.E., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Andersson, B., Wentland, M.A., Ricafrente, J.Y., Liu, W., and Gibbs, R.A. 1996. A “double adaptor” method for improved shotgun library construction. Anal. Biochem. 236: 107–113. [DOI] [PubMed] [Google Scholar]
- Banerjee, T., Basak, S., Gupta, S.K., and Ghosh, T.C. 2004. Evolutionary forces in shaping the codon and amino acid usages in Blochmannia floridanus. J. Biomol. Struct. Dyn. 22: 13–24. [DOI] [PubMed] [Google Scholar]
- Baranov, P.V., Gesteland, R.F., and Atkins, J.F. 2002. Recoding: Translational bifurcations in gene expression. Gene 286: 187–201. [DOI] [PubMed] [Google Scholar]
- Baranov, P.V., Gurvich, O.L., Hammer, A.W., Gesteland, R.F., and Atkins, J.F. 2003. Recode 2003. Nucleic Acids Res. 31: 87–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranov, P.V., Hammer, A.W., Zhou, J., Gesteland, R.F., and Atkins, J.F. 2005. Transcriptional slippage in bacteria: Distribution in sequenced genomes and utilization in IS element gene expression. Genome Biol. 6: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E.L., et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32: D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann, P., Moran, N.A., and Baumann, L. 2000. Bacteriocyte-associated endosymbionts of insects. In The prokaryotes, a handbook on the biology of bacteria; ecophysiology, isolation, identification, applications (ed. M. Dworkin). Springer-Verlag, New York, http://141.150.157.117:8080/prokPUB/index.htm.
- Beckenbach, A.T., Robson, S.K.A., and Crozier, R.H. 2005. Single nucleotide +1 frameshifts in an apparently functional mitochondrial cytochrome b gene in ants of the genus Polyrhachis. J. Mol. Evol. 60: 141–152. [DOI] [PubMed] [Google Scholar]
- Bolton, B. 1995. Identification guide to the ant genera of the world. Harvard University Press, Cambridge, MA.
- Brudno, M., Do, C.B., Cooper, G.M., Kim, M.F., Davydov, E., Green, E.D., Sidow, A., and Batzoglou, S. 2003. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13: 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canbäck, B., Tamas, I., and Andersson, S.G. 2004. A phylogenomic study of endosymbiotic bacteria. Mol. Biol. Evol. 21: 1110–1122. [DOI] [PubMed] [Google Scholar]
- Chamberlin, M. and Berg, P. 1962. Deoxyribonucleic acid-directed synthesis of ribonucleic acid by an enzyme from Escherichia coli. Proc. Natl. Acad. Sci. 48: 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles, H. and Ishikawa, H. 1999. Physical and genetic map of the genome of Buchnera, the primary endosymbiont of the pea aphid Acyrthosiphon pisum. J. Mol. Evol. 48: 142–150. [DOI] [PubMed] [Google Scholar]
- Clark, M.A., Moran, N.A., and Baumann, P. 1999. Sequence evolution in bacterial endosymbionts having extreme base compositions. Mol. Biol. Evol. 16: 1586–1598. [DOI] [PubMed] [Google Scholar]
- Cobucci-Ponzano, B., Rossi, M., and Moracci, M. 2005. Recoding in archaea. Mol. Microbiol. 55: 339–348. [DOI] [PubMed] [Google Scholar]
- Currie, C.R. 2001. A community of ants, fungi, and bacteria: A multilateral approach to studying symbiosis. Annu. Rev. Microbiol. 55: 357–380. [DOI] [PubMed] [Google Scholar]
- Dasch, G.A. 1975. Morphological and molecular studies on intracellular bacterial symbiotes of insects, pp. 329. Yale University, New Haven, CT.
- Dasch, G., Weiss, E., and Chang, K. 1984. Endosymbionts of insects. In Bergy's manual of systematic bacteriology (eds. J. Holt and N. Krieg), pp. 811–833. Williams & Williams, Baltimore.
- Davidson, D. 1997. The role of resource imbalances in the evolutionary ecology of tropical arboreal ants. Biol. J. Linn. Soc. Lond. 61: 153–181. [Google Scholar]
- ———. 1998. Resource discovery versus resource domination in ants: A functional mechanism for breaking the trade-off. Ecol. Entomol. 23: 484–490. [Google Scholar]
- Degnan, P.H., Lazarus, A.B., Brock, C., and Wernegreen, J.J. 2004. Host–symbiont stability and fast evolutionary rates in an ant–bacterium association: Cospeciation of Camponotus species and their endosymbionts, Candidatus Blochmannia. Syst. Biol. 53: 95–110. [DOI] [PubMed] [Google Scholar]
- Delcher, A.L., Harmon, D., Kasif, S., White, O., and Salzberg, S.L. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27: 4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ghachi, M., Bouhss, A., Blanot, D., and Mengin-Lecreulx, D. 2004. The bacA gene of Escherichia coli encodes an undecaprenyl pyrophosphate phosphatase activity. J. Biol. Chem. 279: 30106–30113. [DOI] [PubMed] [Google Scholar]
- Ewing, B. and Green, P. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8: 186–194. [PubMed] [Google Scholar]
- Ewing, B., Hillier, L., Wendl, M.C., and Green, P. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8: 175–185. [DOI] [PubMed] [Google Scholar]
- Fry, A.J. and Wernegreen, J.J. 2005. The roles of positive and negative selection in the molecular evolution of insect endosymbionts. Gene (in press). [DOI] [PubMed]
- Gao, D. and McHenry, C.S. 2001. τ binds and organizes Escherichia coli replication proteins through distinct domains. Domain IV, located within the unique C terminus of τ, binds the replication fork, helicase, DnaB. J. Biol. Chem. 276: 4441–4446. [DOI] [PubMed] [Google Scholar]
- Gil, R., Silva, F.J., Zientz, E., Delmotte, F., González-Candelas, F., Latorre, A., Rausell, C., Kamerbeek, J., Gadau, J., Hölldobler, B., et al. 2003. The genome sequence of Blochmannia floridanus: Comparative analysis of reduced genomes. Proc. Natl. Acad. Sci. 100: 9388–9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, D., Abajian, C., and Green, P. 1998. Consed: A graphical tool for sequence finishing. Genome Res. 8: 195–202. [DOI] [PubMed] [Google Scholar]
- Gurvich, O.L., Baranov, P.V., Zhou, J., Hammer, A.W., Gesteland, R.F.M., and Atkins, J.F. 2003. Sequences that direct significant levels of frameshifting are frequent in coding regions of Escherichia coli. EMBO J. 22: 5941–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, T.M., Baranov, P.V., Ivanov, I.P., Gesteland, R.F., and Atkins, J.F. 2003. Maintenance of the correct open reading frame by the ribosome. EMBO Rep. 4: 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms, J., Schluenzen, F., Zarivach, R., Bashan, A., Gat, S., Agmon, I., Bartels, H., Franceschi, F., and Yonath, A. 2001. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell 107: 679–688. [DOI] [PubMed] [Google Scholar]
- Herbeck, J.T., Wall, D.P., and Wernegreen, J.J. 2003. Gene expression level influences amino acid usage, but not codon usage, in the tsetse fly endosymbiont Wigglesworthia. Microbiology 149: 2585–2596. [DOI] [PubMed] [Google Scholar]
- Hölldobler, B. and Wilson, E.O. 1990. The ants. Belknap Press of Harvard University Press, Cambridge, MA.
- Jaffe, D.B., Butler, J., Gnerre, S., Mauceli, E., Lindblad-Toh, K., Mesirov, J.P., Zody, M.C., and Lander, E.S. 2003. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 13: 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thorton, J.M. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8: 275–282. [DOI] [PubMed] [Google Scholar]
- Kainou, T., Okada, K., Suzuki, K., Nakagawa, T., Matsuda, H., and Kawamukai, M. 2001. Dimer formation of octaprenyl-diphosphate synthase (IspB) is essential for chain length determination of ubiquinone. J. Biol. Chem. 276: 7876–7883. [DOI] [PubMed] [Google Scholar]
- Kanehisa, M. and Goto, S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp, P.D., Arnaud, M., Collado-Vides, J., Ingraham, J., Paulsen, I.T., and Saier, M.H.J. 2004. The E. coli EcoCyc database: No longer just a metabolic pathway database. ASM News 70: 25–30. [Google Scholar]
- Lawrence, J. and Roth, J. 1999. Genomic flux: Genome evolution by gene loss and acquisition. In Organization of the prokaryotic genome (ed. R. Charlesbois), pp. 263–289. ASM Press, Washington, DC.
- Lerat, E. and Ochman, H. 2004. Psi–Phi: Exploring the outer limits of bacterial pseudogenes. Genome Res. 14: 2273–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira, A., Ochman, H., and Moran, N.A. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17: 589–596. [DOI] [PubMed] [Google Scholar]
- Moran, N.A. and Plague, G.R. 2004. Genomic changes following host restriction in bacteria. Curr. Opin. Genet. Dev. 14: 627–633. [DOI] [PubMed] [Google Scholar]
- Mushegian, A.R. and Koonin, E.V. 1996. Sequence analysis of eukaryotic developmental proteins: Ancient and novel domains. Genetics 144: 817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., Xu., P., and Galzko, G. 2001. Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. Proc. Natl. Acad. Sci. 98: 2497–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura, K., Koyama, T., and Sagami, H. 1997. Polyprenyl diphosphate synthases. Subcell. Biochem. 28: 57–87. [DOI] [PubMed] [Google Scholar]
- Park, J.T. 1996. The murein sacculus. In Escherichia coli and Salmonella typhimurium: Cellular and molecular biology (eds. F.C. Neidhardt et al.), pp. 48–57. American Society for Microbiology, Washington, DC.
- Parkhill, J., Wren, B.W., Mungall, K., Ketley, J.M., Churcher, C., Basham, D., Chillingworth, T., Davies, R.M., Feltwell, T., Holroyd, S., et al. 2000. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403: 665–668. [DOI] [PubMed] [Google Scholar]
- Peterson, J.D., Umayam, L.A., Dickinson, T., Hickey, E.K., and White, O. 2001. The Comprehensive Microbial Resource. Nucleic Acids Res. 29: 123–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop, M., Kosack, D.S., and Salzberg, S.L. 2004. Hierarchical scaffolding with Bambus. Genome Res. 14: 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugsley, A.P. 1993. The complete general secretory pathway in Gram-negative bacteria. Microbiol. Rev. 57: 50–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sameshima, S., Hasegawa, E., Kitade, O., Minaka, N., and Matsumoto, T. 1999. Phylogenetic comparison of endosymbionts with their host ants based on molecular evidence. Zoolog. Sci. 16: 993–1000. [Google Scholar]
- Sauer, C., Stackebrandt, E., Gadau, J., Hölldobler, B., and Gross, R. 2000. Systematic relationships and cospeciation of bacterial endosymbionts and their carpenter ant host species: Proposal of the new taxon Candidatus Blochmannia gen. nov. Int. J. Syst. Evol. Microbiol. 50 Pt 5: 1877–1886. [DOI] [PubMed] [Google Scholar]
- Sauer, C., Dudaczek, D., Hölldobler, B., and Gross, R. 2002. Tissue Localization of the endosymbiotic bacterium “Candidatus Blochmannia floridanus” in adults and larvae of the carpenter ant Camponotus floridanus. Appl. Environ. Microbiol. 68: 4187–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, D., Deppisch, H., Obermayer, M., Krohne, G., Stackebrandt, E., Hölldobler, B., Goebel, W., and Gross, R. 1996. Intracellular endosymbiotic bacteria of Camponotus species (carpenter ants): Systematics, evolution and ultrastructural characterization. Mol. Microbiol. 21: 479–489. [DOI] [PubMed] [Google Scholar]
- Serres, M.H. and Riley, M. 2000. MultiFun, a multifunctional classification scheme for Escherichia coli K-12 gene products. Microb. Comp. Genomics 5: 205–222. [DOI] [PubMed] [Google Scholar]
- Serres, M.H., Goswami, S., and Riley, M. 2004. GenProtEC: An updated and improved analysis of functions of Escherichia coli K-12 proteins. Nucleic Acids Res. 32: D300–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigenobu, S., Watanabe, H., Hattori, M., Sakaki, Y., and Ishikawa, H. 2000. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407: 81–86. [DOI] [PubMed] [Google Scholar]
- Tamas, I., Klasson, L., Canbäck, B., Naslund, A.K., Eriksson, A.S., Wernegreen, J.J., Sandström, J.P., Moran, N.A., and Andersson, S.G. 2002. 50 million years of genomic stasis in endosymbiotic bacteria. Science 296: 2376–2379. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin. F., and Higgins, D.G. 1997. The CLUSTAL X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham, R.C., Kamerbeek, J., Palacios, C., Rausell, C., Abascal, F., Bastolla, U., Fernández, J.M., Jiménez, L., Postigo, M., Silva, F.J., et al. 2003. Reductive genome evolution in Buchnera aphidicola. Proc. Natl. Acad. Sci. 100: 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, L.A., Weiss, R.B., Driscoll, R., Dunn, D.S., and Gesteland, R.F. 1990. Transcriptional slippage occurs during elongation at runs of adenine or thymine in Escherichia coli. Nucleic Acids Res. 18: 3529–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, J.R., Hervas, C., Ross, J.D., Blinkova, A., Walbridge, M.J., Pumarega, E.J., Park, M.O., and Neely, H.R. 2000. Escherichia coli DNA polymerase III τ- and γ-subunit conserved residues required for activity in vivo and in vitro. J. Bacteriol. 182: 6106–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, D.P., Fraser, H.B., and Hirsh, A.E. 2003. Detecting putative orthologs. Bioinformatics 19: 1710–1711. [DOI] [PubMed] [Google Scholar]
- Wernegreen, J.J., Lazarus, A.B., and Degnan, P.H. 2002. Small genome of Candidatus Blochmannia, the bacterial endosymbiont of Camponotus, implies irreversible specialization to an intracellular lifestyle. Microbiology 148: 2551–2556. [DOI] [PubMed] [Google Scholar]
- Wernersson, R. and Pedersen, A.G. 2003. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 31: 3537–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler, D.E. and Martinez, T. 1995. Storage proteins in ants (Hymenoptera: Formicidae). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 112: 15–19. [DOI] [PubMed] [Google Scholar]
- Wimberly, B.T., Brodersen, D.E., Clemons Jr., W.M., Morgan-Warren, R.J., Carter, A.P., Vonrhein, C., Hartsch, T., and Ramakrishnan, V. 2000. Structure of the 30S ribosomal subunit. Nature 407: 327–339. [DOI] [PubMed] [Google Scholar]
- Wolschin, F., Hölldobler, B., Gross, R., and Zientz, E. 2004. Replication of the endosymbiotic bacterium Blochmannia floridanus is correlated with the developmental and reproductive stages of its ant host. Appl. Environ. Microbiol. 70: 4096–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. 1997. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]
- Zientz, E., Silva, F.J., and Gross, R. 2001. Genome interdependence in insect–bacterium symbioses. Genome Biol. 2: reviews1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Web site references
- http://ecocyc.org; Encyclopedia of Escherichia coli K12 Genes and Metabolism.
- http://genprotec.mbl.edu; GenProtEC E. coli genome and proteome database.
- http://lagan.stanford.edu/lagan_web/index.shtml; LAGAN.
- http://selab.wustl.edu/cgi-bin/selab.pl?mode=software; tRNAscan-SE.
- http://www.genome.wi.mit.edu/wga; ARACHNE 2.
- http://www.ncbi.nlm.nih.gov; GenBank, NCBI.
- http://www.phrap.org/phredphrapconsed.html; PHRED, CONSED.
- http://www.tigr.org/software/glimmer; GLIMMER.
- http://www.tigr.org/tigr-scripts/CMR2/CMRHomePage.spl; The Institute for Genomic Research: Comprehensive Microbial Resource (CMR) database.