

Abstract
Targeted gene replacement (TGR) in yeast and mammalian cells is initiated by the two free ends of the linear targeting molecule, which invade their respective homologous sequences in the chromosome, leading to replacement of the targeted locus with a selectable gene from the targeting DNA. To study the postinvasion steps in recombination, we examined the effects of DNA structure-specific proteins on TGR frequency and heteroduplex DNA formation. In strains deleted of RAD1, MSH2, or MSH3, we find that the frequency of TGR is reduced and the mechanism of TGR is altered while the reverse is true for deletion of SGS1, suggesting that Rad1 and Msh2:Msh3 facilitate TGR while Sgs1 opposes it. The altered mechanism of TGR in the absence of Msh2:Msh3 and Rad1 reveals a separate role for these proteins in suppressing an alternate gene replacement pathway in which incorporation of both homology regions from a single strand of targeting DNA into heteroduplex with the targeted locus creates a mismatch between the selectable gene on the targeting DNA and the targeted gene in the chromosome.
Keywords: gene targeting, homologous recombination, MSH2, RAD1, SGS1
Introduction
Targeted gene replacement (TGR) has been a fundamental technique in molecular biology for more than 20 years, but little is known about the mechanism by which a targeted sequence in the genome is replaced with a selectable gene from the linear targeting DNA. Without flanking homology sequences to direct it, the marker integrates randomly into the genome, so TGR is clearly homology-driven, but its relationship to other forms of homologous recombination (HR) is unclear. TGR occurs with high efficiency in Saccharomyces cerevisiae, even with very short flanking homologies, but in mammalian cells the efficiency is much lower. Because of the importance of this technique for genome manipulation and its potential use in gene therapy, understanding the mechanisms and limiting steps may result in new methods to improve the efficiency of TGR in mammalian cells.
Repair of DNA double-strand breaks (DSBs) by HR is a high-fidelity process that utilizes an undamaged homologous template to restore missing sequences at the break (Symington, 2002). By contrast, TGR can be considered a corruption of the HR process because recombination leads to permanent loss of the targeted chromosomal region (Rothstein, 1983) rather than restoration of a damaged sequence. In this respect, TGR is similar to another mutagenic HR process, the single-strand annealing (SSA) pathway of recombination between direct repeats (Lin et al, 1984; Ozenberger and Roeder, 1991; Fishman-Lobell et al, 1992). TGR and SSA both involve recombination using interrupted homology sequences and thus share unique genetic dependencies not found in other HR processes. Both pathways require the Msh2:Msh3 mismatch repair complex (Saparbaev et al, 1996) and the heterodimeric Rad1:Rad10 endonuclease (Schiestl and Prakash, 1988, 1990) to process intermediates leading to efficient recombination, which is generally not the case for template-directed repair of DSBs. Msh2:Msh3 and Rad1:Rad10 are also involved in repair of large insertion/deletion (loop) mismatches that can form between heteroalleles during meiotic recombination (Kirkpatrick and Petes, 1997; Kearney et al, 2001). Additionally, Msh2 plays a role in mitotic large loop repair (Clikeman et al, 2001), but the remaining factors have yet to be tested in the same assay system. Msh6, a mismatch repair protein specific for base–base and small insertion/deletion mispairs, is not required for SSA (Sugawara et al, 1997) or meiotic large-loop repair (Kearney et al, 2001), further supporting the idea that the role of mismatch repair factors in SSA is specific for branched DNA structures.
In yeast, SSA, TGR, and template-directed repair of DSBs are all strongly dependent on RAD52 (Symington, 2002), which encodes the founding member of a superfamily of single-strand DNA (ssDNA) binding proteins (Iyer et al, 2002). TGR is most efficient when at least one of the 3′ ssDNA ends of the targeting fragment invades a homologous chromosomal duplex in a reaction mediated by the Rad51 strand exchange protein and its accessory factors Rad55/57 (Schiestl et al, 1994) and Rad54 (Schmuckli-Maurer et al, 2003), but unlike Rad52, none of these proteins is absolutely required for TGR in yeast.
It has been firmly established both in cultured mammalian cells and in yeast that TGR is initiated by separate strand invasions at the two homology ends of the linear targeting molecule accompanied by formation of extensive heteroduplex DNA (hDNA) with the targeted chromosomal locus (Li et al, 2001; Langston and Symington, 2004). The steps that follow strand invasion are less clear, but branched DNA intermediates are likely to be involved because mutant cell lines deficient for proteins that act on these structures show altered frequencies of TGR. A role for RecQ helicases in suppressing TGR has been established in human cells and cultured chicken B cells, both of which have multiple RecQ homologs including three—BLM, WRN, and RECQL4—that are associated with human disease when mutated (Ellis et al, 1995; Yu et al, 1996; Kitao et al, 1999). Inactivation of BLM in a human colorectal cancer cell line increased the frequency of TGR approximately 20-fold at two different loci (Traverso et al, 2003), while disruption of BLM in the chicken DT40 cell line increased the frequency of TGR three- to 10-fold (Wang et al, 2000; Imamura et al, 2002). The human Blm protein binds to and migrates synthetic branched recombination intermediates in vitro, including Holliday junctions (Karow et al, 2000) and D-loops (van Brabant et al, 2000), so it might suppress TGR by unwinding similar intermediates.
The involvement of the structure-specific endonuclease Rad1:Rad10 suggests the formation of branched intermediates during TGR analogous to those found in SSA. The requirement for the mammalian Rad10 homolog, encoded by ERCC1, in TGR is absolute (Niedernhofer et al, 2001), while the requirement for Rad1:Rad10 in yeast is highly variable, depending on the targeted locus and perhaps on the targeting DNA itself. Separate studies have reported TGR defects in yeast rad1 and rad10 mutants ranging from a three- to four-fold reduction (Schiestl et al, 1994; Symington et al, 2000) to a 40-fold reduction (Schiestl and Prakash, 1988, 1990; Saparbaev et al, 1996). The less stringent requirement for Rad1:Rad10, along with the overall higher efficiency of gene targeting in yeast suggests the possibility of a fundamental difference between yeast and mammalian cells in the mechanism of TGR. To investigate this possibility, and to study the role of DNA structure-specific factors in the postinvasion steps of TGR in more detail, we looked for changes in gene targeting frequency and hDNA formation during TGR in strains of S. cerevisiae deleted of the SGS1, RAD1, MSH2, MSH3, and MSH6 genes.
Results
Experimental system
As previously described, we used restriction site polymorphisms in the two homology regions flanking a selectable marker to monitor formation of heteroduplex between corresponding regions of the targeting DNA and the targeted chromosomal locus during TGR (Langston and Symington, 2004) (Figure 1). The restriction enzyme recognition sites were incorporated into 29 bp palindromic inserts that avoid mismatch repair by forming stable, internally complementary loops when paired with DNA lacking the insertion (Nag et al, 1989). Genomic DNA was prepared from isolated single colonies identified as transformants resulting from TGR. Each flanking homology region was analyzed by RFLP analysis after PCR amplification to determine if it contained the restriction enzyme recognition site from the targeting DNA, the chromosomal DNA, or both, the latter indicating a sectored colony resulting from unrepaired hDNA (see Materials and methods for details). Transformants that exhibited hDNA in both flanking homology regions were analyzed further to determine if the same (cis; Figure 2, right) or different (trans; Figure 2, left) strands of targeting DNA at each end interacted with the chromosomal DNA.
Figure 1.
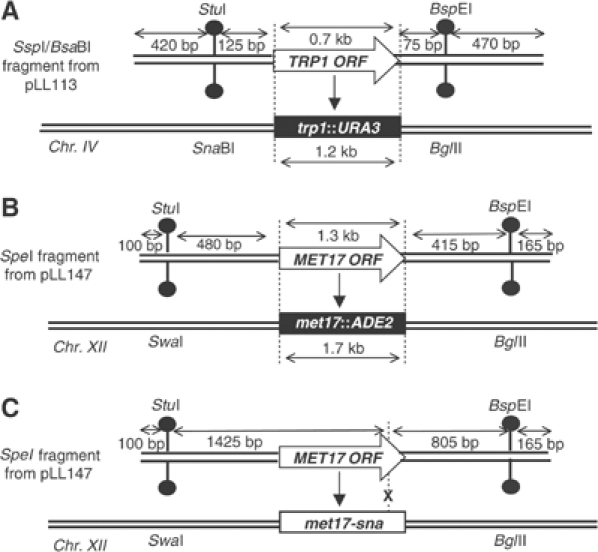
Schematics of targeting DNA and targeted yeast strains. Open arrows show the selectable markers, TRP1 or MET17, and rectangles show the corresponding disrupted allele in the chromosome. (A) Targeting at the TRP1 locus. (B) Targeting at the MET17 locus of a complete disruption or (C) a point mutation. Restriction site polymorphisms in the flanking homology regions are indicated. This figure is adapted from Figure 2 of Langston and Symington (2004).
Figure 2.
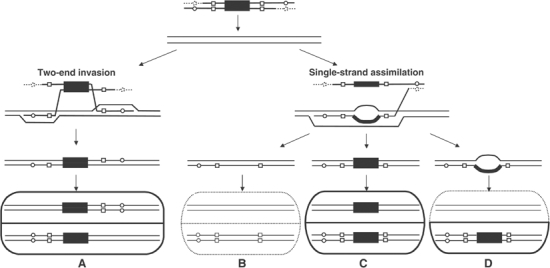
Models of targeted gene replacement. The large black box represents the selectable marker on the targeting DNA; small open boxes and open circles indicate different relative positions of palindromic restriction site inserts in the flanking homology regions of the TRP1 and MET17 targeting DNA, respectively; dotted lines indicate possible nuclease resection of the 5′ ends of the targeting DNA. Uninterrupted lines indicate the targeted chromosomal locus. Bisected ovals at bottom show the different genotypes of the two daughter cells arising from TGR as shown. As indicated by the thick versus dotted lines, only daughter cells containing the targeting marker survive selection to form all or part of a colony. (A) TGR with flanking markers in trans. (B) No TGR. The large bubble mismatch between the selectable marker on the targeting DNA and the targeted chromosomal locus is unwound, or is repaired in favor of chromosomal sequences. (C) TGR with flanking markers in cis. The bubble mismatch is repaired in favor of the targeting DNA. (D) TGR with no hDNA observed. The bubble mismatch is not repaired, and all information from the top, chromosomal strand is lost under selection for the targeting DNA marker on the bottom strand.
We previously examined hDNA formation in wild-type cells during targeted replacement of a chromosomal trp1∷URA3 site by TRP1 (Figure 1A) and a chromosomal met17∷ADE2 site (where ADE2 is under control of the MET17 promoter) with MET17 (Figure 1B). Of the 44 products of TGR that had hDNA in both flanking homology regions, 43 were in the trans configuration (diagrammed in Figure 2A), consistent with a model in which a different strand of targeting DNA at each end invades the chromosome as the dominant mechanism for TGR initiation in wild-type cells (Langston and Symington, 2004).
The mechanism and frequency of TGR are conspicuously altered in the absence of the MSH2 and RAD1 genes
One of the assumptions underlying our experimental system is that palindromic sequence polymorphisms in the flanking homology regions of the targeting DNA escape mismatch repair when hDNA is formed with the native chromosomal DNA. To confirm the validity of this assumption, we analyzed hDNA formation during TGR in strains deleted of MSH2, a homolog of Escherichia coli MutS and a key component of the yeast mismatch repair machinery (Reenan and Kolodner, 1992). Consistent with previous studies (Saparbaev et al, 1996), the frequency of TGR was reduced at both MET17 (decreased four-fold, P=0.01; Figure 3A) and TRP1 (decreased three-fold, P=0.004; Figure 3B) in the msh2 strain. If hDNA in the flanking homology regions was undergoing mismatch repair, we would have expected to see an increased frequency of hDNA in msh2 mutants. To our surprise, we found that the proportion of hDNA was reduced in the msh2 mutant at both MET17 (44/95, or 46% in msh2 versus 65/94, or 69% for the isogenic MSH2 strain, P=0.002; Figure 4A) and TRP1 (43/94, or 46% in msh2 versus 70/103, or 68% for MSH2, P=0.002; Figure 4B). More importantly, we observed that hDNA was altered significantly in the configuration of flanking markers, with eight out of 12 transformants (six of seven at TRP1 and two of five at MET17) with the cis configuration in msh2 versus one cis out of a total of 18 in the MSH2 controls (P=0.0006; Figure 4, bottom). The reduced frequency of TGR and the considerable reduction in the number of transformants with hDNA in the trans configuration in the absence of Msh2 suggests that this protein is important for facilitating the type of TGR found in wild-type cells. The shift to the cis configuration of flanking markers suggests that Msh2 also plays a role in restricting an alternate mechanism of TGR.
Figure 3.
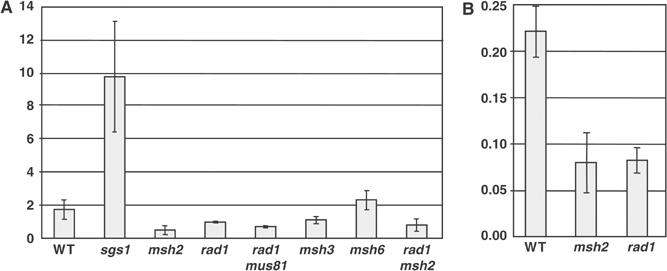
Frequency of TGR. Number of TGR events per 100 ng of targeting DNA compared to the number of transformants per 1 ng of uncut control plasmid DNA. (A) Targeting of met17∷ADE2 in the indicated genetic background. (B) Targeting of trp1∷URA3. Shown are the means and standard deviations of at least three experiments except for rad1 mus81, which is from two experiments.
Figure 4.
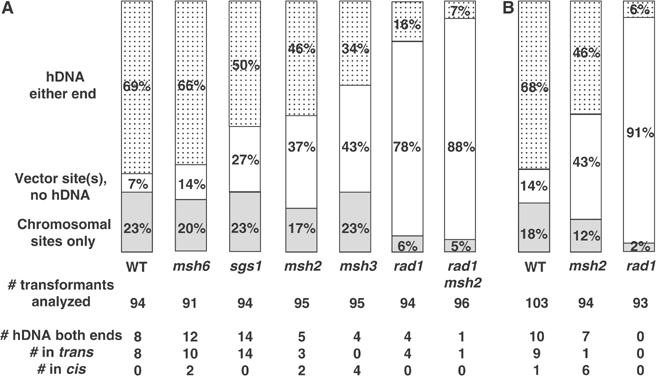
hDNA formation during TGR. (Top) For each strain, the percentage of colonies analyzed that had both vector and chromosomal restriction sites at one or both flanking markers is indicated by the dotted bars, those with vector restriction sites only at one or both flanking markers by open bars, and those with only chromosomal restriction sites at both flanking markers by gray bars. The number of colonies analyzed for each strain is indicated below the stacked bars. (Bottom) For each strain, the number of colonies that had both vector and chromosomal restriction sites at both flanking markers is indicated. Subclone analysis determined how many of these were in the trans or cis configurations. (A) Targeting of met17∷ADE2 in the indicated genetic background. (B) Targeting of trp1∷URA3. Data for wild type in both (A) and (B) are adapted from Langston and Symington (2004).
The structure-specific endonuclease encoded by the ERCC1 and XPF genes is indispensable for TGR in mammalian cells (Niedernhofer et al, 2001). To investigate the role of the corresponding endonuclease in yeast, we examined TGR in strains deleted of RAD1, the homolog of XPF. In accordance with previously published results (Schiestl et al, 1994; Symington et al, 2000), we observed a modest decline in TGR frequency in rad1 mutants, reduced three-fold at TRP1 (P=0.001; Figure 3B) and two-fold at MET17 (P=0.06; Figure 3A) compared to wild-type controls. However, the proportion of transformants with hDNA was radically reduced in rad1 mutants, with only 6/93 (6%) at TRP1 and 15/94 (16%) at MET17 showing hDNA formation in either flanking homology region compared to 70/103 (68%, P<0.0001) and 65/94 (69%, P<0.0001) in the corresponding RAD1 strains (Figure 4). In rad1 strains, only four transformants, all at MET17, formed hDNA at both ends, and all four of these were in the trans configuration (Figure 4A, bottom), suggesting that a small portion of two-end invasion intermediates can still be processed in the absence of Rad1. This alternative processing does not appear to be mediated by the Rad1-related protein Mus81, as the frequency of TGR in a strain mutated for both MUS81 and RAD1 was reduced only slightly compared to the rad1 single mutant (Figure 3A).
The role of Msh2 in TGR is mediated by its interaction with Msh3 but not Msh6
Msh2 forms separate complexes with Msh3 and Msh6 to process different types of intermediates in mismatch repair and HR (Johnson et al, 1996; Marsischky et al, 1996; Saparbaev et al, 1996). To determine if either of these proteins is involved in gene targeting, we analyzed TGR frequency and hDNA formation at MET17 in msh3 and msh6 strains. As expected, deletion of MSH6, which is specific for base–base or very small insertion–deletion mispairs, had no effect on either the frequency (Figure 3A) or the mechanism (Figure 4A) of TGR. By contrast, deletion of MSH3, which is specific for larger insertion/deletion mispairs, reduced the frequency of TGR two-fold when compared with either wild type (P=0.12) or msh6 (P=0.03; Figure 3A) and altered the mechanics of hDNA formation in a manner similar to that of msh2, with 32/95 (34%) transformants showing hDNA in one or both flanking homology regions compared to 44/95 (46%, P=0.10) in msh2, 60/91 (66%, P<0.0001) in msh6, and 65/94 (69%, P<0.0001) in the wild-type MSH3 strain (Figure 4A). Analysis of transformants with hDNA at both flanking markers showed 10/12 in the trans configuration in the msh6 mutant while 4/4 were in the cis configuration in the msh3 mutant (P=0.008, Figure 4A, bottom), confirming that the role of Msh2 in TGR is mediated by its interaction with Msh3, not Msh6.
Msh2:Msh3 and Rad1 operate together to prevent single-strand assimilation
Because the hDNA patterns observed in the msh2 and msh3 mutants were considerably different from those in rad1, we sought to determine whether Msh2:Msh3 and Rad1 affect the mechanism of TGR by the same or different pathways. To do so, we repeated the TGR experiments at MET17 in an msh2 rad1 double mutant. Differences in the frequency of TGR between the double mutant and the respective single mutants were not significant, but hDNA formation in the msh2 rad1 strain closely resembled that of the rad1 single mutant and was significantly different from the msh2 single mutant. In all, 7/96 (7%) transformants showed hDNA in one or both flanking homology regions in the msh2 rad1 double mutant compared to 15/94 (16%) in the rad1 single mutant (P=0.07) and 44/95 (46%) in the msh2 single mutant (P<0.0001) (Figure 4A). As discussed more fully below, these findings are best explained by a model in which Msh2:Msh3 detect the formation of a large unpaired bubble during TGR and direct nicking of the targeting DNA by Rad1, leading to preferential repair of the bubble mismatch in favor of the chromosomal sequences (Figure 2B).
Patterns of flanking markers support the model of an alternate mechanism for TGR in the absence of RAD1
Previously, it was shown that TGR in wild-type yeast occurs by invasion of separate strands of targeting DNA at the two ends, followed by distinct, apparently uncoordinated processing events at the 5′ and 3′ ends of a given strand (Langston and Symington, 2004) (Figure 2A). A model to account for TGR in the absence of Rad1 is that the process frequently occurs by assimilation of a single strand of targeting DNA into a long, unrepaired hDNA tract encompassing portions of both flanking homology regions (Figure 2D). A detailed analysis of MET17 transformants in rad1 and msh2 rad1 shows that the majority of transformants incorporated the flanking marker from one end of the targeting DNA but not the other (Figure 5). By contrast, at TRP1, more than two-thirds of transformants incorporated both flanking markers in the rad1 mutant (P<0.0001). The flanking markers on the MET17 targeting fragment are much farther apart (2.2 kbp; Figure 1B; depicted by circles in Figure 2) than those on the TRP1 targeting fragment (0.9 kbp; Figure 1A; depicted by boxes in Figure 2) and are less likely to be found in a common hDNA tract. These results strongly support a model for TGR in the absence of RAD1 in which strand invasion by one 3′ end of the targeting DNA is accompanied by heteroduplex extension sufficient to incorporate some or all of the 5′ flanking sequences from the same strand, as shown on the right side of Figure 2. Sequences more distant from the invading end are less likely to be incorporated into the single hDNA tract, especially if the opposite, 5′ end of the invading strand has undergone nucleolytic processing analogous to that at a DSB (depicted by dotted lines in Figure 2).
Figure 5.
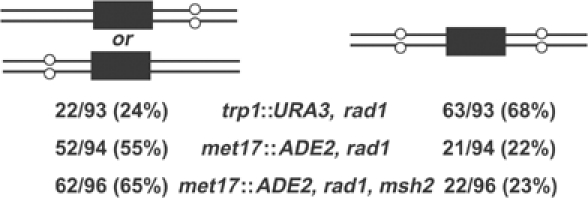
Evidence for single-strand assimilation in the rad1 strain. For the indicated strains, the data from the bars marked ‘vector sites, no hDNA' in Figure 3 are analyzed further to show the percentage of colonies that contained the vector restriction site at only one flanking marker (left) or both flanking markers (right).
RAD1 is not required for correction of a point mutation by gene targeting
In TGR, the two ends of the linear targeting fragment are normally identical in sequence to the chromosomal regions flanking the targeted gene, but the gene itself is replaced by a selectable marker. Thus, after strand invasion, base pairing between the targeting DNA and the targeted gene can extend only as far as the homology/heterology boundary represented by the selectable marker. To explain the requirement for Ercc1 during TGR in mammalian cells, Niedernhofer et al (2001) suggested that the Ercc1:Xpf endonuclease cleaves at the junction of homologous and heterologous sequences formed by strand invasion and branch migration (Niedernhofer et al, 2001). If this model is correct, deletion of ERCC1 or XPF (RAD10 or RAD1) should have no effect when the targeting DNA and the targeted gene are nearly identical.
To test this hypothesis, we examined the role of RAD1 during gene targeting of a chromosomal frameshift mutation, met17-sna, using the MET17 targeting fragment (Figure 1C). Like TGR, this type of gene targeting is initiated by separate strand invasions at the two ends in wild-type cells (Langston and Symington, 2004). As shown in Figure 6, deletion of RAD1 had no significant effect on hDNA formation at met17-sna (P=0.37), and all eight transformants recovered from the rad1 strain with hDNA in both flanking homology regions were in the trans configuration. Furthermore, deletion of RAD1 had no effect on targeting frequency at met17-sna (data not shown), all of which suggest that correction of small point mutations by gene targeting occurs in a RAD1-independent manner. These results differ greatly from those during TGR at met17∷ADE2 and trp1∷URA3, where deletion of RAD1 had a significant effect on both hDNA formation and targeting frequency (Figures 3 and 4), suggesting that RAD1 is only required during TGR when replacing a segment of chromosomal DNA with a large heterologous sequence.
Figure 6.
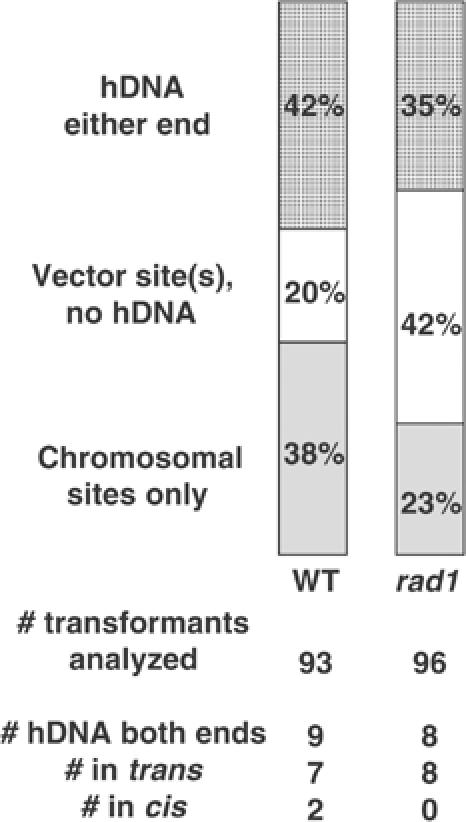
hDNA formation during TGR at met17-sna locus is unaffected by deletion of RAD1. hDNA formation and subclone analysis, as in Figure 4, for targeting of a chromosomal frameshift mutation, met17-sna (Figure 1C), are presented.
The frequency of TGR increases significantly in the absence of the SGS1 gene with no apparent change in the mechanism of TGR
The RecQ family helicase Sgs1 was recently shown to suppress crossover recombination during DSB repair in yeast, possibly through a Holliday junction dissolution mechanism (Ira et al, 2003). With separate strand invasion at each end as the initiating event of TGR, integration of recombinant DNA into the genome requires covalent joining of both the invading and noninvading strands of targeting DNA to the chromosomal DNA. In principle, one strand at each end could be connected through a concerted breakage and rejoining reaction like that postulated to occur during resolution of a Holliday junction. To determine whether Sgs1 plays a role in reversing branched intermediates formed during TGR, we determined the frequency of TGR at the MET17 locus in the presence and absence of the SGS1 gene. In the sgs1 strain, TGR was almost six-fold higher than in the SGS1 control (P=0.0005), showing that the suppression of mitotic crossover recombination by Sgs1 extends to TGR (Figure 3A).
To determine if this increase was attributable to an altered mechanism of TGR, we examined hDNA formation at the MET17 locus in the sgs1 strain. There was a modest although statistically significant reduction in hDNA formation, with 47 of 94 transformants (50%) showing hDNA at either end in the sgs1 strain compared to 65/94 (69%) in SGS1 (P=0.01; Figure 4A). However, among 14 transformants in the sgs1 strain that showed hDNA in both flanking homology regions, subclone analysis showed that all 14 were in the trans configuration, as in the SGS1 control (Figure 4A, bottom), suggesting that TGR is still initiated by separate strand invasions at the two ends of the targeting DNA in the absence of Sgs1. Thus, the increase in TGR frequency seen in the strain deleted of sgs1 does not appear to be attributable to use of an alternate TGR pathway.
Discussion
This study was designed to investigate the mechanism of gene targeting and how it differs from other recombination processes. Although TGR efficiency varies widely in different organisms, it is extremely productive and efficient in S. cerevisiae; however, the basis for this proficiency, like the mechanism of TGR itself, is poorly understood. It was previously established that TGR in wild-type yeast and mammalian cells is initiated by separate strand invasions at the two ends of the targeting DNA (Figure 2A) (Li et al, 2001; Langston and Symington, 2004) and not by single-strand assimilation as suggested earlier (Leung et al, 1997). However, even though the early steps in TGR are the same, yeast and mammals differ greatly in the apparent requirement for their respective homologous Rad1:Rad10 and Ercc1:Xpf complexes, the latter of which is essential for TGR in mammals (Niedernhofer et al, 2001). As discussed below, we find that Rad1 is also important for two-end invasion in yeast but, paradoxically, deletion of RAD1 potentiates TGR by the single-strand assimilation pathway, so the effect of RAD1 deletion appears less severe in yeast than in mammals.
Msh2:Msh3 and Rad1 form the boundary between two distinct mechanisms of TGR
If single-strand assimilation occurs during TGR in wild-type cells, it is extremely unproductive because, among 44 transformants with hDNA in both flanking homology regions, only one was in the cis configuration (Langston and Symington, 2004). The best evidence that this mechanism is suppressed during TGR in wild-type cells comes from msh2 and msh3 mismatch repair mutants, where we find a significant shift to the cis configuration of flanking markers (Figure 4, bottom). Assimilation of both flanking homology regions from a single strand of targeting DNA into a long hDNA tract with the homologous chromosomal locus results in a large bubble mismatch between the selectable marker on the targeting DNA and the targeted gene on the chromosome, as shown on the right-hand side of Figure 2. In wild-type cells, we suggest that Msh2 and Msh3 direct repair of the bubble mismatch in favor of the chromosomal sequences, so this pathway does not lead to productive TGR (Figure 2B). In msh2 and msh3 mutants, by contrast, the bubble mismatch is still repaired, but the bias in favor of chromosomal sequences is relaxed (Figure 2C), resulting in more transformants with the cis configuration of flanking markers (Figure 4, bottom). The fact that TGR frequency declines in msh2 and msh3 strains even though these mutations open up a secondary pathway for TGR strongly suggests that Msh2 and Msh3 facilitate targeted integration by the two-end invasion pathway in wild-type yeast (Figure 2, left) while simultaneously inhibiting the single-strand assimilation pathway.
Our data suggest a similar dual role for Rad1, but the effect of rad1 mutation is a highly significant reduction in the proportion of flanking marker hDNA rather than a shift to the cis configuration (Figure 4). Combined with a two- to three-fold reduction in TGR frequency in rad1 mutants (Figure 3), the four- to 10-fold reduction in hDNA proportion is consistent with a significantly reduced ability to complete targeted integration by the two-end invasion pathway in the absence of Rad1. As with msh2 and msh3, we suggest that the bulk of TGR in rad1 mutants occurs by single-strand assimilation; however, unlike msh2 and msh3, rad1 mutants are unable to repair large bubble mismatches formed by the single-strand assimilation pathway (Figure 2D). Thus, after the next round of DNA replication and cell division, one daughter cell fails to grow because it lacks the selectable gene on the targeting DNA leading to the extremely low levels of hDNA (and the lack of cis transformants) observed in rad1 (Figure 4). The failure of rad1 mutants to repair large bubble mismatches has previously been shown during meiotic recombination (Kirkpatrick and Petes, 1997; Kearney et al, 2001), but this is the first evidence for a similar role in large loop repair during mitotic recombination.
Although other explanations are possible for the observed reduction in hDNA, several results support the hypothesis that deletion of RAD1 shifts the balance of TGR from the two-end invasion pathway to the single-strand assimilation pathway. First, as shown in Figure 6, the defect in hDNA formation in rad1 mutants is specific to TGR with a heterologous selectable marker and not a general defect in heteroduplex formation. Second, as indicated by the boxes and circles in Figure 2, the single-strand assimilation model for TGR predicts that markers near the homology/heterology boundary are more likely to be found in a common hDNA tract, and the data presented for rad1 mutants in Figure 5 support this expectation. Finally, we previously examined repair of a single base-pair insertion by gene targeting and found that PMS1 prevents single-strand assimilation in this context. As with the rad1 mutants presented here, deletion of PMS1 was accompanied by a reduction in flanking marker hDNA, consistent with the model presented in Figure 2D (Langston and Symington, 2004).
How do Msh2:Msh3 and Rad1 facilitate TGR in wild-type cells?
The dual activities of Msh2:Msh3 and Rad1 in TGR are strongly reminiscent of the role these same factors play in other types of recombination involving interrupted homology sequences. For example, recombination between directly repeated sequences by single-strand annealing (SSA) leaves unpaired 3′-ended sequences that are stabilized by Msh2:Msh3 and cleaved by Rad1:Rad10 to permit completion of recombination (Schiestl and Prakash, 1988, 1990; Saparbaev et al, 1996). Rad1 and Msh2:Msh3 are also required for the removal of terminal heterologies during Rad51-dependent strand invasion (Fishman-Lobell and Haber, 1992; Sugawara et al, 1997). Similarly, in TGR, annealing between the targeting DNA and the targeted chromosomal locus is limited to the flanking homology sequences and pairing does not occur between the selectable marker and the targeted gene. Rad1 is not required when correcting a single base-pair insertion mutation by gene targeting (Figure 6), so clearly the requirement for Rad1 in TGR is related to the interrupted homology, but its exact role is unclear.
Based on the known substrate specificities of the Rad1:Rad10 and Ercc1/Xpf endonucleases (Bardwell et al, 1994; Davies et al, 1995; de Laat et al, 1998), there are various ways that Rad1 may be acting to facilitate TGR, as shown in Figure 7. Although data presented here support the hypothesis of Niedernhofer et al (2001), that these enzymes act at the homology/heterology border, replacement of the targeted locus requires cleavage at all four positions indicated in Figure 7A or additional processing by unidentified factors to remove the intervening chromosomal DNA. An alternative hypothesis, presented in Figure 7B, involves pairing of the D-loop with the noninvading strand of targeting DNA, resolution of the Holliday junction, and cleavage of the D-loop by Rad1:Rad10, as shown. While an enzyme for processing Holliday junctions has yet to be identified in eukaryotes, a resolvase activity with specificity similar to that of E. coli RuvC has been demonstrated in fractionated extracts of mammalian cells (Constantinou et al, 2001, 2002), and its ultimate identification should allow us to examine its role in TGR. An alternate pathway for gene targeting that is independent of the need for Holliday junction resolution has been suggested involving strand invasion and duplication of the entire donor chromosome onto the two ends of the targeting fragment (Morrow et al, 1997). It is unclear how this pathway would contribute to TGR because the targeted locus is not altered by break-induced replication (BIR). Furthermore, RAD51 is required for BIR (Davis and Symington, 2004) but is less important for gene targeting (Schiestl et al, 1994) and rad1 mutants are defective for TGR but not for BIR (A Davis and LS Symington, unpublished data).
Figure 7.
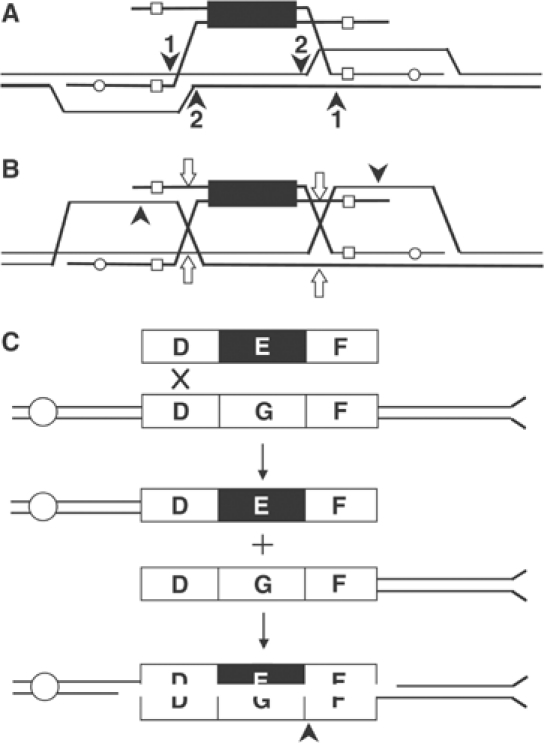
Possible roles for the Rad1:Rad10 nuclease in TGR. (A) Two different positions at the homology/heterology boundary where Rad1:Rad10 could nick the chromosome to facilitate TGR are indicated by arrows labeled 1 and 2. (B) D-loop pairs with noninvading strand; Rad1:Rad10 (black arrows) works in combination with a Holliday junction resolvase (open arrows). (C) One end of targeting DNA integrates into the chromosome in the flanking homology region labeled D, creating two chromosome fragments; 5′-end resection of both fragments reveals homologous sequences at D and F, interrupted by heterologous sequences at E and G; processing by Rad1:Rad10 at the homology/heterology boundary (indicated by an arrow) leads to TGR. For clarity, centromeric and telomeric positions on the targeted chromosome arm are indicated by open circles and flanges, respectively.
Finally, as shown in Figure 7C, homologous integration of one end of targeting DNA into the chromosome creates two chromosome fragments with directly repeated sequences at both free ends, as in SSA. Repair could proceed by SSA using the overlapping homologies present on the two chromosome fragments. Because the regions of homology are interrupted, a branched structure would be formed at the homology/heterology boundary requiring the Rad1:Rad10 nuclease for resolution. Thus, Rad1 might not be required for the initial integration but for later steps in repairing the resulting DSB.
Deletion of Sgs1 increases TGR frequency by the two-end invasion pathway
Sgs1 was recently shown to suppress crossovers during DSB repair, possibly by a Holliday junction dissolution mechanism (Ira et al, 2003), and it also cooperates with Msh2 and other mismatch repair factors to inhibit recombination between diverged sequences (Spell and Jinks-Robertson, 2004; Sugawara et al, 2004). In the present study, deletion of SGS1 led to a significant increase in the frequency of gene replacement (Figure 3A) without a major alteration in the mechanism of TGR (Figure 4A), which is the opposite of msh2 mutants. Thus, it is likely that Sgs1 does not inhibit TGR as part of a heteroduplex rejection complex but by unwinding recombination structures formed in the homologous flanking regions. Whether these structures are D-loops or Holliday junctions remains to be determined, as both intermediates are substrates for the mammalian RecQ homolog BLM (Karow et al, 2000; van Brabant et al, 2000). Because a similar increase in TGR was previously identified in human and chicken cells deficient in BLM (Wang et al, 2000; Traverso et al, 2003) and WRN (Imamura et al, 2002), it appears that deletion or inhibition of RecQ helicases may be a general technique for improving gene targeting efficiency that can be applied to a wide variety of cell types. These results also confirm strong similarities between yeast and mammals in the early steps of TGR.
We now have a developing picture of the means by which linear targeting DNA containing a selectable marker is precisely integrated into the yeast chromosome at a position determined by the flanking homology sequences on the targeting DNA. It is clear that TGR is initiated by strand invasion into homologous sequences by at least one end of the targeting fragment and that this strand invasion is actively opposed or unwound by Sgs1. In most cases, heteroduplex extension of the initial strand invasion intermediate is blocked by the heterology between the selectable marker and the targeted locus, creating a requirement for Msh2:Msh3 and Rad1:Rad10 for further processing, although the exact nature of this processing is unclear. The heterology block also necessitates a second strand invasion by the opposite end of the targeting fragment to complete TGR by this process, known as two-end invasion/resolution (Figure 2, left).
In some cases, the hDNA tract initiated by the first strand invasion extends far enough to include the opposite flanking homology sequence, incorporating the selectable marker and the targeted locus into a large bubble heteroduplex (Figure 2, right). This process, known as single-strand assimilation, is normally reversed by the concerted repair activity of Msh2:Msh3 and Rad1 (Figure 2B) and thus is rarely observed in wild-type cells. Formation and repair of large insertion/deletion mispairs have also been demonstrated in mammalian cells (Raynard and Baker, 2002), but repair of a preformed 216-nucleotide insertion mispair in mammalian cell extracts was independent of Ercc1/Xpf (Littman et al, 1999), which might explain why this pathway is unavailable for TGR in Ercc1−/− ES cells.
Materials and methods
Yeast strains and plasmids
The S. cerevisiae strains used for this study are derivatives of haploid strain W303-1A (Thomas and Rothstein, 1989) with the corrected RAD5 allele (Fan et al, 1996) and are listed in Table I. Parental strains used for gene targeting assays, LSY697, LSY698, and LSY1103, containing the met17-sna, met17∷ADE2, and trp1∷URA3 alleles, respectively, were described previously (Bartsch et al, 2000; Langston and Symington, 2004). Strains containing msh2∷HIS3, msh3∷HIS3, msh6∷TRP1, and mus81∷kanMX6 alleles were made by PCR-mediated gene disruption (Baudin et al, 1993) of LSY1103 or LSY698, and correct integration was confirmed by PCR or Southern blot. Strains containing msh2∷URA3 were derived from crosses to HKY592-11A (a gift from Hannah Klein, New York University School of Medicine, New York). LSY1683 was derived from a cross between LSY698 and W3643-38A (a gift from Rodney Rothstein, Columbia University, New York).
Table 1.
Yeast strains
| Strain no. | Genotypea | Source or reference |
|---|---|---|
| LSY697 | met17-sna ADE2 | Bartsch et al (2000) |
| LSY698 | met17∷ADE2 | Bartsch et al (2000) |
| LSY928-9C | met17∷ADE2 rad1∷LEU2 | Symington et al (2000) |
| LSY960-8B | met17∷ADE2 msh2∷URA3 | This study |
| LSY1103 | trp1∷URA3 | Langston and Symington (2004) |
| LSY1187 | trp1∷URA3 msh2∷HIS3 | This study |
| LSY1256 | trp1∷URA3 rad1∷LEU2 | This study |
| LSY1512 | met17∷ADE2 msh3∷HIS3 | This study |
| LSY1513 | met17∷ADE2 rad1∷LEU2 msh2∷URA3 | This study |
| LSY1542 | met17∷ADE2 msh6∷TRP1 | This study |
| LSY1570 | met17-sna rad1∷LEU2 ADE2 | This study |
| LSY1571 | met17∷ADE2 mus81∷kanMX6 rad1∷LEU2 | This study |
| LSY1683 | met17∷ADE2 sgs1∷HIS3 | This study |
| aAll strains are derived from LSY678, a RAD5 derivative of haploid strain W303-1A (MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 rad5-535) (Thomas and Rothstein, 1989); only differences from this genotype are given. | ||
Plasmids pLL113 (ARS1+) and pLL147 used in gene targeting assays were described previously (Langston and Symington, 2004). The TRP1 linear targeting fragment containing diagnostic restriction site polymorphisms in the flanking homology regions was released from pLL113 by digestion with restriction endonucleases SspI and BsaBI. The MET17 linear targeting fragment, also containing flanking restriction site polymorphisms, was released from pLL147 by SpeI digestion. Uncut transformation control plasmids pSB110 and pRS424, containing the MET17 and TRP1 genes, respectively, were described previously (Sikorski and Hieter, 1989; Bartsch et al, 2000).
Determination of transformation frequency and analysis of hDNA
Transformation of competent yeast cells was carried out by the lithium acetate method using 100–300 ng of linear targeting fragment DNA or 50–100 ng of uncut plasmid control DNA along with 50 μg of salmon sperm carrier DNA. Transformed cells were plated onto synthetic complete (SC) medium lacking Trp (SC−Trp) or Met (SC−Met), as appropriate. To exclude transformants arising by circularization of the linear targeting fragment or random integration, Trp+ colonies were replicated to medium containing 5-fluoroorotic acid (5-FOA) to assure TGR of the chromosomal trp1∷URA3 marker. Similarly, Met+ colonies were replicated to medium lacking both Met and Ade (SC−Met−Ade) to assure TGR of the chromosomal met17∷ADE2 marker.
To determine the frequency of TGR for each strain, colony-forming units (CFU) per 100 ng of transformed linear targeting DNA were compared to CFU/1 ng for the corresponding uncut plasmid for each strain (Figure 3) to normalize for differing transformation competence among strains. Transformation efficiencies at TRP1 were approximately 10-fold lower than those at MET17, reflecting normal variation in transformation efficiencies with different selectable markers and at different locations in the yeast genome. Except where noted, each transformation was performed at least three times and the means and standard deviations from all trials are shown in Figure 3. Statistical comparisons of mean TGR efficiencies between strains were performed using the unpaired t-test. P-values<0.05 were considered statistically significant.
Analysis of hDNA was previously described in detail (Langston and Symington, 2004). Briefly, genomic DNA from ∼96 whole colonies arising from TGR of each strain was amplified by PCR and digested separately with restriction enzymes corresponding to the restriction site on the targeting vector DNA or the site on the chromosomal DNA. PCR products that digested to completion with the enzyme specific to the restriction site on the targeting vector DNA or the site on the chromosomal DNA were scored as ‘vector site only' or ‘chromosomal site only', respectively. PCR products that showed partial digestion with both enzymes were scored as hDNA. For Figure 4 (top), if either flanking homology region showed hDNA, the colony is shown as ‘hDNA either end'; if neither flanking region showed hDNA but at least one region contained vector sequences, the colony is represented as ‘vector site(s), no hDNA'; if both flanking regions showed only chromosomal sequences, the colony is represented as ‘chromosomal sites only'. To compare hDNA formation, differences between strains in the number of hDNA and non-hDNA transformants were treated as categorical variables and analyzed by Fisher's two-tailed exact test. P-values<0.05 were considered statistically significant. Colonies that showed hDNA in both flanking homology regions were subcloned and analyzed by PCR and restriction digestion to determine if hDNA formed between the same strand (cis) or different strands (trans) of the targeting DNA at the two ends, and the results are shown at the bottom of Figure 4.
Acknowledgments
We thank H Klein and R Rothstein for strains, E Alani for helpful suggestions, and WK Holloman, S Jinks-Robertson, VA Murrero, and B Llorente for critical reading of the manuscript. The research described in this article was supported by grants from the National Institutes of Health (GM54099 to LSS, and T32 GM08224 and T32 CA09503 to LDL).
References
- Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC (1994) Specific cleavage of model recombination and repair intermediates by the yeast Rad1–Rad10 DNA endonuclease. Science 265: 2082–2085 [DOI] [PubMed] [Google Scholar]
- Bartsch S, Kang LE, Symington LS (2000) RAD51 is required for the repair of plasmid double-stranded DNA gaps from either plasmid or chromosomal templates. Mol Cell Biol 20: 1194–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C (1993) A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res 21: 3329–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clikeman JA, Wheeler SL, Nickoloff JA (2001) Efficient incorporation of large (>2 kb) heterologies into heteroduplex DNA: Pms1/Msh2-dependent and -independent large loop mismatch repair in Saccharomyces cerevisiae. Genetics 157: 1481–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinou A, Chen XB, McGowan CH, West SC (2002) Holliday junction resolution in human cells: two junction endonucleases with distinct substrate specificities. EMBO J 21: 5577–5585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinou A, Davies AA, West SC (2001) Branch migration and Holliday junction resolution catalyzed by activities from mammalian cells. Cell 104: 259–268 [DOI] [PubMed] [Google Scholar]
- Davies AA, Friedberg EC, Tomkinson AE, Wood RD, West SC (1995) Role of the Rad1 and Rad10 proteins in nucleotide excision repair and recombination. J Biol Chem 270: 24638–24641 [DOI] [PubMed] [Google Scholar]
- Davis AP, Symington LS (2004) RAD51-dependent break-induced replication in yeast. Mol Cell Biol 24: 2344–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat WL, Appeldoorn E, Jaspers NG, Hoeijmakers JH (1998) DNA structural elements required for ERCC1-XPF endonuclease activity. J Biol Chem 273: 7835–7842 [DOI] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J (1995) The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83: 655–666 [DOI] [PubMed] [Google Scholar]
- Fan HY, Cheng KK, Klein HL (1996) Mutations in the RNA polymerase II transcription machinery suppress the hyperrecombination mutant hpr1 delta of Saccharomyces cerevisiae. Genetics 142: 749–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman-Lobell J, Haber JE (1992) Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science 258: 480–484 [DOI] [PubMed] [Google Scholar]
- Fishman-Lobell J, Rudin N, Haber JE (1992) Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol Cell Biol 12: 1292–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura O, Fujita K, Itoh C, Takeda S, Furuichi Y, Matsumoto T (2002) Werner and Bloom helicases are involved in DNA repair in a complementary fashion. Oncogene 21: 954–963 [DOI] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE (2003) Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Koonin EV, Aravind L (2002) Classification and evolutionary history of the single-strand annealing proteins, RecT, Redbeta, ERF and RAD52. BMC Genomics 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Kovvali GK, Prakash L, Prakash S (1996) Requirement of the yeast MSH3 and MSH6 genes for MSH2-dependent genomic stability. J Biol Chem 271: 7285–7288 [DOI] [PubMed] [Google Scholar]
- Karow JK, Constantinou A, Li JL, West SC, Hickson ID (2000) The Bloom's syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci USA 97: 6504–6508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney HM, Kirkpatrick DT, Gerton JL, Petes TD (2001) Meiotic recombination involving heterozygous large insertions in Saccharomyces cerevisiae: formation and repair of large, unpaired DNA loops. Genetics 158: 1457–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick DT, Petes TD (1997) Repair of DNA loops involves DNA-mismatch and nucleotide-excision repair proteins. Nature 387: 929–931 [DOI] [PubMed] [Google Scholar]
- Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y (1999) Mutations in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nat Genet 22: 82–84 [DOI] [PubMed] [Google Scholar]
- Langston LD, Symington LS (2004) Gene targeting in yeast is initiated by two independent strand invasions. Proc Natl Acad Sci USA 101: 15392–15397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung W, Malkova A, Haber JE (1997) Gene targeting by linear duplex DNA frequently occurs by assimilation of a single strand that is subject to preferential mismatch correction. Proc Natl Acad Sci USA 94: 6851–6856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Read LR, Baker MD (2001) The mechanism of mammalian gene replacement is consistent with the formation of long regions of heteroduplex DNA associated with two crossing-over events. Mol Cell Biol 21: 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FL, Sperle K, Sternberg N (1984) Model for homologous recombination during transfer of DNA into mouse L cells: role for DNA ends in the recombination process. Mol Cell Biol 4: 1020–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littman SJ, Fang W-h, Modrich P (1999) Repair of large insertion/deletion heterologies in human nuclear extracts is directed by a 5′ single-strand break and is independent of the mismatch repair system. J Biol Chem 274: 7474–7481 [DOI] [PubMed] [Google Scholar]
- Marsischky GT, Filosi N, Kane MF, Kolodner R (1996) Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev 10: 407–420 [DOI] [PubMed] [Google Scholar]
- Morrow DM, Connelly C, Hieter P (1997) ‘Break copy' duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 147: 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag DK, White MA, Petes TD (1989) Palindromic sequences in heteroduplex DNA inhibit mismatch repair in yeast. Nature 340: 318–320 [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH, Kanaar R (2001) The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J 20: 6540–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozenberger BA, Roeder GS (1991) A unique pathway of double-strand break repair operates in tandemly repeated genes. Mol Cell Biol 11: 1222–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynard SJ, Baker MD (2002) Incorporation of large heterologies into heteroduplex DNA during double-strand-break repair in mouse cells. Genetics 162: 977–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reenan RA, Kolodner RD (1992) Isolation and characterization of two Saccharomyces cerevisiae genes encoding homologs of the bacterial HexA and MutS mismatch repair proteins. Genetics 132: 963–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein RJ (1983) One-step gene disruption in yeast. Methods Enzymol 101: 202–211 [DOI] [PubMed] [Google Scholar]
- Saparbaev M, Prakash L, Prakash S (1996) Requirement of mismatch repair genes MSH2 and MSH3 in the RAD1–RAD10 pathway of mitotic recombination in Saccharomyces cerevisiae. Genetics 142: 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Prakash S (1988) RAD1, an excision repair gene of Saccharomyces cerevisiae, is also involved in recombination. Mol Cell Biol 8: 3619–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Prakash S (1990) RAD10, an excision repair gene of Saccharomyces cerevisiae, is involved in the RAD1 pathway of mitotic recombination. Mol Cell Biol 10: 2485–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Zhu J, Petes TD (1994) Effect of mutations in genes affecting homologous recombination on restriction enzyme-mediated and illegitimate recombination in Saccharomyces cerevisiae. Mol Cell Biol 14: 4493–4500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmuckli-Maurer J, Rolfsmeier M, Nguyen H, Heyer WD (2003) Genome instability in rad54 mutants of Saccharomyces cerevisiae. Nucleic Acids Res 31: 1013–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spell RM, Jinks-Robertson S (2004) Examination of the roles of Sgs1 and Srs2 helicases in the enforcement of recombination fidelity in Saccharomyces cerevisiae. Genetics 168: 1855–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE (2004) Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci USA 101: 9315–9320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Paques F, Colaiacovo M, Haber JE (1997) Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc Natl Acad Sci USA 94: 9214–9219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS (2002) Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 66: 630–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Kang LE, Moreau S (2000) Alteration of gene conversion tract length and associated crossing over during plasmid gap repair in nuclease-deficient strains of Saccharomyces cerevisiae. Nucleic Acids Res 28: 4649–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R (1989) The genetic control of direct-repeat recombination in Saccharomyces: the effect of rad52 and rad1 on mitotic recombination at GAL10, a transcriptionally regulated gene. Genetics 123: 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverso G, Bettegowda C, Kraus J, Speicher MR, Kinzler KW, Vogelstein B, Lengauer C (2003) Hyper-recombination and genetic instability in BLM-deficient epithelial cells. Cancer Res 63: 8578–8581 [PubMed] [Google Scholar]
- van Brabant AJ, Ye T, Sanz M, German IJ, Ellis NA, Holloman WK (2000) Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry 39: 14617–14625 [DOI] [PubMed] [Google Scholar]
- Wang W, Seki M, Narita Y, Sonoda E, Takeda S, Yamada K, Masuko T, Katada T, Enomoto T (2000) Possible association of BLM in decreasing DNA double strand breaks during DNA replication. EMBO J 19: 3428–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD (1996) Positional cloning of the Werner's syndrome gene. Science 272: 258–262 [DOI] [PubMed] [Google Scholar]