

Abstract
In Saccharomyces cerevisiae, over twenty mRNAs localize to the bud tip of daughter cells, playing roles in processes as different as mating type switching and plasma membrane targeting. The localization of these transcripts depends on interactions between a cis-acting localization element(s) or zipcodes and the RNA-binding protein She2p. While previous studies identified four different localization elements in the bud-localized ASH1 mRNA, the main determinants for She2p recognition are still unknown. To investigate the RNA-binding specificity of She2p, we isolated She2p-binding RNAs by in vivo selection from libraries of partially randomized ASH1 localization elements. The RNAs isolated contained a similar loop-stem-loop structure with a highly conserved CGA triplet in one loop and a single conserved cytosine in the other loop. Mutating these conserved nucleotides or the stem separating them resulted in the loss of She2p binding and in the delocalization of a reporter mRNA. Using this information, we identified the same RNA motif in two other known bud-localized transcripts, suggesting that this motif is conserved among bud-localized mRNAs. These results show that mRNAs with zipcodes lacking primary sequence similarity can rely on a few conserved nucleotides properly oriented in their three-dimensional structure in order to be recognized by the same localization machinery.
The cytoplasmic transport and localization of mRNAs are used by various eukaryotic polarized cells to control the spatiotemporal expression of specific proteins. This process has been observed and widely studied in Drosophila and Xenopus embryos (2, 19, 24), neurons (22), fibroblasts (31), and yeast (26, 41). The mRNAs sorted by this mechanism contain in their sequences specific localization elements, or zipcodes, which are recognized by the cell localization machinery (23). These localization elements are usually found in the 3′ untranslated region of the mRNAs, but some have been found in the 5′ untranslated region (43) or within the coding sequence of transcripts (7, 14, 36).
The budding yeast Saccharomyces cerevisiae has emerged as a model system for understanding the molecular basis behind cytoplasmic mRNA transport and localization (9). In this organism, more than twenty transcripts have been shown to be transported and localized to the bud tip of yeast cells via the same pathway (38, 40). The core components of the yeast mRNA localization machinery have been identified and were shown to form a complex called the locasome, which includes She2p, She3p, and Myo4p (3, 4, 25, 29, 42).
The current model for the assembly of this localization complex suggests that the RNA-binding protein She2p recognizes the mRNAs to be localized and recruits the type V myosin Myo4p via their common interaction with the adapter protein She3p (4, 25). This ribonucleoprotein (RNP) complex is then transported along the actin cytoskeleton and anchored at the bud tip, where translation occurs. Of these mRNAs, the yeast ASH1 mRNA was the first identified and the most studied (26, 41). The localization of the ASH1 mRNA to the distal tip of daughter cells during anaphase is responsible for the asymmetric sorting of Ash1p to the daughter cell nucleus (8, 26, 41) where it represses mating type switching (17, 39).
The ASH1 mRNA contains four localization elements which are essential for the proper localization of this transcript (7, 8, 14). Of these localization elements, three (E1, E2A, and E2B) are located within the coding region of the ASH1 mRNA, whereas the remaining element (E3) includes the termination codon and is located primarily within the ASH1 3′ untranslated region. These localization elements were predicted to form RNA secondary structures containing stem-loops (7, 8, 14). Each single element is sufficient to localize a reporter mRNA at the bud of yeast cells (7, 14) and can promote the formation of the locasome complex (7).
Other studies have also shown that the four elements interact independently and specifically with the RNA-binding protein She2p (4, 25). Deletion of the SHE2 gene leads to the delocalization of the ASH1 mRNA (26, 41) and SHE2 is essential for the localization function of each one of the four ASH1 localization elements (25). The X-ray structure of She2p, which has been recently published, shows that this protein forms a symmetric homodimer and contains two RNA-binding domains rich in basic residues (30). Niessing et al. also found that one RNA molecule binds per She2p homodimer and suggested that the RNA molecule would bind to both RNA-binding domains (30).
To date, no consensus sequence and/or structure has been identified among the four elements which may act as an RNA recognition motif for the She2 protein. Since the transport and localization of mRNAs at the bud tip of yeast cells have been shown to be She2p dependent (38, 40), the identification of the specific RNA motif recognized by She2p is crucial for understanding the specificity of this pathway.
Here we report the identification of a three-dimensional motif conserved within the four ASH1 mRNA localization elements and in two other bud-localized transcripts. Using an in vivo screen of partially randomized ASH1 RNA localization elements, we have identified the nucleotides essential for She2p recognition. From these screens, a motif constituted of a loop-stem-loop structure with a highly conserved cytosine in one loop and a CGA triplet in the other loop was found to be present in the ASH1 localization elements. Mutation of the conserved CGA triplet and the single cytosine in this motif resulted in the delocalization of a reporter mRNA and in the loss of binding to She2p. A computer search and analysis of this motif revealed a conserved three-dimensional fold in the ASH1 localization elements, especially in the distance separating the two cytosines. Elongation of the stem between the two cytosines showed that the distance separating these two nucleotides and/or their spatial orientation was important for the recognition of the localization elements by the yeast mRNA localization machinery.
Using this information, we identified the same motif in two other bud-localized mRNAs, IST2 and YMR171c, confirming the importance of this motif for the proper sorting of yeast bud-localized mRNAs. Altogether, these data show that the recognition of different mRNAs by a localization machinery can rely on a few conserved nucleotides properly oriented in the three-dimensional structure of these mRNAs.
MATERIALS AND METHODS
Growth media and yeast strains.
Yeast cells were grown either in synthetic growth media lacking the nutrients indicated or in rich media (32). Yeast strains used in this study are listed in Table S8 of the supplemental material. Transformation was performed according to the protocol of Gietz and Schiestl (34). A yeast gene disruption cassette was created by PCR amplification of the loxP-KAN-loxP construct in plasmid pUG6 and primers specific for the gene of interest (15). Specific disruption was confirmed by PCR analysis of genomic DNA.
Plasmid constructions.
All the mutants in the localization elements were generated by PCR using the splicing through overlap extension strategy (7, 8). Mutants in the localization elements E1, E2A and E2B used in the yeast three-hybrid assay were amplified by PCR from YEPlac195-lacZ-ADHII-based plasmids containing the mutated ASH1 localization elements (7, 8) and cloned in the SmaI site of plasmid pIIIA/MS2-2. These mutations are listed in Table S1 of the supplemental material. Point mutations of the cytosines and elongation of the stem in each localization element were generated by PCR using as template plasmids pXR113 (YEPlac195-lacZ-E1), pXR137 (YEPlac195-lacZ-E2A), pXR156 (YEPlac195-lacZ-E2B) and pXR63 (YEPlac195-lacZ-E3) (7) and cloned in the SmaI site of pIIIA/MS2-2. All mutations were confirmed by DNA sequencing. These mutations are listed in Table S1 of the supplemental material. For the localization assays, mutant localization elements were amplified by PCR from their pIIIA/MS2-2-based plasmids and cloned at the SmaI site of YEPlac195-lacZ-ADHII or YCPlac33-lacZ-ADHII plasmids. For in vitro transcription, localization elements E1 (70 nucleotides), E2A (92 nucleotides) and E2B-D1 (60 nucleotides) were cloned in pGEM-4Z between the HindIII and EcoRI sites.
Yeast three-hybrid assay.
Strain YBZ1 was used in the three-hybrid assay (16). This strain was transformed with the appropriate plasmids and grown in synthetic medium lacking uracil and leucine. Dilution assays were made on plates lacking uracil, leucine and histidine, with or without 3-amino-1,2,4-triazole. The β-galactosidase expression level was measured quantitatively using O-nitrophenyl-β-d-galactopyranoside (ONPG) according to the protocol provided by Clontech. The reported β-galactosidase expression levels represent the average of at least three independent experiments. Proper expression of the RNA-MS2 fusions was confirmed by Northern blot (data not shown).
In vivo selection.
Partially degenerated libraries of the localization elements E1, E2A, E2B-D1 and E3 were produced by PCR. The oligonucleotides used to produce the libraries are described in Table S2 of the supplemental material. The pXR113, pXR137, pXR156, and pXR63 plasmids were used as templates. The libraries were cloned in the SmaI site of the pIIIA/MS2-2 plasmid. The randomization of the sequences was verified by sequencing several clones for each library. The partially degenerated libraries were transformed in YBZ1 yeasts and selected for the interaction with She2p by a three-hybrid assay. The yeasts were grown on a −His-Ura-Leu 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) indicator medium (33) containing 0.04 mg/ml of 5-bromo-4-chloro-3-indolyl-β-d-galactoside. The in vivo selection for the E1 and E2A libraries were realized in the presence of 0.1 mM and 0.25 mM 3-amino-1,2,4-triazole, respectively, whereas the selection for the E2B-D1 and E3 libraries were realized in the presence of 1 mM 3-amino-1,2,4-triazole. The sequences positive for the interaction with She2p were finally identified by automated DNA sequencing.
TABLE 2.
Mutagenesis of the She2p binding motif
 |
Taken from reference 8.
Electrophoretic mobility shift assay.
To produce in vitro transcribed RNA, plasmids pPC1 (pGEM-E1), pPC4 (pGEM-E2A), and pPC9 (pGEM-E2B-D1) were linearized with EcoRI and transcribed with T7 RNA polymerase in the presence of [α-32P]CTP (20). The transcripts were purified on 6% denaturing polyacrylamide gels, then extracted, and desalted on G25 Sephadex spin columns (Roche). For unlabeled RNAs, the transcripts were purified, after treatment with DNase I, by phenol-chloroform extraction, ethanol precipitation and desalting on G25 Sephadex spin columns. GST-She2p was expressed and purified according to the protocol published in reference 4.
For electrophoretic mobility shift assays, the 32P-labeled RNA was denatured by heating at 85°C for 2 min in the binding buffer and allowed to fold at room temperature for 10 min. A total of 10,000 cpm of labeled RNA (≈1 ng) were added to the binding buffer (10 mM HEPES, pH. 7.4, 150 mM KCl, 1 mM dithiothreitol, 5 mM MgCl2, 4% glycerol, 15 U RNase inhibitor [Pharmacia]) prior to the addition of various concentrations of recombinant glutathione S-transferase (GST)-She2p, up to a final volume of 20 μl. The reaction mixtures were incubated at 4°C for 30 min, and then 2 μl of 10 mg/ml heparin was added and incubated for 10 more minutes at 4°C to prevent nonspecific interactions. The samples were separated on a 4% nondenaturing gel at 120 V for 4 h at 4°C, dried, and exposed overnight with Kodak films. For competition experiments, unlabeled RNAs were added prior to the addition of GST-She2p.
Immunoprecipitation and reverse transcription-PCR.
Yeast strain K699 she2 transformed with the plasmids YCP22-She2-Myc and YEPlac195-lacZ-E1, YEPlac195-lacZ-E1-M15 or YEPlac195-lacZ-E1-M16 was grown overnight in a selective medium lacking uracil and tryptophan. The hybrid RNA expression was induced by the addition of 3% galactose to the selective medium. Cells were harvested by centrifugation and resuspended at an OD600 of 100 in the extraction buffer (25 mM HEPES-KOH, pH 7.5, 150 mM KCl, 2 mM MgCl2, 0.1% IGEPAL CA-630, 1 mM dithiothreitol, 87.5 μg/ml phenylmethylsulfonyl fluoride, 0.5 μg/ml pepstatin, 0.5 μg/ml leupeptin, 0.5 μg/ml aprotinin; and 23 U/ml of RNAguard). The yeast cells were broken with glass beads and the supernatant was used for immunoprecipitation and Western blot. For the immunoprecipitation, an anti-Myc antibody (9E10) was added to 500 μl of supernatant and incubated at 4°C with agitation for 1 h; 40 μl of protein A-Sepharose beads was then added and the incubation at 4°C was continued for 2 h. The beads were washed four times for 3 min at 4°C with a wash buffer (25 mM HEPES-KOH, pH 7.5, 150 mM KCl, 2 mM MgCl2). The RNA was eluted from the beads in 200 μl of 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 10 mM EDTA and 1% sodium dodecyl sulfate 10 min at 65°C, followed by a phenol-chloroform extraction and ethanol precipitation. For the reverse transcription, 2 μl of mRNA was incubated at 70°C for 5 min in the presence of 0,5 μg of pd(N)6 and quickly chilled on ice. The reverse transcription reaction was performed according to indications in a 1× buffer (50 mM Tris-HCl, pH 8.3, 50 mM KCl, 4 mM MgCl2, 10 mM dithiothreitol) containing 10 mM deoxynucleoside triphosphate and 20 U of RNAguard, with 200 U reverse transcriptase for 1 h at 42°C. The cDNAs were then amplified by PCR using primers in the lacZ sequence.
In situ hybridization and imaging.
Yeasts cells were processed for fluorescent in situ hybridization according to the protocols described in reference 10. For in situ hybridization, yeast spheroplasts were hybridized with a pool of Cy3-conjugated lacZ DNA oligonucleotide probes (7). When quantitative data were obtained, 50 budding yeast cells were counted per experiment.
Pattern search in the PDB database (MC-SEARCH) and RNA motif search (RNAMotif).
Using MC-SEARCH (P. Gendron and F. Major, unpublished data), we scanned all known tertiary structures in the PDB database for occurrences of the three variants of the motif. MC-SEARCH is a computer program, derived from the automated RNA annotation program MC-ANNOTATE (12), that searches RNA structure files for regions that match a user-defined pattern. The search patterns were described only in terms of primary and secondary structure elements: the type of essential nucleotides and the presence of Watson-Crick base pairs in the stems. No constraint was added concerning potential interactions of nonhelical nucleotides with other parts of the patterns or of the scanned structures.
To identify She2p-binding motifs in other bud-localized mRNAs, we used RNAMotif, a RNA secondary structure search algorithm (11). RNAMotif needs two input files: the sequence database and the descriptor of the searched motif. The database was a file with the 22 bud-localized mRNAs sequences (coding sequences, plus 250 bp in 5′ and 3′) in FASTA format. The descriptor defines the criteria required to generate a match: stem, loop sequences and length. The GU base pairs are authorized. For example, the descriptor looking for a 5-base-pair family stem-loop (motif stem 5 in Fig. S2 of the supplemental material) is as follows: parms, wc+ = gu; descriptor, h5(tag = ′1′, len = 4), ss (minlen = 1, maxlen = 4), h5(tag = ′2′, len = 5), ss (minlen = 7, maxlen = 38, seq = ′∧cga′), h3(tag = ′2′), ss (minlen = 3, maxlen = 10, seq = ′∧[atcg]c′), and h3(tag = ′1′) (h5 and h3 mean stem, and ss means single strand). The authorized length of the stem and loop are mentioned by minlen and maxlen. The required nucleotides are mentioned by seq. The output file contains all the sequences and their positions that match the searched motif. Once obtained, the sequences identified were further filtered with the MFOLD software (http://www.bioinfo.rpi.edu/applications/mfold/old/rna/form1.cgi) in order to confirm the predicted secondary structure. Only the RNA sequences that correctly fold like the She2p binding motif RNA were finally selected.
RESULTS
Yeast three-hybrid assay to study the interaction between She2p and the ASH1 mRNA localization elements.
Extensive mutagenesis had previously allowed the mapping of the four ASH1 mRNA localization elements and had shown that each element adopts a specific stem-loop structure (Fig. 1A) (7, 8, 14). In order to identify the motif(s) bound by She2p in each localization element RNA, we first set up a reliable system to study the interaction between She2p and the four ASH1 localization elements in vivo using the yeast three-hybrid assay. In this assay, the formation of an RNA-protein complex leads to the activation of reporter genes (lacZ and HIS3), and therefore large libraries of mutations on the RNA can be screened and selected for maintenance of the RNA-protein interaction (35).
FIG.1.

Four ASH1 localization elements interact with She2p in a yeast three-hybrid assay. (A) Predicted secondary structures of the ASH1 localization elements E1, E2A, E2B, and E3. Secondary structures are described with latin numbered stems (S) and loops (L). For the element E2B, two separate domains have been previously identified (8), but only the secondary structure of domain 1 has been defined. (B) The yeast strain YBZ1 expresses a dimerized bacteriophage MS2 RNA-binding protein fused to a single LexA DNA-binding domain (MS2-LexA), with either wild-type (capital letters) or knockout (lowercase) SHE2 gene. These strains were transformed with a plasmid expressing the GAL4 activation domain fused with the C-terminal domain of She3p (She3-Cterm-AD), along with a plasmid expressing an hybrid RNA containing the indicated RNA fused to two MS2 binding sites. The formation of a She2p-hybrid RNA complex allows the recruitment of the She3-Cterm-AD and MS2-LexA proteins and the activation of the reporter genes. Controls: empty vector (pIIIA/MS2-2) and the iron response element RNA (IRE).
A fusion of She2p with the GAL4 activation domain has already been shown to interact with the E3 localization element in the yeast three-hybrid assay in a previous study (13). However, we could only obtain a weak but specific interaction between this fusion protein and the four ASH1 mRNA localization elements (data not shown; see the supplemental material). In order to increase the sensitivity of our assay, we used a variation of this three-hybrid assay, developed by Long et al. (25), where the C-terminal domain of She3p was fused to the GAL4 activation domain (She3-Cterm-AD). The C-terminal domain of She3p has been shown previously to bind She2p directly in vitro and in vivo (4, 25). The idea behind this system is that the She3-Cterm-AD protein recruits the endogenous She2 protein, whose interaction with a hybrid RNA containing a localization element RNA fused to two MS2 binding sites leads to the recruitment of the MS2-LexA DNA binding domain fusion protein. The formation of this four-hybrid complex results in the activation of the reporter genes. This approach has been successfully used previously to characterize the interaction between She2p and the localization element E3 (25).
As expected, using the C-terminal domain of She3p fused to the GAL4 activation domain in the three-hybrid assay, we observed a strong interaction with all four ASH1 mRNA localization elements (Fig. 1B). Interestingly, we found that only the domain 1 of the localization element E2B (E2B-D1) interacted with She2p, whereas the domain 2 (E2B-D2) did not (Fig. 1B). Since both domains of E2B are important for its localization function (7), the domain 2 may be involved in another aspect of the localization process. These interactions were dependent on the endogenous She2p since the same assay in a she2 strain led to the complete loss of β-galactosidase expression, as reported previously (25) (Fig. 1B). Overall, these results are in agreement with previous data that showed that She2p interacts with the four ASH1 localization elements in vivo (4, 25).
To confirm that She2p binds directly to the four localization elements, we used an electrophoretic mobility shift assay to test the binding of purified recombinant GST-She2 protein to the RNA localization elements in vitro. Previous in vitro studies had mostly focused on the binding of GST-She2p to the element E3 RNA using either GST-She2p pull-down (4), electrophoretic mobility shift assay (4), or cross-linking (13, 25), but little had been done to confirm a direct interaction between She2p and the other three elements. Since this information is crucial for our investigation, we decided to complement these studies and focus only on the binding of GST-She2p to the localization element E1, E2A, and E2B-D1 RNAs.
As shown in Fig. 2, these three RNA localization elements could form a complex with recombinant GST-She2p in vitro. While the E2B-D1 RNA with GST-She2p leads to the formation of only one main shifted complex, a second shift was observed for the element E2A and, at a lower yield, with the element E1 (Fig. 2A and B). Since She2p acts as a dimer (30), the different shifts observed may correspond to the formation of RNA-protein complexes with a 1:1 or 2:1 ratio of the RNA zipcode with the GST-She2p dimer. GST-She2p bound these RNAs with low affinity (in the micromolar range), but the complexes formed were saturable at high concentration of protein (Fig. 2A). These complexes were specific for She2p since GST alone did not bind the localization elements (Fig. 2C and data not shown). Moreover, the IRE (iron response element) RNA, which also folds in a stem-loop structure (1), interacted only weakly with GST-She2p (Fig. 2C), showing that the complex was specific for the ASH1 localization elements.
FIG.2.

Recombinant GST-She2p binds to the ASH1 localization element RNAs in vitro. (A) Electrophoretic mobility shift assays (EMSA) of GST-She2p with the localization elements RNA E1, E2A, and E2B-D1. Increasing concentration of recombinant GST-She2p (0, 0.5, 0.9, 1.8, 3.7, and 7.4 μM) was added to 32P-labeled localization element RNA. The free RNA is indicated by an asterisk. (B) Competition of the RNA-protein complex with unlabeled localization element RNA E1, E2A and E2B-D1. A constant quantity of GST-She2p (3.7 μM for E1 and E2B-D1, 0.9 μM for E2A) was combined to an increasing concentration of unlabeled RNA (5×, 50×, and 500×). The mutant M16 of the element E2B-D1 was used as a competitor (E2D-D1 MUT). (C) Controls using GST or the IRE RNA. For GST, increasing concentration of recombinant GST (0, 0.5, 0.9, 1.8, 3.7, and 7.4 μM) was added to the 32P-labeled localization element E2B-D1 RNA. For the IRE RNA, increasing concentration of recombinant GST-She2p (0, 0.5, 0.9, 1.8, 3.7, and 7.4 μM) was added to the 32P-labeled IRE RNA.
In order to confirm that the observed shifts were caused by the formation of a protein-RNA complex, we performed competition assays with 5×, 50×, and 500× excesses of unlabeled wild-type localization element RNA. As shown in Fig. 2B, these competitions led to the loss of the RNA shift, indicating that this complex was formed by a GST-She2p:RNA species. Unlike what had been reported recently (13), we did not observe the formation of a base-paired RNA duplex between the labeled and the unlabeled RNA. We also found that an excess of mutated E2B-D1 localization element RNA competed with the formation of the complex more poorly than the wild-type RNA (Fig. 2B). However, mutations in other elements competed as efficiently as the wild-type RNAs (data not shown). GST-She2p was still able to bind to the mutated RNAs, possibly because of the low affinity of GST-She2p for RNA and the high concentration of protein used in these assays. Therefore we could not use this assay to detect mutations that disrupt the formation of this protein-RNA complex.
Identification of the She2p-binding domain in each ASH1 localization element.
In order to identify which domain(s) of the localization elements was essential for binding She2p, we cloned several mutants of each ASH1 mRNA localization element in the pIIIA/MS2-2 vector and tested their interaction with She2p in the yeast three-hybrid assay. Since the same localization element mutants had been previously tested for their mRNA localization function (7, 8), we looked for a correlation between the loss of She2p binding and the loss of mRNA localization function for each localization element. A similar study had already been done on the E3 element and showed that the internal loop and the upper stem of this RNA (see Fig. 1A) were both essential for She2p binding and for the localization function of this element (25).
We therefore used mutants of the localization elements E1, E2A, and E2B-D1 that were previously found to have lost their localization function (7, 8) and tested them for She2p binding. As shown in Fig. 3, disruption of specific stems and loops in each localization element resulted in the complete loss of β-galactosidase expression and growth on −His selection medium. To test for the importance of the stems in the binding of She2p to these RNAs, we restored the predicted stems using compensatory mutations. As expected, these compensatory mutations allowed the binding of She2p to near wild-type levels and restored the localization function of the elements, suggesting that the stems, but not their sequences, are essential for She2p recognition (Fig. 3). Altogether, these results showed a direct correlation between She2p binding and the localization function of these elements. Moreover, this analysis supported the predicted secondary structures for each localization element previously established using fluorescence in situ hybridization (7, 8, 14).
FIG. 3.

Identification of the She2p-binding domain in the localization elements E1, E2A, and E2B-D1. Mutated RNAs were fused to the MS2 binding RNA in the pIIIA/MS2-2 plasmid and transformed in yeasts YBZ1 containing the plasmid pGAD-She3-Ct. The regions mutated are indicated by brackets (for stems) or in gray letters (for loops). The strength of the interaction of the RNA mutants with the She3p-Ct/She2p complex in the yeast three-hybrid assay is reported as lacZ expression level (Miller units) or growth on a plate lacking histidine. Values for the percentage of localization of the mutants are from references 7 and 8.
Overall, in each localization element RNA, the specific region or domain previously found to be important for its localization function were also essential for She2p binding. These domains included two or three stems, and two asymmetric internal loops: stems II, III, and IV and internal loops II and III for E1; stems II and III and internal loops II and III for E2A; and stems I and II and loops I and II for E2B-D1 (see Fig. 1A). Only in the E2B-D1 element was a terminal loop shown to be important for She2p binding. An exception is E3, where only the upper stem (stem II) and the internal loop (loop I) were found to be part of the She2p binding domain (25).
Set of specific nucleotides define a conserved She2p-binding motif in each ASH1 mRNA localization element.
Since the domain recognized by She2p in each localization element was constituted of asymmetric loops, we wanted to determine which nucleotides in these loops might be conserved between the four localization elements and might be part of a She2p-binding RNA motif. Using PCR and degenerate primers, we produced libraries for the four ASH1 localization elements in which the sequences of the internal loops were partially randomized, with 40 to 50% mutation at each position depending on the localization element (see Fig. 4A, underlined nucleotides). We did not mutate the nucleotides in the stems of these RNAs since it was the base pairs and not the sequences of these stems that were found to be determinant for She2p binding (see Fig. 3).
FIG. 4.

In vivo selection of partially randomized ASH1 localization element RNAs. (A) Diagram of the mutagenesis performed on the She2p-binding domains of the four ASH1 localization elements. Partially randomized positions are underlined. Nucleotides conserved in >95% of the clones isolated are indicated in capital letters and italicized. The conserved CGA sequence is boxed with large dotted lines, whereas the conserved cytosine in E1, E2A, and E2B-D1 is boxed with small dotted lines. A cytosine at an analogous position on the element E3 is indicated by an asterisk. (B) Fluorescent in situ hybridization on the lacZ mRNA fused to ASH1 localization element variants isolated from the three-hybrid screen. Numbers correspond to clones listed in the Table 1. DAPI, DNA staining; DIC, Nomarski.
These libraries of partially randomized localization element RNAs were used in our yeast three-hybrid screen in order to identify the localization element variants that still interacted with She2p, which lead to the growth of yeast colonies on a -Ura-Leu-His medium with 3-amino-1,2,4-triazole. For each library, 40 or so positive clones were isolated, and their pIIIA/MS2-2 plasmid (which expressed the localization element RNA) was purified and sequenced. In order to confirm the interaction of these localization element variants with She2p and eliminate the false positives, they were retransformed into the yeast strain and tested for their β-galactosidase expression in the three-hybrid system. All the variants isolated, sequenced, and tested for their β-galactosidase activity are listed in Tables S3 to S7 of the supplemental material. Overall, most of the variants had a β-galactosidase activity similar or superior to that of the wild-type localization element.
For each localization element RNA, we selected five or six variants in order to study their localization capabilities. These variants were chosen for the high number of mutations in their sequences and/or their high β-galactosidase expression in the three-hybrid assay (Table 1). Each variant was inserted at the 3′ end of the lacZ reporter gene and the localization of this reporter mRNA was determined by fluorescence in situ hybridization. As shown in Fig. 4B and Table 1, all the variants of the four ASH1 localization elements were able to localize the lacZ reporter mRNA like the wild-type localization elements. The localization element variants isolated from our three-hybrid assay were thus fully functional localization elements.
TABLE 1.
Analysis of the functionality of localization element variants isolated from the three-hybrid screens: interaction with She2p and localization function
| Localization element | Type | Sequencea | LacZb (% of WT) | Localizationc (%) |
|---|---|---|---|---|
| E1 | WT | GACTATGTTAAAATACGCGAAGAAGTGGCTCATTTCAAGCCATTAAGTATACCCAACTTAACTAATAATC | 1 | 75 |
| 8 | GACTATGTTAAGATACACGCACAAGTGGCTCATTTCAAGCCATTAGGTATCCGGAAATTAACTAATAATC | 4.2 | 66 | |
| 12 | GACTATGTTAAGATACGCGAAAAAGTGGCTCATTTCAAGCCATTAAGTATACCCAACTTAACTAATAATC | 10.1 | 70.5 | |
| 26 | GACTATGTTAAGATACGCAAAAGAGTGGCTCATTTCAAGCCATTAAGTATACCCAACTTAACTAATAATC | 3.4 | 76 | |
| 33 | GACTATGTTAACATACACGA TTAAGTGGCTCATTTCAAGCCATTACGTATACTCGACTTAACTAATAATC | 9.5 | 63.5 | |
| 43 | GACTATGTTAACATACGCTAAGGAGTGGCTCATTTCAAGCCATTATGTATTGCTAAATTAACTAATAATC | 3.0 | 65 | |
| E2A | WT | TTGCGAATAGAGACATTCTATCGAACAATTCCAAATCTAATGTAAGGAAACC ATCTAAGAACAAAATCTC | 1 | 72 |
| 10 | TTGCGAATAGAGACATTCTATCGAACAATTCCAAATCTAATGTAAGGAATTT GGG TAAGAACAAAATCTC | 10.1 | 69 | |
| 12 | TTGCGAATAGAGACATTCTAACGAAGGATTCCAAATCTAATGTAAGGAATCGTTG GCAGAACAAAATCTC | 9.2 | 74 | |
| 34 | TTGCGAATAGAGACATTCTACCGAAGAATTCCAAATCTAATGTAAGGAAACC ATC TAAGAACAAAATCTC | 7.0 | 73.5 | |
| 48 | TTGCGAATAGAGACATTCTATCGAACAATTCCAAATCTAATGTAAGGAACGG AATCAAGAACAAAATCTC | 8.6 | 72 | |
| 61 | TTGCGAATAGAGACATTCTATCGAAAACTTCCAAATCTAATGTAAGGAAGTGTGC GTAGAACAAAATCTC | 1.5 | 79 | |
| 73 | TTGCGAATAGAGACATTCTAACGAACACTTCCAAATCTAATGTAAGGAAGTT AGC ATAGAACAAAATCTC | 7.0 | 75 | |
| E2B/D1 | WT | TGCATCTTCATCTCCATCTCCCTCCACACCGACGAAAAGTGGCAAGATGAGATCAAGATCATCCTCACCTG | 1 | 73 |
| 5 | TGCATCTTCATCTCCATCTACATCCACACCGACGAACAGTGGCAAGATGAGATCAAGATCATCCTCACCTG | 1.1 | 68 | |
| 12 | TGCATCTTCATCTCCATCTCCCTCCACACCGACGTTACGTGGCAAGATGAGATCAAGATCATCCTCACCTG | 1.4 | 65 | |
| 69 | TGCATCTTCATCTCCATCTACCTCCACATCGACGAAAAGTGGCAAGATGAGATCAAGATCATCCTCACCTG | 1.2 | 71 | |
| 87 | TGCATCTTCATCTCCATCTCCTTCCACACCGACGAAAAGTGGCGAGATGAGATCAAGATCATCCTCACCTG | 0.7 | 87 | |
| 106 | TGCATCTTCATCTCCATCTCACTCCACCCCGACGTAAAGTGGCAAGATGAGATCAAGATCATCCTCACCTG | 0.4 | 69 | |
| E3 | WT | AGAGAATTGATACATGGATAACTGAATCTCTTTCAACTAATAAGAGACATTATCACG AAA CAA TTGTACA | 1 | 67 |
| 3 | AGAGAATTGATACAAGGATAACTGAATCTCTTTCAACTAATAAGAGACATTATCTCG TAT ATG TTGTACA | 2.9 | 69 | |
| 20 | AGAGAATTGATACACCGATAACTGAATCTCTTTCAACTAATAAGAGACATTATCACG GCG CTT ATGTACA | 1.6 | 87 | |
| 30 | AGAGAATTGATACATGGATAACTGAATCTCTTTCAACTAATAAGAGACATTATCTCA TCA AAT ATGTACA | 1.2 | 68 | |
| 38 | AGAGAATTGATACATTGATAACTGAATCTCTTTCAACTAATAAGAGACATTATCACG AAA CAA TTGTACA | 3.4 | 74 | |
| 42 | AGAGAATTGATACAAGGATAACTGAATCTCTTTCAACTAATAAGAGACATTATCCCG ACA TAC TTGTACA | 6.1 | 64 |
Randomized positions are underlined. Mutated positions are indicated in italics.
β-Galactosidase activity in a three-hybrid assay, normalized to the wild-type (WT) element.
Percentage of localization of a reporter lacZ mRNA fused to the localization element sequence.
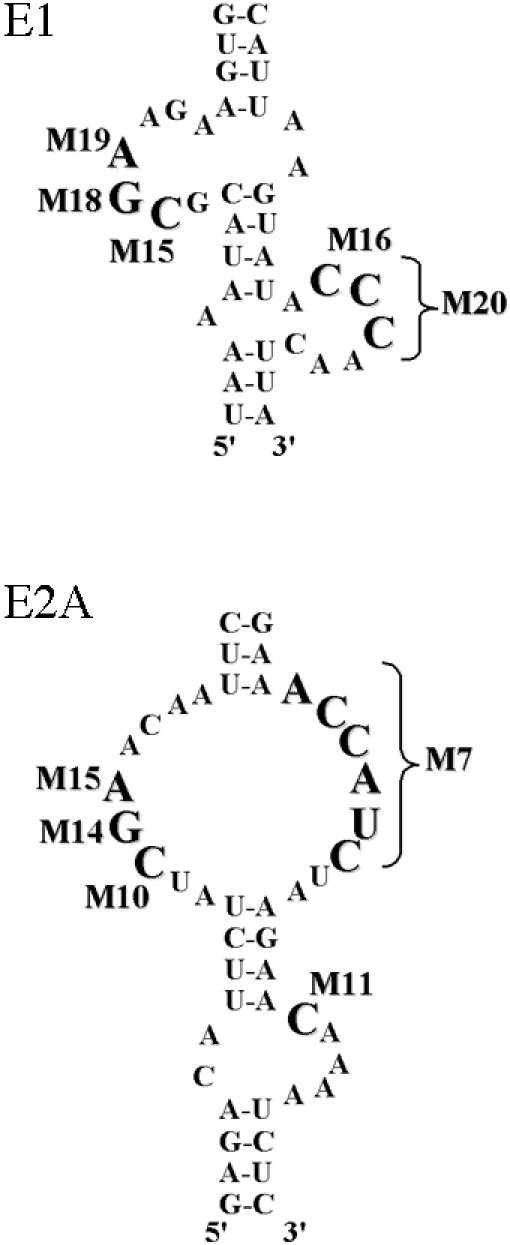
An analysis of the sequences obtained revealed a certain number of highly conserved nucleotides (i.e., present in >95% of the variants isolated) in each localization element RNA. These nucleotides are highlighted in the localization element structures in Fig. 4A. Whereas the nucleotides in some loops were highly conserved between variants, like loop I of the E2A element, other loops, like the internal loop of E3 had only one conserved nucleotide, a C (Fig. 4A). Looking at the nucleotides common between the four localization elements, we found a CGA sequence highly conserved in three of the four elements: E1, E2A, and E2B (solid boxes in Fig. 4A). Interestingly, while the E3 element also has a CGA sequence in its loop I (marked by a solid box in Fig. 4A), the only highly conserved nucleotide is the C of the CGA sequence. Another nucleotide highly conserved between these elements is a single C which is always present in a loop separated from the CGA sequence by a 4-base-pair stem (dotted boxes in Fig. 4A). We also observed a C about 5 base pairs above the loop I of the E3 element (marked by an asterisk in Fig. 4A), suggesting that these cytosines may be involved in She2p binding.
Mutation of the highly conserved nucleotides in the RNA motif resulted in loss of both She2p binding and localization function.
Analysis of the sequences obtained by in vivo selection revealed a possible consensus She2p-binding RNA motif which contains two highly conserved determinants: a CGA triplet and a single cytosine, both in a terminal or an internal loop, and separated by a 4-base-pair stem. These two determinants were conserved in >95% of the sequences obtained for the E1, E2A and E2B localization elements (see Tables S3 to S7 in the supplemental material). Moreover, the 4-base-pair stem separating these two determinants was essential for She2p binding in each of the three localization element (see Fig. 3).
In order to explore the importance of the CGA triplet and the single cytosine for She2p binding, we created point mutations by replacing the single cytosine and the cytosine of the CGA triplet by uracil or adenine and tested the binding of the She2 protein to these mutants using the three-hybrid assay. As shown in Fig. 5A, mutation of the two cytosines in the E1, E2A, and E2B localization elements resulted in loss of the interaction between She2p and the element RNAs, suggesting that these nucleotides are essential for She2p binding. In the case of the E3 localization element, only one highly conserved cytosine was identified in the in vivo selection experiment. Once mutated (mutant E3-M15), the interaction with She2p was lost (see Fig. 5A).
FIG. 5.

Two conserved cytosines in the four ASH1 localization elements define a She2p-binding motif. (A) Mutagenesis of each highly conserved cytosine to uracil or adenine in the ASH1 localization element results in the loss of interaction with She2p in the yeast three-hybrid assay. Mutated cytosines are boxed. lacZ expression is in Miller units. (B) Fluorescent in situ hybridization on the lacZ mRNA fused to ASH1 localizationelements mutated on the conserved cytosines. The presence of these mutations in the localization elements leads to the delocalization of the lacZ-E1, lacZ-E2A, lacZ-E2B, and lacZ-E3 reporter mRNAs. DAPI, nuclear DNA; DIC, Nomarski. (C) Measurement of the percentage of budding yeast cells with bud-localized lacZ mRNA fused to wild-type or mutated localization elements. (D) Coimmunoprecipitation of She2p-Myc and lacZ-E1 mRNAs was disrupted by mutation of the conserved cytosines in the element E1. Reverse transcription-PCR detection of the lacZ mRNA fused to wild-type localization element E1 (lane 1) or mutants M15 (lane 2) and M16 (lane 3) of the element E1 was performed on either total yeast extract (total) or on the pellet of the immunoprecipitated Myc-tagged She2p (IP+RT). A yeast strain expressing the lacZ-E1 mRNA and She2p without a Myc tag (lane 4) or reverse transcription-PCRs on the immunoprecipitates without reverse transcriptase (IP-RT) were used as controls.
Another cytosine is located in the 5-base-pair stem above the internal loop of E3. This cytosine is bulged out of the stem (Fig. 5A). This pattern is reminiscent of the two cytosines separated by a 4-base-pair stem observed in the localization elements E1, E2A, and E2B. Whereas the upper stem of the E3 element had been previously mutated, this cytosine was always maintained in all the mutants (7), so its importance in the function of this localization element was not tested. We replaced this cytosine by a uracil (mutant E3-M16) and tested the interaction of this mutated element E3 RNA with She2p in the three-hybrid assay. Interestingly, this mutation resulted in the loss of interaction with She2p (Fig. 5A).
In order to verify that these mutations affected the localization function of the elements, we inserted all the mutants at the 3′ end of the lacZ reporter gene and the localization of this reporter mRNA was determined by fluorescence in situ hybridization. As shown in Fig. 5B and C, mutation of these cytosines in the four ASH1 localization elements resulted in the delocalization of the reporter mRNA, suggesting that these nucleotides are essential for the proper recognition of the localization elements E1, E2A, E2B and E3 by the localization machinery. To confirm that the observed defect in localization was caused by a decreased interaction between the localization element and She2p in vivo, we immunoprecipitated a Myc-tagged She2p and tested for the presence of the lacZ mRNA reporter by reverse transcription-PCR. As shown in Fig. 5D, we detected the presence of the lacZ mRNA fused to the wild-type element E1 in the precipitate of She2p-Myc. However, when the lacZ mRNA contained E1 with a mutation in the conserved cytosines, this transcript was not detected in the immunoprecipitates, suggesting that these mutations impaired the interaction between the localization element and She2p in vivo.
To further confirm the results from the in vivo selection experiments, we also mutated the guanosine and adenosine moieties of the CGA triplet in two localization elements, E1 and E2A, and compared them to mutants in poorly conserved regions of the asymmetric loops of these localization elements. As shown in Table 2, mutagenesis of the three nucleotides of the CGA triplet and the single cytosine all resulted in the same phenotype: loss of She2p binding and delocalization of the reporter mRNA. Meanwhile, mutations in the poorly conserved nucleotides of the asymmetric loops resulted in functional localization elements.
She2p-binding domains in the four ASH1 localization elements adopt a similar three-dimensional fold.
Out of these analyses emerged a common RNA motif, in the ASH1 localization elements, which is recognized by the RNA-binding protein She2p. This motif consists of two loops separated by a short stem of 4 base pairs, with a conserved cytosine in one loop and a conserved CGA triplet in the other loop, both on opposite strands of the RNA loop-stem-loop structure. However, we observed some variations in this RNA motif among the localization elements. For instance, in the E3 element, the GA of the CGA triplet is not essential, the stem has 5 base pairs instead of 4, and one of the loops is replaced by a single bulged cytosine. Also, the number of nucleotides that separate the two cytosines from the stem varies among the elements from zero (for E2A, E2B-D1, and E3) to one (both cytosines of E1, and E3) or two (for E2A and E2B-D1) nucleotides (see Fig. 5A).
A rule of distance emerged from the inspection of the structures of the She2p-binding motifs: there are always 6 nucleotides between the two cytosines, suggesting that it is the distance between the two cytosines, and not the length of the stem that separates them, that is important for She2p binding. We therefore described the localization elements with the following descriptor: (number of nucleotides that separate the 5′ cytosine from the stem:length of the stem:number of nucleotides that separate the stem from the 3′ cytosine). For element E3, it gives 0:5:1; for E1, 1:4:1; and for E2A and E2B, 2:4:0 (Fig. 6A). The sum of these three numbers is always 6. We therefore predict from these results that other She2p-binding motifs may exist with the 1:5:0 and 0:4:2 descriptors. Even though there are variations in the secondary structures of these RNAs, they may adopt a similar three-dimensional structure which is recognized by She2p.
FIG. 6.

Distance between the two conserved cytosines is important for She2p recognition. (A) Descriptors used in the program MC-SEARCH. (B) Overlap of three structures, one from each descriptor, that were identified by the program. The cytosines are colored according to their descriptor motif in panel A. The distance indicated corresponds to the distance between the 3′ phosphates of the two cytosines. For E3 (yellow), the motif comes from the 23S rRNA, starting at position 295 (PDB 1JZY). For E1 (green), the motif comes from an hairpin similar to the P5Abc region of the group I intron, starting at position 4 (PDB 1EOR). For E2A/B (red), the motif comes from the 23S rRNA, starting at the position 1463 (PDB 1M1K). The root mean square deviation in their common region (the stem structure) is 1.9 Å. (C) Increasing the length of the stem separating the two cytosines in each localization element leads to a decreased interaction with She2p in the yeast three-hybrid assay. The conserved cytosines are labeled in gray. lacZ expression is in Miller units.
In order to explore this possibility, we used an automated program called MC-SEARCH (Gendron and Major, unpublished results) that searches for RNA structure patterns in the tertiary structures of the PDB database. Using secondary structure descriptors of the four ASH1 localization element She2p-binding motifs (see Fig. 6A), this program identified the RNA three-dimensional structures in the PDB database that fit these descriptors. Some 925 structures were thus identified: 123 for E1, 85 for E2A/E2B-D1, and 717 for E3 (this motif is less constrained than the others, mainly because of the absence of the CGA triplet). For each structure that fit the descriptors, the distance between the 3′ phosphates of the two conserved cytosines was measured. Interestingly, this distance was highly similar among all the structures, with an average distance of 28.3 ± 0.9 Å for the structures that fit the E1 descriptor, 28.0 ± 1.0 Å for the structures that fit the E2A/E2B-D1 descriptor and 28.2 ± 0.7 Å for the structures that fit the E3 descriptor. Even when the structures of different descriptors were superimposed, we observed an excellent conservation of the distance between the two cytosines (see Fig. 6B). These results suggest that the variations in the secondary structures of the localization element RNAs can still lead to similar three-dimensional folds.
In order to test this model, we increased the length of the stem separating the two cytosines by adding four additional base pairs (around 1/3 of a helix turn) in each ASH1 localization element RNA and measuring the impact of this modification on their interaction with She2p with the three-hybrid assay. As shown in Fig. 6C, increasing the distance between the two cytosines led to the complete loss of the interaction between the localization elements and She2p, suggesting that the distance between these two cytosines and/or their spatial orientation is important for the recognition of this motif by She2p.
Identification of the She2p-binding RNA motif in other bud-localized mRNAs.
To further confirm the relevance of the She2p-binding RNA motif for the sorting of bud-localized mRNAs, we searched for this motif in the sequences of the other known bud-localized transcripts in yeast (38). To do so, we opted for a computer-based approach with the RNAMotif software (11) that searches for RNA primary and secondary structure patterns in a sequence database using descriptors based on the main determinants identified in the She2p-binding RNA motif (see the supplemental material). With this approach, we identified four potential candidates, cloned these sequences, and tested them in our yeast three-hybrid assay.
Of the four candidate RNA motifs, two of them were found to interact with She2p: one in the IST2 mRNA (positions 2694 to 2785) and one in the YMR171c mRNA (positions 1540 to 1645) (Fig. 7B). Both RNAs could be folded in the loop-stem-loop structure with the conserved C and CGA determinants separated by a distance of six nucleotides (see Fig. 7A). Mutation of the conserved cytosines in the putative localization element from the YMR171c mRNA resulted in loss of interaction with She2p in the three-hybrid assay, further supporting our prediction (Fig. 7C). Interestingly, both cytosines in the YMR171c localization element are part of a CGA triplet.
FIG. 7.

Identification of a She2p-binding motif in the bud-localized IST2 and YMR171C mRNAs. (A) Predicted secondary structures of the IST2 and YMR171C mRNA localization elements. The conserved CGA sequence is boxed with plain lines, whereas the conserved cytosine is boxed with dotted lines. The nucleotides are numbered starting from the adenosine of the start codon as +1. (B) Three-hybrid assays using yeast strain YBZ1 with either wild-type or knockout SHE2 gene, transformed with a plasmid expressing the GAL4 activation domain fused with the C-terminal domain of She3p, along with a plasmid expressing one of the indicated MS2 fusion RNAs. Controls: empty vector (pIIIA/MS2-2) and the iron response element RNA (IRE). (C) Mutagenesis of each highly conserved cytosine in the YMR171c mRNA localization element results in the loss of interaction with She2p in the yeast three-hybrid assay. (D) Fluorescent in situ hybridization on the lacZ mRNA fused to the IST2 and wild-type, or mutated, YMR171C mRNA localization elements. The percentage of budding yeast cells with bud-localized lacZ mRNA is indicated. DAPI, nuclear DNA; DIC, Nomarski.
Finally, to confirm the function of these putative localization elements, they were inserted at the 3′ end of the lacZ reporter gene and the localization of this reporter mRNA was determined by fluorescence in situ hybridization. As shown in Fig. 7D, the RNA motifs from the IST2 and YMR171c mRNAs were able to localize a reporter mRNA to the bud tip, suggesting that both act as functional localization elements. Mutation of the conserved cytosines in the YMR171c localization element resulted in the loss of localization of the lacZ reporter mRNA. Altogether, our results show that the RNA motif identified in this study contains all the necessary determinants that provide the specificity for the recognition of bud-localized transcripts by the yeast mRNA localization machinery.
DISCUSSION
In this study, we have identified a conserved three-dimensional RNA motif, present in the four ASH1 localization elements and in two other bud-localized transcripts, that is essential for She2p recognition and for the localization function of these elements. An in vivo selection approach, using partially randomized libraries of the localization element RNAs coupled with a modified yeast three-hybrid assay, led to the identification of the nucleotides essential for She2p binding in each element. While the interactions detected by the modified three-hybrid assay are indirect (since we used the C-terminal domain of She3p as the bait), we have also shown that more direct assays (like using She2p as the bait or using RNA band-shift assays) did not provide comparable readout and sensibility to measure the effect of mutations in the ASH1 localization elements on their interaction with She2p.
This approach has been vindicated by the consistent results obtained from the screens, where a motif consisting of a CGA triplet and a single conserved cytosine in two loops separated by a short stem was found to be the only conserved feature among the localization elements. Mutation of this motif resulted in a decreased interaction with She2p and loss of the localization function of the elements. Interestingly, a distance of ≈28 Å between the two cytosines must be maintained in order to preserve the interaction with She2p, suggesting that three-dimensional features in these RNAs are essential for She2p recognition. However, our experimental results cannot determine if it is the distance between the cytosines or the spatial orientation of these residues or both that are important, since any insertion or deletion within the helix that separates the cytosines will affect both.
Our results suggest that the RNA motif identified contains the main specificity determinants of the She2p binding domain. These determinants are highly conserved in three of the four ASH1 localization elements (E1, E2A, and E2B). For the element E3, several features of the She2p binding RNA motif are conserved: the two essential cytosines on opposite strands, separated by a stem and at a distance of 28 Å from each other. However, variations from the classic motif are present in this element: one of the loops is replaced by a single bulged cytosine and the GA of the CGA triplet is not essential. In this case, a particularity of this localization element, a single cytosine bulging from a stem instead of being incorporated in a loop, may result in a conformation where She2p binding becomes less dependent on the GA of the CGA triplet. Interestingly, among the variants isolated for the element E1, we found three variants with mutations in the GA of the CGA triplet (mutants 8, 26, and 43). These variants can be folded like the element E3 (with a stem of 5 or 6 base pairs between the two conserved cytosines; data not shown), suggesting that the GA of the CGA triplet may be less important for She2p binding in some structures.
While these determinants are essential for She2p recognition, other elements around this RNA motif have been found to be important. For instance, the stems around the internal loops of the elements E1, E2A, and E2B-D1 were also shown to be important for She2p binding, possibly by participating in the proper folding of these RNAs. From the randomized libraries, we also identified highly conserved nucleotides in each individual element that were not present in the other three elements (see Fig. 4A). The nucleotides surrounding the highly conserved CGA triplet, present in all four localization elements, provide such example (see the elements E1 versus E2B-D1). These nucleotides may be involved in noncanonical base pairs and may improve the accessibility of the single cytosine or of the CGA triplet for She2p binding in the particular context of a given RNA structure. Overall, our results suggest that the She2p-binding motif requires a set of conserved specificity determinants around which several nucleotide sequence combinations are tolerated only if they maintain the proper folding of this motif.
Recently, more than twenty new mRNAs have been found to be specifically localized at the bud tip of yeast cells during mitosis (38). The localization of these mRNAs depends on Myo4p, She3p, and She2p, suggesting that the localization pathway of the ASH1 mRNA is not restricted to this transcript. No specific zipcodes have yet been characterized in any of these transcripts, but since they were identified by coimmunoprecipitation with She2p, it is highly probable that they all contain She2p-binding domains similar to the one found in the ASH1 zipcodes. Indeed, using the She2p binding RNA motif in a computer search, we have been able to identify functional localization elements in two of these transcripts: IST2 and YMR171c. Ist2p is a membrane protein targeted to the plasma membrane via a new trafficking pathway that requires the localization of its mRNA to the daughter cell cortex (21, 40). The function of YMR171c is still unknown. The identification of the same RNA motif in the localization elements of the bud-localized ASH1, IST2, and YMR171c mRNAs strongly supports our conclusion that this motif contains the main determinants required for their recognition by She2p.
These results raise questions about how She2p may recognize these determinants. The recent publication of the three-dimensional structure of She2p revealed that this protein acts as a homodimer and that a series of basic residues essential for the interaction between this protein and the ASH1mRNA localization elements are clustered in a specific region that folds as a basic helical hairpin (30). Interestingly, this region of the protein, from R44 to K57, covers a distance of around 27 Å in length (data not shown), which is very close to the conserved 28 Å distance between the CGA triplet and the single cytosine of the She2p-binding motif. Therefore, we can speculate that this RNA motif may dock into the basic helical hairpin, where amino acid residues at the extremities of this RNA binding motif (R43, R44, K57, and K60) may interact with the conserved CGA triplet and the single cytosine of the RNA motif, while basic residues in the middle (R52 and R63) may interact with the phosphate backbone of the helix that separates the two loops.
While the presence of two RNA-binding domain per homodimer would suggest that two RNA molecules could bind the homodimer (2:1 ratio), Niessing et al. have shown instead that a She2p homodimer binds only one RNA molecule (1:1 ratio) (30). They proposed that the RNA molecule arches over the upper, uncharged surface of the She2p homodimer and binds simultaneously to the RNA-binding domain of both monomers. However, our electrophoretic mobility shift assays suggest the presence of both 1:1 and 2:1 ratios of RNA zipcode versus She2p homodimer. The ratios observed vary with the localization element (E2B-D1 has only one shifted species while E2A has two shifted species of equivalent intensities), and may depend on the size of the RNA zipcode used in the electrophoretic mobility shift assay (the E2B-D1 RNA has 75 nucleotides while the E2A RNA has 103 nucleotides).
The low level of sequence conservation and the dependence on secondary and tertiary structure of their target RNA is a common feature found in RNA-binding proteins involved in the transport of several different mRNAs within the same cell. For instance, in Drosophila, the K10, bcd, and hairy mRNAs are localized during oogenesis and in blastoderm embryos by the Egalitarian (Egl) and Bicaudal-D (BicD) localization machinery (5). The localization of these transcripts depends on zipcodes containing short stem-loop structures without sequence similarities (6, 27, 37), suggesting that structural features in these RNAs are recognized by the Egl-BicD localization pathway (6). Another example is Staufen, an RNA-binding protein containing double-stranded RNA-binding domains, which interacts with the localized mRNAs bicoid, oskar, and prospero in Drosophila and BC1 and CamKIIα mRNAs in neurons (28). Although Staufen interacts with specific mRNAs in vivo, it binds nonspecifically to double-stranded RNA in vitro (18). Our study show that mRNAs with zipcodes lacking primary sequence similarity can rely on a few conserved nucleotides properly oriented in their three-dimensional structure in order to be recognized by the same localization machinery.
Supplementary Material
Acknowledgments
We thank Caroline Martel for technical help.
This work was supported by grants from the Canadian Institutes of Health Research (F.M. and P.C.). F.M. is a CIHR Investigator. P.C. is supported by the Fonds de Recherche sur la Nature et les Technologies du Québec. We are grateful to L. DesGroseillers, G. Ferbeyre, P. Legault, and E. Querido for their critical reading of the manuscript.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Addess, K. J., J. P. Basilion, R. D. Klausner, T. A. Rouault, and A. Pardi. 1997. Structure and dynamics of the iron responsive element RNA: implications for binding of the RNA by iron regulatory binding proteins1. J. Mol. Biol. 274:72-83. [DOI] [PubMed] [Google Scholar]
- 2.Bashirullah, A., R. L. Cooperstock, and H. D. Lipshitz. 1998. RNA localization in development. Annu. Rev. Biochem. 67:335-394. [DOI] [PubMed] [Google Scholar]
- 3.Bertrand, E., P. Chartrand, M. Schaefer, S. M. Shenoy, R. H. Singer, and R. M. Long. 1998. Localization of ASH1 mRNA particles in living yeast. Mol. Cell. 2:437-445. [DOI] [PubMed] [Google Scholar]
- 4.Bohl, F., C. Kruse, A. Frank, D. Ferring, and R.-P. Jansen. 2000. She2p, a novel RNA-binding protein tethers ASH1 mRNA to the Myo4p myosin motor via She3p. EMBO J. 19:5514-5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bullock, S. L., and D. Ish-Horowicz. 2001. Conserved signals and machinery for RNA transport in Drosophila oogenesis and embryogenesis. Nature 414:611-616. [DOI] [PubMed] [Google Scholar]
- 6.Bullock, S. L., D. Zicha, and D. Ish-Horowicz. 2003. The Drosophila hairy RNA localization signal modulates the kinetics of cytoplasmic mRNA transport. EMBO J. 22:2484-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chartrand, P., X.-H. Meng, R. H. Singer, and R. M. Long. 1999. Structural elements required for the localization of ASH1 mRNA and of a green fluorescent protein reporter particle in vivo. Curr. Biol. 9:333-336. [DOI] [PubMed] [Google Scholar]
- 8.Chartrand, P., X. Meng, S. Huttelmaier, D. Donato, and R. H. Singer. 2002. Asymmetric sorting of Ash1p in yeast results from inhibition of translation by localization elements in the mRNA. Mol. Cell 10:1319-1330. [DOI] [PubMed] [Google Scholar]
- 9.Chartrand, P., R. H. Singer, and R. M. Long. 2001. RNP localization and transport in yeast. Annu. Rev. Cell Dev. Biol. 17:297-310. [DOI] [PubMed] [Google Scholar]
- 10.Chartrand, P., R. H. Singer, and R. M. Long. 2000. Sensitive and high-resolution detection of RNA in situ. Methods Enzymol. 318:493-506. [DOI] [PubMed] [Google Scholar]
- 11.Gautheret, D., F. Major, and R. Cedergren. 1990. Pattern searching/alignment with RNA primary and secondary structures: an effective descriptor for tRNA. Comput. Appl. Biosci. 6:325-331. [DOI] [PubMed] [Google Scholar]
- 12.Gendron, P., S. Lemieux, and F. Major. 2001. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 308:919-936. [DOI] [PubMed] [Google Scholar]
- 13.Gonsalvez, G. B., K. A. Lehmann, D. K. Ho, E. S. Stanitsa, J. R. Williamson, and R. M. Long. 2003. RNA-protein interactions promote asymmetric sorting of the ASH1 mRNA ribonucleoprotein complex. RNA. 9:1383-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez, I., S. B. C. Buonomo, K. Nasmyth, and U. von Ahsen. 1999. ASH1 mRNA localization in yeast involves multiple secondary structural elements and Ash1 protein translation. Curr. Biol. 9:337-340. [DOI] [PubMed] [Google Scholar]
- 15.Guldener, U., S. Heck, T. Fielder, J. Beinhauer, and J. Hegemann. 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24:2519-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hook, B., D. Bernstein, B. Zhang, and M. Wickens. 2005. RNA-protein interactions in the yeast three-hybrid system: affinity, sensitivity, and enhanced library screening. RNA. 11:227-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jansen, R.-P., C. Dowzer, C. Michaelis, M. Galova, and K. Nasmyth. 1996. Mother cell-specific HO expression in budding yeast depends on the unconventional myosin Myo4p and other cytoplasmic proteins. Cell 84:687-697. [DOI] [PubMed] [Google Scholar]
- 18.Johnston, D., N. Brown, J. Gall, and M. Jantsch. 1992. A conserved double-stranded RNA-binding domain. Proc. Natl. Acad. Sci. USA 89:10979-10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnstone, O., and P. Lasko. 2001. Translational regulation and RNA localization in Drosophila oocytes and embryos. Annu. Rev. Genet. 35:365-406. [DOI] [PubMed] [Google Scholar]
- 20.Jorgen, K., J. Egebjerg, and J. Christiansen. 1998. Analysis of RNA-protein complexes in vitro. Elsevier, Amsterdam, The Netherlands.
- 21.Juschke, C., D. Ferring, R.-P. Jansen, and M. Seedorf. 2004. A novel transport pathway for a yeast plasma membrane protein encoded by a localized mRNA. Curr. Biol. 14:406-411. [DOI] [PubMed] [Google Scholar]
- 22.Kiebler, M. A., and L. DesGroseillers. 2000. Molecular insights into mRNA transport and local translation in the mammalian nervous system. Neuron 25:19-28. [DOI] [PubMed] [Google Scholar]
- 23.Kislauskis, E. H., and R. H. Singer. 1992. Determinants of mRNA localization. Curr. Opin. Cell Biol. 4:975-978. [DOI] [PubMed] [Google Scholar]
- 24.Kloc, M., S. Bilinski, A. P. Chan, L. H. Allen, N. R. Zearfoss, and L. D. Etkin. 2001. RNA localization and germ cell determination in Xenopus. Int. Rev. Cytol. 203:63-91. [DOI] [PubMed] [Google Scholar]
- 25.Long, R. M., W. Gu, E. Lorimer, R. H. Singer, and P. Chartrand. 2000. She2p is a novel RNA-binding protein that recruits the Myo4p-She3p complex to ASH1 mRNA. EMBO J. 19:6592-6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Long, R. M., R. H. Singer, X. Meng, I. Gonzalez, K. Nasmyth, and R.-P. Jansen. 1997. Mating type switching in yeast controlled by asymmetric localization of ASH1 mRNA. Science 277:383-387. [DOI] [PubMed] [Google Scholar]
- 27.Macdonald, P. M., and K. Kerr. 1998. Mutational analysis of an RNA recognition element that mediates localization of bicoid mRNA. Mol. Cell. Biol. 18:3788-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mallardo, M., A. Deitinghoff, J. Muller, B. Goetze, P. Macchi, C. Peters, and M. A. Kiebler. 2003. Isolation and characterization of Staufen-containing ribonucleoprotein particles from rat brain. Proc. Natl. Acad. Sci. USA 100:2100-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munchow, S., C. Sauter, and R. Jansen. 1999. Association of the class V myosin Myo4p with a localised messenger RNA in budding yeast depends on She proteins. J. Cell Sci. 112:1511-1518. [DOI] [PubMed] [Google Scholar]
- 30.Niessing, D., S. Huttelmaier, D. Zenklusen, R. H. Singer, and S. K. Burley. 2004. She2p Is a novel RNA binding protein with a basic helical hairpin motif. Cell 119:491-502. [DOI] [PubMed] [Google Scholar]
- 31.Palacios, I. M., and D. S. Johnston. 2001. Getting the message across: the intracellular localization of mRNAs in higher eukaryotes. Annu. Rev. Cell Dev. Biol. 17:569-614. [DOI] [PubMed] [Google Scholar]
- 32.Rose, M. D., F. Winston, and P. Hieter. 1990. Methods in yeast genetics: a laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 33.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed., vol. 3. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 34.Schiestl, R., and R. D. Gietz. 1989. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr. Genet. 16:339-346. [DOI] [PubMed] [Google Scholar]
- 35.SenGupta, D. J., B. Zhang, B. Kraemer, P. Pochart, S. Fields, and M. Wickens. 1996. A three-hybrid system to detect RNA-protein interactions in vivo. Proc. Natl. Acad. Sci. USA 93:8496-8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serano, J., and G. M. Rubin. 2003. The Drosophila synaptotagmin-like protein bitesize is required for growth and has mRNA localization sequences within its open reading frame. Proc. Natl. Acad. Sci. USA 100:13368-13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serano, T., and R. Cohen. 1995. A small predicted stem-loop structure mediates oocyte localization of Drosophila K10 mRNA. Development 121:3809-3818. [DOI] [PubMed] [Google Scholar]
- 38.Shepard, K. A., A. P. Gerber, A. Jambhekar, P. A. Takizawa, P. O. Brown, D. Herschlag, J. L. DeRisi, and R. D. Vale. 2003. Widespread cytoplasmic mRNA transport in yeast: identification of 22 bud-localized transcripts using DNA microarray analysis. Proc. Natl. Acad. Sci. USA 100:11429-11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sil, A., and I. Herskowitz. 1996. Identification of asymmetrically localized determinant, Ash1p, required for lineage-specific transcription of the yeast HO gene. Cell 84:711-722. [DOI] [PubMed] [Google Scholar]
- 40.Takizawa, P. A., J. L. DeRisi, J. E. Wilhelm, and R. D. Vale. 2000. Plasma membrane compartmentalization in yeast by messenger RNA transport and a septin diffusion barrier. Science 290:341-344. [DOI] [PubMed] [Google Scholar]
- 41.Takizawa, P. A., A. Sil, J. R. Swedlow, I. Herskowitz, and R. D. Vale. 1997. Actin-dependent localization of an mRNA encoding a cell-fate determinant in yeast. Nature 389:90-93. [DOI] [PubMed] [Google Scholar]
- 42.Takizawa, P. A., and R. D. Vale. 2000. The myosin motor, Myo4p, binds Ash1 mRNA via the adapter protein, She3p. Proc. Natl. Acad. Sci. USA 97:5273-5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thio, G. L., R. P. Ray, G. Barcelo, and T. Schupbach. 2000. Localization of gurken RNA in Drosophila oogenesis requires elements in the 5′ and 3′ regions of the transcript. Dev. Biol. 221:435-446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.