

Abstract
Comparative genomics provides a rapid means of identifying functional DNA elements by their sequence conservation between species. Transcription factor binding sites (TFBSs) may constitute a significant fraction of these conserved sequences, but the annotation of specific TFBSs is complicated by the fact that these short, degenerate sequences may frequently be conserved by chance rather than functional constraint. To identify intergenic sequences that function as TFBSs, we calculated the probability of binding site conservation between Saccharomyces cerevisiae and its two closest relatives under a neutral model of evolution. We found that this probability is <5% for 134 of 163 transcription factor binding motifs, implying that we can reliably annotate binding sites for the majority of these transcription factors by conservation alone. Although our annotation relies on a number of assumptions, mutations in five of five conserved Ume6 binding sites and three of four conserved Ndt80 binding sites show Ume6- and Ndt80-dependent effects on gene expression. We also found that three of five unconserved Ndt80 binding sites show Ndt80-dependent effects on gene expression. Together these data imply that although sequence conservation can be reliably used to predict functional TFBSs, unconserved sequences might also make a significant contribution to a species' biology.
The ability of the cell to tightly control the expression of thousands of genes under a wide array of developmental and environmental conditions is still poorly understood for all except a few well studied processes (e.g., Stanojevic et al. 1991; Yuh et al. 1998; Vershon and Pierce 2000). A major goal of computational biology is to extend these specific examples to the generalized set of rules that explain the complex system of regulation used by the cell. In Saccharomyces cerevisiae, various experimental approaches have been used to identify a large number of regulatory motifs and the transcription factors that bind them (e.g., Bowdish and Mitchell 1993; Strich et al. 1994; Ozsarac et al. 1997; Pierce et al. 2003). Additionally, two comparative genomics approaches have identified many new motifs by their conservation between different yeast species (Cliften et al. 2003; Kellis et al. 2003). The combined experimental and computational approaches provide a sufficient knowledgebase of cis-regulatory motifs in S. cerevisiae, such that it is now possible to begin the next challenge of identifying which transcription factor binding sites (TFBSs) in the genome are functional, and under what circumstances they regulate transcription.
Identifying functional TFBSs is a difficult task because most transcription factor binding motifs are short, degenerate sequences occurring frequently in the genome. Even in the compact S. cerevisiae genome, many instances of these binding sites are likely to be nonfunctional, spurious matches to the motif sequence. One approach to this problem is chromatin immunoprecipitation experiments, which can identify the promoters bound by a particular transcription factor (Lee et al. 2002; Martone et al. 2003; Harbison et al. 2004). However, these data are limited by the conditions under which they are assayed. Alternatively, it has been shown that conservation of noncoding DNA between genomes is a good indicator of biological function (e.g., Loots et al. 2000; Bergman and Kreitman 2001; Boffelli et al. 2003; Frazer et al. 2004; Johnson et al. 2004; Woolfe et al. 2004), so it is plausible that functional and nonfunctional TFBSs may be distinguished by sequence conservation alone. One difficulty is that TFBSs may frequently be conserved by chance, rather than functional constraint. This frequency will depend on the amount of divergence between species. The observation that TFBSs are as conserved as their adjacent sequences (Cliften et al. 2003) implies that the Saccharomyces species are too closely related to identify functional TFBSs. However, an alternative explanation that must be considered is that both the TFBS and its flanking sequences are under functional constraint.
This alternative can be tested using molecular evolutionary models, which provide a probabilistic framework in which constrained and unconstrained sequences can be distinguished (Li 1997). In these models, sequences that are functionally constrained between species by purifying selection can be identified as those with fewer substitutions than expected in the absence of any constraint. Using these methods, we estimated that ∼40% of S. cerevisiae intergenic sequences are functionally constrained. Because of this, we developed a probabilistic method for calculating how often a particular TFBS would be conserved in the absence of any functional constraint. Using the synonymous rate to estimate the neutral rate of evolution, we found that the majority of TFBSs have a very low probability of being conserved among S. cerevisiae and its two closest relatives, S. paradoxus and S. mikatae. Experimental validation of multiple TFBSs illustrates that conservation among three closely related species is sufficient to predict functional TFBSs, making it possible to annotate the genome for functionally constrained binding sites for the majority of known transcription factors. Surprisingly, our annotation suggests that the TFBSs account for less than half of the functional constraint in noncoding sequences.
Results
Functional constraint in intergenic sequences
The identification of TFBSs by their sequence conservation between species requires that nonfunctional or unconstrained TFBSs are rarely conserved by chance. The probability that a nonfunctional binding site is conserved depends on the neutral substitution rate. We estimated the neutral substitution rate along the lineages leading to S. cerevisiae, S. paradoxus, and S. mikatae from the median synonymous substitution rate of 2098 coding sequence alignments of these species (Table 1). The total rate across the phylogeny is 0.83 substitutions per site and is a conservative estimate of the neutral rate, since synonymous sites have been shown to be under weak selective constraint (Akashi 2001). At this distance, the probability that a 10-base pair sequence is identical across the three species is 0.002 (Kimura 1980). This implies that TFBSs may rarely be conserved between these three species by chance.
Table 1.
Median substitution rates in coding and noncoding DNA
|
Substitution rate
|
|||||
|---|---|---|---|---|---|
| Lineage | dSa | dNb | dIc | dN/dS | dI/dS |
| S. cerevisiae | 0.23 | 0.02 | 0.12 | 0.07 | 0.52 |
| S. paradoxus | 0.13 | 0.01 | 0.08 | 0.09 | 0.59 |
| S. mikatae | 0.46 | 0.04 | 0.27 | 0.08 | 0.58 |
| Three species | 0.83 | 0.07 | 0.47 | 0.08 | 0.57 |
Synonymous substitution rate.
Nonsynonymous substitution rate.
Intergenic substitution rate.
The fraction of functionally constrained intergenic sequences can be estimated from the ratio of the intergenic to synonymous substitution rate (Table 1) (Wong and Nielsen 2004). From 4188 alignments, the median intergenic substitution rate across the three yeast species is 0.57, which implies that 43% of intergenic sequences are functionally constrained. The extent to which conserved TFBSs can account for this constraint is discussed below.
Identification of functionally constrained Ndt80 and Ume6 binding sites using a neutral model of molecular evolution
The neutral substitution rate implies that functional TFBSs can be identified by sequence conservation alone, regardless of the conservation of the flanking sequences. We tested this hypothesis using two well characterized transcription factors, Ndt80 and Ume6. Both proteins regulate the expression of meiosis-specific genes in S. cerevisiae. Ume6 is known to repress genes during vegetative growth and activate genes during early meiosis (Bowdish and Mitchell 1993; Steber and Esposito 1995). Ume6 affects gene expression by binding to the consensus sequence TSGGC GGCTAW (Williams et al. 2002). Ndt80 activates genes expressed in the middle stages of meiosis (Chu and Herskowitz 1998) by binding to the consensus sequence YGNCACAAAW (Pierce et al. 2003). We used a position weight matrix (PWM) representation of these TFBSs creating a probabilistic description of the nucleotide frequencies at each position of the motif. Our goal was to determine which sites matching these sequences function in Ndt80 or Ume6 transcriptional regulation. If functional Ndt80 and Ume6 binding sites are also functional in other species, then they are likely to be under purifying selection and can be identified in S. cerevisiae by their conservation at orthologous positions in other Saccharomyces species.
For each site identified as a match to the Ndt80 or Ume6 PWM (Hertz and Stormo 1999), we counted the number of differences observed between S. cerevisiae, S. paradoxus, and S. mikatae. The neutral model predicts that there will be an average of 4.8 and 5.3 differences across the three species for Ndt80 and Ume6, respectively, and that less than 1.5% of the Ndt80 and 0.8% of the Ume6 sites should have 0 or 1 difference between the three species. For both transcription factors, we found a substantial overrepresentation of sites with 0 or 1 difference, 63% of Ndt80 sites and 97% of Ume6 sites, suggesting that only a small number of the Ndt80 or Ume6 sites are consistent with a neutral model (Fig. 1). When we restricted the analysis to only those sites occurring in promoters of genes that are likely targets of these transcription factors (Chu et al. 1998; Williams et al. 2002), there was an even further enrichment for constrained sites (Fig. 1). These results imply that we can readily distinguish constrained and unconstrained sites using the evolutionary data from these three species.
Figure 1.

The observed and expected numbers of differences among S. cerevisiae, S. paradoxus, and S. mikatae in (A) 101 Ndt80 binding sites and (B) 82 Ume6 binding sites. The expected number of differences occurring within the TFBS was calculated using the synonymous substitution rate of 0.83 substitutions per site. The observed number of differences between the species was inferred by parsimony. The observed number of differences in Ndt80 sites occurring in the promoters of 39 middle meiosis genes (Chu et al. 1998) is shown in A. The observed number of differences in 23 Ume6 binding sites occurring in Ume6 regulated genes (Williams et al. 2002) is shown in B.
To find the conserved TFBSs in the genome, we identified all sites that had a significant match to the PWM at orthologous positions in the three species. The advantage of this conservation test is that it does not rely on sequence conservation, which is not necessarily equal to TFBS conservation due to the degenerate nature of TFBSs (Moses et al. 2003). For example, we found that six of 20 Ndt80 sites and eight of 21 Ume6 sites with a single difference were not actually conserved sites, in that the sequence in either the S. paradoxus or S. mikatae genome was no longer a significant match to the PWM. Additionally, we identified a match to the Ndt80 site in the GAS4 promoter of S. cerevisiae that has three differences across the three species, but the sequence in each species is a match to the Ndt80 PWM. In total, we identified 59 conserved Ndt80 TFBSs and 63 conserved Ume6 TFBSs. The list of genes with conserved Ndt80 and Ume6 TFBSs can be found in the Supplemental material (Tables S1 and S2).
The fraction of conserved Ndt80 and Ume6 TFBSs that are functional depends on how often a TFBS is conserved in the absence of selective constraint. We modeled the probability of TFBS conservation as the likelihood that a TFBS observed in S. cerevisiae remains a match to the PWM given the neutral rate of evolution of 0.83 substitutions per site. Because there can be a number of sequences that match a PWM, we enumerated all possible evolutionary descendents of the observed sequence and tested each sequence for a significant match to the PWM. If it matched, we calculated the probability of it occurring given the neutral substitution rate. The total probability that a TFBS is conserved is the sum of the probability of all sequences that maintain the TFBS (Fig. 2).
Figure 2.

Diagram of the method used to calculate the probability of TFBS conservation. (A) A hypothetical two-base pair-long motif, which specifies either T or C in the first position andaGinthe second position. (B) A schematic of how the probability of TFBS conservation is calculated. In this case, two of the 16 possible descendents are conserved instances of the initial TFBS sequence. The probability of TFBS conservation is the cumulative probability of both evolutionary paths, conditioned on the initial sequence and the evolutionary time between them.
For Ndt80 and Ume6, the probability that a single TFBS is conserved is 0.0058 and 0.0023, respectively. Given that 59 Ndt80 and 63 Ume6 sites are conserved, we expect that less than one of these conserved sites occurred by chance (Table 2). This low probability of TFBS conservation implies that conservation of Ndt80 and Ume6 across the three Saccharomyces species is a strong indicator that the binding site is under functional constraint, and we can therefore confidently predict that these conserved sites are functional in S. cerevisiae.
Table 2.
Binding site conservation for five known transcription factors
| Name | Consensus | S. cer. sitesa | S. par. sitesa | S. mik. sitesa | Conserved sitesb | Percent conservedc | P(cons)d | False discovery rate |
|---|---|---|---|---|---|---|---|---|
| ABF1 | RTCRYNNNNNACG | 671 | 670 | 666 | 470 | 70.0 | 0.017 | 0.024 |
| GAL4 | CGGNNNNNNNNNNNCCG | 144 | 152 | 134 | 21 | 14.6 | 0.022 | 0.153 |
| MCM1 | CCNNNWWRGG | 963 | 1019 | 911 | 194 | 20.1 | 0.023 | 0.112 |
| NDT80 | YGNCACAAAW | 141 | 133 | 141 | 59 | 41.8 | 0.006 | 0.015 |
| UME6 | TSGGCGGCTAW | 82 | 86 | 86 | 63 | 76.8 | 0.002 | 0.003 |
The number of matches to the PWM in each genome.
The number of conserved matches to the PWM.
The percentage of matches to the PWM that are conserved.
The probability of motif conservation.
Mutation of conserved Ndt80 and Ume6 binding sites produces Ndt80- and Ume6-dependent effects on gene expression
The conservation of Ndt80 and Ume6 binding sites suggests that they are true TFBSs and should therefore affect the expression levels of their adjacent genes. To test our predictions, we randomly selected four promoters containing conserved Ndt80 sites and four promoters containing conserved Ume6 sites (Fig. 3). We cloned each intergenic sequence, averaging 625 base pairs, and made mutations in the conserved TFBSs, changing the Ndt80 sites from YGNCACAAAW to YGNCTTCAAAW, and the Ume6 sites from TSGGCGGCTAW to TSGATTGCTAW. The expression of the mutant and wild-type promoters were compared using a β-gal reporter construct expressed from a yeast episomal plasmid. One of the promoters selected, the region upstream of ADY2, contains two conserved Ume6 TFBSs, and mutations were made in both and tested independently.
Figure 3.

Multiple sequence alignments of (A) four experimentally tested Ndt80 or (B) five experimentally tested Ume6 binding sites. The binding sites are highlighted in gray. Ten nucleotides flanking the 5′ and 3′ ends of the TFBS are also shown.
Eight of nine mutants produced a significant change in expression levels during meiosis compared to the wild-type promoters from which they were derived (Fig. 4). Mutations in the four Ndt80 sites in sporulation media resulted in a 50-fold decrease in gene expression on average, consistent with previously characterized Ndt80 binding sites (Ozsarac et al. 1997; Chu et al. 1998). Mutations in two of the Ume6 sites resulted in an average eightfold increase in expression from the mutant promoter, whereas mutations in the three others produced an average 100-fold decrease in gene expression levels. These data are consistent with previous studies that showed that Ume6 can act as both a repressor and activator of gene expression (Bowdish and Mitchell 1993). Interestingly, the two Ume6 binding sites within the ADY2 promoter have an opposite effect on expression levels, despite having identical sequences.
Figure 4.

(A) Expression levels from five S. cerevisiae promoters containing wild-type (white) or mutant (black) Ume6 sites after8hin sporulation media. (B) Expression levels from four promoters containing wildtype (white) or mutant (black) Ndt80 sites after8hin sporulation media. Expression levels from wild-type and mutant promoters in an ume6Δ (C) or ndt80Δ (D) strain after8hin sporulation media. The data displayed are the log2 of the mean β-gal expression intensity of five biological replicates. * = P < 0.05, ** = P < 0.01, by Student's t-test.
If the conserved Ndt80 and Ume6 binding sites are functional Ndt80 and Ume6 binding sites, the effects of mutations within these sites should be Ndt80- and Ume6-dependent. We measured expression levels in either an ndt80Δ or ume6Δ strain where appropriate (Fig. 4). Seven of the nine mutant promoters show no significant difference compared to wild type in the absence of the transcription factor. Surprisingly, the mutant Ndt80 site in YFL040W, the one promoter that showed no significant difference in the S288C background, showed a significant increase in gene expression in the ndt80Δ strain in complete media (Supplemental Fig. 2). Further examination of this promoter revealed that there is a conserved Sum1 binding site overlapping the Ndt80 site. Ndt80 and Sum1 compete for binding to similar sites in the genome, and Sum1 acts to repress meiotic genes during vegetative growth (Pierce et al. 2003). The difference between the mutant and wild-type promoters may be caused by disruption of Sum1 binding, but only in the absence of Ndt80. The two additional sites which showed a significant difference in the deletion strains are the Ume6 binding sites in the ADY2 promoter. These mutant sites, which showed opposing effects in the S288C background, showed similar small effects in the absence of Ume6.
Mutations in Ndt80 binding sites that are not conserved affect gene expression
One assumption underlying the comparative genomic approach is that functional TFBSs will be conserved between closely related species (Cliften et al. 2003; Kellis et al. 2003). To test the importance of conservation in identifying functional regulatory sites, we tested five Ndt80 binding sites that are not conserved between the three species for Ndt80-specific regulatory effects (Supplemental Fig. 1).
Of five unconserved sites, two (URA4 and YMR111C) showed an Ndt80-specific effect in sporulation medium, suggesting that these sites are functional Ndt80 binding sites in S. cerevisiae, despite their absence in the other species (Fig. 5). One additional site, YBR255C-A showed a significant effect on gene expression in complete media, but showed no significant effect in sporulation media (Supplemental Fig. 3). The remaining two sites did not alter transcription when mutated in the S288C background.
Figure 5.

Expression levels from five unconserved Ndt80 sites that are present in S. cerevisiae promoters. Wild type is shown in white, mutant in black. Expression levels are from wild-type and mutant promoters in the S288C strain (A) and ndt80Δ strain (B) after8hin sporulation media. The data displayed are the log2 of the mean β-gal expression intensity of five biological replicates. * = P < 0.05, ** = P < 0.01, by Student's t-test.
Genome annotation of transcription factor binding sites
The low probability of TFBS conservation under a neutral model combined with the experimental validation of multiple Ndt80 and Ume6 binding sites suggests that the same method may be applied to all other known S. cerevisiae transcription factor binding motifs. We compiled a list of 163 unique motifs from three sources (Zhu and Zhang 1999; Cliften et al. 2003; Kellis et al. 2003). To increase the sensitivity of our analysis, we derived a PWM for each of these published motifs (see Methods).
We calculated the probability of each of the 163 motifs being conserved in neutrally evolving sequence. The probability of a TFBS being conserved across the three species is <5% for 134 of 163 motifs, and <1% for 69 of 163 motifs (e.g., Table 2). As expected, the neutral probability was well correlated with information content, which is a function of the size and degeneracy of the TFBS (Fig. 6). For those TFBSs of ≥8 base pairs, 98 of 101 TFBSs had a neutral probability <5%, and 68 of 101 had a neutral probability <1%. Thus, despite their short degenerate nature, most of the TFBSs we tested had a very low probability of being conserved among S. cerevisiae, S. paradoxus, and S. mikatae.
Figure 6.

The probability of TFBS conservation is correlated with the information content of the PWM. The information content and probability of TFBS conservation is plotted for the 163 motifs used in this analysis. The dotted line indicates the P = 0.05 cutoff.
We annotated the S. cerevisiae genome for conserved instances of the 134 TFBSs that have a low probability of conservation. Annotation of 2 Mb of aligned intergenic sequences produced 27,225 predicted TFBSs or 6.5 sites per intergenic sequence. However, most intergenic sequences showed much higher levels of sequence conservation than could be explained by a handful of conserved TFBSs. The ratio of intergenic to synonymous substitution rate implies that 43% of all intergenic sites are more functionally constrained than synonymous sites (Table 1). The 27,225 conserved TFBSs identified in this study only cover 17% of the intergenic sequences examined after accounting for overlap between sites. The function of the remaining conserved intergenic sequences has yet to be determined.
The complete results for the 163 motifs are in Supplemental Table 4. GBrowse (Stein et al. 2002) formatted files containing TFBS annotation are available as Supplemental files.
Discussion
We estimated that ∼40% of intergenic sequences in S. cerevisiae are functionally constrained. These sequences may function in transcriptional regulation, translational regulation, or may function as noncoding RNA genes. This estimate is comparable to a previous estimate that 34% of the intergenic sequences are functionally constrained between S. cerevisiae, S. paradoxus, S. mikatae, and S. bayanus (Chin et al. 2005). In the present study we examined the portion of this constraint that can be attributed to conservation of TFBSs. We found that 134 of 163 regulatory motifs should rarely be conserved by chance, and we identified 27,225 conserved instances of these sites in 4188 intergenic sequences. Together these conserved TFBSs account for less than half of the functionally constrained intergenic sequences. Mutations in eight of nine predicted Ume6 and Ndt80 binding sites confirm our prediction that functional TFBSs can be identified through sequence conservation alone. However, as we discuss below, not all functional TFBSs may be identified. Additionally, we found that three of five Ndt80 binding sites that are not conserved produce Ndt80-dependent effects on expression when mutated. Whether or not these sites confer any biological differences between species remains an important unanswered question.
Identifying novel Ndt80 and Ume6 binding sites by conservation alone
We experimentally verified five novel Ume6 binding sites and three novel Ndt80 binding sites that were identified by their conservation among three closely related species. However, comparison of the predicted sites to those reported in the literature indicates that some previously known Ndt80 and Ume6 binding sites were not identified. As discussed below, there are a number of explanations for these false negatives.
Our search of the literature found four experimentally identified Ume6 sites (Bowdish and Mitchell 1993, Bowdish et al. 1995; Strich et al. 1994), and eight Ndt80 sites (Ozsarac et al. 1997). For Ume6, we correctly identified the site in the HOP1 promoter. The IME2 promoter contains two conserved sites that fall just below the threshold for a match to the Ume6 PWM. The SPO13 promoter contains a Ume6 site, but there is no alignment available for its promoter. Of the eight Ndt80 sites, we correctly identified four (SPR3, SPS19, SMK1, and NDT80); SPS4 and SPS1 were not in our alignment data set, and DIT2 and CDC10 do not match the Ndt80 PWM, despite matching the middle sporulation element (CRCAAAW). In total, we correctly identified five of eight known Ndt80 or Ume6 TFBSs for which we have alignments. Thus, our strict motif model and stringent filter for alignment quality make our predictions a conservative estimate of the total Ndt80 and Ume6 regulons.
Annotation of transcription factor binding sites throughout the genome
We found that 133 of 163 known or putative transcription factor binding motifs have a sufficiently low probability of conservation to identify single sites that are under functional constraint. Annotating these constrained sites in the S. cerevisiae genome results in the prediction of 6.5 functional TFBSs per intergenic sequence. However, this annotation relies on a number of assumptions. First, we must assume mutational homogeneity across the genome. Direct estimates of mutation rates are consistent with a uniform mutation rate (Drake 1991), but it is also possible that rare mutational hot-spot or cold-spots may exist (Chuang and Li 2004). In yeast the synonymous substitution rate is nearly uniformly distributed across the genome (Chin et al. 2005). This suggests that mutational heterogeneity does not contribute to the divergence rate among the Saccharomyces genomes.
A second assumption is that substitutions are independent of other positions within a TFBS. Although position independence is widely assumed within TFBSs, this has been proven false for some binding sites (Man and Stormo 2001; Bulyk et al. 2002). When calculating the probability that a TFBS is conserved between species, any dependencies between columns will reduce the number of ways in which a site could be maintained as a TFBS. Therefore, the position-independence assumption makes the probabilities calculated here a conservative estimate of the true probability that a binding site will be conserved in neutral sequence.
A third assumption required for TFBS annotation is that TFBS conservation is due entirely to its functional importance in transcriptional regulation. This assumption may break down under conditions where overlap exists between TFBSs and other functional noncoding sequences. However, only 659 of the conserved TFBSs (2%) were found in 106 noncoding RNA genes (including tRNAs) present in the 4188 intergenic alignments. It is also possible that TFBSs overlap with each other, creating ambiguity about which TFBS is responsible for the functional constraint. The 27,225 significantly conserved TFBSs identified in the three yeast genomes account for ∼470 kb of noncoding sequence, of which ∼122 kb (27%) is shared between two or more binding sites. Therefore, in many cases we cannot uniquely determine which TFBS is responsible for the functional constraint.
Finally, we assume that the multiple sequence alignments are correct. Simulations have shown that when the number of substitutions is less than one per site, the multiple sequence alignment programs perform quite well (Pollard et al. 2004). We avoided misalignments by choosing closely related yeast species and filtering out any dubious alignments (see Methods). However, some errors due to misalignment may be unavoidable. Even if some misaligned sequences are included in our analysis, this will likely result in missing conserved TFBSs (false negatives), rather than identifying false positives.
The limitations of comparative genomics for identifying functional elements in noncoding DNA
The comparative genomic method applied here and in many other studies relies on the assumption that functional elements will be shared between species. However, it is hypothesized that the majority of biological differences between species are due to changes in gene regulation (Wilson et al. 1974), suggesting that there may be species-specific regulatory signals. The fraction of genes that are differentially regulated between species is not known, but there is evidence to suggest that this fraction may be significant (Dermitzakis and Clark 2002; Moses et al. 2003). Furthermore, many conserved noncoding sequences are only conserved in a subset of taxa (Frazer et al. 2004), suggesting that very closely related species are needed to identify more recently evolved functional sequences (Boffelli et al. 2003). In addition, TFBS turnover (Ludwig et al. 2000; Dermitzakis and Clark 2002; Costas et al. 2003) and misalignment (Pollard et al. 2004) limit the comparative method even when the biology of the two organisms is the same (Ludwig et al. 1998). Our data (Fig. 5) suggest that species-specific regulatory sequences may be common in S. cerevisiae. These sites may be newly evolved Ndt80 sites that serve an S. cerevisiae-specific function, or they may also exist if there are no deleterious consequences of their presence in the promoter sequence. In Drosophila, there is evidence of negative selection against spurious TFBSs (Hahn et al. 2003). Although many Ndt80 binding sites are not conserved, the paucity of unconserved Ume6 binding sites could be explained by negative selection against mutations that create spurious Ume6 binding sites (Fig. 1). The ability to identify TFBS gain, loss, and turnover between species will be crucial to our understanding of the role transcriptional regulation plays in evolution.
The cis-regulatory code
The cis-regulatory code is the set of rules that enables a cell to direct the expression program of each gene based on its cis-regulatory sequences. Identifying functional cis-regulatory sites in the S. cerevisiae genome will be a significant step towards describing this regulatory code. However, our results also suggest that although we may be able to annotate the location of the TFBSs, their effects on gene regulation may often be context-dependent, as shown by the two identical Ume6 sites with opposite effects on expression (Fig. 4). It has been suggested that the presence of Abf1 is required for Ume6 activation (Bowdish and Mitchell 1993), and we found a highly significant concurrence of the conserved Ume6 and Abf1 binding sites (hypergeometric P < 10–18). Further experiments will be required to determine in what context (e.g., location from the transcription start site, adjacent regulatory sites, orientation) Ume6 acts as a repressor or an activator. The cis-regulatory code hypothesis also requires that the majority of regulatory elements are not unique to a single promoter. Our estimation that 43% of intergenic sequences are functionally constrained (Table 1) but that only 17% of intergenic sequences match conserved TFBSs suggests that a large fraction of conserved intergenic sequences may serve promoter-specific functions. This is supported by the observation that many experimentally verified regulatory motifs are often flanked by conserved sequences (Fig. 3). A similar pattern is found in Drosophila, where experimentally verified enhancers are as conserved as their flanking sequences (Berman et al. 2004). The role of promoter-specific sequences in transcriptional regulation needs to be further explored.
Methods
Substitutions in coding and noncoding sequences
The alignments used for this analysis were downloaded from http://www.broad.mit.edu/annotation/fungi/comp_yeasts/downloads.html (Kellis et al. 2003). The intergenic alignments were filtered to remove any alignment with >10% Ns or 10% missing data in any one species. In the TFBS analysis described here, 4188 alignments were used; 2098 coding sequence alignments were used to generate the synonymous and nonsynonymous rate estimates. The synonymous and nonsynonymous and intergenic substitution rates were estimated using PAML software (Yang 1997). The intergenic substitution rate was estimated using the Hasegawa, Kishino, and Yano substitution model (Hasegawa et al. 1985).
Motif models
The initial motif models were obtained from three sources. Cliften et al. (2003) published 65 motifs that were perfectly conserved at least five times between multiple yeast species. Kellis et al. (2003) also identified 71 motifs by their sequence conservation between species. The Kellis et al. (2003) motifs were downloaded from http://www.broad.mit.edu/annotation/fungi/comp_yeasts/motiflist.html. The Saccharomyces cerevisiae Promoter Database (SCPD) (Zhu and Zhang 1999) contains 42 motifs identified by various experimental methods. There were a total of 163 motifs after we removed motifs that were exactly identical, as well as shorter motifs that were contained within longer motifs.
A position weight matrix (PWM) was used to describe all motif models. Because the published motif models were only available as consensus sequences, we generated substitution-derived PWMs, based on the observation that the same positions that are degenerate within a species are degenerate between species (Moses et al. 2003). The substitution PWM is based on all instances of the consensus sequence with 0 or 1 difference across the three species (Supplemental Fig. 4). To avoid adding too much noise to the motif model, we tested whether the differences were uniformly distributed across the positions within the motif using a χ2 test (d.f. equal to the motif width – 1). If the differences were uniformly distributed, we used a PWM representing the consensus sequence. The substitution-derived PWMs were found to be very similar to the original motif model except that we were able to refine the frequency of each base at degenerate positions for 91 of the 163 of the motifs. The PWMs used are available in Supplemental File 1.
We used Patser (Hertz and Stormo 1999), with equal base frequencies, to scan all sequences for matches to a PWM. The use of local base frequencies may have biased the results, as 43% of the intergenic sequences are under functional constraint. Furthermore, using local base frequencies may lead to missing weak sites in regions of biased nucleotide composition (Dermitzakis et al. 2003). For a motif of width w, Patser scored each w-mer as the log-likelihood ratio of observing the sequence under the motif model or the background nucleotide frequencies. If S was greater than the default threshold score, then the w-mer was a match to the motif. The identification of TFBSs and the probability of TFBS conservation were dependent on this cutoff for PWM matches. As shown in Supplemental Figure 5, the choice of cutoff does not affect the overall percentage of TFBSs estimated to be functional.
Calculating the neutral expectation for TFBS conservation
We calculated the neutral expectation for TFBS conservation as the probability that a sequence matching the PWM in S. cerevisiae will remain a match to the PWM given a specified amount of evolutionary time. Starting from the highest possible scoring sequence for a particular PWM, we tested all possible sequences to which this initial sequence could evolve. For each descendant sequence, we tested whether or not this sequence was a match to the motif. If it was a match, we calculated the probability of observing this sequence (Fig. 2). The probability of TFBS conservation can be written as:
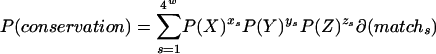 |
(1) |
where x is the number of identical bases between sequence s and the starting TFBS sequence, y is the number of transitions, and z is the number of transversions. P(X) is the probability of a base remaining the same over the given evolutionary distance, P(Y) is the probability of a transition, and P(Z) is the probability of a transversion. The probabilities were calculated using Kimura's two-parameter model (Kimura 1980) with a transition/transversion ratio of two, and the synonymous substitution rate. W is the width of the motif, and δ(match) is a binary function that returns 1 if the sequence was a significant match to the PWM. The probability that a TFBS is conserved across the three-species phylogeny depends on the ancestral state of the three species, which is not known. To avoid this difficulty, we calculated the probability from the sum of the three branch lengths. This simplification results in a conservative estimate of the probability of TFBS conservation.
Strains, media, and plasmids
Escherichia coli DH5α was used for all plasmid manipulations. All yeast strains were derivatives of S288C. DBY8268 (a/a, Δura3 EcoRV-StuI/ura3-52, ho/ho) was used to measure wild-type expression levels. Expression levels were also measured in ndt80Δ and ume6Δ strains obtained from the yeast deletion collection (Giaever et al. 2002), present in BY4743 (a/a, his3D1/his3D1, leu2D0/leu2D0, lys2D0/LYS2, MET15/met15D0, ura3D0/ura3D0). YEp357R is a yeast-bacteria shuttle vector maintained in yeast as an episomal plasmid carrying the β-galactosidase gene and the URA3 gene for selection in yeast (Myers et al. 1986). Yeast cultures were grown in complete minimal medium, minus Uracil. Expression was measured in complete minimal medium or sporulation medium (1% potassium acetate).
Measuring expression from wild-type and mutant promoters
Promoters containing Ume6 or Ndt80 binding sites were cloned from S. cerevisiae strain S288C into YEp357R to generate a β-GAL fusion construct. The Ume6 or Ndt80 motifs were mutated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The alignments of these binding sites and their flanking sequences can be found in Figure 3. Both mutant sequences score well below the Patser threshold and were confirmed by sequencing. Wild-type and mutant promoters were transformed into S288C using lithium acetate (Gietz et al. 1995). Yeast cultures were started at an OD600 of 0.2 in complete medium and an OD600 of 0.5 in sporulation medium. Expression was measured after 4, 6, 8, and 24 h of growth. The data for growth in complete media can be found in Supplemental Figures 2 and 3. The 8-h timepoint is shown in Figures 4 and 5. Data from the other timepoints agree with the 8-h data.
Acknowledgments
We thank Maia Dorsett and Hyun Seok Kim for helpful discussions about the experimental and computational design and procedures, Mark Johnston, Linda Riles, and Jim Dover for providing the deletion strains and plasmids, and Ting Wang, Sudhir Nayak, Sean Eddy, Mark Johnston, and Gary Stormo for helpful comments about the manuscript. S.W.D. is supported by NSF graduate fellowship #DGE-0202737.
[Supplemental material is available online at www.genome.org.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3578205. Article published online ahead of print in April 2005. Freely available online through the Genome Research Immediate Open Access option.
References
- Akashi, H. 2001. Gene expression and molecular evolution. Curr. Opin. Genet Dev. 11: 660–666. [DOI] [PubMed] [Google Scholar]
- Bergman, C.M. and Kreitman, M. 2001. Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genome Res. 11: 1335–1345. [DOI] [PubMed] [Google Scholar]
- Berman, B.P., Pfeiffer, B.D., Laverty, T.R., Salzberg, S.L., Rubin, G.M., Eisen, M.B., and Celniker, S.E. 2004. Computational identification of developmental enhancers: Conservation and function of transcription factor binding-site clusters in Drosophila melanogaster and Drosophila pseudoobscura. Genome Biol. 5: R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffelli, D., McAuliffe, J., Ovcharenko, D., Lewis, K.D., Ovcharenko, I., Pachter, L., and Rubin, E.M. 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299: 1391–1394. [DOI] [PubMed] [Google Scholar]
- Bowdish, K.S. and Mitchell, A.P. 1993. Bipartite structure of an early meiotic upstream activation sequence from Saccharomyces cerevisiae. Mol. Cell Biol. 13: 2172–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowdish, K.S., Yuan, H.E., and Mitchell, A.P. 1995. Positive control of yeast meiotic genes by the negative regulator UME6. Mol. Cell Biol. 15: 2955–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulyk, M.L., Johnson, P.L., and Church, G.M. 2002. Nucleotides of transcription factor binding sites exert interdependent effects on the binding affinities of transcription factors. Nucleic Acids Res. 30: 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C.S., Chuang, J.H., and Li, H. 2005. Genome-wide regulatory complexity in yeast promoters: Separation of functionally conserved and neutral sequence. Genome Res. 15: 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, S. and Herskowitz, I. 1998. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol. Cell 1: 685–696. [DOI] [PubMed] [Google Scholar]
- Chu, S., DeRisi, J., Eisen, M., Mulholland, J., Botstein, D., Brown, P.O., and Herskowitz, I. 1998. The transcriptional program of sporulation in budding yeast. Science 282: 699–705. [DOI] [PubMed] [Google Scholar]
- Chuang, J.H. and Li, H. 2004. Functional bias and spatial organization of genes in mutational hot and cold regions in the human genome. PLoS Biol. 2: E29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliften, P., Sudarsanam, P., Desikan, A., Fulton, L., Fulton, B., Majors, J., Waterston, R., Cohen, B.A., and Johnston, M. 2003. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 301: 71–76. [DOI] [PubMed] [Google Scholar]
- Costas, J., Casares, F., and Vieira, J. 2003. Turnover of binding sites for transcription factors involved in early Drosophila development. Gene 310: 215–220. [DOI] [PubMed] [Google Scholar]
- Dermitzakis, E.T. and Clark, A.G. 2002. Evolution of transcription factor binding sites in Mammalian gene regulatory regions: Conservation and turnover. Mol. Biol. Evol. 19: 1114–1121. [DOI] [PubMed] [Google Scholar]
- Dermitzakis, E.T., Bergman, C.M., and Clark, A.G. 2003. Tracing the evolutionary history of Drosophila regulatory regions with models that identify transcription factor binding sites. Mol. Biol. Evol. 20: 703–714. [DOI] [PubMed] [Google Scholar]
- Drake, J.W. 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. 88: 7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer, K.A., Tao, H., Osoegawa, K., de Jong, P.J., Chen, X., Doherty, M.F., and Cox, D.R. 2004. Noncoding sequences conserved in a limited number of mammals in the SIM2 interval are frequently functional. Genome Res. 14: 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever, G., Chu, A.M., Ni, L., Connelly, C., Riles, L., Veronneau, S., Dow, S., Lucau-Danila, A., Anderson, K., Andre, B., et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391. [DOI] [PubMed] [Google Scholar]
- Gietz, R.D., Schiestl, R.H., Willems, A.R., and Woods, R.A. 1995. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11: 355–360. [DOI] [PubMed] [Google Scholar]
- Hahn, M.W., Stajich, J.E., and Wray, G.A. 2003. The effects of selection against spurious transcription factor binding sites. Mol. Biol. Evol. 20: 901–906. [DOI] [PubMed] [Google Scholar]
- Harbison, C.T., Gordon, D.B., Lee, T.I., Rinaldi, N.J., Macisaac, K.D., Danford, T.W., Hannett, N.M., Tagne, J.B., Reynolds, D.B., Yoo, J., et al. 2004. Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, M., Kishino, H., and Yano, T. 1985. Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22: 160–174. [DOI] [PubMed] [Google Scholar]
- Hertz, G.Z. and Stormo, G.D. 1999. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563–577. [DOI] [PubMed] [Google Scholar]
- Johnson, D.S., Davidson, B., Brown, C.D., Smith, W.C., and Sidow, A. 2004. Noncoding regulatory sequences of Ciona exhibit strong correspondence between evolutionary constraint and functional importance. Genome Res. 14: 2448–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, M., Patterson, N., Endrizzi, M., Birren, B., and Lander, E.S. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241–254. [DOI] [PubMed] [Google Scholar]
- Kimura, M. 1980. A simple model for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16: 111–120. [DOI] [PubMed] [Google Scholar]
- Lee, T.I., Rinaldi, N.J., Robert, F., Odom, D.T., Bar-Joseph, Z., Gerber, G.K., Hannett, N.M., Harbison, C.T., Thompson, C.M., Simon, I., et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804. [DOI] [PubMed] [Google Scholar]
- Li, W.H. 1997. Molecular evolution. Sinauer Associates, Inc., Sunderland, MA.
- Loots, G.G., Locksley, R.M., Blankespoor, C.M., Wang, Z.E., Miller, W., Rubin, E.M., and Frazer, K.A. 2000. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 288: 136–140. [DOI] [PubMed] [Google Scholar]
- Ludwig, M.Z., Patel, N.H., and Kreitman, M. 1998. Functional analysis of eve stripe 2 enhancer evolution in Drosophila: Rules governing conservation and change. Development 125: 949–958. [DOI] [PubMed] [Google Scholar]
- Ludwig, M.Z., Bergman, C., Patel, N.H., and Kreitman, M. 2000. Evidence for stabilizing selection in a eukaryotic enhancer element. Nature 403: 564–567. [DOI] [PubMed] [Google Scholar]
- Man, T.K. and Stormo, G.D. 2001. Non-independence of Mnt repressor-operator interaction determined by a new quantitative multiple fluorescence relative affinity (QuMFRA) assay. Nucleic Acids Res. 29: 2471–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martone, R., Euskirchen, G., Bertone, P., Hartman, S., Royce, T.E., Luscombe, N.M., Rinn, J.L., Nelson, F.K., Miller, P., Gerstein, M., et al. 2003. Distribution of NF-κB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. 100: 12247–12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses, A.M., Chiang, D.Y., Kellis, M., Lander, E.S., and Eisen, M.B. 2003. Position specific variation in the rate of evolution in transcription factor binding sites. BMC Evol. Biol. 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, A.M., Tzagoloff, A., Kinney, D.M., and Lusty, C.J. 1986. Yeast shuttle and integrative vectors with multiple cloning sites suitable for construction of lacZ fusions. Gene 45: 299–310. [DOI] [PubMed] [Google Scholar]
- Ozsarac, N., Straffon, M.J., Dalton, H.E., and Dawes, I.W. 1997. Regulation of gene expression during meiosis in Saccharomyces cerevisiae: SPR3 is controlled by both ABFI and a new sporulation control element. Mol. Cell Biol. 17: 1152–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce, M., Benjamin, K.R., Montano, S.P., Georgiadis, M.M., Winter, E., and Vershon, A.K. 2003. Sum1 and Ndt80 proteins compete for binding to middle sporulation element sequences that control meiotic gene expression. Mol. Cell Biol. 23: 4814–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard, D.A., Bergman, C.M., Stoye, J., Celniker, S.E., and Eisen, M.B. 2004. Benchmarking tools for the alignment of functional noncoding DNA. BMC Bioinformatics 5: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanojevic, D., Small, S., and Levine, M. 1991. Regulation of a segmentation stripe by overlapping activators and repressors in the Drosophila embryo. Science 254: 1385–1387. [DOI] [PubMed] [Google Scholar]
- Steber, C.M. and Esposito, R.E. 1995. UME6 is a central component of a developmental regulatory switch controlling meiosis-specific gene expression. Proc. Natl. Acad. Sci. 92: 12490–12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, L.D., Mungall, C., Shu, S., Caudy, M., Mangone, M., Day, A., Nickerson, E., Stajich, J.E., Harris, T.W., Arva, A., et al. 2002. The generic genome browser: A building block for a model organism system database. Genome Res. 12: 1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strich, R., Surosky, R.T., Steber, C., Dubois, E., Messenguy, F., and Esposito, R.E. 1994. UME6 is a key regulator of nitrogen repression and meiotic development. Genes & Dev. 8: 796–810. [DOI] [PubMed] [Google Scholar]
- Vershon, A.K. and Pierce, M. 2000. Transcriptional regulation of meiosis in yeast. Curr. Opin. Cell Biol. 12: 334–339. [DOI] [PubMed] [Google Scholar]
- Williams, R.M., Primig, M., Washburn, B.K., Winzeler, E.A., Bellis, M., Sarrauste de Menthiere, C., Davis, R.W., and Esposito, R.E. 2002. The Ume6 regulon coordinates metabolic and meiotic gene expression in yeast. Proc. Natl. Acad. Sci. 99: 13431–13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, A.C., Maxson, L.R., and Sarich, V.M. 1974. Two types of molecular evolution. Evidence from studies of interspecific hybridization. Proc. Natl. Acad. Sci. 71: 2843–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, W.S. and Nielsen, R. 2004. Detecting selection in noncoding regions of nucleotide sequences. Genetics 167: 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfe, A., Goodson, M., Goode, D.K., Snell, P., McEwen, G.K., Vavouri, T., Smith, S.F., North, P., Callaway, H., Kelly, K., et al. 2004. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3: e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. 1997. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]
- Yuh, C.H., Bolouri, H., and Davidson, E.H. 1998. Genomic cis-regulatory logic: Experimental and computational analysis of a sea urchin gene. Science 279: 1896–1902. [DOI] [PubMed] [Google Scholar]
- Zhu, J. and Zhang, M.Q. 1999. SCPD: A promoter database of the yeast Saccharomyces cerevisiae. Bioinformatics 15: 607–611. [DOI] [PubMed] [Google Scholar]
Web site references
- http://www.broad.mit.edu/annotation/fungi/comp_yeasts/downloads.html; yeast intergenic alignments used in this article's analysis.
- http://www.broad.mit.edu/annotation/fungi/comp_yeasts/motiflist.html cis-regulatory motifs used in this article's analysis.