

Abstract
The NCCN Guidelines for Colon Cancer provide recommendations regarding diagnosis, pathologic staging, surgical management, perioperative treatment, surveillance, management of recurrent and metastatic disease, and survivorship. These NCCN Guidelines Insights summarize the NCCN Colon Cancer Panel discussions for the 2018 update of the guidelines regarding risk stratification and adjuvant treatment for patients with stage III colon cancer, and treatment of BRAF V600E mutation–positive metastatic colorectal cancer with regimens containing vemurafenib.
Overview
Colorectal cancer (CRC) is the fourth most frequently diagnosed cancer and the second leading cause of cancer death in the United States. In 2018, an estimated 97,220 new cases of colon cancer and approximately 43,030 cases of rectal cancer will be diagnosed. During the same year, an estimated 50,630 people will die of colon and rectal cancers combined.1 The incidence of CRC has been declining; incidence per 100,000 people decreased from 60.5 in 1976 to 46.4 in 2005, and to 40.7 in 2009–2013.2,3 In fact, the incidence of CRC decreased at a rate of approximately 3% per year between 2003–2012.4 In addition, mortality from CRC decreased by almost 35% from 1990–2007,5 and is currently reduced by 51% from peak mortality rates—from 28.6 per 100,000 in 1976 to 14.1 in 2014.3 These improvements in incidence of and mortality from CRC are thought to be a result of shifting patterns of CRC risk factors, cancer prevention and earlier diagnosis through screening, and better treatment modalities.6
Despite the observed improvements in the overall CRC incidence rate, its incidence in patients aged <50 years has been increasing.3,7 In 2017, approximately 7,550 cases of CRC were diagnosed in this population.3 In a retrospective cohort study of the SEER CRC registry, it was estimated that the incidence rates for colon and rectal cancers in patients aged 20 to 34 years will increase by 90.0% and 124.2%, respectively, by 2030.7 The cause of this trend is currently unknown.
2018 Updates to the NCCN Guidelines for Colon Cancer
During the meeting to update the guidelines for 2018, the panel discussed many issues. Most notable were the results of the IDEA collaboration and their impact on adjuvant treatment in patients with stage III colon cancer,8 and results of the SWOG S1406 trial and their impact on treatment of patients with BRAF-mutant metastases.9
Risk Stratification and Adjuvant Treatment for Stage III Colon Cancer
Nonmetastatic colon cancer is generally treated with curative intent by colectomy and, in some cases, adjuvant chemotherapy.10 Population and institutional studies have shown that adjuvant therapy may confer a survival advantage in some patients with resected colon cancer (eg, those with stage III or high-risk stage II disease).11–14 Furthermore, randomized controlled trials have shown that the addition of oxaliplatin to these adjuvant regimens benefits some patients.15–19 However, adjuvant treatment, especially with regimens containing oxaliplatin, is associated with considerable toxicity (notably chemotherapy-induced peripheral neuropathy),16,20 and not all patients derive benefit. Consideration of disease stage and pathologic features, microsatellite instability (MSI) status, possible efficacy and toxicity profiles associated with treatment choice, and patient age, comorbidities, and preferences aid in decision-making regarding the use of adjuvant therapy for patients
The IDEA collaboration investigated whether a shortened duration of adjuvant therapy would be a feasible way to avoid or lessen toxicities associated with oxaliplatin-containing adjuvant therapy in some patients with locoregional colon cancer, without impairing oncologic outcomes. IDEA included >12,000 patients in an international effort that pooled data from 6 concurrently conducted randomized phase III trials to assess the noninferiority of 3 months compared with 6 months of adjuvant FOLFOX or CapeOX in patients with stage III colon cancer.8 Median follow-up was 39 months. Importantly, grade ≥3 neurotoxicity rates were lower in the 3 months’ versus 6 months’ treatment arms (3% vs 16% for FOLFOX; 3% vs 9% for CapeOX; P<.0001), as were grade 2 neurotoxicity rates (14% vs 32% for FOLFOX; 12% vs 36% for CapeOX; P<.0001). Rates of grades 2 and 3/4 diarrhea were also lower with the shorter duration of therapy (P<.0001 for FOLFOX; P=.01 for CapeOX). The primary end point of 3-year disease-free survival (DFS) did not meet the pre-specified cutoff for noninferiority, despite the small absolute difference of 0.9% (74.6% for 3 months vs 75.5% for 6 months; hazard ratio [HR], 1.07; 95% CI, 1.00–1.15), which is of questionable clinical significance. Notable differences in 3-year DFS were seen between FOLFOX and CapeOX and between patients with T1–3N1 (low-risk) versus T4 or N2 (high-risk) disease. Specifically, in the T1–3N1 subset, DFS with 3 months of CapeOX was noninferior to that with 6 months of CapeOX (HR, 0.85; 95% CI, 0.71–1.01), whereas noninferiority could not be proven for 3 versus 6 months of FOLFOX (HR, 1.10; 95% CI, 0.96–1.26). In the T4 or N2 subset, DFS with 3 months of FOLFOX was inferior to that of 6 months with FOLFOX (HR, 1.20; 95% CI, 1.07– 1.35), whereas noninferiority could not be proven for 3 versus 6 months of CapeOX (HR, 1.02; 95% CI, 0.89–1.17).
The panel discussed several details of the trial and how they believed the results should be translated into guideline recommendations. First, the panel discussed what might account for the unexpected differences seen in DFS between FOLFOX and CapeOX. Oxaliplatin doses are different between the regimens (85 mg/m2 every 2 weeks for mFOLFOX6; 130 mg/m2 every 3 weeks for CapeOX), which might partially account for the observed difference in outcomes. Although total oxaliplatin exposure is similar, more oxaliplatin is received in the first 4 weeks with CapeOX (260 mg/m2) compared with FOLFOX (170 mg/m2). Also, the panel noted that capecitabine may result in more continuous fluorouracil exposure than 5-FU/leucovorin. One panel member noted that some sites used FOLFOX4; however, panel consensus was that this difference would only account for differences in toxicity, not in efficacy. Treatment compliance was similar between the CapeOX and FOLFOX arms (percent reaching planned last cycle was 90% for 3-month FOLFOX, 86% for 3-month CapeOX, 71% for 6-month FOLFOX, and 65% for 6-month CapeOX), and therefore compliance is unlikely to account for the differences between the regimens. Importantly, the panel noted that no significant differences have been seen between CapeOX and FOLFOX in randomized adjuvant or metastatic studies.26,27 Therefore, the panel discussed the possibility that selection bias may account for the difference in DFS seen between FOLFOX and CapeOX in IDEA. The use of FOLFOX versus CapeOX was by physician’s choice, not by randomization; choice of regimen was used as a stratification factor for randomization to 3 or 6 months of treatment duration. The proportion of patients treated with CapeOX varied from 0% to 75% between trial sites.8 In the US/Canadian study that used only mFOLFOX6, in fact, 3 months of FOLFOX achieved noninferiority to 6 months of FOLFOX, with an HR for DFS of 1.10. However, in the French trial,8 most patients (90%) received FOLFOX, and the HR for DFS between 3 and 6 months of FOLFOX was the worst of all trial sites at 1.27. Though the reasons for the different outcomes based on regimen choice are not completely clear, the panel agreed that the data are convincing and that the difference cannot be ignored.
Another point that the NCCN Panel discussed was whether DFS data formed a sufficient basis for guideline recommendations. The panel was unified in its opinion that, although DFS effects have been shown to be larger than overall survival (OS) effects in other trials, the almost complete elimination of oxaliplatin toxicity with a shorter treatment duration justified changes to the guidelines at this time.
The data were sufficient to warrant division of stage III colon cancer into a low-risk (T1-3N1) and a high-risk group (T4N1-2 or TanyN2; see COL-3, page 361. For the low-risk group, the recommended duration of adjuvant therapy is 3 months if CapeOX is chosen, because noninferiority for DFS was proven in IDEA and patients should be spared the increased toxicity, cost, and inconvenience of longer therapy. If FOLFOX is chosen for these patients, then 3 to 6 months of FOLFOX is recommended. The panel believes the shorter duration can be considered to reduce toxicity, cost, and inconvenience, but noninferiority could not be proven for 3 versus 6 months of FOLFOX in this subset. For patients with high-risk stage III colon cancer, 3 to 6 months of adjuvant therapy is recommended if CapeOX is chosen; noninferiority of the shorter duration was not proven in this subset of patients, but the panel believes the shorter duration can be considered to minimize toxicity, cost, and inconvenience. Furthermore, DFS with 3 months of therapy was noninferior to DFS with 6 months for the entire cohort (TanyN1-2) who received CapeOX (HR, 0.95; 95% CI, 0.85–1.06).8 Finally, FOLFOX, if chosen, should be given for a full 6 months in the high-risk group because 3 months was shown to be inferior to 6 months for DFS. With these updated and nuanced recommendations, careful discussion and review of the available data between the patient and clinician are important to guide the choice of approach.
The panel then discussed some concerns about the dosing of capecitabine in CapeOX, which they believe is critical if the decision is to give 3 months of CapeOX. Many oncologists, especially in the United States, routinely reduce the starting dose of capecitabine, but the panel strongly believes that the starting dose used in IDEA8 (1,000 mg/m2 twice daily) should be used because it is the only validated dose. The panel also discussed that, in practice, oxaliplatin is dropped from FOLFOX and CapeOX after 6 to 10 doses to reduce the risk of severe neurotoxicity. Therefore, it may be reasonable to complete 3 months of CapeOX for patients with high-risk stage III colon cancer and then drop oxaliplatin and continue capecitabine. However, the panel was divided on whether to recommend this approach.
Because of the methodologies and results of IDEA, some of the findings are open to interpretation. However, it will be difficult to generate data that are any more definitive than those from the IDEA analysis. Therefore, the NCCN Guidelines list FOLFOX and CapeOX as equally preferred options without strongly recommending one over the other. Efficacy is similar, FOLFOX is less expensive for US patients, and CapeOX is more convenient for most patients (especially 3 months of CapeOX vs 6 months of FOLFOX). Both regimens are preferred over the other options for adjuvant treatment of patients with stage III colon cancer—6 months of either capecitabine or 5-FU/leucovorin—which are generally only recommended for patients with stage III colon cancer if they cannot tolerate oxaliplatin.
Despite the large number of patients in IDEA, the results were not as definitive as the panel would require for these new recommendations to be listed as category 1. Therefore, these shorter durations are included in the guidelines as category 2A recommendations. The 6-month durations of FOLFOX or CapeOX for patients with stage III colon cancer remain as category 1 recommendations based on older trials.15–19
Finally, the panel also briefly discussed the limited data from IDEA on stage II colon cancer or stage II/III rectal cancer. Only one site included patients with rectal cancer, none of whom had preoperative chemotherapy, and only 2 sites included patients with stage II colon cancer, one of which included mostly those with high-risk stage II. Overall, the panel believed there are not enough data from IDEA to draw any conclusions for patients with rectal or stage II colon cancer.
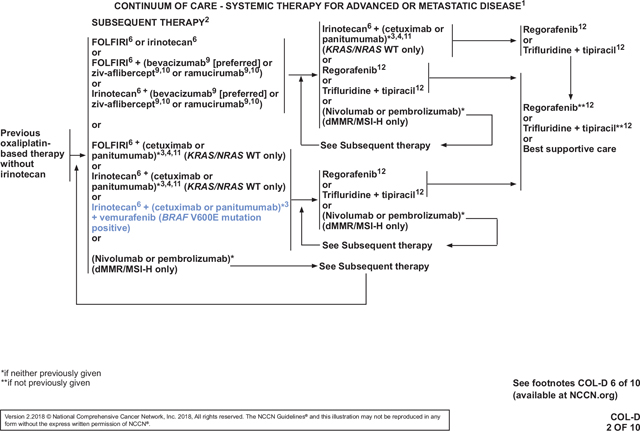
Treatment of RAS Wild-Type/BRAF Mutation–Positive Metastatic CRC
In the 2017 version of the NCCN Guidelines, patients with unresectable, advanced, or metastatic CRC (mCRC) were managed with a continuum of care that included 20 first-line systemic treatment options, 33 second-line options, and 13 options for subsequent therapies in up to 7 lines of treatment.28,29 A growing list of factors are considered when choosing therapies for each patient, including the goals of treatment, type and timing of prior therapy, different efficacy and toxicity profiles of the regimens, KRAS and NRAS mutational tumor status, and patient comorbidities and preferences. MSI status and location of the primary tumor were recently added as additional considerations.30–34 Although survival for patients with advanced CRC has improved dramatically over the past decades, the most recently reported 5-year survival rate for patients with stage IV CRC was only 14%.1,35 Moreover, patients with BRAF-mutated mCRC have a particularly poor prognosis. BRAF V600E mutations are present in approximately 8% of mCRC cases and are associated with a more aggressive biology, shorter OS, and decreased response to chemotherapy compared with BRAF wild-type tumors.36 Thus, additional treatment options are still needed in mCRC, especially for the BRAF mutation–positive subset of patients.
BRAF is downstream of EGFR and RAS in the MAPK signaling pathway. The BRAF V600E mutation results in constitutive MAPK signaling that encourages cellular proliferation.37 Vemurafenib selectively inhibits the V600E-mutated form of the BRAF kinase, thereby reducing aberrant MAPK signaling.38 It has an FDA indication for treatment of patients with BRAF V600E–mutated, unresectable, or metastatic melanoma.39 However, vemurafenib monotherapy has shown limited activity in mCRC.40 Preclinical data suggest that BRAF V600E inhibition alone is ineffective because feedback activation of EGFR occurs.41,42 Therefore, blockage of EGFR and BRAF V600E together has been speculated to be more effective than BRAF V600E inhibition alone. In fact, preclinical studies show a synergistic effect of such dual inhibition.41,42 However, vemurafenib in combination with cetuximab- or panitumumab-based therapy has been ineffective in early clinical trials.43 Results from preclinical and early clinical studies suggest that the addition of irinotecan to BRAF and EGFR inhibition may improve antitumor activity.44,45
The combination of vemurafenib, cetuximab, and irinotecan was thus tested in patients with BRAF V600E–mutated mCRC in the recent phase II SWOG S1406 trial. In this trial, 99 patients with BRAF-mutant, RAS wild-type tumors who received 1 or 2 prior regimens were randomized to irinotecan and cetuximab with or without vemurafenib.9 The NCCN Panel reviewed updated results of this trial that were presented at the 2017 ASCO annual meeting. The primary end point of median progression-free survival (PFS) was improved in the vemurafenib arm (4.3 vs 2.0 months; HR, 0.48; 95% CI, 0.31–0.75; P=.001). Response was also improved, with response rates (only partial responses [PRs] were seen) of 4% vs 16% (P=.08) and disease control rates (partial responses + stable disease) of 22% vs 67% (P=.001). Grade 3/4 adverse events (AEs) that were higher in the vemurafenib arm included neutropenia (33% vs 7%), anemia (13% vs 0%), and nausea (20% vs 2%). Crossover was allowed for the cetuximab/irinotecan group, and 48% of those patients received vemurafenib at time of progression. Of those who crossed over, the partial response rate was 17% and the stable disease rate was 55%, for a disease control rate of 72%. The HR for the secondary end point of OS, which would be attenuated by the crossover, was 0.73 (95% CI, 0.45–1.17; P=.19).
After the panel reviewed these data, one member pointed out that results of the safety lead-in phase of the BEACON CRC trial (ClinicalTrials.gov Identifier: NCT02928224) were to be presented at the ESMO 2017 Congress. This international phase III trial is comparing (1) encorafenib with cetuximab plus or minus binimetinib versus (2) investigator choice of irinotecan/cetuximab or FOLFIRI/cetuximab in patients with BRAF V600E–mutant mCRC whose disease has progressed on 1 or 2 prior metastatic regimens. Encorafenib is another inhibitor of mutant BRAF, and binimetinib inhibits MEK, which is downstream from BRAF in the MAPK pathway.37,46 The combination of these 2 drugs has been shown to improve PFS in BRAF-mutant melanoma compared with encorafenib monotherapy.47 Although the BEACON CRC data had not been presented at the time of panel discussion, several panel members were aware that the results would be better than those seen in SWOG S1406. In fact, the now-presented data show that the triplet regimen (encorafenib/cetuximab/binimetinib) was fairly well tolerated in 30 patients, with dose-limiting toxicities in 5 patients (cetuximab infusion reaction, n=2; grade 2 retinopathy, n=2; grade 2 decreased ejection fraction, n=1). Of the 30 patients, 19 (63%) experienced a grade 3 or 4 AE, including fatigue (grade 3 in 4 patients), urinary tract infection (grade 3 in 3 patients), increased aspartate aminotransferase (grade 3 in 2 patients; grade 4 in 1 patient), and increased blood creatine phosphokinase (grade 3 in 3 patients). The most common AEs of any grade were diarrhea (77%), dermatitis acneiform (67%), nausea (63%), and fatigue (63%).48,49 In 29 patients with BRAF V600E–mutated mCRC, the overall response rate (3 complete responses + 11 partial responses) was 48%. All remaining patients had stable disease, for a disease control rate of 100%. The preliminary estimated median PFS was 8 months. One panel member reasoned that vemurafenib/cetuximab/irinotecan should not be added based on the SWOG S1406 results, but rather the panel should wait for BEACON CRC results and FDA approval of encorafenib and binimetinib. Although the panel members eagerly await more mature data from BEACON CRC and forthcoming FDA approvals, the vast majority of the panel felt that the data from SWOG S1406 were strong enough to warrant the addition of vemurafenib/cetuximab/irinotecan for patients with BRAF V600E–mutated mCRC at this time. They believed that this subset of patients is in great need of additional treatment options and saw no justification for waiting for better regimens.
The panel then discussed whether to recommend vemurafenib/panitumumab/irinotecan for these patients by extrapolation from the SWOG S1406 data. In general, the panel believes that panitumumab and cetuximab are interchangeable, and some clinic formularies may only list one or the other agent. Furthermore, panel members pointed out that some patients may experience a severe reaction to cetuximab. Safety of vemurafenib/panitumumab/irinotecan has been shown in a case report of a patient with BRAF-mutant cholangiocarcinoma.50 Therefore, the panel voted to add this regimen as an additional option for patients with BRAF V600E–mutant mCRC.
Next, the panel discussed whether dabrafenib could be substituted for vemurafenib by extrapolation of SWOG S1406 data. However, they do not believe that dabrafenib and vemurafenib are interchangeable. Dabrafenib was studied in patients with BRAF V600E–mutant mCRC in a case report and a few early clinical trials.51–54 The largest was a single-arm trial that included 43 patients with BRAF V600E–mutant mCRC who received dabrafenib with trametinib.51 Trametinib is another MEK inhibitor with an FDA indication in BRAF-mutated melanoma and non–small cell lung cancer in combination with dabrafenib.55 The complete response rate was 2% (1 patient), partial response rate was 9% (4 patients), and stable disease rate was 56% (24 patients). Grade 3 AEs occurred in 58% of patients, with nausea, pyrexia, and fatigue being the most common events of any grade. Four patients (9%) discontinued treatment because of AEs. To the best of the panel’s knowledge, however, dabrafenib has not been studied in combination with irinotecan or an EGFR inhibitor, and therefore no safety data for the proposed combination exist. Therefore, the panel declined to add dabrafenib/(cetuximab or panitumumab)/irinotecan to the continuum of care for patients with BRAF-mutant mCRC. The panel noted that trametinib/dabrafenib/panitumumab is currently being studied in an open-label phase I/II trial of patients with BRAF V600E–mutated mCRC (ClinicalTrials.gov identifier: NCT01750918).
Overall, the panel is gratified by the addition of new treatment options for patients with BRAF-mutated mCRC that has progressed on 1 or 2 previous metastatic regimens (see COL-11 and COL-D 2 of 10, pages 362 and 364, respectively, as examples). They are hopeful that ongoing trials will lead to even better options for this difficult-to-treat subset of patients. With the addition of vemurafenib/(cetuximab or panitumumab)/irinotecan to the mCRC continuum of care, the BRAF status of the tumor is now another factor that must be considered when choosing therapies for patients with mCRC. BRAF status can be determined using any methodology, including next-generation sequencing, which can be used to simultaneously test for MSI and mutations in RAS (COL-B 4 of 5, page 363). The panel further notes that BRAF and RAS mutations are mutually exclusive. Therefore, although the guidelines only indicate that the new regimens are for “BRAF V600E mutation–positive” disease, these patients’ tumors contain wild-type RAS.
Conclusions
Personalizing treatment allows patients to maximize benefits while minimizing harms, thus providing optimal survival and quality of life. In stage III colon cancer, data from the IDEA trial have led to more refined risk stratification that allows some patients to opt for a shorter duration of adjuvant therapy. Thus, they can be spared the associated toxicity, cost, and inconvenience of longer adjuvant treatment without jeopardizing their oncologic outcomes. In mCRC, data from the SWOG S1406 trial has led to new treatment options specifically for patients with BRAF V600E–mutated tumors. These patients have derived little benefit from EGFR-targeted agents in the past and have had poor prognoses. The addition of an inhibitor of the specific BRAF mutation provides additional treatment options that include EGFR inhibitors and gives these patients a chance for delayed disease progression.
NCCN Categories of Evidence and Consensus.
Category 1: Based upon high-level evidence, there is uniform NCCN consensus that the intervention is appropriate.
Category 2A: Based upon lower-level evidence, there is uniform NCCN consensus that the intervention is appropriate.
Category 2B: Based upon lower-level evidence, there is NCCN consensus that the intervention is appropriate.
Category 3: Based upon any level of evidence, there is major NCCN disagreement that the intervention is appropriate.
All recommendations are category 2A unless otherwise noted.
Clinical trials: NCCN believes that the best management for any patient with cancer is in a clinical trial. Participation in clinical trials is especially encouraged.



Footnotes
Provided content development and/or authorship assistance.
The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) are a statement of consensus of the authors regarding their views of currently accepted approaches to treatment. The NCCN Guidelines® Insights highlight important changes to the NCCN Guidelines® recommendations from previous versions. Colored markings in the algorithm show changes and the discussion aims to further the understanding of these changes by summarizing salient portions of the NCCN Guideline Panel discussion, including the literature reviewed.
These NCCN Guidelines Insights do not represent the full NCCN Guidelines; further, the National Comprehensive Cancer Network® (NCCN®) makes no representation or warranties of any kind regarding the content, use, or application of the NCCN Guidelines and NCCN Guidelines Insights and disclaims any responsibility for their applications or use in any way.
The full and most current version of these NCCN Guidelines are available at NCCN.org.
© National Comprehensive Cancer Network, Inc. 2018, All rights reserved. The NCCN Guidelines and the illustrations herein may not be reproduced in any form without the express written permission of NCCN.
Disclosure of Relevant Financial Relationships
The NCCN staff listed below discloses no relevant financial relationships:
Kerrin M. Rosenthal, MA; Kimberly Callan, MS; Genevieve Emberger Hartzman, MA; Erin Hesler; Kristina M. Gregory, RN, MSN, OCN; Rashmi Kumar, PhD; Karen Kanefield; and Kathy Smith.
Individuals Who Provided Content Development and/or Authorship Assistance:
Al B. Benson III, MD, Panel Chair, has disclosed that he receives grant/research support from Acerta Pharma; Bristol-Myers Squibb Company; Celgene Corporation; Infinity Pharmaceuticals; MedImmune Inc.; Merck & Co., Inc.; Novartis Pharmaceuticals Corporation; and Taiho Parmaceuticals Co., Ltd. He also receives consulting fees/honoraria from and serves as a scientific advisor for AstraZeneca Pharmaceuticals LP; Boehringer Ingelheim GmbH; Boston Biomedical; Bristol-Myers Squibb Company; Celgene Corporation; Eli Lilly and Company; EMO Serono; Exelixis Inc.; Genentech, Inc.; Guidant Corporation; Halozyme, Inc.; Helsinn Therapeutics; ImmunoGen, Inc.; Lexicon Pharmaceuticals, Inc.; Novartis Pharmaceuticals Corporation; Opsona Therapeutics; Pfizer Inc.; Purdue Pharma LP; and Taiho Parmaceuticals Co., Ltd.
Alan P. Venook, MD, Panel Vice Chair, has disclosed that he serves as a scientific advisor for Bayer HealthCare; Bristol-Myers Squibb Company; Genentech, Inc.; Merck & Co., Inc.; and Taiho Pharmaceuticals Co., Ltd. He also receives grant/research support from Genentech, Inc., and Merck & Co., Inc.
Stacey Cohen, MD, Panel Member, has disclosed that she has no relevant financial relationships.
Deborah A. Freedman-Cass, PhD, Oncology Scientist/Senior Medical Writer, NCCN, has disclosed that she has no relevant financial relationships. This activity is supported by educational grants from AstraZeneca, Celldex Therapeutics, Celgene Corporation, Genentech, Jazz Pharmaceuticals, Inc., Novartis Pharmaceuticals Corporation, and Seattle Genetics, Inc. This
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Cheng L, Eng C, Nieman LZ, et al. Trends in colorectal cancer incidence by anatomic site and disease stage in the United States from 1976 to 2005. Am J Clin Oncol 2011;34:573–580. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin 2017;67:177–193. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 5.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011;61:212–236. [DOI] [PubMed] [Google Scholar]
- 6.Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010;116:544–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey CE, Hu CY, You YN, et al. Increasing disparities in the age-related incidences of colon and rectal cancers in the United States, 1975–2010. JAMA Surg 2015;150:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuv) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): the IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration [abstract]. J Clin Oncol 2017;35(Suppl):Abstract LBA1. [Google Scholar]
- 9.Kopetz S, McDonough SL, Lenz HJ, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406) [abstract]. J Clin Oncol 2017;35(Suppl):Abstract 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benson AB III, Venook AP, Al-Hawary MM, et al. NCCN Guidelines for Colon Cancer. Version 2.2018. Accessed March 17, 2018. To view the most recent version of these guidelines, visit NCCN.org. [DOI] [PMC free article] [PubMed]
- 11.Boland GM, Chang GJ, Haynes AB, et al. Association between adherence to National Comprehensive Cancer Network treatment guidelines and improved survival in patients with colon cancer. Cancer 2013;119:1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Booth CM, Nanji S, Wei X, et al. Use and effectiveness of adjuvant chemotherapy for stage III colon cancer: a population-based study. J Natl Compr Canc Netw 2016;14:47–56. [DOI] [PubMed] [Google Scholar]
- 13.Casadaban L, Rauscher G, Aklilu M, et al. Adjuvant chemotherapy is associated with improved survival in patients with stage II colon cancer. Cancer 2016;122:3277–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hines RB, Barrett A, Twumasi-Ankrah P, et al. Predictors of guideline treatment nonadherence and the impact on survival in patients with colorectal cancer. J Natl Compr Canc Netw 2015;13:51–60. [DOI] [PubMed] [Google Scholar]
- 15.Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–2351. [DOI] [PubMed] [Google Scholar]
- 16.Andre T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: updated 10-year survival and outcomes according to BRAF mutation and mismatch repair status of the MOSAIC study. J Clin Oncol 2015;33:4176–4187. [DOI] [PubMed] [Google Scholar]
- 17.Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–1471. [DOI] [PubMed] [Google Scholar]
- 18.Schmoll HJ, Cartwright T, Tabernero J, et al. Phase III trial of capecitabine plus oxaliplatin as adjuvant therapy for stage III colon cancer: a planned safety analysis in 1,864 patients. J Clin Oncol 2007;25:102–109. [DOI] [PubMed] [Google Scholar]
- 19.Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–2204. [DOI] [PubMed] [Google Scholar]
- 20.Andre T, Boni C, Navarro M, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol 2009;27:3109–3116. [DOI] [PubMed] [Google Scholar]
- 21.Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 2010;28:3219–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JE, Hong YS, Kim HJ, et al. Defective mismatch repair status was not associated with DFS and OS in stage II colon cancer treated with adjuvant chemotherapy. Ann Surg Oncol 2015;22(Suppl 3):S630–637. [DOI] [PubMed] [Google Scholar]
- 23.McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses. J Clin Oncol 2011;29:3768–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tournigand C, Andre T, Bonnetain F, et al. Adjuvant therapy with fluorouracil and oxaliplatin in stage II and elderly patients (between ages 70 and 75 years) with colon cancer: subgroup analyses of the Multicenter International Study of Oxaliplatin, Fluorouracil, and Leucovorin in the Adjuvant Treatment of Colon Cancer trial. J Clin Oncol 2012;30:3353–3360. [DOI] [PubMed] [Google Scholar]
- 26.Pectasides D, Karavasilis V, Papaxoinis G, et al. Randomized phase III clinical trial comparing the combination of capecitabine and oxaliplatin (CAPOX) with the combination of 5-fluorouracil, leucovorin and oxaliplatin (modified FOLFOX6) as adjuvant therapy in patients with operated high-risk stage II or stage III colorectal cancer. BMC Cancer 2015;15:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cassidy J, Clarke S, Diaz-Rubio E, et al. Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. J Clin Oncol 2008;26:2006–2012. [DOI] [PubMed] [Google Scholar]
- 28.Benson AB III, Venook AP, Cederquist L, et al. NCCN Guidelines for Colon Cancer. Version 2.2017. Accessed June 26, 2017. To view the most recent version of these guidelines, visit NCCN.org.
- 29.Benson AB III, Venook AP, Cederquist L, et al. NCCN Guidelines for Rectal Cancer. Version 3.2017. Accessed July 21, 2017. To view the most recent version of these guidelines, visit NCCN.org.
- 30.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol 2018;36:773–779. [DOI] [PubMed] [Google Scholar]
- 32.Moretto R, Cremolini C, Rossini D, et al. Location of primary tumor and benefit from anti-epidermal growth factor receptor monoclonal antibodies in patients with RAS and BRAF wild-type metastatic colorectal cancer. Oncologist 2016;21:988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tejpar S, Stintzing S, Ciardiello F, et al. Prognostic and predictive relevance of primary tumor location in patients with RAS wild-type metastatic colorectal cancer: retrospective analyses of the CRYSTAL and FIRE-3 trials [published online October 10, 2016]. JAMA Oncol, doi: 10.1001/jamaoncol.2016.3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Venook AP, Niedzwiecki D, Innocenti F, et al. Impact of primary (1°) tumor location on overall survival (OS) and progression free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): analysis of all RAS wt patients on CALGB/SWOG 80405 (Alliance) [abstract]. J Clin Oncol 2016;34(Suppl):Abstract 3504. [Google Scholar]
- 35.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin 2014;64:104–117. [DOI] [PubMed] [Google Scholar]
- 36.Morris V, Overman MJ, Jiang ZQ, et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin Colorectal Cancer 2014;13:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol 2005;23:6771–6790. [DOI] [PubMed] [Google Scholar]
- 38.Sharma A, Shah SR, Illum H, Dowell J. Vemurafenib: targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs 2012;72:2207–2222. [DOI] [PubMed] [Google Scholar]
- 39.Zelboraf [package insert]. South San Francisco, CA: Genentech USA, Inc.; 2017. [Google Scholar]
- 40.Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol 2015;33:4032–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100–103. [DOI] [PubMed] [Google Scholar]
- 42.Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012;2:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pietrantonio F, Petrelli F, Coinu A, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer 2015;51:587–594. [DOI] [PubMed] [Google Scholar]
- 44.Yang H, Higgins B, Kolinsky K, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res 2012;72:779–789. [DOI] [PubMed] [Google Scholar]
- 45.Hong DS, Morris VK, El Osta B, et al. Phase IB study of vemurafenib in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discov 2016;6:1352–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sullivan R, LoRusso P, Boerner S, Dummer R. Achievements and challenges of molecular targeted therapy in melanoma. Am Soc Clin Oncol Educ Book 2015:177–186. [DOI] [PubMed] [Google Scholar]
- 47.Dummer R, Ascierto PA, Gogas H, et al. Results of COLUMBUS part 2: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus ENCO in BRAF-mutant melanoma [abstract]. Ann Oncol 2017;28(Suppl 5):Abstract 1215O. [Google Scholar]
- 48.Huijberts S, Schellens JH, Elez E, et al. BEACON CRC: safety lead-in (SLI) for the combination of binimetinib (BINI), encorafenib (ENCO), and cetuximab (CTX) in patients (Pts) with BRAF-V600E metastatic colorectal cancer (mCRC) [abstract]. Ann Oncol 2017;28(Suppl 5):Abstract 517P. [Google Scholar]
- 49.Cutsem EV, Cuyle PJ, Huijberts S, et al. BEACON CRC study safety lead-in (SLI) in patients with BRAFV600E metastatic colorectal cancer (mCRC): efficacy and tumor markers [abstract]. J Clin Oncol 2018;36(Suppl):Abstract 627.29283749 [Google Scholar]
- 50.Silkin SV, Startsev SS, Krasnova ME, et al. Complete clinical response of BRAF-mutated cholangiocarcinoma to vemurafenib, panitumumab, and irinotecan. J Gastrointest Cancer 2016;47:502–505. [DOI] [PubMed] [Google Scholar]
- 51.Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol 2015;33:4023–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 2012;379:1893–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong DS, Vence L, Falchook G, et al. BRAF(V600) inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency. Clin Cancer Res 2012;18:2326–2335. [DOI] [PubMed] [Google Scholar]
- 54.Williams CB, McMahon C, Ali SM, et al. A metastatic colon adenocarcinoma harboring BRAF V600E has a durable major response to dabrafenib/trametinib and chemotherapy. Onco Targets Ther 2015;8:3561–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mekinist [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2017. [Google Scholar]