

Abstract
BACKGROUND & AIMS:
Advanced colorectal carcinoma (CRC) is characterized by a high frequency of primary immune evasion and refractoriness to immunotherapy. Given the importance of interferon (IFN)-γ in CRC immunosurveillance, we investigated whether and how acquired IFN-γ resistance in tumor cells would promote tumor growth, and whether IFN-γ sensitivity could be restored.
METHODS:
Spontaneous and colitis-associated CRC development was induced in mice with a specific IFN-γ pathway inhibition in intestinal epithelial cells. The influence of IFN-γ pathway gene status and expression on survival was assessed in patients with CRC. The mechanisms underlying IFN-γ resistance were investigated in CRC cell lines.
RESULTS:
The conditional knockout of the IFN-γ receptor in intestinal epithelial cells enhanced spontaneous and colitis-associated colon tumorigenesis in mice, and the loss of IFN-γ receptor α (IFNγRα) expression by tumor cells predicted poor prognosis in patients with CRC. IFNγRα expression was repressed in human CRC cells through changes in N-glycosylation, which decreased protein stability via proteasome-dependent degradation, inhibiting IFNγR-signaling. Downregulation of the bisecting N-acetylglucosaminyltransferase III (MGAT3) expression was associated with IFN-γ resistance in all IFN-γ–resistant cells, and highly correlated with low IFNγRα expression in CRC tissues. Both ectopic and pharmacological reconstitution of MGAT3 expression with all-trans retinoic acid increased bisecting N-glycosylation, as well as IFNγRα protein stability and signaling.
CONCLUSIONS:
Together, our results demonstrated that tumor-associated changes in N-glycosylation destabilize IFNγRα, causing IFN-γ resistance in CRC. IFN-γ sensitivity could be reestablished through the increase in MGAT3 expression, notably via all-trans retinoic acid treatment, providing new prospects for the treatment of immune-resistant CRC.
Keywords: Colon Cancer, Immune Evasion, IFNGR1
Graphical Abstract
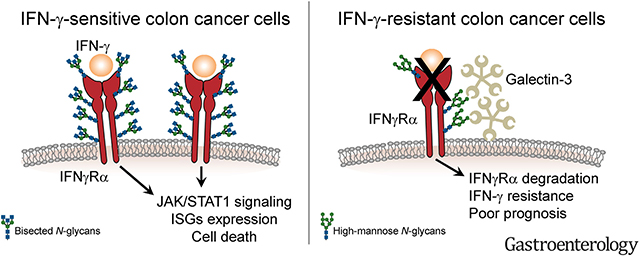
The primary host immune response is an important determinant of the evolution of solid tumors. In colorectal cancer (CRC), the infiltration of T helper 1 and cytotoxic T cells correlates with an increased patient survival and the presence of an interferon (IFN)-γ–driven expression signature.1,2 The crucial role of IFN-γ in tumor immunosurveillance has been amply documented in preclinical models.3 IFN-γ is secreted by immune cells, mostly cytotoxic T cells, and can exert long-distance effects in the tumor microenvironment, inducing a sustained IFN-γ response in tumor cells.4,5 Tumor cell–specific effects of IFN-γ include the inhibition of cell proliferation, the induction of cell death, and an increased immunogenicity.3,6 IFN-γ binds the ubiquitously expressed IFN-γ receptor (IFNγR) α/β complex and signals through the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway. Activation of STAT1 and interferon regulatory factor 1 induces the transcription of interferon-stimulated genes (ISGs).7 In CRC, the expression of various ISGs has been associated with less aggressive disease.8–10
Tumor cells can develop strategies to escape detection and destruction by immune cells. Metastatic CRC is characterized by an increased immune resistance.11 Enhanced expression of immune checkpoint molecules, such as programmed death ligand 1, is infrequent in CRC. Accordingly, immunotherapy with anti-programmed death 1 (anti-PD-1) antibodies only led to an objective response in a small subgroup of microsatellite instable CRCs.12,13 This indicates that, in the large majority of CRCs, primary immune escape involves mechanisms other than immune checkpoint activation. We previously observed the selective loss of ISG expression in CRC tumor cells compared with the stroma,14 suggesting that resistance to IFN-γ might be involved in primary immune evasion of CRC. This is supported by the fact that some colorectal cell lines are insensitive to IFN-γ treatment.14,15 In addition, the treatment of patients with CRC with IFN-γ has yielded a low response rate.16–18
Inactivating frameshift mutations of JAK1 have been reported in CRC with microsatellite instability (microsatellite instability–high, MSI).19,20 On the contrary, microsatellite-stable (MSS) tumors, which represent 85% of sporadic CRCs, show a much lower frequency of JAK1 mutations.19,21,22 Investigations of an association between CRC prognosis and the absence of expression of either Stat-1 or IFNγRα, at the protein or messenger RNA (mRNA) levels, have yielded contradictory results.10,23,24 Hence, in the large majority of CRCs, the mechanisms of IFN-γ resistance remain poorly characterized. In the present study, we investigated the mechanisms by which tumor cells inactivate the IFN-γ response, as well as the consequences of such inactivation in terms of tumorigenesis, and whether it is possible to restore IFN-γ sensitivity in CRC tumor cells.
Materials and Methods
Mouse Models of Colon Carcinogenesis
Colitis-associated carcinogenesis was induced in 6- to 8-week-old mice in 2 independent facilities using azoxymethane (AOM) and dextran-sulfate sodium (DSS) with slight protocol variations. Intraperitoneal injection of AOM was performed on day 1 (Ifngr2ΔIEC: 10 mg/kg body weight Ifngr2ΔIEC−2: 12.5 mg/kg body weight) and DSS-containing drinking water (Ifnγr2ΔIEC: 2% Ifngr2ΔIEC−2: 2.5% DSS solution) was given for 5 (Ifngr2ΔIEC−2) to 7 (Ifngr2ΔIEC) days. DSS supplementation was repeated twice, separated by 14 days of normal drinking water. Animals were killed around day 80. Spontaneous colon carcinogenesis was evaluated in Ifngr2ΔIEC−2Apc; CPC mice or control mice (Ifngr2fl/wtApc; CPC). Mice were killed at 5 months old. Colon length, tumor number, and diameter were determined macroscopically, and the tumor load was calculated as the cumulative tumor diameter in millimeters per mouse.
Patients
Tissue array.
The cohort included patients (n = 416) undergoing surgery at the Universitätsklinikum Erlangen from 1991 to 2001 with follow-up until 2006. Patient and tumor characteristics are given in Supplementary Table 1. Inclusion criteria of the patients were as follows: solitary invasive colon carcinoma (invasion at least of the submucosa) in Union for International Cancer Control stage II–IV, localization >16 cm from the anal verge, no appendix carcinoma, no other previous or synchronous malignant tumor except squamous and basal cell carcinoma of the skin and carcinoma in situ of the cervix uteri, treatment by colon resection with formal regional lymph node dissection, and residual tumor classification R0 (no residual tumor, clinical and histopathological examination). Patients suffering from hereditary CRC or inflammatory bowel disease, patients who died postoperatively, and patients with unknown tumor status (with respect to local and distant recurrence) at the end of the study were excluded.
CRISPR-Cas9 Gene Editing
Single-guide RNAs were designed using Benchling (Biology Software; 2017; retrieved from https://benchling.com). Cloning was performed using the pSpCas9(BB)-2a-Puro (PX459) V2.0 vector (Addgene #62988) and pL-CRISPR.EFS.tRFP vector (Addgene #57819), and confirmed by sequencing. HT-29 cells were transfected with the PX459 plasmid by Lipofectamine 2000 and, 24 hours later, were selected with puromycin for another 72 hours. Afterward, single-cell expansion was performed in a 96-well plate to grow clonal cells. HT-29 cells were then transduced by viral particles generated from the transient transfection of HEK293TN cells with 3 different plasmids encoding VSV-G (pMD2.6, Addgene plasmid 12259), packaging genes (psPAX2, Addgene plasmid 12260) and the pL-CRISPR.EFS.tRFP plasmid. After 72 hours of transduction, the cells were sorted by fluorescence-activated cell sorting for RFP-positivity and single cells were seeded in 96-well plates for further clonal expansion. The clones were screened for knockout of the IFNγRα via Western blotting.
Results
Inhibition of the IFN-γ Response in Intestinal Epithelial Cells Promotes Tumorigenesis
To determine whether the specific loss of sensitivity to IFN-γ by tumor cells influences intestinal tumorigenesis, we generated a mouse model with conditional deletion of the IFN-γ receptor beta chain (IFNγRβ encoded by Ifngr2) in intestinal epithelial cells by crossing Ifngr2fl/fl mice with Villin-Cre mice (Supplementary Figure 1A). The resulting Ifngr2ΔIEC mice showed a reduction in Ifngr2 mRNA expression in colon tissue, and an absence of murine IFNγRβ in colon epithelial cells while the protein was still present in stromal cells (arrows), confirming the specificity of the knockout (Figure 1A and B). Colon carcinogenesis was induced in Ifngr2ΔIEC and Ifngr2fl/fl control mice by treatment with AOM and DSS. Ifngr2ΔIEC mice developed more and larger tumors than Ifngr2fl/fl control mice (Figure 1C–F). This increase in tumorigenesis was not due to an increase in inflammation because Ifngr2ΔIEC mice treated solely with DSS showed less colonic inflammation than control Ifngr2fl/fl mice (Supplementary Figure 1B–E). These results could be replicated independently in another mouse facility using an independent strain (Ifngr2ΔIEC−2, Supplementary Figure 1F and G). The conditional deletion of Ifngr2 in a sporadic CRC mouse model (Apc; CPC mice, which spontaneously develop colon tumors) similarly increased tumor number and load compared with controls (Figure 1G and H). Hence, genetic mouse models demonstrated that tumor cell intrinsic resistance to IFN-γ increases both colitis-associated and spontaneous colon tumorigenesis.
Figure 1.
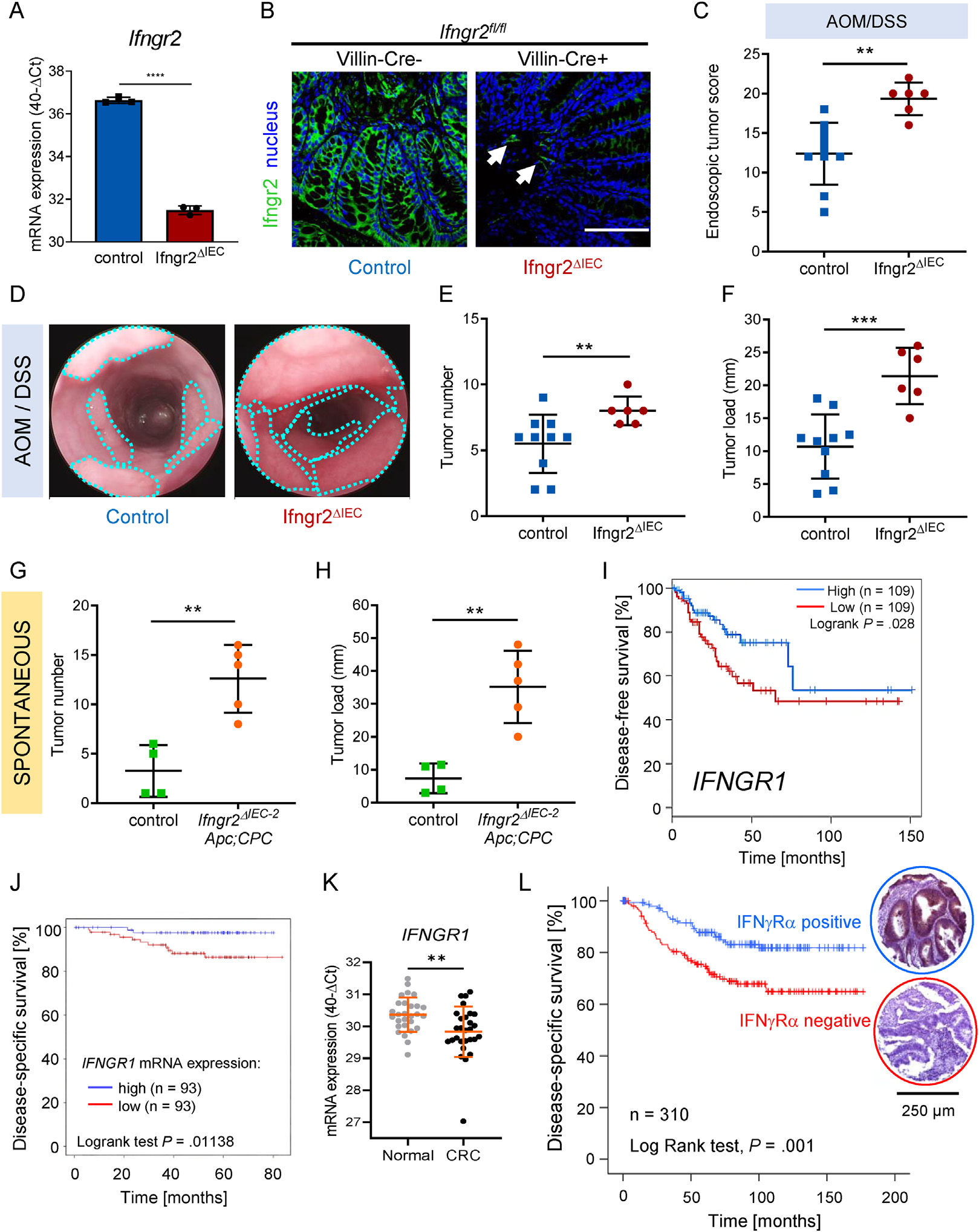
Absence of IFN-γ-receptor expression in colon tumor cells promotes tumorigenesis in mice and correlates with poor prognosis in patients with CRC. (A) Ifngr2 mRNA expression was measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in mouse colon tissue in triplicates. Results are given as mean ± SD of 40-ΔCt (CtIfngr2-CtGapdh) values. Two-tailed unpaired Student t test was used for statistical evaluation (****P < .0001). (B) Representative micrographs of fluorescent immunostaining of IFNγRβ (green) in mouse colon tissues. Nuclei were stained with DRAQ5 (blue). Arrows point at stromal expression of IFNγRβ in otherwise negative colon sections. Scale bar = 100 μm. (C–F) Colitis-associated colon carcinogenesis was induced in Ifngr2ΔIEC (n = 6) and control mice (n = 10) by AOM-DSS treatment. Endoscopic scoring (C, D) and macroscopic evaluation of tumor number (E) and tumor load (F) are given. Bars represent means±SD (C, E, F). Two-tailed unpaired Student t test (C, **P = .0014 F, ***P= .0006) or 2-tailed Mann-Whitney test (E, **P = .0079) were used for statistical evaluation. (D) Representative endoscopic pictures showing colonic tumors (turquoise dotted lines). (G and H) Spontaneous colon carcinogenesis was monitored in Apc; CPC mice either heterozygous for Ifngr2 (control, Ifngr2+/−, n = 4) or devoid of Ifngr2 in intestinal epithelial cells (Ifngr2ΔIEC−2, n = 5). Two-tailed unpaired Student t test was used for statistical evaluation of differences in tumor number (G **P = .0029) and tumor load (H **P= .0022). (I) Kaplan-Meier disease-free survival curve of patients with CRC comparing the 30% highest IFNGR1 mRNA gene expression samples (blue, n = 109) with the 30% lowest (red, n = 109 P = .028). (J) Prognostic value of IFNGR1 mRNA expression for human patients with CRC (Polyprobe cohort, n = 410). Kaplan-Meier plots of disease-specific survival comparing the 25% highest (red, n = 93) and the 25% lowest (blue, n = 93) expressing samples (P = .01138). (K) IFNGR1 mRNA expression was determined in triplicate by qRT-PCR in corresponding tumor and normal tissues (n = 28). Results are given as 40-ΔCt (CtIFNGR1-CtRPL37A) (mean ± SD Mann-Whitney test, **P = .0041). (L) Disease-specific survival of patients with colon carcinoma with positive (blue, n = 152) and negative (red, n = 158) tumor cell IFNγRα protein expression (Kaplan-Meier plot, P = .001).
Loss of IFNγRα Expression Correlates With Decreased Disease-specific Survival in Patients With CRC
The prognostic value of IFN-γ pathway gene expression was assessed in patients with CRC. The mRNA expression of IFNGR1, but not IFNGR2, STAT1, JAK1, or JAK2, was associated with disease-free survival in The Cancer Genome Atlas cohort (Figure 1I and Supplementary Figure 2A). These results were confirmed using an independent cohort of patients with CRC, where low IFNGR1 mRNA expression correlated with a reduced disease-specific survival (Figure 1J, Supplementary Table 2). We also observed a modest reduction (less than 2-fold) in IFNGR1 mRNA expression in CRC tissues compared with matching patient normal tissues (Figure 1K, Supplementary Table 3). We could confirm that mutations in the IFN-γ response pathway genes are rare in CRC (Supplementary Figure 2B) and not related to disease-specific survival (Supplementary Figure 2C).
Gene expression analysis in whole tissue mRNA samples is not systematically predictive of protein levels, nor does it allow the specific evaluation of tumor cell–associated expression. Therefore, we analyzed the protein expression of the IFN-γ receptor α chain (IFNγRα, encoded by IFNGR1) by immunohistochemistry on a tumor tissue microarray from a cohort of 416 patients with colon cancer (Figure 1L, Supplementary Table 1). Overall, the quality criteria for analysis were met in 310 of the 416 tumors. a loss of IFNγRα expression in tumor cells was observed in approximately half of the cases (158 of 310), and was associated with increased tumor size (χ2 test, P = .04), lymph node invasion (χ2 test, P < .0001), extramural venous invasion (χ2 test, P < .0001), distant metastasis (χ2 test, P = .019), and Union for International Cancer Control stage (χ2 test, P < .0001). Furthermore, the absence of IFNγRα expression in tumor cells (n = 158, 50.9%) correlated with a shorter disease-specific survival (Figure 1L).
IFNγRα Expression Level and Pattern Correlate With IFN-γ Resistance in CRC Cells
To investigate whether IFNγRα loss correlates with IFN-γ resistance in CRC tumor cells, we examined the induction of ISG expression and cell death by IFN-γ in human CRC cell lines together with IFNγRα expression. Among the 11 cell lines tested, 6 were resistant to IFN-γ–induced cell death, Stat1 phosphorylation and ISG expression (GBP1, IDO1, CASP1) (Figure 2A and B, and Supplementary Figure 3A and 4B; resistant cell lines highlighted in red). In the other 5 cell lines (in blue), the induction of cell death by IFN-γ correlated with Stat-1 phosphorylation and ISG expression at the protein and mRNA levels (Figure 2A and B, and Supplementary Figures 3A and 4B).
Figure 2.
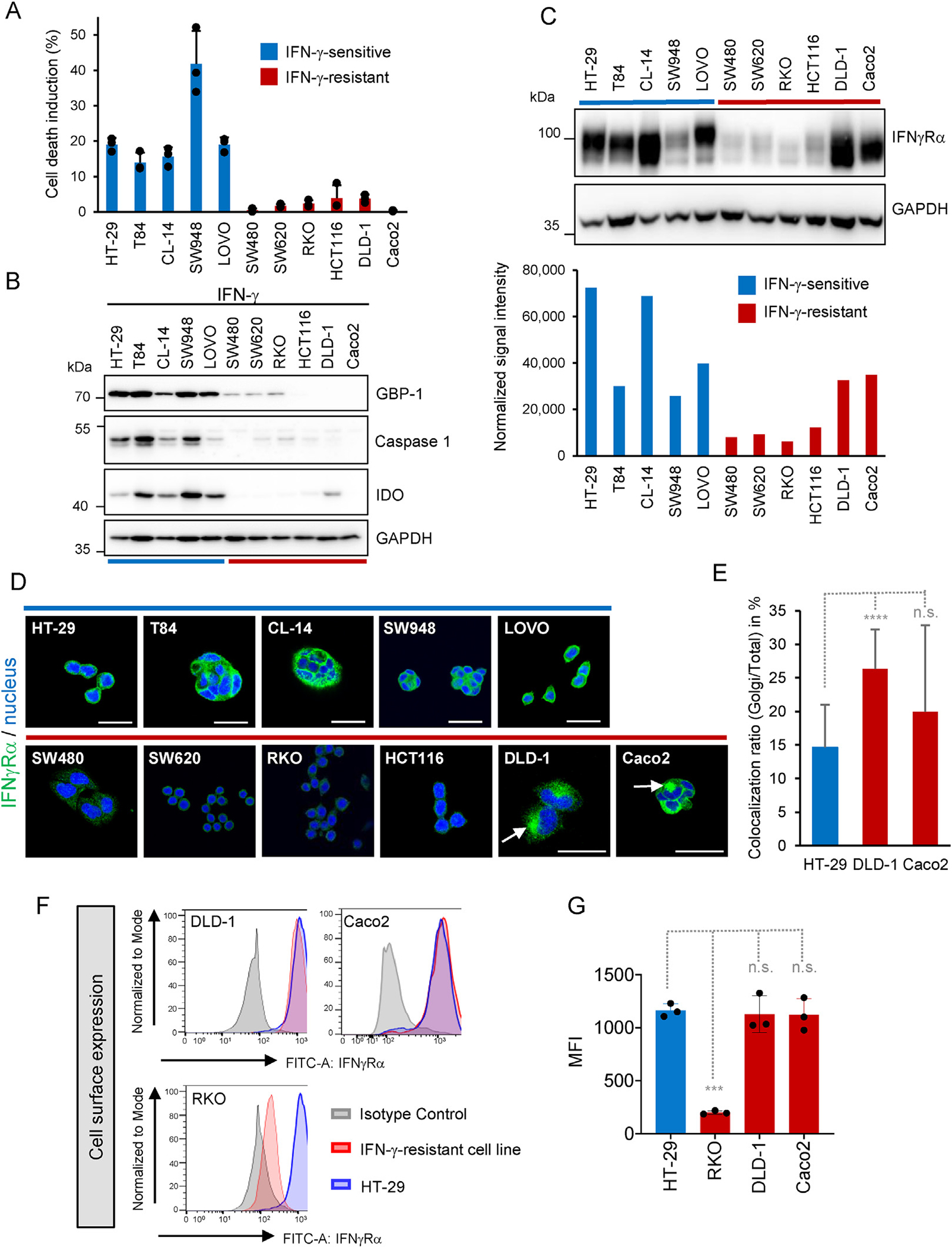
INγRα expression is down-regulated in IFN-γ–resistant cells. IFN-γ–resistant cells are highlighted in red, and IFN-γ–sensitive cells in blue. GAPDH was used as loading control for Western blots. (A) Cell death induction determined 72 hours after IFN-γ treatment (100 U/mL) by flow cytometry. Results are given in percent as the difference between IFN-γ–treated and mock-treated controls (mean ± SD, n = 3 distinct samples). (B) ISG expression in CRC cell lines. (C) IFNγRα expression in CRC cell lines. (D) Intracellular staining of IFNγRα in CRC cells. Nuclei were counterstained with DRAQ5 (blue). Scale bars = 25 μm arrows show perinuclear accumulation of IFNγRα. (E) Quantification of IFNγRα Golgi localization in HT-29 (n = 14), DLD-1 (n = 19), and Caco-2 (n = 19) cells. (F) Cell surface expression of IFNγRα analyzed by flow cytometry in CRC cell lines. IFN-γ–resistant cells (red), isotype staining (negative control, gray) and HT-29 (positive control, blue). (G) Mean fluorescence intensity of IFNγRα (MFI) ± SD (n = 3 distinct samples) measured by fluorescence-activated cell sorting in CRC cell lines. Two-tailed unpaired Student t test: ***PRKO = .0009 n.s. = not significant.
Four of 6 IFN-γ–resistant cell lines (RKO, HCT116, SW480, SW620) displayed a strong reduction in IFNγRα mRNA and protein expression compared with the IFN-γ–sensitive cell lines as shown by Western blotting, intracellular and surface immunofluorescence staining, or flow cytometry (Figure 2C–G, Supplementary Figures 3C, 4A and B, and 5C). The 2 remaining resistant lines (DLD-1 and Caco2) expressed IFNγRα, but the protein was detected with a reduced apparent molecular weight (Figure 2C and Supplementary Figure 3C). In these cells, intracellular staining revealed a perinuclear accumulation of IFNγRα (Figure 2D, arrows), associated with the Golgi apparatus (GM130) but not with the endoplasmic reticulum (calnexin) (Supplementary Figure 5D and E). The quantification of Golgi-associated IFNγRα localization showed an increase in DLD-1 (P < .0001) and Caco2 (P = .1124) cells compared with HT-29 cells (Figure 2E). Although this finding was indicative of some intracellular retention, the cell surface expression of IFNγRα in both cell lines was similar to that in IFN-γ–sensitive cells (Figure 2F and G, Supplementary Figure 5B). Similar differences in the pattern of IFNγRα expression were observed in CRC tissue extracts (Supplementary Figure 4C), including both differences in expression level and apparent molecular size shifts toward lower molecular weight.
In contrast, mRNA and protein expression of the other mediators of the IFN-γ response (IFNGR2, STAT1, JAK1, and JAK2) was observed in all cell lines except Caco-2. In the latter, no STAT1 expression was detected at the protein level, and JAK2 mRNA expression was reduced (Supplementary Figures 3C and 4A). Therefore, Caco-2 cells were excluded from further functional tests with IFNγRα because of the presence of additional defects in the JAK-STAT pathway. According to the data retrieved from the Cancer Cell Line Encyclopedia, few mutations were found for IFNGR1, IFNGR2, STAT1, JAK1, and JAK2 in the 11 cell lines investigated, and they did not correlate with IFN-γ resistance (Supplementary Table 4). Furthermore, independent sequencing of IFNGR1 mRNA from IFN-γ–resistant cells showed no alteration, indicating that the IFNγRα size shift observed in DLD-1 and Caco-2 cells was not due to a truncating mutation or alternative splicing (Supplementary Table 4). Of note, among the 11 CRC cell lines investigated here, none harbored a frameshift JAK mutation and only 1 resistant line (RKO) exhibited overexpression of programmed death ligand 1 (PD-L1) (Supplementary Figure 3B), which was not mutually exclusive with down-regulated IFNγRα expression. Overall, these results suggested that defects in IFNγRα expression or post-translational maturation correlate with IFN-γ resistance in CRC cell lines, whereas other signaling components of the pathway are either not or only marginally involved.
IFNγRα Is Aberrantly Glycosylated in IFN-γ–resistant CRC Cells
To determine whether epigenetic gene silencing may be responsible for the downregulation of IFNGR1 expression in RKO, HCT116, SW480, and SW620 cells, cells were treated with the DNA methylation inhibitor decitabine. Decitabine treatment led to a modest but statistically significant increase in IFNGR1 mRNA expression in all 4 cell lines (Figure 3A), which was, however, not accompanied by an increase at the protein level (Figure 3B).
Figure 3.
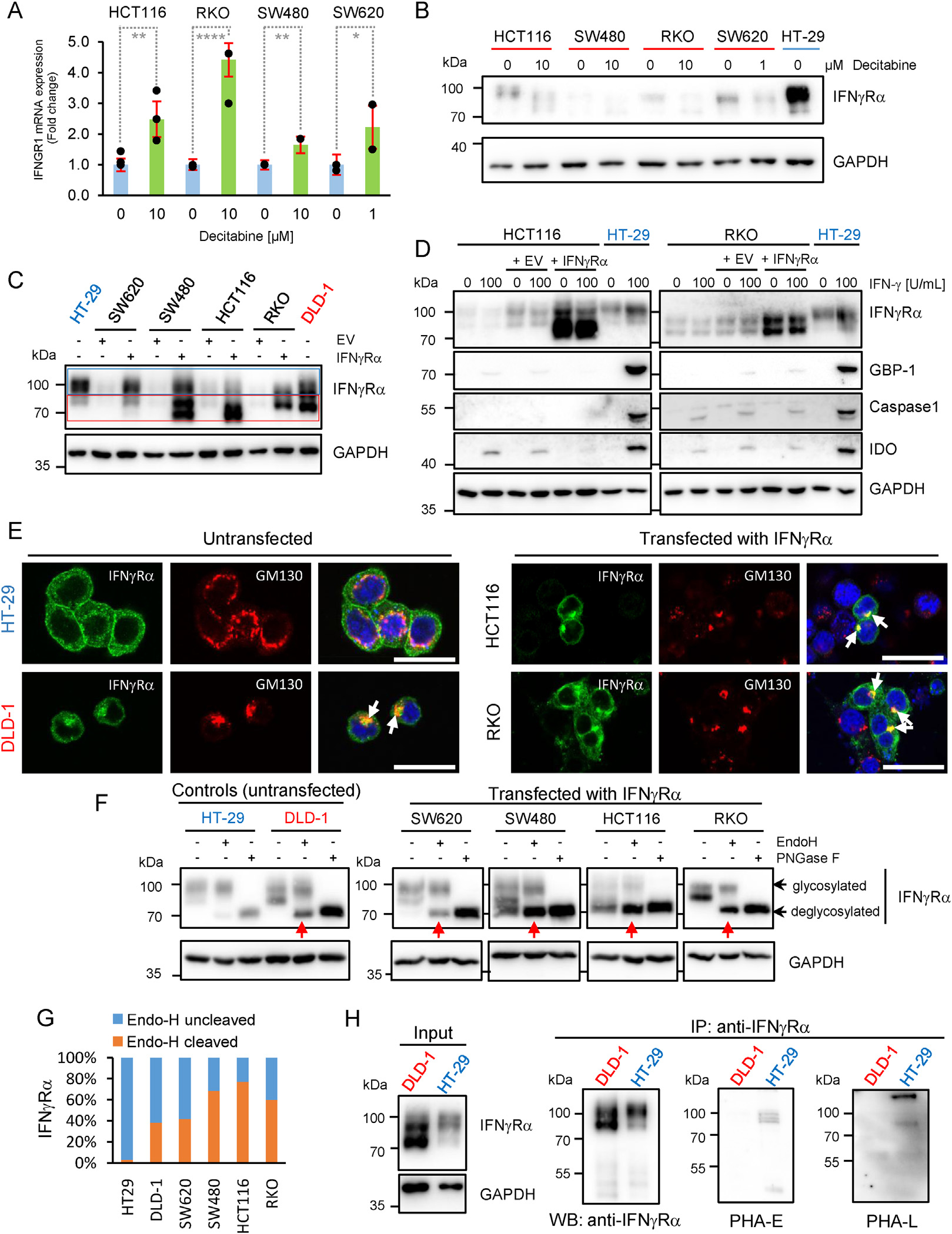
IFNγRα is aberrantly glycosylated in IFN-γ-resistant CRC cell lines. GAPDH was used as loading control for Western blots. (A) IFNGR1 mRNA expression measured in triplicate by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in IFN-γ–resistant cell lines treated with decitabine (1–10 μM) or dimethyl sulfoxide (DMSO) as control for 96 hours. Results are given as fold-change ± SD compared to DMSO-treated control Student t test was performed using ΔCt (CtIFNGR1-CtRPL37A) values with **PHCT116 = .0059, ****PRKO < .0001, **PSW480 = .0046, and *PSW620 = .0346. (B) IFNγRα protein expression in CRC cell lines treated with decitabine (1–10 μM) or DMSO as control for 96 hours. (C and D) IFNγRα and ISG protein expression in SW620, SW480, HCT116, and RKO after transfection with empty vector or IFNγRα-expressing construct. HT29 and DLD-1 cells were used as expression controls. Rectangles highlight high (blue) and low (red) migrating bands. (E) Staining of IFNγRα (green) and GM130 (red) in CRC cells after reconstitution of IFNγRα expression. Nuclei were counterstained with DRAQ5 (blue). Scale bar = 25 μm arrows indicate colocalization of IFNγRα and GM130. (F) IFNγRα expression in protein lysates from transfected IFN-γ–resistant CRC cells digested with either Endo-H or PNGase-F (each at 1 U/μg protein), lysates processed in absence of enzyme being used as controls. EV, empty vector IFNγRα, IFNγRα expression plasmid. Red arrows indicate digestion of IFNγRα by Endo-H. (G) Signal intensity ratio of cleaved and uncleaved IFNγRα in percent of total. (H) IFNγRα expression and lectin binding (PHA-E and PHA-L) in immunoprecipitated protein lysates from HT-29 and DLD-1 cells. Input samples (10 μg) were analyzed for IFNγRα expression. WB, Western blot.
The expression of IFNγRα was then reconstituted by transfection in RKO, HCT116, SW480, and SW620 cells to explore whether this could restore the IFN-γ response. Although ectopic IFNγRα expression achieved by transfection in RKO, HCT116, SW480, and SW620 cells yielded protein levels comparable to those of the IFN-γ–sensitive control cells (Figure 3C), and increased IFNγRα expression at the cell surface (Supplementary Figure 6A), it failed to restore the response to IFN-γ (Figure 3D and Supplementary Figure 6B). In addition, IFNγRα showed a reduced apparent molecular weight (Figure 3C and D and Supplementary Figure 6B and C) together with an intracellular accumulation at the Golgi apparatus, similar to what was observed in DLD-1 and Caco2 cells (Figure 3E, arrows, and Supplementary Figure 6D and E). The altered migratory pattern and Golgi retention of IFNγRα observed in all IFN-γ–resistant lines, either endogenously or after ectopic expression, suggested differences in N-glycosylation.25 This was supported by the observation of a similar apparent molecular weight shift after ectopic expression of the endothelial surface protein Endoglin in IFN-γ–resistant, but not in IFN-γ–sensitive CRC cells (Supplementary Figure 6F).
To assess whether IFNγRα was differently glycosylated in IFN-γ–sensitive and –resistant cell lines, we used 2 endoglycosidases with different specificity, protein-N-glycosidase F (PNGase-F) and endoglycosidase-H (Endo-H). PNGase-F cleaves all N-glycans, whereas Endo-H specifically removes high-mannose N-glycan chains (but not mature complex N-glycans). PNGase-F treatment resulted in a shift of IFNγRα band size to approximately 70 kDa in all cell lines tested, indicating that the receptor undergoes N-glycosylation to some extent in both IFN-γ–sensitive and IFN-γ–resistant cell lines (Figure 3F). In IFN-γ–sensitive cells (HT29), IFNγRα was resistant to Endo-H digestion, indicating a mature complex N-glycosylation (Figure 3F and G). In all IFN-γ–resistant CRC cell lines, IFNγRα was highly sensitive to Endo-H digestion, regardless of whether ectopically (RKO, HCT116, SW480, SW620) or endogenously (DLD-1) expressed, indicating the presence of high-mannose N-glycans characteristic of an immature, low-complexity glycosylation.
Endogenously expressed IFNγRα was then immunoprecipitated from HT-29 (IFN-γ–sensitive) and DLD-1 (IFN-γ–resistant) cells, and its modification with complex N-glycans was detected by Western blotting using the specific lectins Phaseolus vulgaris erythroagglutinin (PHA-E) and Phaseolus vulgaris phytohemagglutinin-L (PHA-L). Binding of both PHAE and PHA-L to IFNγRα was reduced in DLD-1 cells compared with HT-29 cells, indicating a decreased complexity of IFNγRα N-glycosylation in IFN-γ–resistant cells (Figure 3H).
IFNγRα Stability and Signaling Are Regulated by N-glycosylation
In the next step, we investigated whether changes in N-glycosylation were indeed able to affect IFNγRα function. Treatment of IFN-γ–sensitive HT-29 cells with the N-glycosylation inhibitors tunicamycin and 2-deoxyglucose induced a shift in the IFNγRα apparent molecular weight and inhibited IFN-γ signaling (Figure 4A, Supplementary Figure 7A–C). We then investigated whether a non-glycosylated form of IFNγRα obtained by mutation of its 5 putative glycosylation sites25,26 (ΔG-IFNγRα) could signal in response to IFN-γ (Figure 4B). As recipient cells, we chose N-glycosylation-competent HT-29 cells with a CRISPR-Cas9–mediated knockout of IFNGR1 (IFNGR1-KO HT-29, clone sg 2.21) that exhibited a complete inhibition of IFNγRα protein expression, IFN-γ signaling, and ISG expression compared with controls (NTC) (Figure 4C and Supplementary Figure 8A–C). Ectopic expression of wild-type IFNγRα in IFNGR1-KO HT-29 cells resulted in electrophoretic migration at the expected molecular weight (~90 kDa), as well as induction of pStat-1 phosphorylation, ISG expression, membrane localization, and cell death upon treatment with IFN-γ (Figure 4D and Supplementary Figure 8D–H). In contrast, ectopically expressed ΔG-IFNγRα was detected at approximately 70 kDa by Western blot, below the molecular weight of wild-type IFNγRα and similar to the PNGase-F-treated fully deglycosylated receptor (Figure 4D). Contrary to wild-type IFNγRα, ΔG-IFNγRα had a strongly reduced IFN-γ response, both in pools of transduced cells and in single clones (Figure 4D and Supplementary Figure 8D–G). Furthermore, a dose-response analysis suggested that ligand binding of ΔG-IFNγRα is impaired compared with that of wild-type IFNγRα (Supplementary Figure 8F and G).
Figure 4.
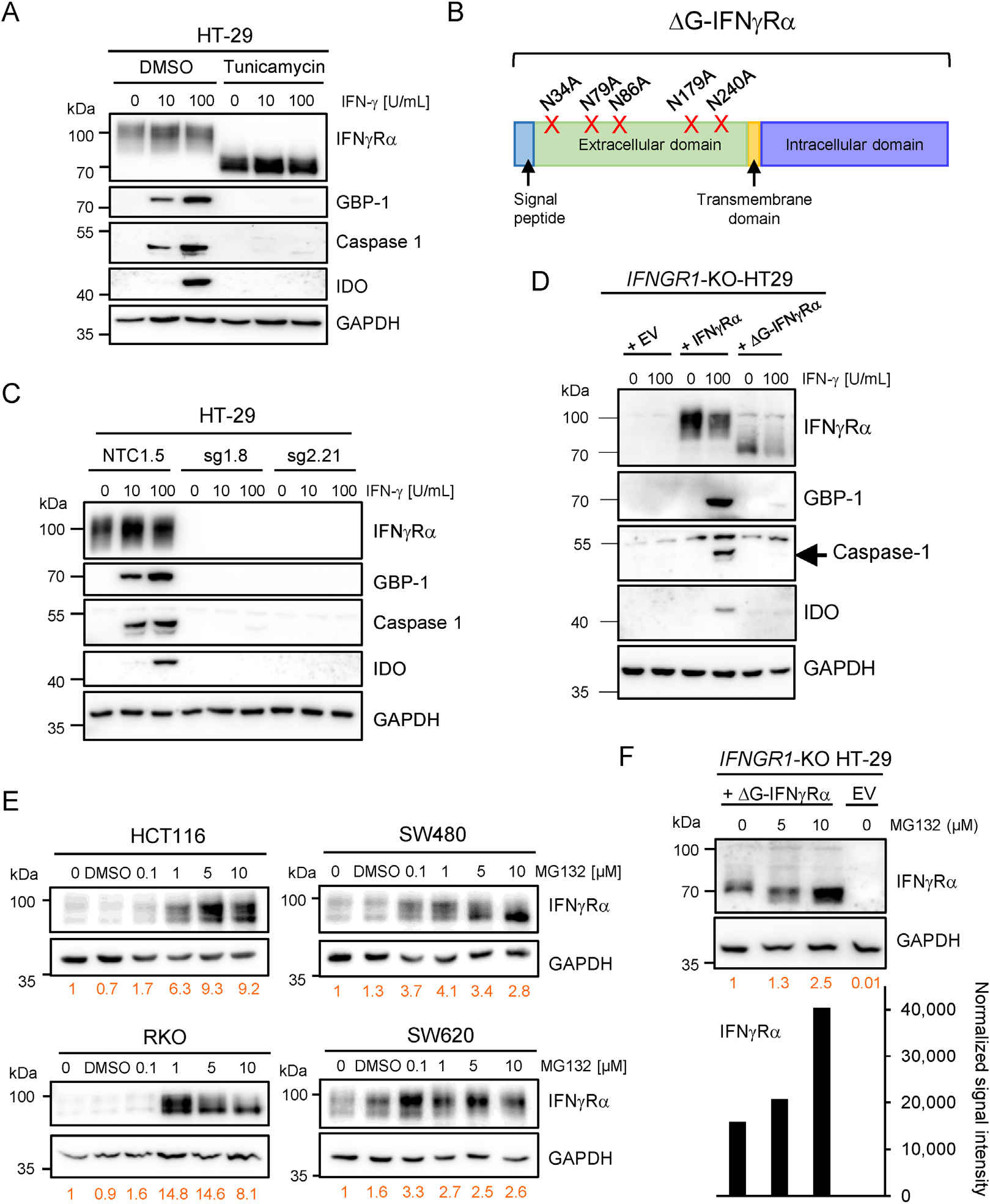
N-glycosylation regulates IFNγRα signaling and protein stability in IFN-γ–resistant cells. GAPDH was used as loading control for Western blots. (A) IFNγRα and ISG protein expression in HT-29 cells treated with tunicamycin or dimethyl sulfoxide (DMSO) as control for 24 hours before stimulation with IFN-γ. (B) Schematic representation of the point mutations inserted in the IFNGR1 sequence to generate a glycosylation-defective mutant (ΔG-IFNγRα). N, asparagine A, alanine. (C) IFNγRα and ISGs protein expression in IFNGR1-KO HT-29 clone sg1.8, sg2.21 and control cells (NTC1.5) after 24 hours of IFN-γ stimulation. (D) IFNγRα and ISG expression in IFNGR1-KO HT-29 cells transduced with empty virus (EV), wild-type IFNγRα, or the ΔG-IFNγRα. Cells were stimulated for 24 hours with indicated amounts of IFN-γ. (E) IFNγRα expression in HCT116, RKO, SW480, and SW620 cell lines treated with increasing concentrations of MG132 (24 hours). The signal intensity for IFNγRα (in orange) was normalized to GAPDH intensity and is given relatively to untreated control (set to 1). (F) IFNγRα expression in IFNΓR1-CRISPR KO HT-29 clone sg 2.21 transduced with EV or ΔG-IFNγRα ± increasing concentrations of MG132 (24 hours). IFNγRα signal intensity (graph, text in orange) was normalized and calculated as in (E).
To determine whether immature N-glycosylation of IFNγRα might increase protein degradation, we treated RKO, HCT116, SW480, and SW620 cells with the proteasome inhibitor MG132. MG132 induced a strong dose-dependent increase in IFNγRα protein levels in all 4 IFN-γ–resistant cell lines (Figure 4E), which was, in comparison, not observed in HT-29 cells (Supplementary Figure 8I). The protein expression level of ΔG-IFNγRα was also enhanced by treatment with MG132 (Figure 4F), confirming that N-glycosylation defects can trigger proteasome-dependent degradation of IFNγRα.
Reconstitution of MGAT3 Expression Rescues IFNγRα N-glycosylation and Signaling
To determine which enzymes of the N-glycan synthesis pathway might be responsible for the downregulation of IFNγRα complex glycosylation in IFN-γ–resistant CRC cells, a quantitative reverse transcription polymerase chain reaction array was performed (Figure 5A). Multiple genes displayed differential expression patterns between IFN-γ–sensitive and –resistant cells. MGAT3 (N-acetylglucosaminyltransferase III, or GnT-III) was the only gene consistently down-regulated in all IFN-γ–resistant cells compared with IFN-γ–sensitive control cells (HT-29 and SW948; Figure 5A, arrow). MGAT3/GnT-III catalyzes the addition of β1,4-linked GlcNAc on the central mannose of the trimannosyl core of N-linked oligosaccharides, generating so-called “bisected” N-glycans. The downregulation of MGAT3 expression in IFN-γ–resistant cell lines was confirmed at the mRNA and protein levels (Figure 5C and Supplementary Figure 9A and B). In addition, the level of bisected N-glycans detected by PHA-E lectin binding correlated with MGAT3 expression, and was reduced in all IFN-γ–resistant cell lines (Figure 5B–D).
Figure 5.
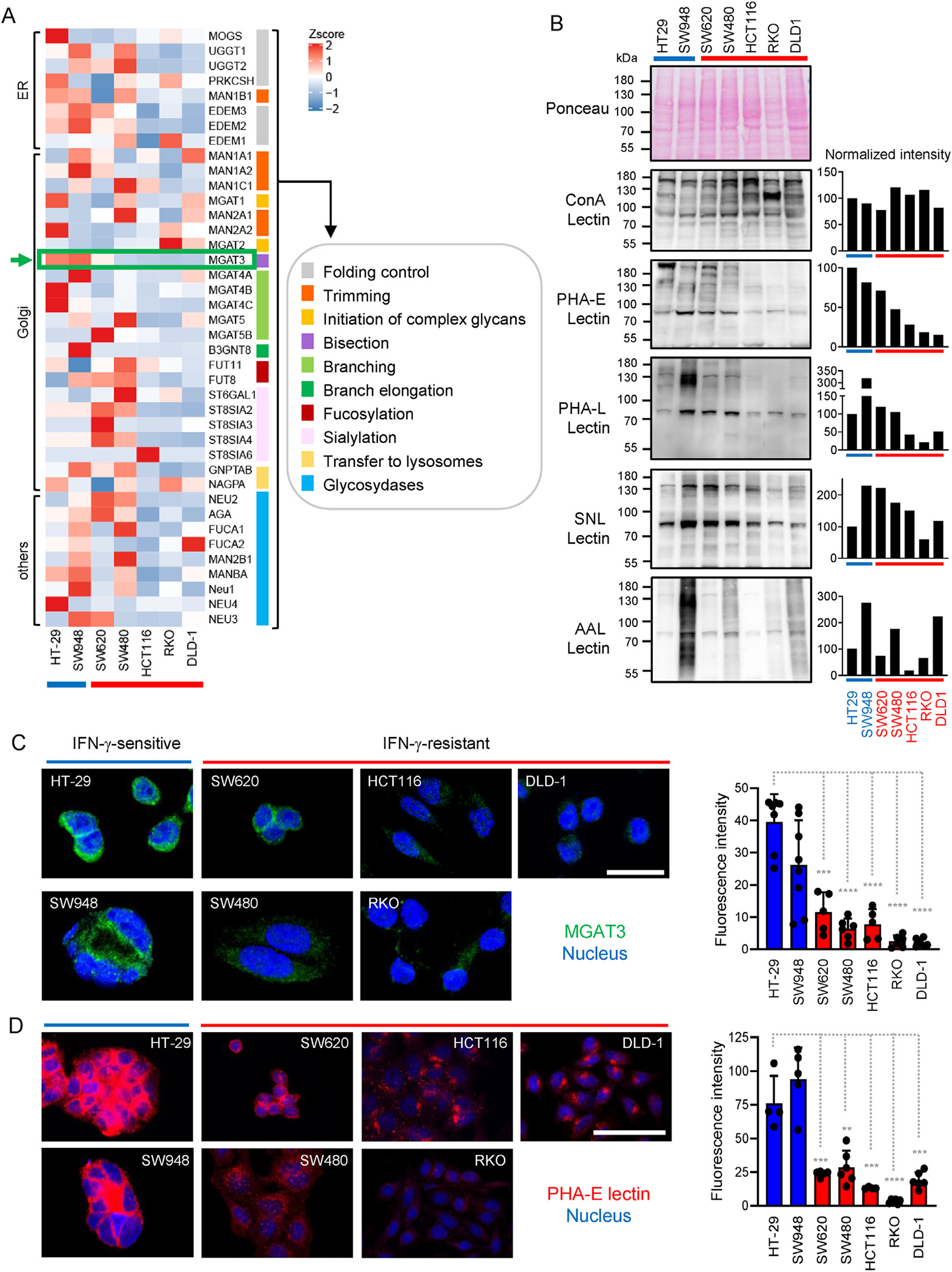
IFN-γ–sensitive and –resistant cells exhibit different N-glycosylation profiles. (A) Heatmap representation of N-glycosylation gene expression data of IFN-γ–sensitive (blue) and IFN-γ–resistant (red) CRC cells. Results are depicted as 2-(ΔCtGOI - ΔCtMean GOI). (B) Lectin blotting analysis of CRC cells (25 μg of protein lysates/lane) was performed using PHA-E, PHA-L, Sambucus nigra lectin, and Aleuria auraentia lectin lectins. Concanavalin A (ConA) detected the overall level of mannose and glucose residues and was used as control. Ponceau staining was used to verify equal loading. Bar diagrams depict intensity values normalized to HT-29 (in percent). (C) Staining of MGAT3 (green) in CRC cell lines. Nuclei were counterstained with DRAQ5 (blue). Scale bar = 25 μm. Bar diagram shows mean ± SD of single cell-corrected fluorescence intensity. Two-tailed unpaired Student t test was used for comparison between HT-29 (blue) and IFN-γ–resistant cell lines (red), with ***PSW620 = .000105, ****PSW480 < .0001, ****PHCT116 < .0001, ****PRKO < .0001, ****PDLD-1 < .0001. (D) CRC cell lines were stained with PHA-E lectin (red) and nuclei were counterstained with DRAQ5 (blue). Scale bar = 50 μm. Bar diagram shows mean ± SD of single cell-corrected fluorescence intensity. Two-tailed unpaired Student t test was used for comparison between HT-29 (blue) and IFN-γ–resistant cell lines (red), with ***PSW620 = .0007, **PSW480 = .0017, ***PHCT116 = .0008, ****PRKO < .0001, ***PDLD-1 = .00017.
Therefore, we examined whether the modulation of MGAT3 expression influences IFNγRα complex N-glycosylation. Chinese hamster ovary (CHO) cells without MGAT3 activity (Pro-5) or with an MGAT3 gain-of-function mutant (Lec10B cells) were used to ectopically express the IFNγRα protein.27 Western blot analysis revealed an increased apparent molecular weight and expression level of IFNγRα in Lec10B CHO cells compared with control Pro5 CHO cells (Supplementary Figure 9C), suggesting that MGAT3 activity increases IFNγRα bisected glycosylation and protein levels. All-trans retinoic acid (ATRA) has been shown to increase MGAT3 expression levels and to enhance the addition of bisecting N-glycans to proteins in vitro.28 In the IFN-γ–resistant/MGAT3-low RKO cells, ATRA induced a dose-dependent increase in MGAT3 and IFNγRα protein expression (Supplementary Figure 9D; quantification of IFNγRα: middle panel, and MGAT3: lower panel), accompanied by a higher IFNγRα molecular weight (Supplementary Figure 9D; middle panel; green: IFNγRα upper band, gray: IFNγRα lower band), confirming that MGAT3 promotes bisected glycosylation and stabilization of the IFNγRα protein.
The addition of bisecting N-glycans by MGAT3 has been shown to reduce the affinity of cell surface proteins to the galectin lattice, particularly to galectin-3.29 Consistent with this, the level of bisected N-glycans observed on immunoprecipitated endogenous IFNγRα inversely correlated with galectin-3 binding (Figure 3H and Supplementary Figure 9E), indicating that a reduced MGAT3-dependent addition of bisecting N-glycans to IFNγRα increases its association with the galectin lattice.
To assess whether the restoration of MGAT3 expression would improve IFNγRα signaling in IFN-γ–resistant cells, we stably expressed MGAT3 in IFN-γ–resistant RKO cells, resulting in a higher level of bisected N-glycans (Figure 6A, PHA-E). The increase in MGAT3 expression correlated in a dose-dependent manner with an increase in IFNγRα protein levels and a shift toward higher molecular weights as shown in 2 independent clones (cl. 1 and 2) (Figure 6B). Most importantly, responsiveness to IFN-γ was restored in the MGAT3-positive cells (cl. 2) compared with the RKO cells transfected with the empty vector (Figure 6C–E).
Figure 6.
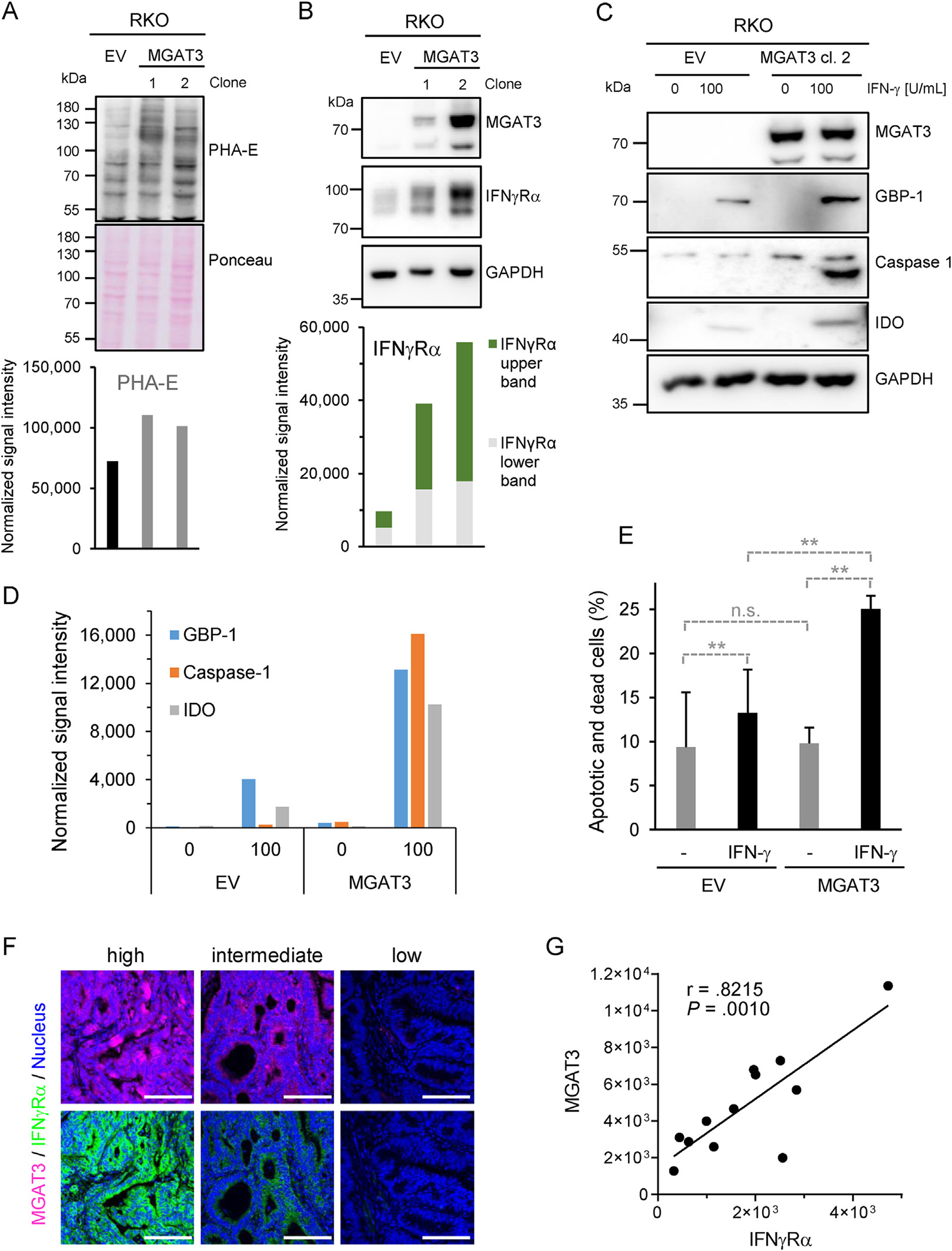
MGAT3 downregulation reduces IFNγRα bisected N-glycosylation and signaling. (A) PHA-E lectin blotting in RKO cells after stable transfection of MGAT3 (clone 1 and 2) or empty vector (EV) (25 μg of proteins/lane). Ponceau staining served as loading control and for normalization. (B) MGAT3 and IFNγRα protein expression in MGAT3-reconstituted RKO clones or RKO-EV. GAPDH was used as loading control and for normalization. Normalized signal intensity IFNγRα is given with upper and lower bands highlighted in green and gray, respectively. (C) MGAT3 and ISG expression in IFN-γ stimulated RKO cells stably transfected with either MGAT3-expressing or empty vector (EV). Cells were stimulated with indicated amounts of IFN-γ for 24 h. (D) Normalized signal intensity of Western blot in (C). (E) Cell death induction was determined 72 hours after treatment of RKO-EV/-MGAT3 cells with IFN-γ (0–100 U/ml) by flow cytometry. Results are given in percent of apoptotic and necrotic cells (mean ± SD, n = 3 distinct samples). Student P test: n.s., not significant **PEV+/−IFN-γ = .009, **PMGAT3+/−IFN-γ = .0031, **PEV+IFN-γ/MAGT3++IFN-γ = .0077. (F) Staining of MGAT3 (pink) and IFNγRα (green) in consecutive human CRC sections. Scale bars = 100μ. (G) Single tumor-corrected fluorescence intensity of MGAT3 and IFNγRα consecutive staining (n = 12) with Pearson’s correlation coefficient r and P value.
To confirm the relevance of our results at the clinical level, we investigated protein expression of MGAT3 and IFNγRα in tumor tissues by immunohistochemistry. a strong positive correlation between MGAT3 and IFNγRα protein levels was observed in human CRC samples (Pearson r = .8215, P = .001), which supported our in vitro data (Figure 6F and G and Supplementary Figure 9F).
Finally, we investigated whether the modulation of bisected N-glycosylation might influence colon tumor growth and checkpoint inhibitor therapy. Using a syngeneic colon tumor mouse model, we observed that ATRA treatment reduced tumor growth in 3 of 5 animals (Supplementary Figure 10A). The mean tumor diameter was decreased by 28.5% in the ATRA-treated group after 30 days but did not reach statistical significance (Supplementary Figure 10B and C, P = .0823). Anti-PD-1 treatment resulted in complete shrinkage of tumors within 2 weeks, and addition of ATRA reduced the half-life of anti-PD-1 treatment from 3.141 to 1.617 day (Supplementary Figure 10D). These results suggested that the addition of ATRA might increase the efficacy of checkpoint inhibitor treatment. Further investigations (eg, using lower anti-PD-1 concentrations) are needed to establish whether ATRA can synergistically improve the efficacy of checkpoint inhibitors.
Discussion
In CRC, the development of an IFN-γ–driven host antitumor immune response positively influences patient survival.1 However, the clinical benefit of IFN-γ treatment or second-generation immunotherapies, such as immune checkpoint inhibitors, remains limited in CRC, suggesting that primary immune escape is a common event.16–18,21 Immune evasion driven by frameshift mutations of JAK1/2 or PDL-1 overexpression, has been described in CRC in association with the microsatellite instability–high subtype, which represents only 15% of sporadic CRCs.12,13,21 In agreement with our results, both kinds of events remain rare in MSS CRCs, and there is no clinical benefit for anti-PD1 treatment due to the low frequency of PDL-1 overexpression.19,21,22 In MSS CRCs, up-regulated PDL-1 expression is even associated with high PD-1–negative TIL infiltration, IFN-γ production, and improved survival.30 Here we described an alternative mechanism of immune evasion mediated by the loss of IFNγRα expression, which occurred more frequently than PDL-1 overexpression or JAK mutations, and correlated with poor survival in patients with CRC. Interestingly, the down-regulation of Ifngr1 in murine colon cancer cells resulted in resistance to checkpoint inhibitor treatment (anti-PD-1) in a syngeneic mouse model,31 suggesting that the loss of IFNγRα expression also contributes to the intrinsic resistance to checkpoint inhibitor therapy in patients with MSS-CRC.
In our study, all IFN-γ–resistant cells were also N-glycosylation deficient. At the cellular level, we found that immature/lack of N-glycosylation of IFNγRα increased proteasome-dependent degradation. IFNγRα harbors several ubiquitin acceptor sites and undergoes endogenous ubiquitin-dependent proteasomal degradation.32 This basal proteasome-dependent protein turnover is not physiologically increased after ligand binding, but is enhanced on Toll-like receptor engagement or in case of viral infection,32–34 supporting our results and suggesting that IFNγRα expression is down-regulated under pathologic conditions. a similar link between N-glycosylation status and proteasomal degradation has been described for several transmembrane proteins including the epidermal growth factor receptor and PDL-1.35,36 At the molecular level, N-glycosylation-deficient IFNγRα could reach the cell surface, but its function was inhibited, in agreement with radioligand binding assays showing that N-glycosylation of IFNγRα is necessary for ligand binding, but does not affect membrane transport.26 Furthermore, we observed that immaturely glycosylated IFNγRα had a much stronger affinity for galectin-3 than the functional receptor, indicative of an association with the galectin lattice that could impair ligand accessibility.37,38
In CRC cell lines, downregulation of MGAT3 expression was systematically associated with IFN-γ resistance. In addition, MGAT3 expression correlated with IFNγRα expression in human CRC. The downregulation of MGAT3 expression along with the reduction of bisected N-glycans are indeed commonly observed in CRC.39,40 The reconstitution of MGAT3 expression after either transfection or by treatment with ATRA was sufficient to increase both the bisected N-glycan modification and the protein stability of IFNγRα in IFN-γ–resistant cells, restoring its signaling ability. These results are supported by the fact that manipulation of bisected N-glycan levels was previously shown to regulate protein turnover, function, and membrane subdomain localization of a number of transmembrane proteins, including the epidermal growth factor receptor, IFNγRβ, and E-cadherin.38,41,42 In the MC38 xenograft syngeneic tumor model, the downregulation of Ifngr1 in CRC cells induced resistance to anti-PD-1 treatment,31 whereas treatment with ATRA resulted in a modest inhibition of tumor growth when applied alone, and reduced the half-life of the anti-PD-1 response by half. Although the use of the MC38 xenograft tumor model to assess a synergistic effect was limited by the very strong and rapid effect of the anti-PD-1 treatment, our data suggested that ATRA might improve the efficacy of checkpoint inhibitor treatment in CRC. This is supported by reports showing that ATRA cooperates synergistically with IFN-γ to induce apoptosis in tumor cells,43 and enhances the effects of immunotherapy in metastatic renal cell carcinoma and melanoma models.44,45 Further studies are warranted to assess the positive impact of ATRA on the response to checkpoint inhibitor therapy in CRC, notably in conditions of immune resistance.
Taken together, our study provides evidence for a new pathway of immune escape in CRC involving decreased bisecting N-glycosylation and degradation of IFNγRα. This mechanism, more frequent than PD-L1 overexpression or JAK1/2 mutations, might explain, at least partially, the low response to IFN-γ treatment or checkpoint inhibitors in patients with CRC.16–18,21 The modulation of MGAT3 activity represented a 1-step approach to restore sensitivity to IFN-γ, suggesting a new strategy to overcome primary immune evasion and allow a broader use of immunotherapy in CRC, for example through application of ATRA, a molecule already included in the treatment of various solid cancers.46
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
During tumor progression, colorectal carcinomas develop intrinsic immune evasion. We investigated to which extent a tumor cell acquired interferon-γ resistance participates in tumor development.
NEW FINDINGS
Interferon-γ receptor expression knockout in intestinal tumor cells fosters colon tumorigenesis in mice. MGAT3/GnT-III–mediated bisecting N-glycosylation regulates interferon-γ receptor α protein stability and function, modulating interferon-γ sensitivity in colorectal cancer cells.
LIMITATIONS
A direct in-situ measure of interferon-γ receptor α N-glycosylation could not be performed because of technical limitations.
CLINICAL RESEARCH RELEVANCE
Interferon-γ receptor expression correlated with MGAT3/GnT-III expression and poor prognosis in patients with colorectal cancer. Interferon-γ receptor α downregulation was more frequent than programmed death ligand 1 overexpression or JAK1/2 mutations, and might contribute to refractoriness to checkpoint inhibitors in patients with colorectal cancer. MGAT3 and interferon-γ receptor a expression can be increased in interferon-γ–resistant colorectal cancer cells by pharmacological treatment with all-trans retinoic acid, providing new perspectives to overcome primary immune evasion.
BASIC RESEARCH RELEVANCE
Our results demonstrated the importance of N-glycosylation for the interferon-γ response in cancer cells. Defective N-glycosylation of interferon-γ receptor α was systematically observed in interferon-γ-resistant colorectal cancer cells, resulting in its proteasome-dependent degradation. The downregulation of MGAT3/GnT-III expression was consistently associated with interferon-γ resistance in colorectal cancer cells, and bisecting glycosylation catalyzed by MGAT3/GnT-III was crucial for the stability and function of interferon-γ receptor α.
Acknowledgments
The present work was performed in (partial) fulfillment of the requirements for obtaining the degree of Dr. rer. nat. by Julia Krug (née Straube). The authors thank C. Christoph, G. Hoffmann, T. Gass, and C. Flierl for excellent technical assistance and Werner Muller (Division of Infection, Immunity & Respiratory Medicine, University of Manchester, Manchester, United Kingdom) for providing C57BL/6J Ifngr2fl/fl mice. We are also very thankful to Pamela Stanley for generously providing the Pro-5 and LEC10B CHO cells. Special thanks to Regine Schneider-Stock (Institute of Pathology, Friedrich-Alexander University Erlangen-Nuremberg, Erlangen, Germany) and Clemens Neufert (Medical Clinic I, Friedrich-Alexander University Erlangen-Nuremberg, Erlangen, Germany), for providing access to the mini-endoscope (founded by the Rudolf-Bartling Stiftung, Hannover, Germany).
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 292410854 - BR 5196/2-1 to N.B.L., Project-ID 375876048 – TRR 241 subproject A06 to N.B.L. and M.St., Project-ID 429280966 – TRR305 subproject B07 to N.B.L. and B08 to E.N., Project-ID STU 238/10-1 to M.St., Project-ID 190140969 - KFO 257-4 to M.St. and M.W., Project-ID 280163318 - FOR 2438-2 to E.N. and M.St.; by the Interdisciplinary Center for Clinical Research (IZKF) of the Clinical Center Erlangen (ELAN-Fonds project P027 to N.B.L., Project J19 to N.B.L., and D34 to M.St.); by the W. Lutz Stiftung, the Forschungsstiftung Medizin am Universitätsklinikum Erlangen and the Corona-funding initiative of the Bavarian Ministry of Science and Arts (to M.St.); by the National Institutes of Health (NIH) to S.G. (Project ID NIH R01CA227629 and CA218133).
Abbreviations used in this paper:
- AOM
azoxymethane
- ATRA
all-trans retinoic acid
- CHO
Chinese hamster ovary
- CRC
colorectal carcinoma
- DSS
dextran-sulfate sodium
- Endo-H
endoglycosidase-H
- IFN
interferon
- IFNγR
interferon-gamma receptor
- ISG
interferon-stimulated gene
- JAK
Janus kinase
- mRNA
messenger RNA
- MSS
microsatellite-stable
- PD-1
programmed death 1
- PD-L1
programmed death ligand 1
- PHA-E
Phaseolus vulgaris erythroagglutinin
- PHA-L
Phaseolus vulgaris phytohemagglutinin-L
- PNGase-F
protein-N-glycosidase F
- STAT
signal transducer and activator of transcription
Footnotes
Conflicts of interest
The authors disclose no conflicts.
CRediT Authorship Contributions
Julia Krug, PhD (Conceptualization: Equal; Formal analysis: Equal; Investigation: Lead; Methodology: Equal; Visualization: Lead; Writing – original draft: Supporting).
Gabriele Rodrian, BSc (Investigation: Equal; Methodology: Equal).
Katja Petter, MSc (Investigation: Equal; Methodology: Equal).
Hai Yang, MD (Methodology: Equal; Resources: Equal).
Svetlana Khoziainova, PhD (Investigation: Equal; Methodology: Equal).
Wei Guo, PhD (Investigation: Equal; Methodology: Equal).
Alan Bénard, PhD (Methodology: Equal; Resources: Equal).
Susanne Merkel, MD (Data curation: Equal; Formal analysis: Equal).
Susan Gellert, MSc (Investigation: Equal; Methodology: Equal).
Simone Maschauer, PhD (Investigation: Equal; Methodology: Equal).
Monika Spermann, MSc (Investigation: Equal).
Maximilian Waldner, MD (Methodology: Equal; Resources: Equal).
Peter Bailey, PhD (Formal analysis: Equal; Methodology: Equal).
Christian Pilarsky, PhD (Methodology: Equal; Resources: Equal; Writing – review & editing: Equal).
Andrea Liebl, PhD (Methodology: Equal; Resources: Equal).
Philipp Tripal, PhD (Methodology: Equal; Resources: Equal).
Jan Christoph, PhD (Data curation: Equal; Methodology: Equal).
Elisabeth Naschberger, PhD (Formal analysis: Equal; Methodology: Equal; Resources: Equal; Writing – review & editing: Equal).
Roland S. Croner, MD (Resources: Equal).
Vera S. Schellerer, MD (Resources: Equal).
Christoph Becker, PhD (Methodology: Equal; Resources: Equal; Writing – review & editing: Equal).
Arndt Hartmann, MD (Resources: Equal).
Thomas Tüting, PhD (Conceptualization: Equal; Formal analysis: Equal; Methodology: Equal; Resources: Equal).
Olaf Prante, PhD (Conceptualization: Equal; Formal analysis: Equal; Methodology: Equal; Resources: Equal).
Robert Grützmann, MD (Resources: Equal).
Sergei I. Grivennikov, PhD (Conceptualization: Equal; Formal analysis: Equal; Funding acquisition: Equal; Methodology: Equal; Resources: Equal; Supervision: Equal).
Michael Stürzl, PhD (Conceptualization: Lead; Funding acquisition: Lead; Methodology: Equal; Project administration: Lead; Resources: Lead; Supervision: Lead; Writing – review & editing: Lead).
Nathalie Britzen-Laurent, PhD (Conceptualization: Lead; Formal analysis: Equal; Funding acquisition: Lead; Investigation: Lead; Methodology: Equal; Project administration: Lead; Supervision: Lead; Visualization: Lead; Writing – original draft: Lead; Writing – review & editing: Lead).
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2022.11.018.
Data Availability
All material and data requests should be submitted to the corresponding author for consideration.
References
- 1.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006. 313:1960–1964. [DOI] [PubMed] [Google Scholar]
- 2.Fridman WH, Pages F, Sautes-Fridman C, et al. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012. 12:298–306. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011. 331:1565–1570. [DOI] [PubMed] [Google Scholar]
- 4.Hoekstra ME, Bornes L, Dijkgraaf FE, et al. Long-distance modulation of bystander tumor cells by CD8+ T-cell-secreted IFN-γ. Nat Cancer 2020. 1:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thibaut R, Bost P, Milo I, et al. Bystander IFN-γ activity promotes widespread and sustained cytokine signaling altering the tumor microenvironment. Nat Cancer 2020. 1:302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol 2006. 6:836–848. [DOI] [PubMed] [Google Scholar]
- 7.Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol 2019. 20:1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naschberger E, Croner RS, Merkel S, et al. Angiostatic immune reaction in colorectal carcinoma: Impact on survival and perspectives for antiangiogenic therapy. Int J Cancer 2008. 123:2120–2129. [DOI] [PubMed] [Google Scholar]
- 9.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012. 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson JA, Al-Attar A, Watson NF, et al. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut 2010. 59:926–933. [DOI] [PubMed] [Google Scholar]
- 11.Mlecnik B, Bindea G, Kirilovsky A, et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med 2016. 8:327ra326. [DOI] [PubMed] [Google Scholar]
- 12.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015. 372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mandal R, Samstein RM, Lee KW, et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019. 364:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Britzen-Laurent N, Lipnik K, Ocker M, et al. GBP-1 acts as a tumor suppressor in colorectal cancer cells. Carcinogenesis 2013. 34:153–162. [DOI] [PubMed] [Google Scholar]
- 15.Pfizenmaier K, Bartsch H, Scheurich P, et al. Differential gamma-interferon response of human colon carcinoma cells: inhibition of proliferation and modulation of immunogenicity as independent effects of gamma-interferon on tumor cell growth. Cancer Res 1985. 45:3503–3509. [PubMed] [Google Scholar]
- 16.Pavlidis N, Nicolaides C, Athanassiadis A, et al. Phase II study of 5-fluorouracil and interferon-gamma in patients with metastatic colorectal cancer. a Hellenic Cooperative Oncology Group Study. Oncology 1996. 53:159–162. [DOI] [PubMed] [Google Scholar]
- 17.Turner PK, Houghton JA, Petak I, et al. Interferon-gamma pharmacokinetics and pharmacodynamics in patients with colorectal cancer. Cancer Chemother Pharmacol 2004. 53:253–260. [DOI] [PubMed] [Google Scholar]
- 18.Zeller W, Garbrecht M, Dierlamm J, et al. Phase I-II study of interferon-gamma and eflornithine (DFMO) in patients with advanced renal cell carcinoma, malignant melanoma and colorectal carcinoma. Oncol Rep 1996. 3:447–451. [DOI] [PubMed] [Google Scholar]
- 19.Albacker LA, Wu J, Smith P, et al. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS One 2017. 12:e0176181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berg KCG, Eide PW, Eilertsen IA, et al. Multi-omics of 34 colorectal cancer cell lines—a resource for biomedical studies. Mol Cancer 2017. 16:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 2017. 7:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sveen A, Johannessen B, Tengs T, et al. Multilevel genomics of colorectal cancers with microsatellite instability-clinical impact of JAK1 mutations and consensus molecular subtype 1. Genome Med 2017. 9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang C, Hou D, Wei H, et al. Lack of interferon-γ receptor results in a microenvironment favorable for intestinal tumorigenesis. Oncotarget 2016. 7:42099–42109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du W, Hua F, Li X, et al. Loss of optineurin drives cancer immune evasion via palmitoylation-dependent IFNGR1 lysosomal sorting and degradation. Cancer Discov 2021. 11:1826–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hershey GK, Schreiber RD. Biosynthetic analysis of the human interferon-gamma receptor. Identification of N-linked glycosylation intermediates. J Biol Chem 1989. 264:11981–11988. [PubMed] [Google Scholar]
- 26.Fischer T, Thoma B, Scheurich P, et al. Glycosylation of the human interferon-gamma receptor. N-linked carbohydrates contribute to structural heterogeneity and are required for ligand binding. J Biol Chem 1990. 265:1710–1717. [PubMed] [Google Scholar]
- 27.Stanley P, Sundaram S, Tang J, et al. Molecular analysis of three gain-of-function CHO mutants that add the bisecting GlcNAc to N-glycans. Glycobiology 2005. 15:43–53. [DOI] [PubMed] [Google Scholar]
- 28.Chen C, Diao D, Guo L, et al. All-trans-retinoic acid modulates ICAM-1 N-glycan composition by influencing GnT-III levels and inhibits cell adhesion and transendothelial migration. PLoS One 2012. 7:e52975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miwa HE, Song Y, Alvarez R, et al. The bisecting GlcNAc in cell growth control and tumor progression. Glycoconj J 2012. 29:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Droeser RA, Hirt C, Viehl CT, et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur J Cancer 2013. 49:2233–2242. [DOI] [PubMed] [Google Scholar]
- 31.Lv C, Yuan D, Cao Y. Downregulation of interferon-γ receptor expression endows resistance to anti-programmed death protein 1 therapy in colorectal cancer. J Pharmacol Exp Ther 2021. 376:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Londino JD, Gulick DL, Lear TB, et al. Post-translational modification of the interferon-gamma receptor alters its stability and signaling. Biochem J 2017. 474:3543–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skrenta H, Yang Y, Pestka S, et al. Ligand-independent down-regulation of IFN-γ receptor 1 following TCR engagement. J Immunol 2000. 164:3506–3511. [DOI] [PubMed] [Google Scholar]
- 34.Blouin CM, Lamaze C. Interferon gamma receptor: the beginning of the journey. Front Immunol 2013. 4:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gabius HJ, van de Wouwer M, André S, et al. Downregulation of the epidermal growth factor receptor by altering N-glycosylation: emerging role of β1,4-galactosyltransferases. Anticancer Res 2012. 32:1565–1572. [PubMed] [Google Scholar]
- 36.Li CW, Lim SO, Xia W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun 2016. 7:12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferreira IG, Pucci M, Venturi G, et al. Glycosylation as a main regulator of growth and death factor receptors signaling. Int J Mol Sci 2018. 19:580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blouin CM, Hamon Y, Gonnord P, et al. Glycosylation-dependent IFN-γR partitioning in lipid and actin nanodomains is critical for JAK activation. Cell 2016. 166:920–934. [DOI] [PubMed] [Google Scholar]
- 39.Balog CI, Stavenhagen K, Fung WL, et al. N-glycosylation of colorectal cancer tissues: a liquid chromatography and mass spectrometry-based investigation. Mol Cell Proteomics 2012. 11:571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gebert J, Kloor M, Lee J, et al. Colonic carcinogenesis along different genetic routes: glycophenotyping of tumor cases separated by microsatellite instability/stability. Histochem Cell Biol 2012. 138:339–350. [DOI] [PubMed] [Google Scholar]
- 41.Partridge EA, Le Roy C, Di Guglielmo GM, et al. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 2004. 306:120–124. [DOI] [PubMed] [Google Scholar]
- 42.Xu Q, Isaji T, Lu Y, et al. Roles of N-acetylglucosaminyltransferase III in epithelial-to-mesenchymal transition induced by transforming growth factor beta1 (TGF-beta1) in epithelial cell lines. J Biol Chem 2012. 287:16563–16574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karmakar S, Walle T, Banik N, et al. Treatment with combination of all-trans retinoic acid and interferon-gamma regressed human glioblastoma T98G xenografts in nude mice. Cancer Res 2008. 68:4035–4035. [Google Scholar]
- 44.Mirza N, Fishman M, Fricke I, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res 2006. 66:9299–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tobin RP, Jordan KR, Robinson WA, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int Immunopharmacol 2018. 63:282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen MC, Hsu SL, Lin H, et al. Retinoic acid and cancer treatment. Biomedicine (Taipei) 2014. 4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All material and data requests should be submitted to the corresponding author for consideration.