
Abstract
Duchenne muscular dystrophy (DMD) is the most common childhood form of muscular dystrophy. It is caused by mutations in the DMD gene, leading to reduced or absent expression of the dystrophin protein. Clinically, this results in loss of ambulation, cardiomyopathy, respiratory failure, and eventually death. In the past decades, the use of corticosteroids has slowed down the disease progression. More recently, the development of genetically mediated therapies has emerged as the most promising treatment for DMD. These strategies include exon skipping with antisense oligonucleotides, gene replacement therapy with adeno-associated virus, and gene editing with CRISPR (clustered regularly interspaced short palindromic repeats) technology. In this review, we highlight the most up-to-date therapeutic progresses in the field, with emphasis on past and recent experiences, as well as the latest clinical results of DMD micro-dystrophin gene therapy. Additionally, we discuss the lessons learned along the way and the challenges encountered, all of which have helped advance the field, with the potential to finally alleviate such a devastating disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
DMD is a rare, X-linked recessive, fatal, degenerative disease of the muscle caused by DMD gene mutations. The estimated incidence worldwide is 1 in 5000 live male births [1, 2]. The DMD gene (OMIM 300377) encodes for dystrophin, a 427-kDa cytoskeletal protein required for sarcolemmal stability. Protein loss leads to susceptibility to repeated cycles of necrosis and regeneration. Over time, there is diminished regenerative capacity, resulting in fat and connective tissue replacement (fibrosis).
The disease begins in utero. The movement of the unborn fetus in utero damages the muscle fiber membrane. The result being that DMD boys are born with a creatinine kinase (CK) over 2000 U/L. Early development is delayed, head control is poor, and DMD boys have trouble sitting unsupported. They are late in standing and cruising and may not walk until 18 months of age or after. Cognition may be affected, and delayed speech is common. Large calf muscles are noticeable by age 3–4; by age 5–7, toe walking is more apparent, and stairs are more difficult; by age 10–11, falling is frequent, and boys fatigue more easily. In early teens, DMD boys usually require a wheelchair. In late teenage years, respiratory muscles are compromised with loss of intercostal and diaphragm muscles. Cardiac muscle fiber loss becomes clinically significant when dystrophin is lost around each cardiomyocyte, resulting in a gradual substitution of fibrous and adipose tissue. This process ultimately leads to premature mortality.
In its current status, DMD is clearly a disease of unmet needs. Fortunately, the landscape is changing, and treatment, as reviewed in the discussion to follow, shows how scientists and clinicians are collaborating to make a difference for boys with DMD. The purpose of this review is to define, clarify, and describe the approaches that can make a difference for boys with this disorder. Approaches that are both nonmolecular (corticosteroids) and molecular (exon skipping and gene therapy). Comments on the very promising gene editing approach, CRISPR, are briefly described but the methodology has only been applied to a single DMD patient without success.
Glucocorticoids
Background
Glucocorticoids (representing a class of corticosteroids) embody the most common treatment and the standard of care for DMD [3]. Glucocorticoids prolong independent walking, survival, and quality of life [4, 5]. Prednisone is an anti-inflammatory glucocorticoid that must be converted to prednisolone in the liver to become biologically active. First attempts to use prednisone or prednisolone for DMD were initially described in modest terms, as a “useful palliative treatment” [6]. Over the past three decades, the role of prednisone and other steroids in the treatment of DMD has extensively evolved. Glucocorticoids play an important role in regulating inflammatory and immune responses benefitting DMD in many ways. Glucocorticoid-anti-inflammatory properties are mediated by the inhibition of NF-kB, a protein complex transcription factor that reduces T-cell proliferation and modulates humoral immunity by reduction of B cells and antibody production (Fig. 1A). Evidence for this is seen in muscle biopsies (reduced inflammation and fibrosis) from steroid-treated DMD patients [7, 8]. The improvement in skeletal muscle function observed with glucocorticoids directly correlates with the preservation of cardiac and pulmonary function as well, according to retrospective observational studies [9, 10]. The growing use of glucocorticoids in clinics has delayed the onset of dilated cardiomyopathy, which remains the leading cause of death in DMD patients [11, 12].

A, B Mechanism of action of prednisolone/prednisone versus vamorolone. Top of form. On the left, the diagram illustrates how prednisolone binds to a specific cytoplasmic nuclear hormone receptor [glucocorticoid receptor (GR)]. This receptor-ligand interaction plays a crucial role in maintaining the inhibitory complex (consisting of p65, p50, IkB) that keeps NF-kB in its inactive form. Following translocation to the nucleus, GR further suppresses NF-kB activity by interacting with specific DNA sequences. This process, called transrepression, contributes to the anti-inflammatory effects of prednisolone. Within the nucleus, the GR complex also facilitates gene transcription, a process called transactivation, which is responsible for the side effects commonly associated with prednisolone and other glucocorticoids. The vamorolone model is shown on the right and it is uniquely different. Upon entering the cell, the GR complex exhibits anti-inflammatory properties by inhibiting NF-kB, both in the cytoplasm and in the nucleus (transrepression). However, unlike glucocorticoids, vamorolone does not possess transactivating properties that up-regulate gene transcription; thus, the side effect profile is minimalized. Additionally, vamorolone is known to protect against membrane damage in animal models. This figure is an adaptation from ReveraGen BioPharma with the permission of Dr. Eric Hoffman
DMD Adopts Glucocorticoids
The first attempt to treat DMD reporting the possible benefit of glucocorticoids is credited to an open-label trial of prednisone in 1974 [6]. Despite the credibility of researchers, skepticism remained dominant. Seigel, a prominent orthopedic surgeon, reported no steroid-related value when comparing treated versus untreated, age-matched, DMD boys [13]. The status of steroids remained stagnant for over a decade, until an open-label trial was reported in 1987. Thirty-three DMD patients, ages 5–15 years, were reported to show improvement using prednisone, 1.5 mg/kg daily for 6 months [14]. Two years later, the efficacy of prednisone was definitively confirmed in a multiclinic, randomized, double-blind, controlled 6-month study of prednisone in 103 DMD boys ages 5–15 years [15]. Three prednisone-dosed cohorts were compared: prednisone 0.75 mg/kg/day (n = 33); prednisone 1.5 mg/kg/day (n = 34); and placebo (n = 36). Both prednisone doses showed benefits without clinical differences. DMD boys improved strength [4], and functional tests, including time to climb stairs, time to stand from supine, and time to walk 9 m. Improvement was seen as early as 1 month and peaked at 3 months. Even forced vital capacity was modestly improved. There was a mild decrease in CK levels (p < 0.03) and an increase in urinary creatine excretion (p < 0.0001), indicative of increased muscle mass. Anticipated steroid-related side effects were seen, including weight gain, cushingoid appearance, hair growth, and behavioral changes.
Speculations about the mechanism of action of the observed findings were addressed from muscle biopsies from the participants in the trial [7]. Total T-cell (CD2 +) numbers were reduced compared to the placebo-treated. Similarly, the number of CD8 + cytotoxic/suppressor T cells was significantly decreased.
As the interest in glucocorticoids expanded, attention switched to exploring glucocorticoid benefits using different regimens and different products. Given that azathioprine had shown benefits for immune-mediated diseases, an attempt to explore efficacy in DMD was done, but failed to show benefit [16]. In 2002, Connolly et al. [17] reported the results of twice-weekly oral prednisone (5 mg/kg/dose) in 20 boys (8.0 ± 1.2 years). At 6 months, strength and timed functional testing improved in proximal upper limbs and in quadriceps muscles. Longer time points up to 2 years supported benefit. A major advantage of weekend dosing was the improved side effect profile, especially linear growth, and less weight gain. Favorable findings of weekend dosing forced a comparative double-blind, randomized study of daily versus weekend prednisone in DMD [18]. In a 12-center, 1-year trial, including 64 boys, there was no difference in efficacy between weekend dosing (5 mg/kg/dose) and daily prednisone (0.75 mg/kg).
Alternative Glucocorticoids
Deflazacort (DFZ), also known as Emflaza®, is a glucocorticoid derived from prednisolone. It was patented in 1965, belonging to the oxazoline-steroid family. DFZ was introduced to treat DMD in 1991 in a controlled trial with the intent of reducing prednisone side effects [19]. Improvement was seen in muscle strength and function in a randomized, double-blind protocol [20] and confirmed in a 2-year follow-up trial. Improvement in rising from supine, stair climbing, and walking 10 m was seen and there were suggestions that left ventricular ejection fraction improved [21].
Multiple confirmatory trials were done over a span of two decades. Most published studies agreed on a consensus dosing of 0.9 mg/kg daily [22,23,24,25,26]. The higher dose of DFZ, 1.2 mg daily, caused more side effects. DFZ fulfilled one goal, it controlled appetite better than prednisone and treatment caused less weight gain, but shared other side effects, including a cushingoid appearance, hirsutism, and growth delay. One deterrent of DFZ is a greater likelihood of cataracts accompanying its long-term use. In 2017, the Food and Drug Administration (FDA) approved DFZ (Emflaza®) for DMD patients 2 years and older. Currently, this is the only corticosteroid with defined FDA approval for DMD.
Vamorolone
Researchers have proposed vamorolone, an anti-inflammatory drug that targets the same receptor as corticosteroids. Vamorolone takes advantage of distinctive downstream transcription properties, accounting for its designation as “dissociative” steroid [27]. It is a synthetic steroidal drug with potent anti-inflammatory properties, blocking NF-κB–associated proinflammatory signals (transrepression) and dissociating these efficacy actions from those that cause the familiar side effects of steroids (transactivation) (Fig. 1B). This makes vamorolone an attractive alternative to other corticosteroids currently available. Testing the dissociative benefit has been addressed in a series of clinical trials.
Safety was the first goal in the clinical translation. In a multi-dose ranging, short-term trial, lack of toxicity was established in adult volunteers and in DMD patients [28, 29]. Efficacy was then tested in DMD boys 4 to < 7 years of age (n = 48) in an open-label, dose-ascending trial and its long-term extension, confirming safety and efficacy (time to stand improved from baseline). Questions were raised in this trial related to a possible positive effect of vamorolone on bone loss [30, 31]. In January 2022, a nonrandomized controlled trial was reported [32]. Participants included 41 boys, 4–7 years of age, who completed a long-term 30-month trial. The mean time-to-stand failed to show a change in velocity. Strength was maintained similarly to matched participants taking glucocorticoids. In addition, there was no statistically significant difference in body mass index, or timed function tests in matched controls receiving glucocorticoids. One possible advantage reported was a statistically significant difference in growth “deceleration” in vamorolone-treated patients. These findings were confirmed by a randomized, double-blind, placebo versus prednisone-controlled trial [33]. Further studies are underway, and the pharmacologic sponsor has requested accelerated approval from the FDA while a phase 3 clinical trial is ongoing.
Conclusions Regarding Steroid Use in Clinical Practice (Table. 1)
Glucocorticoids, both prednisone/prednisolone and deflazacort (Emflaza) are necessary anti-inflammatory pharmacologic agents for DMD. The benefits are approximately equal in motor function. The side effect profile seems to differ mainly in more weight gain with prednisone when these drugs are taken daily. Both, however, cause cushingoid appearance and short stature from bone growth delay. It is also undisputed that cataracts are more common with deflazacort.
It is unequivocal that the main advantages of weekend dosing of prednisone/prednisolone include preserved normal bone growth and less weight gain. Functional outcomes are the same for weekend dosing compared to daily dosing. Cataracts are also very rare with weekend dosing. The potential downside of high weekend dosing is the change in behavior that often occurs on the days the steroids are given.
Vamorolone has appeal as a dissociative steroid; however, it is premature to recommend this drug over the standard of care with glucocorticoids. The drug is currently still being evaluated in a phase 3 study, and results have not yet been reported. Overall, it seems reasonable to conclude that it is equal in efficacy to glucocorticoids, and for that reason, it can be considered an alternative corticosteroid, but not superior in efficacy or safety.
Exon Skipping
Background
Small clusters of dystrophin-positive fibers on muscle biopsies called “revertant fibers” are well described in muscle biopsies of DMD boys [34]. They appear to arise from activated precursor cells (satellite cells) that clonally expand, skip the mutant exon(s), and restore the open reading frame [35] (Fig. 2). This naturally occurring event was replicated for clinical applications using synthetic phosphorodiamidate morpholino oligomers (PMOs), a subclass of antisense oligonucleotides (ASOs) designed to bind to pre-mRNA, alter the splicing process, and skip the targeted exon. This restores the open reading frame of the mature mRNA sequence and allows the production of a truncated dystrophin transcript that can produce a functional protein [36]. The PMOs have a low risk for immune activation.

Pathway to revertant fiber(s). An out-of-frame deletion of exon 50 in the DMD gene (A) results in the absence of dystrophin as shown by the lack of immunofluorescence staining with antibodies directed against exon 50 (B). There must be spontaneous exon skipping to restore the reading frame and establish positive fibers (revertant fibers) showing IF staining at the C-terminal. The arrow (C) illustrates the exons skipped to restore the in-frame sequence. In this instance, there is in-frame restoration at exon 57 by skipping exons 50 to 56 (C); confirmation of restoration at exon 57 is established by monoclonal antibody directed against exon 57 (D)
DMD patients with out-of-frame deletions or duplications are potentially amenable to exon skipping, with estimated applicability to as many as 83% of DMD mutations (54% of which are deletions, 23% small mutations, 6% duplications) [36,37,38].
Eteplirsen-Exon 51 Skipping
Exon skipping was well described pre-clinically with efficacy and safety and applied to this exon capturing the single most common mutation [39]. Skipping of exon 51 in DMD patients results in the restoration of the open reading frame, which allows the production of partially functional dystrophin in approximately 13% of the affected individuals. This particular exon-skipping process includes or incorporates exons 50, 52, 52–63, 45–50, 47–50, and 49–50 in the final dystrophin mRNA product.
Eteplirsen (Exondys 51®, Sarepta Pharmaceuticals) was the first exon-skipping pharmacologic treatment approved by the FDA in 2016. The trial supporting this approval was a double-blind placebo-controlled trial enrolling 12 DMD boys aged 7 to 13 years, randomized to either an ascending dose schedule (30 mg/kg or 50 mg/kg) or placebo [40]. Dystrophin increases were small as quantified by western blot (1%). Patients exhibited a slowing of disease progression (measured by the 6-min walk test, 6MWT) relative to matched historical controls [41] and an improvement in their respiratory function relative to a comparable cohort of patients from a natural history database [42]. A long-term follow-up of patients in the initial cohort confirmed a delay in disease progression and prolonged ambulation (by 2.1 years) [43, 44]. Finally, a phase 3 post-approval trial of eteplirsen (30 mg/kg) demonstrated a statistically significant increase in dystrophin expression (from 0.24 to 0.63%, p < 0.001) in addition to a notable attenuation of decline in the forced vital capacity and 6MWT over 96 weeks compared to external controls [45].
Eteplirsen is well tolerated, and side effects are minimal with reports of vomiting, difficulty with balance, and contact dermatitis. A major limitation of eteplirsen (and of exon-skipping treatment in general) is the need for weekly infusions that could extend over the lifetime of the patient.
Peptide-Conjugated Phosphorodiamidate Morpholino Oligomers (PPMOs)-Exon 51 Skipping
To improve clinical efficacy using eteplirsen, one strategy currently under investigation is a peptide-conjugated PMO (PPMO). This has been introduced into clinical trials under the designation SRP-5051, also known as vesleteplirsen. The intravenous dosing is monthly and dystrophin expression looks promising with exon skipping enhanced (1.6 times greater) and a fivefold increase in dystrophin compared to the regular PMO. However, adverse events have delayed entry to the clinic and include hypomagnesemia, hypokalemia, muscle cramps, and mild to moderate tingling of the extremities. The trial was put on hold by the FDA in June 2022 and after a thorough review, the hold was lifted in September 2022. Vesleteplirsen currently continues in clinical trials under close scrutiny.
Golodirsen-Exon 53 Skipping
Eteplirsen paved the way for the introduction of PMOs targeting other exons to restore the open reading frame. The next target was exon 53, golodirsen (Vyondys 53®, Sarepta Pharmaceuticals, company sponsor), increasing the eligible DMD population for reading frame correction by 8%. Twenty-five boys with DMD gene mutations amenable to skipping exon 53 received golodirsen (30 mg/kg) in a 2-part study [46]. The muscle biopsies at the 1-year timepoint showed a mean dystrophin protein increase of 1.019% of normal, representing a 16-fold increase compared to baseline. These results exceed the level of dystrophin detected using eteplirsen. Dystrophin expression was documented to be correctly localized to the sarcolemma. The detailed results based on high-throughput digital image analysis assessing regeneration and levels of dystrophin-associated protein via immunofluorescent analysis support improvement [47, 48]. Muscle biopsies at week 48 showed showing increased muscle regeneration providing evidence of functionality from golodirsen treatment [48]. In addition, at 3 years, the change from baseline in the 6MWT was − 99.0 m, significantly better than − 181.4 m (P < 0.001) for external controls, and loss of ambulation occurred in only 9% versus 26% (p = 0.21). Based on these findings Vyondys53 was FDA approved in December 2019.
Casimersen-Exon 45 Skipping
Casimersen (Amondys 45®, Sarepta Pharmaceuticals) received accelerated approval for use in the USA by the FDA on February 25, 2021. Dystrophin production was reported to increase by skipping exon 45 [49]. In DMD boys 7–13 years, 30 mg/kg casimersen modestly increased dystrophin on muscle biopsies at week 48. The mean dystrophin levels increased from 0.93 at baseline to 1.74 as a percentage of normal adults (mean change from baseline, 0.081; p < 0.001). Side effects reported after 2 years of treatment included mild increases in upper respiratory tract infection, cough, fever, headache, and arthralgias. A warning alert, released with packaging upon drug approval, provided animal data that showed possible kidney toxicity. Despite not being observed in clinical trials, cystatin C levels are monitored in follow-up studies and in ongoing phase 3 clinical trials with expected completion in 2024. Continued approval for this indication is contingent upon the validation of clinical benefits in confirmatory trials.
Viltolarsen-Exon 53 Skipping
Viltolarsen a PMO targeting exon 53 (Viltepso®, NS Pharma, parent company Nippon Shinyaku), different from those released by Sarepta, has also been approved for clinical use [50]. Sixteen DMD boys were infused with this PMO-variant that increased dystrophin by 5.7% and 5.9%, at 40 mg/kg (n = 8) and 80 mg/kg (n = 8), respectively. Compared with treatment-matched natural history controls (n = 65), all 16 participants treated with viltolarsen showed significant improvements in timed function tests from baseline, including time to stand from supine (viltolarsen: − 0.19 s; control: 0.66 s), time to run/walk 10 m (viltolarsen: 0.23 m/s; control: − 0.04 m/s), and 6-min walk test (viltolarsen: 28.9 m; control: − 65.3 m). Treatment was well tolerated with common side effects of cough and upper respiratory infection. Additional safety and efficacy data have been collected in the long-term extension study, confirming the stabilization of motor function and slowing of disease progression, with only mild to moderate adverse events [51]. Currently recommended dosing for clinical use is weekly, 80 mg/kg IV infusion.
Conclusions Regarding Exon Skipping in Clinical Practice (Table. 2)
Exon skipping using any of the 4 available products is safe and results show modest efficacy. Treatment is well tolerated with few adverse events. Original concerns regarding renal toxicity from casimersen have not been encountered clinically, but the FDA continues to recommend following cystatin C levels while taking this PMO. Exon skipping was introduced to the clinic as an exciting molecular therapy; enthusiasm has been calmed by experience because of its modest efficacy. As molecular approaches mature, combinational therapies with gene therapy might have a more satisfactory role [52]. There is a potential place for exon skipping for DMD boys with pre-existing immunity to AAV vectors, precluding gene therapy. In addition, next-generation PMOs could potentially enhance dystrophin expression. Those strategies include modifying the chemistry of the backbone to allow for greater stability and conjugation with peptides to improve brain and cardiac muscle expression [53].
Nonsense Mutation Suppression
Ataluren
Approximately 13% of DMD patients have a nonsense mutation in the dystrophin gene, resulting in a premature stop codon. Ataluren (Translarna®) was developed to enable ribosomal readthrough of stop codons, allowing the incorporation of an amino acid into the growing protein chain, rather than terminating protein translation. Between 20 and 25% of dystrophin-positive fibers were observed in mdx mice treated with ataluren [54]. Moreover, in a group of 38 boys treated in a phase 2a trial, 23 (61%) exhibited an increase in dystrophin expression assessed by immunofluorescence in the extensor digitorum brevis muscle [55] Despite these initial encouraging results, in a phase 3 trial, the drug failed to meet its primary endpoint of improving 6MWT, resulting in its rejection by the FDA. Nevertheless, a subgroup analysis of the study suggested that ataluren might be beneficial for specific patients, the ones with a baseline 6MWD of ≥ 300 m to < 400 m [56]. On the basis of this data, conditional approval for ataluren was granted in Europe in 2016 for ambulatory patients over the age of 5. A subsequent registry analysis further supported the safety profile of ataluren and its benefits in patients treated with the drug compared to matched historical controls. The long-term study demonstrated that ataluren plus standard of care significantly delayed the age at loss of ambulation by 4 years. Additionally, it delayed the age at decline to %-predicted forced vital capacity of < 60% and < 50% by 1.8 and 2.3 years, respectively [57]. Ongoing investigations continue to explore the potential of ataluren in treating DMD, including a randomized, double-blinded trial, expected to conclude in 2023 [58].
Gene Therapy
Background
Overcoming the obstacles for translational gene therapy as a clinical treatment for DMD seemed an impossible dream just a short time ago. Sequential, stepwise advances over the past 2–3 decades have made this feasible. A major contribution is the well-known report that ambulation with mild dystrophinopathy persisted beyond 60 years of age [59]. The patient had a large, in-frame deletion of exons 17–48 that removed 48% of the coding region of the gene. The translated protein contained approximately one-third of the 24 spectrin repeats normally found in the rod domain of the DMD gene.
Viral Vectors
To find the vehicle of choice for transfer, adenovirus, a common cause of upper respiratory illness, seemed to be the best choice. As a nonenveloped icosahedral (geometric shape with 20 sides) virus, 65–80 nm in diameter, the size offered the potential for gene transfer without the need for minimizing the gene. However, when adenovirus was chosen as the transfer vehicle to treat X-linked ornithine transcarbamylase deficiency, an unfortunate set of events led to the death of an 18-year-old, mildly affected patient. An overwhelming inflammatory response followed gene transfer and could not be effectively suppressed [60]. From that point forward, adeno-associated virus (AAV) was considered to be a more favorable vehicle for gene transfer in human trials. Despite similarities in their names, the viral species adenovirus and adeno-associated virus are completely different. Their names are linked dating back more than 50 years when AAV was isolated as a contaminant from an adenovirus preparation [61, 62].
AAV is a small nonenveloped icosahedral virus, approximately 25 nm in diameter, belonging to the Dependovirus genus of the Parvoviridae family containing a linear single-stranded DNA genome of about 4.7 kb [63]. It is replication-defective and dependent on helper functions provided by other viruses, especially adenoviruses and herpesviruses. AAV is attractive for human gene transfer because it causes no known human disease. In addition, the design of self-complementary AAV (scAAV) significantly improves gene transfer removing the need for second-strand synthesis and shortening the lag time for gene expression [64]. AAV has vastly improved the safety of clinical gene transfer, without compromising efficacy, although careful oversight and monitoring are required in clinical trials.
Miniaturizing the Gene
The path to DMD gene transfer was established in the millennium with the proof that a gene the size of DMD could be minimized, packaged in AAV, and expressed using a muscle-specific promoter, AAV1-MCK-D3990 [65]. This small cDNA minidystrophin gene (designated “mini” because of the presence of 5 spectrin repeats), one-third the size of the 14-kb full-length dystrophin coding sequence, was transferred to the gastrocnemius muscle of the mdx mouse. Widespread dystrophin expression at the sarcolemma was accompanied by protection from leakage of Evans blue dye. The muscle from mice treated at 10 days of age showed minimal central nucleation, normal myofiber size, and protection against fibrosis. For DMD, this was highly significant showing that the 14 kb dystrophin cDNA could be reduced to one-third of its size, packaged, and delivered to muscle by AAV. This observation, combined with findings from the Chamberlain lab, showed that multiple regions of the gene could be deleted with the preservation of functional dystrophins [66].
Micro-dystrophin and the First Clinical Trial
For translational gene delivery, a cassette with a good pre-clinical record, DR4-R23, stood out in its capacity to rescue the mdx phenotype. We chose this DMD cDNA for clinical gene transfer (Fig. 3). Working with the support of our industry partner, Sarepta Therapeutics, we have now completed three preliminary micro-dystrophin clinical trials and entered a confirmatory phase 3 controlled trial, currently ongoing. The vector is designated SRP-9001. A safe dose of a virus with demonstrated efficacy was previously established in our SMA clinical trial and adopted for the DMD trial (2 × 1014 vg/kg converted to 1.33 × 1014 vg/kg by digital droplet PCR titering to promote accuracy) [67]. In addition, in the SMA trial, we learned that the immune response could be modified by starting patients on a prednisone regimen of 1 mg/kg 1 day before gene delivery. For DMD, boys were already on a glucocorticoid treatment regimen as part of their standard of care and the steroid regimen also included additional dosing of 1 mg/kg/day, maintained for 30–60 days.

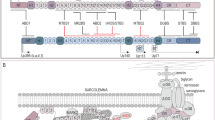
Building the micro-dystrophin cassette for gene transfer. The micro-dystrophin construct possesses the R4-R23/D71-78 transgene. The cDNA is codon-optimized, contains the N-terminus (ABD) for binding to f-actin, spectrin repeats SR1, SR2, SR3, and SR24, and hinges 1, 2, and 4. The cysteine-rich (CR) domain links to the b-dystroglycan complex in the sarcolemma. The micro-dystrophin is under the control of the MHCK7 promoter driving expression in skeletal muscle and heart. The consensus Kozak sequence, an SV40 intron, and a synthetic polyadenylation site (53 bp) are important components of the transgene. The miniaturized construct is packaged in adeno-associated virus rh74 (AAVrh74)
On January 4, 2018, we prepared a micro-dystrophin cassette for DMD gene transfer further modified by using an MHCK7 promoter specifically intended to achieve expression in both heart and skeletal muscle (Fig. 3). The promoter and micro-dystrophin transgene were packaged in AAVrh74, a serotype with demonstrated tropism to skeletal and cardiac muscle. This first trial was an open-label, phase 1/2, single-site study. Ambulatory boys aged 4 to 7 (n = 4) with confirmed frameshift DMD mutations were selected to be treated with AAVrh74.MHCK7.micro-dystrophin. Eligible patients were required to be on a stable dose of corticosteroids for at least 12 weeks before entry. By patient choice, all in this first trial were on weekend dosing prior to gene delivery and the protocol mandated added prednisone/prednisolone 1 mg/kg/day starting 24 h prior to gene transfer and maintained for 30 days. AAVrh74.MHCK7.micro-dystrophin was infused intravenously over 1 h and 15 min. Also, during viral delivery, the muscles of the arms bilaterally (shoulder abductor, elbow flexor/extensor, wrist flexor/extensor) and legs (hip flexors, knee extensors, ankle dorsiflexors/plantar flexors) were exercised every 10 min to increase blood flow and divert potential gene delivery away from the liver.
Pretreatment and post-treatment (90-day) needle muscle biopsies were obtained under general anesthesia from gastrocnemius muscles through ultrasound-guided needle placement. Gene expression was documented in all 4 patients by showing 3.3 vector genome copies per nucleus, indicating successful gene delivery. At 12 weeks, immunohistochemistry of muscle biopsy specimens showed a mean of > 80% micro-dystrophin-positive fibers in the gastrocnemius muscle, with a mean intensity of 96.0%. Findings were confirmed by western blot, in which mean micro-dystrophin expression in the sample was 74% of normal dystrophin expression level without adjustment for fat or fibrosis. b-Sarcoglycan also showed a robust increase indicative of restoration of the dystrophin-associated protein complex. Gene expression was further validated as significant as assessed by improvement (mean 5.5 points) in the North Star Ambulatory Assessment (NSAA) and reductions in the CK levels. The full impact of the success of this trial has been validated from data 4 years post-gene transfer [68].
Subsequent Micro-dystrophin Clinical Trials
The benefits of DMD gene therapy using SRP-9001 have been demonstrated in subsequent trials that included more than 80 boys since our first treated candidate in 2018. Efficacy has been maintained in our first open-label trial (Study 101) for 4 years with persistence of gene expression, no significant loss of function, and an excellent safety profile (see discussion below). In two subsequent trials (Studies 102 and 103), effectual levels of gene expression followed systemic delivery of AAV-delivered micro-dystrophin demonstrated at high levels (> 50% increase from baseline) post-gene transfer. In Study 102, a double-blind, randomized controlled trial was initiated. Forty-one subjects were stratified by age, 4 to 5 years old and 6–7 years of age. In the younger cohort, NSAA improved by > 2 points at 1 year compared to placebo. In the older cohort, the initial stratification by age alone and not function led to an unequal match of placebo versus treated. To provide comparative data an external control group matched at baseline for corticosteroids, NSAA, time to rise, and 10MWR was analyzed using propensity weighting. This comparison with an outside perfectly matched control group supported findings seen in the first micro-dystrophin trial. Efficacy was comparable to the younger cohort (p = 0.0001). A third trial, Study 103, included data from a 20-patient, open-label trial, and again confirmed efficacy for 4–7-year-old ambulatory subjects. In study 103, the NSAA at week 52 improved by 3.9 points from baseline and by 3.2 points compared to an external control group (p < 0.0001).
Based on findings from these three trials, Sarepta Therapeutics requested a status of accelerated approval for SRP-9001 (Delandistrogene Moxeparvovec®) from the FDA Center of Biologics Evaluation and Research (CBER). This FDA-instituted program allows for earlier approval of products to treat serious conditions and fill an unmet need. As of June 2023, the first commercial product using micro-dystrophin delivered intravenously as SRP-9001 has been approved for DMD. At Sarepta, a phase 3 (n = 120) randomized, double-blind, controlled trial is underway as a final confirmatory trial projected for completion in 2024.
Safety Issues That Have Arisen from Clinical Trials
A review of the safety profile of SRP-9001 (Delandistrogene Moxeparvovec®) and the risk–benefit ratio of this gene therapy product as a therapeutic option is invaluable. Two examples of serious adverse events were encountered using SRP-9001, both related to the treatment of patients with large mutations and deletions of exons 3–43 and exons 8–21. In both patients, inflammatory myositis was encountered with the addition of myocarditis (elevated troponin I) that was treated by added immune suppression using prednisone, plasmapheresis, and tacrolimus. Both patients recovered. It had been previously established that expression of the gene into a deleted domain of the patient’s mutation was immunogenic [69]. Therefore, patients with such mutations have been excluded from Sarepta-sponsored micro-dystrophin gene therapy trials. It is also of interest that similar safety concerns from other industry-sponsored micro-dystrophin trials were presented at the ASGCT Meeting in 2022 (including Pfizer, Solid, and Genethon) [68] and may be found on internet news agency websites (e.g., BioPharma Dive®, Fierce Biotech®). Overall, in SRP-9001, most other adverse events were ones typical of gene therapy trials, including elevated transaminases and gamma-glutamyl transferase (GGT) from liver injury, frequent vomiting, and transient low platelet counts. These adverse events may extend prednisone treatment as determined by providers of the designated treatment. Safety concerns have been of relatively limited concern and have not put Delandistrogene Moxeparvovec on clinical hold.
Conclusions Regarding Gene Therapy in Clinical Practice
Gene therapy using AAVrh74.MHCK7.micro-dystrophin has achieved accelerated approval by FDA in the spring of 2023. The final decision for full approval is in the hands of the FDA and will depend on confirmation in the ongoing phase 3 clinical trial. The side effect profile for Delandistrogene Moxeparvovec®, sponsored by Sarepta Therapeutics, has been very manageable. The most serious adverse events have occurred when the transgene is expressed directly in the domain of the patient’s identified DMD deletion. There is a consideration that this domain is restricted to DMD deletions inclusive of exons 9–13, which will be part of the product label for Delandistrogene Moxeparvovec.
Other questions persist regarding gene therapy. One commonly asked is how long will benefits persist following single-dose gene delivery. The answer is unknown, but from SRP-9001–101, we have data now out beyond 4 years. Longer-term results will be required for more definitive answers. Another question relates to the potential need for a second dose of gene delivery. Tolerance is unknown presently, but preliminary answers will be addressed by plasmapheresis studies, with the removal of pre-existing AAV antibody levels for patients who are candidates for gene transfer. This will be done in the very near future at Nationwide Children’s Hospital sponsored by Sarepta Therapeutics.
CRISPR Clinical Trials for DMD
The final topic for discussion and comparison is gene editing via clustered regularly interspaced short palindromic repeats (CRISPR). The technology has emerged as a new strategy to directly edit the human genome, correcting the disease-causing pathogenic variant. Applying CRISPR technology to DMD is attractive, based on the potential for single administration genome editing to correct the disease for a long duration, possibly the lifetime of the patient [70]. This approach utilizes Cas9 nuclease, guided by a single guide RNA (sgRNA) to target a specific genomic locus followed by a protospacer adjacent motif (PAM). Cas9 generates DNA-double-stranded breaks (DSBs). In post-mitotic tissue, nonhomologous end joining (NHEJ) is the primary pathway for the repair of DSBs; thus, allowing the broken DNA ends to ligate together with little or no homology, generating deletions or insertions. Pre-clinical studies in the mdx mouse (mdx4cv) [71, 72] and in the canine model for DMD [73] achieved high levels of dystrophin in skeletal and cardiac muscle.
Thus far, the single DMD patient treated using CRISPR gene editing died shortly after gene transfer. The treatment was designed to upregulate an alternate form of the dystrophin protein by targeting the 5’ domain of the gene in the region of exon 1 and its promoter. The patient was in the early 20 s and had severe muscle weakness, reduced pulmonary function, and reduced left ventricular systolic dysfunction. He had no evidence of pre-existing immunity to AAV9 or transgene. He was treated with prophylactic immune suppression (tacrolimus, sirolimus, methylprednisolone) starting 13 days prior to dosing. Cardiac manifestations were suspected 1-day post-gene delivery and cardiac function worsened with acute respiratory distress and death 8-day post-gene transfer. The cause of death following autopsy was hypothesized to be cytokine-mediated capillary leak syndrome without evidence of thrombotic microangiopathy or adaptive humoral or cell-mediated immunity to AAV or transgene products [74]. This was the first attempt to apply CRISPR technology for the treatment of DMD. Further clinical trials are in the planning stages and will address the challenges including improved systemic delivery of CRISPR-editing components, suppression of possible immune responses, and optimizing gene editing.

Current and investigational therapies for Duchenne muscular dystrophy (DMD). Corticosteroids: for DMD, corticosteroids are the standard of care. The choice of corticosteroid is dependent on the provider. The target is muscle inflammation and sarcolemma repair. Their most important attribute is prolonging ambulation. Corticosteroids are usually started at about age 4. At the time of gene transfer, additional prednisone is given for 30–60 days to protect against the immune response elicited by AAV. Exon-skipping strategies: exon skipping restores the reading frame of the DMD gene. The example shows eteplirsen (Exondys 51®) treatment for a subject with deletion of exon 50. Deleting exon 51 restores the reading frame and translates a smaller yet functional dystrophin product. Exon-skipping products currently available and approved by the FDA include those listed: casimersen skips exon 45, eteplirsen skips exon 51, golodirsen skips exon 53, and viltolarsen skips exon 51. Gene replacement therapy: this approach necessitates miniaturing the DMD gene, building a cassette with components most likely to achieve safety and efficacy and small enough to be packaged (≤ 4.7 kb) in AAV. Infusion into the circulation of the DMD patient, followed by muscle transduction, and micro-dystrophin expression leads to gene expression shown by sarcolemmal staining surrounding individual muscle fibers seen on muscle biopsy post-treatment. Gene editing: clustered regularly interspaced short palindromic repeats (CRISPR) is a promising technique having the potential to target specific pathogenic variants in the DMD gene, offering a precise approach to treatment. The endonuclease, Cas9, recognizes and cuts specific DNA sequences guided by an RNA synthetic guide that provides precise targeting at the DNA site that requires editing of the existing gene
One particular example of modification relatively easily achieved is the adaptation of self-complementary AAV (scAAV) to deliver CRISPR sgRNAs for gene editing [75, 76]. scAAV is a well-established platform for clinical systemic gene delivery with proven efficacy [77]. The use of ssAAV-delivered CRISPR leads to preferential depletion of the sgRNA compared to the Cas9 nuclease [78, 79]. In contrast, scAAV is more resistant to DNA degradation providing more copies of stable episomes after viral transduction [64]. In mouse CRISPR experiments, systemic injection of scAAV-sgRNAs showed greater gene editing and dystrophin restoration compared to ssAAV-treated cohorts [64, 80].
Final Comments
This is an exciting time in the field of rapidly evolving potential therapies for DMD (Fig. 4). Each of the major approaches including corticosteroids, exon skipping, and systemic gene delivery is described in detail with a historical perspective. Where comparisons are possible, they are discussed but it seems clear that each has its own indications and benefits. CRISPR has great potential and is on the cusp of clinical trials; results will be of interest.
References
Mendell JR, Shilling C, Leslie ND, Flanigan KM, al-Dahhak R, Gastier-Foster J, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71(3):304–13.
Mendell JR, Lloyd-Puryear M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve. 2013;48(1):21–6.
Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;(1):CD003725.
Mendell JR, Province MA, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, et al. Clinical investigation of Duchenne muscular dystrophy. A methodology for therapeutic trials based on natural history controls. Arch Neurol. 1987;44(8):808–11.
McDonald CM, Henricson EK, Abresch RT, Duong T, Joyce NC, Hu F, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet Lond Engl. 2018;391(10119):451–61.
Drachman DB, Toyka KV, Myer E. Prednisone in Duchenne muscular dystrophy. Lancet Lond Engl. 1974;2(7894):1409–12.
Kissel JT, Lynn DJ, Rammohan KW, Klein JP, Griggs RC, Moxley RT, et al. Mononuclear cell analysis of muscle biopsies in prednisone- and azathioprine-treated Duchenne muscular dystrophy. Neurology. 1993;43(3 Pt 1):532–6.
Peverelli, Lorenzo et al. Histologic muscular history in steroid-treated and untreated patients with Duchenne dystrophy. Neurology. 2015; 85:1886–1893.
Silversides CK, Webb GD, Harris VA, Biggar DW. Effects of deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. Am J Cardiol. 2003;91(6):769–72.
Markham LW, Spicer RL, Khoury PR, Wong BL, Mathews KD, Cripe LH. Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr Cardiol. 2005;26(6):768–71.
Schram G, Fournier A, Leduc H, Dahdah N, Therien J, Vanasse M, et al. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol. 2013;61(9):948–54.
Van Ruiten HJA, Marini Bettolo C, Cheetham T, Eagle M, Lochmuller H, Straub V, et al. Why are some patients with Duchenne muscular dystrophy dying young: an analysis of causes of death in North East England. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2016;20(6):904–9.
Siegel IM, Miller JE, Ray RD. Failure of corticosteroid in the treatment of Duchenne (pseudo-hypertrophic) muscular dystrophy. Report of a clinically matched three year double-blind study. IMJ Ill Med J. 1974;145(1):32–33 passim.
Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley RT, Miller JP, et al. Clinical investigation of Duchenne muscular dystrophy. Interesting results in a trial of prednisone. Arch Neurol. 1987;44(8):812–7.
Mendell JR, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, Miller JP, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320(24):1592–7.
Griggs RC, Moxley RT, Mendell JR, Fenichel GM, Brooke MH, Pestronk A, et al. Duchenne dystrophy: randomized, controlled trial of prednisone (18 months) and azathioprine (12 months). Neurology. 1993;43(3 Pt 1):520–7.
Connolly AM, Schierbecker J, Renna R, Florence J. High dose weekly oral prednisone improves strength in boys with Duchenne muscular dystrophy. Neuromuscul Disord NMD. 2002;12(10):917–25.
Escolar DM, Hache LP, Clemens PR, Cnaan A, McDonald CM, Viswanathan V, et al. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology. 2011;77(5):444–52.
Mesa LE, Dubrovsky AL, Corderi J, Marco P, Flores D. Steroids in Duchenne muscular dystrophy–deflazacort trial. Neuromuscul Disord NMD. 1991;1(4):261–6.
Angelini C, Pegoraro E, Turella E, Intino MT, Pini A, Costa C. Deflazacort in Duchenne dystrophy: study of long-term effect. Muscle Nerve. 1994;17(4):386–91.
Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord NMD. 2006;16(4):249–55.
Bonifati MD, Ruzza G, Bonometto P, Berardinelli A, Gorni K, Orcesi S, et al. A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. 2000;23(9):1344–7.
Houde S, Filiatrault M, Fournier A, Dubé J, D’Arcy S, Bérubé D, et al. Deflazacort use in Duchenne muscular dystrophy: an 8-year follow-up. Pediatr Neurol. 2008;38(3):200–6.
Bello L, Gordish-Dressman H, Morgenroth LP, Henricson EK, Duong T, Hoffman EP, et al. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology. 2015;85(12):1048–55.
Griggs RC, Herr BE, Reha A, Elfring G, Atkinson L, Cwik V, et al. Corticosteroids in Duchenne muscular dystrophy: major variations in practice. Muscle Nerve. 2013;48(1):27–31.
Griggs RC, Miller JP, Greenberg CR, Fehlings DL, Pestronk A, Mendell JR, et al. Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology. 2016;87(20):2123–31.
Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther. 2012;134(1):54–67.
Hoffman EP, Riddle V, Siegler MA, Dickerson D, Backonja M, Kramer WG, et al. Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes. Steroids. 2018;134:43–52.
Conklin LS, Damsker JM, Hoffman EP, Jusko WJ, Mavroudis PD, Schwartz BD, et al. Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug. Pharmacol Res. 2018;136:140–50.
Hoffman EP, Schwartz BD, Mengle-Gaw LJ, Smith EC, Castro D, Mah JK, et al. Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function. Neurology. 2019;93(13):e1312–23.
Smith EC, Conklin LS, Hoffman EP, Clemens PR, Mah JK, Finkel RS, et al. Efficacy and safety of vamorolone in Duchenne muscular dystrophy: an 18-month interim analysis of a non-randomized open-label extension study. PLoS Med. 2020;17(9): e1003222.
Mah JK, Clemens PR, Guglieri M, Smith EC, Finkel RS, Tulinius M, et al. Efficacy and safety of vamorolone in Duchenne muscular dystrophy: a 30-month nonrandomized controlled open-label extension trial. JAMA Netw Open. 2022;5(1): e2144178.
Guglieri M, Clemens PR, Perlman SJ, Smith EC, Horrocks I, Finkel RS, et al. Efficacy and safety of vamorolone vs placebo and prednisone among boys with Duchenne muscular dystrophy: a randomized clinical trial. JAMA Neurol. 2022;79(10):1005–14.
Klein CJ, Coovert DD, Bulman DE, Ray PN, Mendell JR, Burghes AH. Somatic reversion/suppression in Duchenne muscular dystrophy (DMD): evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. Am J Hum Genet. 1992;50(5):950–9.
Lu QL, Morris GE, Wilton SD, Ly T, Artem’yeva OV, Strong P, et al. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J Cell Biol. 2000;148(5):985–96.
Amanat M, Nemeth CL, Fine AS, Leung DG, Fatemi A. Antisense oligonucleotide therapy for the nervous system: from bench to bedside with emphasis on pediatric neurology. Pharmaceutics. 2022;14(11):2389.
Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30(3):293–9.
Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2(12):731–40.
Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8(10):918–28.
Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74(5):637–47.
Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. 2016;79(2):257–71.
Kinane TB, Mayer OH, Duda PW, Lowes LP, Moody SL, Mendell JR. Long-term pulmonary function in Duchenne muscular dystrophy: comparison of eteplirsen-treated patients to natural history. J Neuromuscul Dis. 2018;5(1):47–58.
Mitelman O, Abdel-Hamid HZ, Byrne BJ, Connolly AM, Heydemann P, Proud C, et al. A combined prospective and retrospective comparison of long-term functional outcomes suggests delayed loss of ambulation and pulmonary decline with long-term eteplirsen treatment. J Neuromuscul Dis. 2022;9(1):39–52.
Alfano LN, Charleston JS, Connolly AM, Cripe L, Donoghue C, Dracker R, et al. Long-term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine (Baltimore). 2019;98(26): e15858.
McDonald CM, Shieh PB, Abdel-Hamid HZ, Connolly AM, Ciafaloni E, Wagner KR, et al. Open-label evaluation of eteplirsen in patients with Duchenne muscular dystrophy amenable to exon 51 skipping: PROMOVI trial. J Neuromuscul Dis. 2021;8(6):989–1001.
Frank DE, Schnell FJ, Akana C, El-Husayni SH, Desjardins CA, Morgan J, et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. 2020;94(21):e2270–82.
Servais L, Mercuri E, Straub V, Guglieri M, Seferian AM, Scoto M, et al. Long-term safety and efficacy data of golodirsen in ambulatory patients with duchenne muscular dystrophy amenable to exon 53 skipping: a first-in-human, multicenter, two-part, open-label, phase 1/2 trial. Nucleic Acid Ther. 2022;32(1):29–39.
Scaglioni D, Catapano F, Ellis M, Torelli S, Chambers D, Feng L, et al. The administration of antisense oligonucleotide golodirsen reduces pathological regeneration in patients with Duchenne muscular dystrophy. Acta Neuropathol Commun. 2021;9(1):7.
Wagner KR, Kuntz NL, Koenig E, East L, Upadhyay S, Han B, et al. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: a randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve. 2021;64(3):285–92.
Clemens PR, Rao VK, Connolly AM, Harper AD, Mah JK, Smith EC, et al. Safety, tolerability, and efficacy of viltolarsen in boys with Duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol. 2020;77(8):982–91.
Clemens PR, Rao VK, Connolly AM, Harper AD, Mah JK, McDonald CM, et al. Efficacy and safety of viltolarsen in boys with Duchenne muscular dystrophy: results from the phase 2, open-label, 4-year extension study. J Neuromuscul Dis. 2023;
Oechsel KF, Cartwright MS. Combination therapy with onasemnogene and risdiplam in spinal muscular atrophy type 1. Muscle Nerve. 2021;64(4):487–90.
Wu B, Lu P, Cloer C, Shaban M, Grewal S, Milazi S, et al. Long-term rescue of dystrophin expression and improvement in muscle pathology and function in dystrophic mdx mice by peptide-conjugated morpholino. Am J Pathol. 2012;181(2):392–400.
Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91.
Finkel RS, Flanigan KM, Wong B, Bönnemann C, Sampson J, Sweeney HL, et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE. 2013;8(12): e81302.
McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Lond Engl. 2017;390(10101):1489–98.
Mercuri E, Osorio AN, Muntoni F, Buccella F, Desguerre I, Kirschner J, et al. Safety and effectiveness of ataluren in patients with nonsense mutation DMD in the STRIDE Registry compared with the CINRG Duchenne Natural History Study (2015–2022): 2022 interim analysis. J Neurol. 2020;270(8):3896–913.
Long-term outcomes of ataluren in Duchenne muscular dystrophy. Available at: https://clinicaltrials.gov/ct2/show/NCT03179631. Accessed August 23, 2020.
England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343(6254):180–2.
Marshall E. Gene therapy death prompts review of adenovirus vector. Science. 1999;286(5448):2244–5.
Atchison RW, Casto BC, Hammon WM. Adenovirus-associated defective virus particles. Science. 1965;149(3685):754–6.
Hoggan MD, Blacklow NR, Rowe WP. Studies of small DNA viruses found in various adenovirus preparations: physical, biological, and immunological characteristics. Proc Natl Acad Sci U S A. 1966;55(6):1467–74.
Naso MF, Tomkowicz B, Perry WL, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs Clin Immunother Biopharm Gene Ther. 2017;31(4):317–34.
McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8(16):1248–54.
Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci U S A. 2000;97(25):13714–9.
Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2008;8(3):253–61.
Mendell JR, Sahenk Z, Lehman K, Nease C, Lowes LP, Miller NF, et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 2020;77(9):1122–31.
Bönnemann CG, Belluscio BA, Braun S, et al. A collaborative analysis by clinical trial sponsors and academic experts of anti-transgene SAEs in studies of gene therapy for DMD. Presented at: ASGCT 25th Annual Meeting, May 16–19, 2022; Washington DC.
Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363(15):1429–37.
Chen G, Wei T, Yang H, Li G, Li H. CRISPR-based therapeutic gene editing for Duchenne muscular dystrophy: advances, challenges and perspectives. Cells. 2022;11(19):2964.
Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun. 2017;8:14454.
Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351(6271):400–3.
Amoasii L, Hildyard JCW, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362(6410):86–91.
Angela Lek, Brenda Wong, Allison Keeler, Meghan Blackwood, Kaiyue Ma, Shushu Huang, et al. Unexpected death of a Duchenne muscular dystrophy patient in an N-of-1 trial of rAAV9-delivered CRISPR-transactivator. medRxiv. 2023;2023.05.16.23289881
Min YL, Chemello F, Li H, Rodriguez-Caycedo C, Sanchez-Ortiz E, Mireault AA, et al. Correction of three prominent mutations in mouse and human models of Duchenne muscular dystrophy by single-cut genome editing. Mol Ther J Am Soc Gene Ther. 2020;28(9):2044–55.
Moretti A, Fonteyne L, Giesert F, Hoppmann P, Meier AB, Bozoglu T, et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat Med. 2020;26(2):207–14.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–22.
Hakim CH, Wasala NB, Nelson CE, Wasala LP, Yue Y, Louderman JA, et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight. 2018;3(23):e124297, 124297.
Min YL, Li H, Rodriguez-Caycedo C, Mireault AA, Huang J, Shelton JM, et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci Adv. 2019;5(3):eaav4324.
Ren C, Kumar S, Shaw DR, Ponnazhagan S. Genomic stability of self-complementary adeno-associated virus 2 during early stages of transduction in mouse muscle in vivo. Hum Gene Ther. 2005;16(9):1047–57.
Acknowledgements
The micro-dystrophin gene therapy clinical trials have been done with Sarepta Therapeutics as the Industry Sponsor.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
D’Ambrosio, E.S., Mendell, J.R. Evolving Therapeutic Options for the Treatment of Duchenne Muscular Dystrophy. Neurotherapeutics 20, 1669–1681 (2023). https://doi.org/10.1007/s13311-023-01423-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-023-01423-y