
Abstract
Human cytomegalovirus (HCMV) latency and reactivation is regulated by the chromatin structure at the major immediate early promoter (MIEP) within myeloid cells. Both cellular and viral factors are known to control this promoter during latency, here we will review the known mechanisms for MIEP regulation during latency. We will then focus on the virally encoded G-protein coupled receptor, US28, which suppresses the MIEP in early myeloid lineage cells. The importance of this function is underlined by the fact that US28 is essential for HCMV latency in CD34+ progenitor cells and CD14+ monocytes. We will describe cellular signalling pathways modulated by US28 to direct MIEP suppression during latency and demonstrate how US28 is able to ‘regulate the regulators’ of HCMV latency. Finally, we will describe how cell-surface US28 can be a target for antiviral therapies directed at the latent viral reservoir.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human cytomegalovirus (HCMV) persists for the lifetime of the host, a process underpinned by the establishment of latency in specific cell types. Sporadic reactivation of HCMV is thought to be well-controlled by host immune responses resulting in subclinical events, but HCMV reactivation poses a grave risk to immunocompromised individuals, especially immunosuppressed organ transplant recipients. All current therapies for HCMV disease target the lytic phase of infection, and therefore cannot reduce or remove latent reservoirs in either the donor organ or recipient. Here, we discuss our molecular understanding of latency and reactivation and how our insights have yielded novel ways to target the latent reservoir.
HCMV latency and reactivation is regulated by chromatin structure at the major immediate early promoter
Latent carriage of HCMV requires the maintenance of the viral genome in the absence of the production of infectious virus particles; however, under certain conditions, virus is able to reactivate and produce new virus particles. This ability to reactivate sets latency apart from abortive infection and cellular differentiation is intimately linked with both latency and reactivation.
One important site of human cytomegalovirus latency is in cells of the early myeloid lineage. CD34+ progenitors and their derivatives, including granulocyte–macrophage progenitors and CD14+ monocytes, are latently infected in seropositive individuals [1,2,3,4]. Reactivation of HCMV has been observed in vitro and ex vivo upon differentiation of CD34+ progenitor cells into mature dendritic cells or macrophages [5,6,7,8]. While differentiation-independent virus reactivation has been recently reported in an immortal myeloid cell line [9], the mechanism of reactivation from latency has only been extensively described during myeloid cell differentiation.
A key hallmark of latency is the suppression of immediate early (IE) gene expression, and conversely, the earliest events in reactivation are the activation of IE gene expression. The absence of IE1/IE72 and IE2/IE86 transcripts during latency is a common theme throughout the results of multiple analyses of viral gene expression in latently infected cells, both ex vivo and in in vitro models [3, 4, 7, 10,11,12]. It follows that control of IE gene expression is an important determinant of latency and reactivation. IE gene expression is regulated by the major immediate early promoter and enhancer regions, which will be referred to, here, simply as the MIEP. Encompassing over 1 kb of DNA, regions within the MIEP can be bound by activatory or repressive transcription factors, and since HCMV DNA is rapidly chromatinised upon entry into the nucleus, the MIEP is subject to regulation by chromatin structure [13,14,15,16,17].
Analyses of chromatin structure at the major immediate early promoter reveals that latency coincides with a repressive chromatin structure around the MIEP, including the presence of the heterochromatin marker HP1 [7, 8, 18], as well as the histone modifications histone-H3-lysine-27-trimethylation (H3K27me3) and histone-H3-lysine-9-trimethylation (H3K9me3) [19, 20] (see also Fig. 1). Histone deacetylase (HDAC) activity is also important for maintaining a repressed chromatin state; treatment of latently infected monocytes with HDAC inhibitors leads to transient activation of IE gene expression [21].

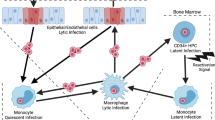
Regulation of HCMV latency and reactivation during myeloid differentiation. HCMV infects CD34+ progenitor cells and establishes latency (top left). The HCMV genome is maintained in the nucleus as an episome (blue circle) and is chromatinised. The MIEP (represented bottom left) is prevented from driving IE gene expression by a repressive chromatin state. Histones (purple) are trimethylated (me3) at H3K9 and H3K27. The repressive factor HP1 associates with the MIEP, as do ERF and YY1, and KAP1 acts to suppress the MIEP from distal binding sites. Latency-associated viral factors (listed) contribute to MIEP suppression, and the activatory factor pp71 is excluded from the nucleus. During differentiation-induced reactivation in mature dendritic cells or macrophages (top right), transcription of IE genes is activated leading to full lytic replication and release of infectious virions. As a result of differentiation, the chromatin structure around the MIEP is more open (bottom right), and activatory histone marks including histone acetylation (Ac) and H3-serine-10-phosphorylation (S10P) are present. Activated CREB and NF-κB become associated with the MIEP, as do histone acetyl transferases (HATs). Several viral factors are reported to be important for reactivation in myeloid cells, including LUNA, UL7, and certain members of the ULb’ family
The differentiation of CD34+ progenitor cells, which can carry latent HCMV in vivo, into mature dendritic cells results in the removal of repressive H3K27me3 and H3K9me3 marks and associated HP1 from the MIEP [7, 8, 19, 20]. Additionally, phosphorylation of histone H3-serine-10 (H3S10P) at the MIEP has been shown to precede the removal of repressive marks during the differentiation of experimentally infected monocytes into immature dendritic cells [22]. Acetylation of histone H4 has also been demonstrated during reactivation from latency in maturing dendritic cells [7, 8]. As such, an open chromatin structure around the MIEP permits the initiation of IE transcription which is necessary for reactivation.
Cellular and viral factors control the MIEP during latency
Clearly, a repressive chromatin structure around the MIEP must be established during latency in myeloid progenitors and then modified during reactivation to permit efficient IE gene expression in differentiated dendritic cells and macrophages (Fig. 1). We know that this process relies upon both cellular and viral factors; these can function by direct binding to the MIEP or by indirect mechanisms and have either activatory or repressive functions. A long-standing hypothesis states that it is the balance of these activatory or repressive factors that then controls whether or not the MIEP drives IE gene transcription, and that cellular differentiation must alter this balance [23,24,25].
Some host cell transcription factors bind directly to the overlapping 18, 19, and 21 bp repeats within the MIEP as well as other motifs in more upstream sequences (direct acting factors) [17]. This includes the repressive factors YY1 and ERF, and the activatory factors NF-κB and CREB, which have been discussed in the context of latency and reactivation previously [17]. In brief, in undifferentiated, non-permissive cells, the repressive factors YY1 [26] and ERF [27, 28] bind to the 21 bp repeats. ERF is thought to recruit HDAC1 to the MIEP, thus providing a link between transcription factor binding to specific DNA sequence motifs and epigenetic modification. Interestingly, absolute levels of YY1 decreased during differentiation of the non-permissive NT2 cell line [26].
KAP1 was more recently identified as a chromatin organiser that can mediate repression during latency [20]. While not strictly a DNA-binding protein, KAP1 was found to associate with a number of sites on the HCMV genome in CD34+ progenitor cells, and KAP1 deposition at these sites correlated with the presence of the KAP1 effector SETDB1, as well as HP1 and H3K9me marks at the MIEP. When KAP1 was depleted, these marks were lost and the virus entered lytic replication in the absence of cellular differentiation. Furthermore, KAP1 activity was shown to be repressed during lytic infection by mTOR-mediated phosphorylation, thus providing a potential mechanism for exiting latency.
Other host factors which do not, themselves, bind to viral DNA are thought to control the presence or activation of other direct-acting factors. As discussed, mTOR-mediated phosphorylation of KAP1 abrogates the repressive activity of KAP1, implying that mTOR is important for regulating latency. Other host kinases are also important. Linking reactivation with cellular differentiation, IL-6/LPS-stimulated activation of ERK-MAPK pathways was shown to be crucial for inducing MIEP activity in maturing dendritic cells [22, 29]. CREB is phosphorylated by the downstream kinase MSK, which is required for its activation at the MIEP. The absence of this signalling during latency in myeloid progenitors may, therefore, prevent CREB activity.
The role of viral factors during latency is becoming more appreciated (Fig. 1) [30]. Viral gene expression during experimental and natural latency, as measured by RNAseq, has recently been found to be rather broad and complex [19, 31, 32] than when compared with earlier microarray or targeted RT-PCR studies of latently infected cells [10, 33,34,35]. In addition, it is important to consider the numerous viral factors that may enter myeloid cells as components of the virion. For example, the viral long non-coding RNA 4.9 has been reported to bind the MIEP and recruit the repressor complex PRC2 to the MIEP [19]. The viral transactivator pp71 is excluded from the nucleus of non-permissive cells, and since pp71 has been shown to be important for antagonising the functions of PML bodies during lytic infection, exclusion of pp71 may help mediate PML-mediated repression of the MIEP [36]. However, other reports in different systems note that knockdown of PML components had no effect on the establishment of latency [37, 38] and, furthermore, a recent study found that the viral factor LUNA actually disperses PML bodies during latent infection in CD34+ cells [39].
The latency-associated gene product UL138 does not localise to the nucleus but instead manipulates cellular signalling pathways from the ER, probably in concert and in opposition with other members of the ULb’ region [40]. In brief, UL138 has been reported to repress MIEP activity, in part by blocking histone lysine-demethylase activity during latency [41] and also likely via manipulation of EGFR signalling [41, 42]. Meanwhile, other viral factors promote reactivation from latency, including LUNA and UL7 [39, 43, 44].
The virally-encoded G-protein coupled receptor US28 is expressed during lytic and latent infections, as well as coming in with the virion [45] and has recently gained prominence as an essential protein for latency. In the remainder of this review, we will discuss how US28 is able to alter cell signalling in a differentiation dependent manner, and thus promote latency in myeloid progenitor cells.
US28 is essential for HCMV latency in CD34+ progenitor cells and CD14+ monocytes
It has been known for some time that US28 is expressed during latent infection of myeloid cells [45,46,47,48,49] but the functions of US28 have mostly been described for lytic infection. During the replication cycle of HCMV, US28 acts as a chemokine receptor homologue, binding CXXXC and CC chemokines [50, 51], but US28 can also signal constitutively [52, 53]. A comprehensive summary of US28 signalling functions during lytic infection, including cell type specificity, ligand interactions, and G-protein usage, was recently published [54].
However, intriguingly, US28 gene deletion viruses (ΔUS28) fail to establish latency in CD34+ progenitor cells [45, 49] and CD14+ monocytes [55], instead they fail to repress the MIEP, driving IE expression and full lytic cycle with the eventual release of infectious viral particles. Removing US28 uncouples permissiveness from cellular differentiation, since monocytes infected with ΔUS28 HCMV undergo lytic infection but do not show differentiation-specific cell surface markers [55]. US28 was shown to be expressed and translated de novo as well as entering the cell with the virion [45] and it has now become clear that both incoming US28 and de novo expressed US28 are important for the establishment and ongoing maintenance of latency in myeloid progenitor cells [56]. Other sites of HCMV latency or low level persistence, such as neuronal cells and endothelial cells, have been suggested but, as yet, these have not been confirmed in vivo and there is no evidence that US28 is required to negatively regulate the MIEP during latent/persistent infections of these cell types in vitro [54].
Use of characterised mutants of US28 has elucidated some US28-mediated functions that are important for latency. The Y16F mutant removes some ligand binding activity [57] and the R129A mutant abrogates coupling of G-proteins to the DRY box motif of US28 [58,59,60,61]. Expression of US28-WT in trans rescues latency-establishment in cell line models with the ΔUS28 virus. Similarly, expression of US28-Y16F in trans could also complement the US28 deletion virus suggesting that certain modes of ligand binding may not be necessary for the latency-associated function of US28 [55]. However, deletion of the entire ligand binding domain of US28 in the virus causes lytic infection in myeloid cells [56], which is perhaps explained by recent work demonstrating the multiple modes by which US28 can bind a wide array of ligands [62]. It is clear, however, that either expression of US28-R129A in trans or within the virus, fails to lead to latency establishment, providing clear evidence that US28-signalling via G proteins is essential for latency [55, 56]. The way that US28 signalling manipulates the host environment to support latency is therefore of great interest and under intense study.
US28 alters cellular signalling within myeloid cells
Analysis of the activation states of cellular kinases during latency with US28 expressed in isolation in myeloid cells has revealed several signalling pathways that are important for latency (summarised in Fig. 2). Infection of CD34+ progenitor cells with WT virus, but not ΔUS28 HCMV, drives activation of the STAT3-iNOS pathway, and the resultant nitric oxide production was shown to suppress the MIEP [49]. Furthermore, these authors showed that presence of US28 in the context of latent infection may reprogramme infected cells to become immunosuppressive monocytes akin to myeloid-derived suppressor cells, rather than conventional monocytes or, indeed, parts of other myeloid or lymphoid lineages.

US28 controls several signaling pathways to suppress the MIEP in early myeloid lineage cells. US28 is present at the cell surface, and probably other membranes, of latently infected cells. Here, it attenuates several signaling pathways and transcription factors, including NF-κB, c-fos, and ERK1/2. NF-κB can no longer enter the nucleus (dashed line), nor bind and activate the MIEP. c-fos typically forms a dimer with c-jun to form the AP1 complex; US28 causes loss of c-fos, the AP1 complex does not form and thus cannot activate the MIEP. Attenuation of ERK1/2 causes loss of ERK1/2 phosphorylation (P) and subsequent activation of MSK and, therefore, MSK does not phosphorylate and activate CREB. Inactive CREB cannot activate the MIEP. US28 is also reported to activate the STAT3-iNOS signaling axis, leading to nitric oxide (NO) production. NO suppresses the MIEP in myeloid cells by unknown mechanisms. By these, and probably other pathways, US28 helps establish and maintain a repressive chromatin structure at the MIEP, and a lack of IE gene expression
Additionally, US28 has been found to attenuate several cellular signalling pathways, such as ERK, MSK, NF-κB, and STAT5 [55] when expressed in isolation in undifferentiated myeloid cells. It is interesting to note that ERK signalling is crucial for CREB phosphorylation at the MIEP and subsequent deposition of the activatory mark H3S10P on the MIEP upon differentiation-induced reactivation [22]. Consistent with this, and the ability of US28 to attenuate ERK signalling, infection of monocytes with ΔUS28 HCMV (which no longer suppresses the MIEP) is also associated with activated CREB and phosphorylated H3S10 on the MIEP. Furthermore, pharmacological inhibition of ERK in combination with NF-κB could prevent lytic replication of ΔUS28 HCMV in monocytes and, conversely, treatment of monocytes with small molecule inhibitors of US28 also results in a lytic infection rather than latency [55].
Attenuation of these cellular signalling pathways is reversed when US28-expressing cells are differentiated into macrophage-like cells using phorbol esters [55]. The implication then is that US28 helps to maintain latency via the attenuation of MIEP-activatory cascades but does not block signalling from these pathways during reactivation, and may even support their function during cellular differentiation. Indeed, in reporter systems, US28 represses the MIEP in undifferentiated THP-1 monocytes, but activates the MIEP in PMA-differentiated THP-1 derived macrophages [55].
Recent work has also shown that US28 decreases c-fos levels during latency. Binding to the AP-1 site within the MIEP by fos/jun dimers activates the MIEP, and so, in decreasing c-fos, US28 enacts MIEP suppression via an additional mechanism. As such, treatment of myeloid cells with a c-fos inhibitor reduced lytic gene expression when infecting with ΔUS28 HCMV [56].
Taken together, a key mechanism by which US28 supports latency in undifferentiated myeloid cells is to modulate multiple cellular signalling pathways, which alters the balance of activatory and repressive factors at the MIEP, the result of which is to promote a repressive chromatin structure at the MIEP and thus suppress IE gene expression. In contrast, this suppressive function of US28 does not occur in differentiated myeloid cells and so US28 does not promote a repressive chromatin structure at the MIEP during differentiation-induced reactivation.
Targeting US28 represents a novel way to target the latent viral reservoir
Latent carriage of HCMV in myeloid progenitor cells provides a reservoir of reactivatable virus that cannot be cleared with current therapeutics. CMV reactivation events in immunocompromised patients can cause serious morbidity and mortality, particularly in the organ transplant setting. Clearing or reducing the latent reservoir in patients or donors may, therefore, be a way to reduce the burden of CMV-disease in transplant recipients.
Understanding the role of latency-associated gene products identifies viral genes that must be expressed during latency and thus represent potential strategies for targeting latently infected cells. Proof of principle for this came with the observation that UL138 reduces MRP1 on the cell surface of latently infected cells, and therefore the toxin vincristine was selectively taken up by latently infected cells [63].
US28 represents excellent potential for targeting the latent reservoir because (1) US28 is expressed on the cell surface during latency [48]; (2) G-protein coupled receptors are well-appreciated pharmacological targets; (3) US28 controls latency via the MIEP. Indeed, US28 on the surface of latently infected cells may be a target for antibody-dependent killing by autologous neutrophils, but HCMV evades this killing in part by downregulating neutrophil chemoattractants [64].
One strategy considered for US28-targetting was to link a high-affinity ligand for US28 with a toxin. Upon binding ligand, US28 is internalised, and thus would deliver the toxin into latently infected cell. By fusing part of the Pseudomonas Endotoxin A with CX3CL1 (also known as fractalkine), such a fusion toxin protein (F49A-FTP) was devised [65]. Since US28 has a higher affinity for CX3CL1 than the native receptor, F49A-FTP could selectively kill experimentally and naturally latent monocytes and reduce reactivation events in vitro [48]. Using F49A-FTP to flush out the latent reservoir in normothermic solid organs for transplant is currently under investigation.
A second strategy relies on the known function of US28 during latency. US28 suppresses the MIEP in myeloid cells, and the inverse agonist VUF2274 inhibits US28 function during latency, leading to reactivation [55]. Full reactivation of HCMV may not be desirable since HCMV encodes many immune evasins at later time points [66, 67] and would thus evade natural immune control by T and NK cells. Transient induction of IE gene expression might be considered preferable [68], since up to 5% of a seropositive individual’s CD8+ T cells are capable of recognising lytic IE antigen [69]. Furthermore, striking evidence from studies of murine cytomegalovirus indicates that IE antigen is recognised by cytotoxic CD8+ T cells [70, 71]. Interestingly, sporadic induction of IE gene expression is observed in vivo in the lungs of infected mice [72, 73], and these events have been linked to the T cell “memory inflation” phenomenon [74]. In vitro analyses of primary human cells have shown that HDAC inhibitors can transiently induce IE expression in latently infected monocytes, thus allowing autologous cytotoxic T cells from seropositive donors to recognise and kill these infected cells. The result is a reduction in latent carriage in this experimental model of latency [21]. Perhaps an US28 inhibitor that partially blocks US28-mediated suppression of the MIEP would transiently induce IE and allow recognition by cytotoxic T cells. This is currently under study in our own laboratory. Several groups are also developing alternative US28 inhibitors [75,76,77] which might provide a highly-selective US28-based shock and kill strategy in the transplant setting.
Concluding remarks
A molecular understanding of human cytomegalovirus latency has revealed pathways and mechanisms which may be therapeutically targeted to reduce the burden of reactivation-associated CMV disease. Chromatin structure at the MIEP is crucial for the control of latency and reactivation, and targeting the cellular and viral factors, including US28, which regulate the MIEP directly or indirectly, is a strategy for potential reduction of the latent viral reservoir within patients.
References
Taylor-Wiedeman J, Sissons JGP, Borysiewicz LK, Sinclair JH (1991) Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol 72:2059–2064. https://doi.org/10.1099/0022-1317-72-9-2059
Mendelson M, Monard S, Sissons P, Sinclair J (1996) Detection of endogenous human cytomegalovirus in CD34 + bone marrow progenitors. J Gen Virol 77:3099–3102. https://doi.org/10.1099/0022-1317-77-12-3099
Kondo K, Xu J, Mocarski ES (1996) Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc Natl Acad Sci USA 93:11137–11142
Hahn G, Jores R, Mocarski ES (1998) Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc Natl Acad Sci USA 95:3937–3942
Reeves MB, Sinclair JH (2013) Circulating dendritic cells isolated from healthy seropositive donors are sites of human cytomegalovirus reactivation in vivo. J Virol 87:10660–10667. https://doi.org/10.1128/JVI.01539-13
Poole E, Juss JK, Krishna B, Herre J, Chilvers ER, Sinclair J (2015) Alveolar macrophages isolated directly from human cytomegalovirus (HCMV)-seropositive individuals are sites of HCMV reactivation in vivo. J Infect Dis 211:1936–1942. https://doi.org/10.1093/infdis/jiu837
Reeves MB, MacAry PA, Lehner PJ, Sissons JGP, Sinclair JH (2005) Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci 102:4140–4145. https://doi.org/10.1073/pnas.0408994102
Reeves MB, Lehner PJ, Sissons JGP, Sinclair JH (2005) An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J Gen Virol 86:2949–2954. https://doi.org/10.1099/vir.0.81161-0
Forte E, Swaminathan S, Schroeder MW, Kim JY, Terhune SS, Hummel M (2018) Tumor necrosis factor alpha induces reactivation of human cytomegalovirus independently of myeloid cell differentiation following posttranscriptional establishment of latency. MBio. https://doi.org/10.1128/mBio.01560-18
Goodrum FD, Jordan CT, High K, Shenk T (2002) Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci USA 99:16255–16260. https://doi.org/10.1073/pnas.252630899
Reeves MB, Sinclair JH (2010) Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J Gen Virol 91:599–604. https://doi.org/10.1099/vir.0.015602-0
Söderberg-Nauclér C, Fish KN, Nelson JA (1997) Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119–126. https://doi.org/10.1016/S0092-8674(01)80014-3
Groves IJ, Reeves MB, Sinclair JH (2009) Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic “pre-immediate-early” repression of viral gene expression mediated by histone post-translational modification. J Gen Virol 90:2364–2374. https://doi.org/10.1099/vir.0.012526-0
Boshart M, Weber F, Jahn G, Dorsch-Häsler K, Fleckenstein B, Schaffner W (1985) A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 41:521–530
Nelson JA, Groudine M (1986) Transcriptional regulation of the human cytomegalovirus major immediate-early gene is associated with induction of DNase I-hypersensitive sites. Mol Cell Biol 6:452–461
Stinski MF, Isomura H (2008) Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med Microbiol Immunol 197:223–231. https://doi.org/10.1007/s00430-007-0069-7
Sinclair J (2010) Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim Biophys Acta Gene Regul Mech 1799:286–295. https://doi.org/10.1016/J.BBAGRM.2009.08.001
Murphy JC, Fischle W, Verdin E, Sinclair JH (2002) Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J 21:1112–1120. https://doi.org/10.1093/emboj/21.5.1112
Rossetto CC, Tarrant-Elorza M, Pari GS (2013) Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) Cells. PLoS Pathog 9:e1003366. https://doi.org/10.1371/journal.ppat.1003366
Rauwel B, Jang SM, Cassano M, Kapopoulou A, Barde I, Trono D (2015) Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. Elife. https://doi.org/10.7554/eLife.06068
Krishna BA, Lau B, Jackson SE, Wills MR, Sinclair JH, Poole E (2016) Transient activation of human cytomegalovirus lytic gene expression during latency allows cytotoxic T cell killing of latently infected cells. Sci Rep 6:24674. https://doi.org/10.1038/srep24674
Kew V, Yuan J, Meier J, Reeves M (2014) Mitogen and stress activated kinases act co-operatively with creb during the induction of human cytomegalovirus immediate-early gene expression from latency. PLoS Pathog 10:e1004195. https://doi.org/10.1371/journal.ppat.1004195
Nelson JA, Reynolds-Kohler C, Smith BA (1987) Negative and positive regulation by a short segment in the 5′-flanking region of the human cytomegalovirus major immediate-early gene. Mol Cell Biol 7:4125–4129
Sinclair J, Sissons P (1996) Latent and persistent infections of monocytes and macrophages. Intervirology 39:293–301. https://doi.org/10.1159/000150501
Sissons JGP, Bain M, Wills MR, Sinclair JH (2002) Latency and reactivation of human cytomegalovirus. J Infect 44:73–77. https://doi.org/10.1053/jinf.2001.0948
Liu R, Baillie J, Sissons JG, Sinclair JH (1994) The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic Acids Res 22:2453–2459
Wright E, Bain M, Teague L, Murphy J, Sinclair J (2005) Ets-2 repressor factor recruits histone deacetylase to silence human cytomegalovirus immediate-early gene expression in non-permissive cells. J Gen Virol 86:535–544. https://doi.org/10.1099/vir.0.80352-0
Bain M, Mendelson M, Sinclair J (2003) Ets-2 Repressor Factor (ERF) mediates repression of the human cytomegalovirus major immediate-early promoter in undifferentiated non-permissive cells. J Gen Virol 84:41–49. https://doi.org/10.1099/vir.0.18633-0
Reeves MB, Compton T (2011) Inhibition of inflammatory interleukin-6 activity via extracellular signal-regulated kinase-mitogen-activated protein kinase signaling antagonizes human cytomegalovirus reactivation from dendritic cells. J Virol 85:12750–12758. https://doi.org/10.1128/JVI.05878-11
Poole E, Sinclair J (2015) Sleepless latency of human cytomegalovirus. Med Microbiol Immunol 204:421–429. https://doi.org/10.1007/s00430-015-0401-6
Shnayder M, Nachshon A, Krishna B, Poole E, Boshkov A, Binyamin A et al (2018) Defining the transcriptional landscape during cytomegalovirus latency with single-cell RNA sequencing. MBio. https://doi.org/10.1128/mBio.00013-18
Cheng S, Caviness K, Buehler J, Smithey M, Nikolich-Žugich J, Goodrum F (2017) Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc Natl Acad Sci USA 114:E10586–E10595. https://doi.org/10.1073/pnas.1710522114
Jenkins C, Abendroth A, Slobedman B (2004) A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J Virol 78:1440–1447
Cheung AKL, Abendroth A, Cunningham AL, Slobedman B (2006) Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood. https://doi.org/10.1182/blood-2005-12-026682
Goodrum F, Reeves M, Sinclair J, High K, Shenk T (2007) Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945. https://doi.org/10.1182/blood-2007-01-070078
Saffert RT, Penkert RR, Kalejta RF (2010) Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34 + cells. J Virol 84:5594–5604. https://doi.org/10.1128/JVI.00348-10
Groves IJ, Sinclair JH (2007) Knockdown of hDaxx in normally non-permissive undifferentiated cells does not permit human cytomegalovirus immediate-early gene expression. J Gen Virol 88:2935–2940. https://doi.org/10.1099/vir.0.83019-0
Wagenknecht N, Reuter N, Scherer M, Reichel A, Müller R, Stamminger T (2015) Contribution of the major ND10 proteins PML, hDaxx and Sp100 to the regulation of human cytomegalovirus latency and lytic replication in the monocytic cell line THP-1. Viruses 7:2884–2907. https://doi.org/10.3390/v7062751
Poole EL, Kew VG, Lau JCH, Murray MJ, Stamminger T, Sinclair JH et al (2018) A virally encoded DeSUMOylase activity is required for cytomegalovirus reactivation from latency. Cell Rep 24:594–606. https://doi.org/10.1016/j.celrep.2018.06.048
Goodrum F (2016) Human cytomegalovirus latency: approaching the Gordian knot. Annu Rev Virol 3:333–357. https://doi.org/10.1146/annurev-virology-110615-042422
Lee SH, Albright ER, Lee J-H, Jacobs D, Kalejta RF (2015) Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci Adv 1:e1501164. https://doi.org/10.1126/sciadv.1501164
Kim JH, Collins-McMillen D, Buehler JC, Goodrum FD, Yurochko AD (2017) Human cytomegalovirus requires epidermal growth factor receptor signaling to enter and initiate the early steps in the establishment of latency in CD34 + human progenitor cells. J Virol. https://doi.org/10.1128/JVI.01206-16
Keyes LR, Hargett D, Soland M, Bego MG, Rossetto CC, Almeida-Porada G et al (2012) HCMV protein LUNA Is required for viral reactivation from latently infected primary CD14 + cells. PLoS One 7:e52827. https://doi.org/10.1371/journal.pone.0052827
Crawford LB, Kim JH, Collins-McMillen D, Lee B-J, Landais I, Held C et al (2018) Human cytomegalovirus encodes a novel FLT3 receptor ligand necessary for hematopoietic cell differentiation and viral reactivation. MBio. https://doi.org/10.1128/mBio.00682-18
Humby MS, O’Connor CM (2016) Human Cytomegalovirus US28 Is Important for Latent Infection of Hematopoietic Progenitor Cells. J Virol 90:2959–2970. https://doi.org/10.1128/JVI.02507-15
Beisser PS, Laurent L, Virelizier J-LL, Michelson S (2001) Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J Virol 75:5949–5957. https://doi.org/10.1128/JVI.75.13.5949-5957.2001
Hargett D, Shenk TE (2010) Experimental human cytomegalovirus latency in CD14 + monocytes. Proc Natl Acad Sci USA 107:20039–20044. https://doi.org/10.1073/pnas.1014509107
Krishna BA, Spiess K, Poole EL, Lau B, Voigt S, Kledal TN et al (2017) Targeting the latent cytomegalovirus reservoir with an antiviral fusion toxin protein. Nat Commun. https://doi.org/10.1038/ncomms14321
Zhu D, Pan C, Sheng J, Liang H, Bian Z, Liu Y et al (2018) Human cytomegalovirus reprogrammes haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency. Nat Microbiol 3:503–513. https://doi.org/10.1038/s41564-018-0131-9
Vomaske J, Nelson JA, Streblow DN (2009) Human Cytomegalovirus US28: a functionally selective chemokine binding receptor. Infect Disord Drug Targets 9:548–556. https://doi.org/10.2174/187152609789105696
Lee S, Chung YH, Lee C (2017) US28, a virally-encoded GPCR as an antiviral target for human cytomegalovirus infection. Biomol Ther 25:69–79. https://doi.org/10.4062/biomolther.2016.208
Casarosa P, Bakker RA, Verzijl D, Navis M, Timmerman H, Leurs R et al (2001) Constitutive signaling of the human cytomegalovirus-encoded chemokine receptor US28. J Biol Chem 276:1133–1137. https://doi.org/10.1074/jbc.M008965200
Minisini R, Tulone C, Lüske A, Michel D, Mertens T, Gierschik P et al (2003) Constitutive inositol phosphate formation in cytomegalovirus-infected human fibroblasts is due to expression of the chemokine receptor homologue pUS28. J Virol 77:4489–4501. https://doi.org/10.1128/jvi.77.8.4489-4501.2003
Krishna BA, Miller WE, O’Connor CM (2018) US28: HCMV’s Swiss Army Knife. Viruses. https://doi.org/10.3390/v10080445
Krishna BA, Poole EL, Jackson SE, Smit MJ, Wills MR, Sinclair JH (2017) Latency-associated expression of human cytomegalovirus US28 attenuates cell signaling pathways to maintain latent infection. MBio 8:e01754–e01717. https://doi.org/10.1128/mBio.01754-17
Krishna BA, Humby MS, Miller WE, O’Connor CM (2019) Human cytomegalovirus G protein-coupled receptor US28 promotes latency by attenuating c-fos. Proc Natl Acad Sci 2019. https://doi.org/10.1073/pnas.1816933116
Casarosa P, Waldhoer M, LiWang PJ, Vischer HF, Kledal T, Timmerman H et al (2005) CC and CX3C chemokines differentially interact with the N terminus of the human cytomegalovirus-encoded US28 receptor. J Biol Chem 280:3275–3285. https://doi.org/10.1074/jbc.M407536200
Miller WE, Zagorski WA, Brenneman JD, Avery D, Miller JLCC, O’Connor CM (2012) US28 is a potent activator of phospholipase C during HCMV infection of clinically relevant target cells. PLoS One 7:e50524. https://doi.org/10.1371/journal.pone.0050524
Waldhoer M, Kledal TN, Farrell H, Schwartz TW (2002) Murine cytomegalovirus (CMV) M33 and human CMV US28 receptors exhibit similar constitutive signaling activities. J Virol 76:8161–8168
Maussang D, Langemeijer E, Fitzsimons CP, Stigter-van Walsum M, Dijkman R, Borg MK et al (2009) The human cytomegalovirus-encoded chemokine receptor US28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res 69:2861–2869. https://doi.org/10.1158/0008-5472.CAN-08-2487
DeYoung KL, Ray ME, Su YA, Anzick SL, Johnstone RW, Trapani JA et al (1997) Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene 15:453–457. https://doi.org/10.1038/sj.onc.1201206
Miles TF, Spiess K, Jude KM, Tsutsumi N, Burg JS, Ingram JR et al (2018) Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. Elife. https://doi.org/10.7554/eLife.35850
Weekes MP, Tan SYL, Poole E, Talbot S, Antrobus R, Smith DL et al (2013) Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 340:199–202. https://doi.org/10.1126/science.1235047
Elder E, Krishna B, Williamson J, Aslam Y, Farahi N, Wood A et al (2019) Monocytes latently infected with human cytomegalovirus evade neutrophil killing. IScience https://doi.org/10.1016/J.ISCI.2019.01.007
Spiess K, Jeppesen MG, Malmgaard-Clausen M, Krzywkowski K, Dulal K, Cheng T et al (2015) Rationally designed chemokine-based toxin targeting the viral G protein-coupled receptor US28 potently inhibits cytomegalovirus infection in vivo. Proc Natl Acad Sci USA 112:8427–8432. https://doi.org/10.1073/pnas.1509392112
Patel M, Vlahava V-M, Forbes SK, Fielding CA, Stanton RJ, Wang ECY. HCMV-Encoded NK (2018) Modulators: lessons from in vitro and in vivo genetic variation. Front Immunol 9:2214. https://doi.org/10.3389/fimmu.2018.02214
Noriega V, Redmann V, Gardner T, Tortorella D (2012) Diverse immune evasion strategies by human cytomegalovirus. Immunol Res 54:140–151. https://doi.org/10.1007/s12026-012-8304-8
Wills MR, Poole E, Lau B, Krishna B, Sinclair JH (2015) The immunology of human cytomegalovirus latency: Could latent infection be cleared by novel immunotherapeutic strategies? Cell Mol Immunol 12:128–138. https://doi.org/10.1038/cmi.2014.75
Khan N, Cobbold M, Keenan R, Moss PAH (2002) Comparative analysis of CD8 + T Cell responses against human cytomegalovirus proteins pp65 and immediate early 1 shows similarities in precursor frequency, oligoclonality, and phenotype. J Infect Dis 185:1025–1034. https://doi.org/10.1086/339963
Reddehase MJ, Koszinowski UH (1984) Significance of herpesvirus immediate early gene expression in cellular immunity to cytomegalovirus infection. Nature 312:369–371. https://doi.org/10.1038/312369a0
Simon CO, Holtappels R, Tervo H-M, Bohm V, Daubner T, Oehrlein-Karpi SA et al (2006) CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J Virol 80:10436–10456. https://doi.org/10.1128/JVI.01248-06
Grzimek NKA, Dreis D, Schmalz S, Reddehase MJ (2001) Random, asynchronous, and asymmetric transcriptional activity of enhancer-flanking major immediate-early genes ie1/3 and ie2 during murine cytomegalovirus latency in the lungs. J Virol 75:2692–2705. https://doi.org/10.1128/JVI.75.6.2692-2705.2001
Kurz SK, Rapp M, Steffens HP, Grzimek NK, Schmalz S, Reddehase MJ (1999) Focal transcriptional activity of murine cytomegalovirus during latency in the lungs. J Virol 73:482–494
Seckert CK, Griessl M, Büttner JK, Scheller S, Simon CO, Kropp KA et al (2012) Viral latency drives “memory inflation”: a unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med Microbiol Immunol 201:551–566. https://doi.org/10.1007/s00430-012-0273-y
Heukers R, Fan TS, de Wit RH, van Senten JR, De Groof TWM, Bebelman MP et al (2018) The constitutive activity of the virally encoded chemokine receptor US28 accelerates glioblastoma growth. Oncogene 37:4110–4121. https://doi.org/10.1038/s41388-018-0255-7
Lückmann M, Amarandi RM, Papargyri N, Jakobsen MH, Christiansen E, Jensen LJ et al (2017) Structure-based discovery of novel US28 small molecule ligands with different modes of action. Chem Biol Drug Des 89:289–296. https://doi.org/10.1111/cbdd.12848
Vischer HF, Hulshof JW, Hulscher S, Fratantoni SA, Verheij MHP, Victorina J et al (2010) Identification of novel allosteric nonpeptidergic inhibitors of the human cytomegalovirus-encoded chemokine receptor US28. Bioorg Med Chem 18:675–688. https://doi.org/10.1016/j.bmc.2009.11.060
Acknowledgements
We thank past and present members of our research group as well as Linda Teague and Roy Whiston for technical support. This research was funded by the Wellcome Trust (Grant 109075/Z/15/A) and MRC (Grant G0701279) and supported by the Cambridge NIHR BRC cell phenotyping hub.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Special Issue on Immunological Imprinting during Chronic Viral Infection.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Elder, E., Sinclair, J. HCMV latency: what regulates the regulators?. Med Microbiol Immunol 208, 431–438 (2019). https://doi.org/10.1007/s00430-019-00581-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-019-00581-1