
Abstract
Pancreatic cancer is highly resistant to the currently available chemotherapeutic agents. Less than 5% of patients diagnosed with this disease could survive beyond 5 years. Thus, there is an urgent need for the development of novel, efficacious drugs that can treat pancreatic cancer. Herein we report the identification of artesunate (ART), a derivative of artemisinin, as a potent and selective antitumor agent against human pancreatic cancer cells in vitro and in vivo. ART exhibits selective cytotoxic activity against Panc-1, BxPC-3 and CFPAC-1 pancreatic cancer cells with IC50 values that are 2.3- to 24-fold less than that of the normal human hepatic cells (HL-7702). The pan caspase inhibitor zVAD-fmk did not inhibit the cytotoxic activity of ART. Electron microscopy of ART-treated cells revealed severe cytoplasmic swelling and vacuolization, swollen and internally disorganized mitochondria, dilation (but not fragmentation) of the nuclei without chromatin condensation, and cell lysis, yielding a morphotype that is typical of oncosis. The ART-treated cells exhibited a loss of mitochondrial membrane potential (ΔΨm) and ART-induced cell death was inhibited in the presence of the reactive oxygen species (ROS) scavenger N-acetyl-cysteine (NAC). Importantly, ART produced a dose-dependent tumor regression in an in vivo pancreatic cancer xenografts model. The in vivo antitumor activity of ART was similar to that of gemcitabine. Taken together, our study suggests that ART exhibits antitumor activity against human pancreatic cancer via a novel form of oncosis-like cell death, and that ART should be considered a potential therapeutic candidate for treating pancreatic cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer is one of the most malignant tumors with a very poor prognosis. Its high mortality can be attributed to late diagnosis and poor response to chemo- and radiation therapies [1]. The 5-year survival rate is <5%, and only approximately one in five of the 10% of patients with resectable disease survive 5 years [2]. Systemic chemotherapy remains the principle treatment option for patients with advanced pancreatic cancer. However, pancreatic cancer is extraordinarily resistant to genotoxic therapy [3]. Currently, gemcitabine-based therapy is the standard treatment for unresectable, locally advanced/metastatic pancreatic cancer, but yields a median survival duration of only 6 months [4]. Hence, there is an urgent need to develop effective chemotherapeutic agents for pancreatic cancer.
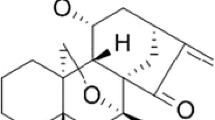
Artesunate (ART) is a derivative of artemisinin (Fig. 1A), the principle active component of the Chinese herb Artemisia annua L. Artesunate, which is approved for the treatment of multidrug-resistant malaria and has an excellent safety profile [5]. In addition to its anti-malarial activity, ART has been shown to have cytotoxic effects on a number of human cancer cell types, including leukemia, colon cancer, and melanoma cells [6]. Moreover, ART has recently been shown to possess cytotoxicity against hepatoma cells and ovarian cancer cells in vitro and in vivo [7, 8]. Mechanisms that might explain the antitumor activity of ART include induction of apoptosis of T leukemia cells [9], anti-proliferative or anti-angiogenic effects in xenografted Kaposi’s sarcoma in mice [10], inhibition of growth of C6 glioma cells by increasing levels of reactive oxygen species [11] or DNA damage [12]. However, little is known about the effects of ART on pancreatic cancers. Therefore, it is worth determining whether the known cytotoxic activity of ART extends to pancreatic cancer.

Cytotoxic effects of ART on human pancreatic cancer cells. A Chemical structure of ART. B Panc-1, BxPC-3, CFPAC-1, and normal (HL-7702) hepatic cells were treated with the indicated concentrations of ART for 48 h. Mean values for three independent cell viability MTT assays are plotted. C Phase-contrast micrographs of Panc-1 cells treated with vehicle (a, control), 50 μmol/l ART for 24 h (b), 50 μmol/l ART for 48 h (c) or 50 μmol/l cisplatin for 48 h (d) are shown. All images captured at 100× magnification
Here we first evaluated the therapeutic potential of ART against pancreatic cancer cells in vitro and in vivo, and then proceeded to examine the underlying mechanism involved in ART-mediated cell death. Flow cytometry and electron microscopy were employed to characterize ART’s mechanism of action related to mitochondrial dysfunction and reactive oxygen species (ROS) generation. In particular, we examined whether ART-treated cancer cells exhibit a profile consistent with an oncosis-like (non-apoptotic) mechanism.
Materials and methods
Cell culture and reagents
Panc-1 (ATCC No. CRL-1469), BxPC-3 (ATCC No. CRL-1687) and CFPAC-1 (ATCC No. CRL-1918) human pancreatic cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA). Normal human hepatic cells HL-7702 (GNHu 6) were obtained from the Cell Collection of Chinese Academy of Science (Shanghai, China). Panc-1 and BxPC-3 cell lines were maintained in RMPI1640, the CFPAC-1 cell line was grown in IMDM, and HL-7702 cells were cultured in DMEM. All media were supplemented with 10% fetal bovine serum (GIBCO, Invitrogen Corp., Carlsbad, CA), 100 U/ml penicillin and 100 μg/ml streptomycin and maintained at 37°C in the presence of 5% CO2.
ART (Guilin Pharmaceutical Co. Ltd, Guangxi, China) were freshly prepared in 5% sodium bicarbonate solution and diluted in cell cultures to the indicated final concentrations. Cisplatin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Hoechst 33258, 6-carboxy-2′,7′-dichlorofluorescein diacetate (DCFH-DA), the antioxidant N-acetyl-cysteine (NAC), and the pan caspase inhibitor zVAD-fmk were purchased from Sigma Chemical Co. (St. Louis, MO). Gemcitabine was purchased from Lilly France S.A.S (Fegersheim, France).
Cytotoxicity assay
Cells were plated in triplicate in 96-well plates (Falcon, BD, Franklin Lakes, NJ) at 104 cells per well. Twenty-four hours later, the cells were treated with ART for 48 h at the concentrations indicated. Cell viability was measured using an MTT assay. Cell viability numbers were determined by calculating the average counts from three wells and the experiment was repeated in triplicate.
Determination of apoptosis
Cells were plated in triplicate and treated with ART or cisplatin for the indicated periods of time. Apoptotic cell death was examined by two parameters: cell and nuclear morphology by fluorescence microscopy (Axiovert-200M microscope, Zeiss) after intravital staining with 5 μg/ml Hoechst 33258 and DNA fragmentation analysis by flow cytometry according to Nicoletti’s method [13]. Briefly, cells were resuspended in a buffer containing 0.1% (w/v) sodium citrate, 0.1% (v/v) Triton X-100, and 50 μg/ml propidium iodide (Sigma, St. Louis, MO). After incubation at 4°C in the dark for at least 16 h, apoptotic nuclei were quantified by a flow cytometer (Epics XL, Beckman Coulter).
Electron microscopy
Cells were fixed with 3% glutaraldehyde in 0.1 M cacodylate buffer for 15 min at room temperature. The cells were scraped, transferred to an Eppendorf tube, and centrifuged at 400×g for 5 min. The pellets were washed with 0.1 M cacodylate buffer, postfixed in 2% osmium tetroxide, dehydrated in acetone, and embedded in araldite. Ultra-thin sections stained with uranyl acetate and lead citrate were examined using a transmission electron microscope (CM-10, Philips).
Caspase inhibition assay
To determine whether caspase is involved in the effects of ART, a caspase inhibition assay was carried out with the pan-caspase inhibitor zVAD-fmk dissolved in DMSO, according to the manufacturer’s instructions. Briefly, Panc-1 cells (3 × 105 cells per well) were seeded in a 60-mm dish and incubated overnight. zVAD-fmk (40 μmol/l) was then added to the cells, and 2 h later, the ART treatment was administered. After 24 h, cell viability was determined by MTT assay as described above. The viability of untreated cells in the presence of diluted DMSO was regarded as 100%.
Mitochondrial membrane potential (ΔΨm) assays
Mitochondria were selectively probed with potential-sensitive JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine iodide; Molecular Probes, Invitrogen Corp., Carlsbad, CA). Cells were harvested after a 24-h exposure to ART and centrifuged at 400×g for 5 min, and the cell pellet was resuspended in 0.5 ml of JC-1 solution (10 μg/ml) for 10 min. The cells were then washed and resuspended in PBS for flow cytometry analysis. JC-1 exhibits ΔΨm-dependent accumulation in mitochondria, indicated by a shift in its fluorescence emission from green to red as J-aggregates form. Mitochondrial depolarization is thus indicated by a decrease in the red/green fluorescence ratio [14].
Measurement of reactive oxygen species
Production of ROS was determined using DCFH-DA and flow cytometry analysis as described previously [15]. This cell-permeating, non-fluorescent probe is oxidized by ROS and converted into a fluorescent product, 2′,7′-dichlorofluorescein, which can be measured by flow cytometry. Panc-1 cells (3 × 105 cells per well) were seeded into 60-mm plates and incubated overnight. The cells were incubated for 1 h in DCFH-DA (10 μmol/l) in FCS-free medium. The probe was then removed and the cells were rinsed with PBS. Rinsed cells were then incubated for 5 h in fresh medium with ART at the final indicated concentrations. Finally, the cells were trypsinized, resuspended in PBS, and subjected to flow cytometry analysis.
In vivo xenograft studies
Female BALB/c athymic nude mice (4–6 weeks old) were purchased from the Shanghai Laboratory Animal Center of the Chinese Academy Sciences. Panc-1 cells were grown, harvested, washed with PBS, and resuspended in DMEM. Mice were injected subcutaneously (s.c.) into each posterior flank region with approximately 6.0 × 106 cells. Xenografts were allowed to grow, and treatment was started when the injected cell mass reached a mean volume of 130 mm3. After tumor formation, the mice were randomized into five groups (N = 5 per group), and administered ART (25, 50, or 100 mg/kg/day, i.p.), saline only in the case of the untreated control group, or gemcitabine (100 mg/kg/day i.p. every 3 days) in the case of the chemotherapy control group. Tumor growth was monitored twice weekly. The length (l) and the width (w) of the tumors were measured using a slide caliper and the volume (V) of each tumor was calculated as follows: V = l × w 2/2. The mice were sacrificed and the tumors were excised after 20 days of treatment.
Statistical analysis
All quantitative assays were performed in triplicate. The results are expressed as means ± SD. Pairs of datasets were compared using Student’s t test. Significant differences were considered to exist for those probabilities below 5% (P < 0.05).
Results
ART demonstrates cytotoxic activity against human pancreatic cancer cells
The dose-dependent cell proliferation curves showed that ART exhibited cytotoxicity in all pancreatic cell lines examined (Panc-1, BxPC-3, and CFPAC-1) in a micromolar dose range. The IC50 values for Panc-1, BxPC-3 and CFPAC-1 cells were 26.76, 279.3, and 142.8 μmol/l, respectively (Fig. 1B). Importantly, ART was 2.3- to 24-fold less cytotoxic to normal human hepatic cells (HL-7702) (IC50 = 643.3 μmol/l) than to the cancer cells, suggesting that ART induces selective cytotoxicity in human pancreatic cancer cells.
Under the phase-contrast microscope, pancreatic cancer cells showed a dramatic morphological change 24 h after treatment with 50 μmol/l ART. Specifically, the ART-treated cells had intense swelling and vacuolization, which culminated in cell lysis by 48 h after the treatment. No morphological characteristics of apoptosis were observed in the ART-treated cells. On the contrary, cells exposed to cisplatin (50 μmol/l) for 48 h did exhibit morphological features of apoptosis, such as cell membrane blebbing, cell shrinkage, increased cytoplasm granules, and detachment from the plate (Fig. 1C).
ART induces caspase-independent and non-apoptotic cell death in Panc-1 cells
Hoechst 33258 staining of Panc-1 cells treated with 50 μmol/l ART for 48 h demonstrated that the ART treatment did not induce biochemical features of apoptosis such as nuclear condensation and chromatin fragmentation. In contrast, cells treated with cisplatin showed chromatin fragmentation, a familiar characteristic of apoptosis (Fig. 2a). Consistent with these morphological observations, the propidium iodide-based flow cytometric DNA fragmentation assay indicated that ART-treated cells did not produce a hypodiploid peak (a hallmark of apoptosis), though a hypodiploid peak was observed for the cisplatin-treated cells (Fig. 2b). Furthermore, pretreatment (2 h) of ART-treated Panc-1 cells with the general caspase inhibitor z-VAD-fmk did not inhibit ART’s cytotoxic activity (Fig. 2c), suggesting that ART induced caspase-independent cell death in Panc-1 cells.

Characterization of ART-induced cell death. a Hoechst 33258 fluorescent staining revealed nuclear and chromatin modifications during ART induced cell death (200× magnification). Panc-1 cells were treated with vehicle (control), 50 μmol/l ART, or 50 μmol/l cisplatin for 48 h. b Propidium iodide-based flow cytometric DNA fragmentation assay data indicated that ART (50 μmol/l at indicated times) does not cause DNA fragmentation in Panc-1 cells. c MTT cell viability assay data indicated that ART-induced cell death is not blocked by inhibition of caspase activity with the pan-caspase inhibitor z-VAD-fmk (40 μmol/l, started 2 h before ART treatment for a period of 48 h). Parallel cultures treated with cisplatin (50 μmol/l for 48 h) were used as controls. Values correspond to means ± SD of triplicates obtained in three independent experiments
ART-induced cell death has ultrastructural features of oncosis
Electron microscopy confirmed the control Panc-1 cells have intact cellular morphology with ultrastructurally normal nuclei and organelles (Fig. 3a). In contrast, Panc-1 cells treated with 50 μmol/l ART for 48 h exhibited cytoplasmic swelling with a dramatic disruption of cytoarchitecture, including numerous small and large cytoplasmic vacuoles as well as swollen and internally disorganized mitochondria. The most prominent nuclear changes were dilation of the nuclei, irregular clumping of chromatin, and the appearance of cleared chromatin-free nuclear domains (Fig. 3b). High magnification electron micrographs of ART-treated Panc-1 cells revealed severely damaged mitochondria with decreased matrix density and swelling of cristae, but no evidence of autophagy (Fig. 3c). The swollen cytoplasm contained remnants of vesiculated organelles and some ruptures of the plasma membrane could be observed up to the final stages of cell death (Fig. 3d). This cytoplasmic degradation was not accompanied by an overall chromatin condensation. These morphologic features are consistent with a type of cell death termed oncosis [16–19].

ART-mediated cell death features resemble ultrastructural features of oncosis-like cell death. a Control cells showing intact ultrastructurally normal nuclei (N) and cellular organelles, and an absence of cytoplasmic vacuolation. b Low-magnification survey electron micrograph illustrating numerous large and small cytoplasmic vacuoles, dilation of nuclei with cleared nuclear domains free of chromatin in 50 μmol/l ART-treated cells. c High-magnification electron micrograph illustrating swollen mitochondria (M) with disintegrated cristae and decreased matrix density. d Severe dilation of nuclei, swollen mitochondria, and ruptured plasma membrane late in the process of ART-treated cell death. Magnifications: 1800× (a, b and d); 5000× (c)
ART induced change in ΔΨm and ROS-mediated cell death in Panc-1 cells
Mitochondrial ΔΨm was measured in cells stained with the cationic dye JC-1. As shown in Fig. 4a, Panc-1 cells subjected to the 50 μmol/l ART treatment for 6 h exhibited an increase in green and a decrease in red JC-1 fluorescence, indicating a loss of ΔΨm. A marked increase in the number of cells with decreased red fluorescence indicative of mitochondrial depolarization was found 24 h after ART treatment (Fig. 4a).

ART-induced cell death involves loss of mitochondrial membrane potential and generation of ROS. a Flow cytometry analysis of JC-1-stained Panc-1 cells incubated with 50 μmol/l ART for 6, 12, or 24 h. Mitochondrial depolarization is reflected by a red to green shift in fluorescence emission. Data from one representative experiment out of three are shown. b DCFH-DA assay and flow cytometry analysis revealed a dose-dependent increase in ROS production in response to 50 μmol/l (dark line) or 100 μmol/l (light line) ART treatment for 5 h. The shaded histograms were generated from untreated control data (left). Pretreatment of the cells with the antioxidant NAC (15 mM) for 30 min markedly inhibited ROS production (right). c MTT assays showed that a 48-h incubation with the antioxidant NAC (15 mM) inhibited ART cytotoxicity of Panc-1 cells. Data are shown as means ± SD of three independent experiments
The DCFH-DA assay for ROS generation revealed 1.6-fold and 4.49-fold increases in fluorescence in cells treated with 50 and 200 μmol/l ART, respectively; these findings demonstrate a dose-dependent increase in ROS production in response to ART treatment (Fig. 4b, left). Pretreatment of Panc-1 cells for 30 min with the ROS scavenger NAC after 5 h of exposure to ART almost completely inhibited ROS generation (Fig. 4b, right) and markedly reduced ART’s cytotoxic effect (Fig. 4c), suggesting ROS generation could mediate ART-induced cell death.
In vivo antitumor activity of ART
As shown in Fig. 5a, ART induced a dose-dependent inhibition of Panc-1 tumor growth. Daily administration of 25, 50 and 100 mg/kg ART (i.p.) in established Panc-1 xenografts mode (average tumor volume = 130 mm3) suppressed tumor growth by 33, 44, and 65%, respectively (P < 0.01 versus saline controls), at the end of the study (day 20). Gemcitabine treatment (100 mg/kg i.p., twice weekly) reduced tumor growth on day 20 by 70%; this result did not differ significantly from the result seen with 100 mg/kg ART (Fig. 5a).

Evaluation of therapeutic use of ART against pancreatic cancer tumor xenografts in nude mice. a ART (25, 50 or 100 mg/kg), gemcitabine (100 mg/kg), or saline (N = 5 per group) was administered i.p. when Panc-1 cell tumors (s.c. in the flank) reached an average volume of 130 mm3. Semi-weekly assessment of tumor volume demonstrated that administration of ART suppressed Panc-1 xenograft tumor growth. b The body weights of the ART-treated mice were similar to that of saline controls, while gemcitabine treatment reduced body weight
The body weights of ART-treated and control mice did not differ from each other. ART at the highest dose tested (100 mg/kg) was well tolerated and did not cause any observable toxicity. In comparison, gemcitabine treatment was associated with a 25% reduction in body weight (Fig. 5b). In addition, mice treated with gemcitabine were weak and inactive, and two of the gemcitabine-treated mice died on day 18.
Discussion
The present study demonstrated that micromolar doses of ART can induce selective cytotoxic activity against human pancreatic cancer cells. Electron microscopy observations revealed that ART initiates a death process characterized by marked swelling and a series of profound morphological alterations, including numerous small and large cytoplasmic vacuoles, swollen and internally disorganized mitochondria, dilation of the nuclei without chromatin condensation, and ultimately cell lysis. These morphologic observations in the absence of features characteristic of apoptosis, together with the finding that caspase inhibition did not affect ART-induced cell death, suggest that ART induces a non-apoptosis type of cell death, just like oncosis [16–19]. ART induced cell death was associated with a loss of ΔΨm, but could be blocked by inhibition of ROS generation, suggesting that the mechanism of pancreatic cancer cell death caused by ART may involve mitochondrial dysfunction and generation of ROS. Furthermore, ART produced a dose-dependent tumor regression in an in vivo pancreatic cancer xenograft model. The in vivo antitumor activity of ART was similar to that of gemcitabine. Moreover, ART has been approved by the State Food and Drug Administration of China for the treatment of multidrug-resistant malaria and has an excellent safety profile. Thus, our data suggest that, relative to established drugs, ART could be a safer and more efficacious anticancer agent for treatment of human pancreatic cancer.
The term oncosis was adapted by Majno and Joris [18] to describe a mechanism of cell death distinct from the more extensively studied apoptosis. While apoptosis is characterized by cell shrinkage, chromatin condensation, and formation of apoptotic bodies, oncosis is characterized by rapid cell swelling, organelle swelling, membrane permeability, and cell lysis [18, 19]. Cell death by apoptosis leads to necrosis with karyorrhexis, whereas cell death by oncosis may result in necrosis with karyolysis [17, 18].
It is often assumed that chemotherapy drugs induce cell death through apoptotic mechanisms [20]. Indeed, studies have suggested that ART can trigger apoptotic cell death in various tumor cell lines [7–9, 21, 22], but the effect of ART on pancreatic cancer is still unknown. The present data for the first time indicate that ART induces a unique type of cell death in pancreatic cancer cells via an atypical non-apoptotic pathway. Numerous factors including both a distinct morphological profile (cell swelling instead of shrinking) and biochemical findings support our view that ART induces oncosis-like cell death rather than apoptosis. First, ART did not induce nuclear condensation and chromatin fragmentation. Second, ART-treated cells did not exhibit the hypodiploid peak that is a hallmark of apoptotic cells subjected to the iodide-based flow cytometric DNA fragmentation assay. Third, the pan caspase inhibitor zVAD-fmk did not inhibit the cytotoxic activity of ART.
Although ART was demonstrated to have cytotoxic effects on tumor cells a decade ago [23], the molecular mechanisms by which ART exerts its anti-tumor activity have yet to be elucidated clearly. Given the severe destruction of cell organelles, including mitochondria, observed by electron microscopy, it is reasonable to consider whether ART cytotoxicity involves an alteration of mitochondrial function. On the other hand, ART is a sesquiterpene lactone that exerts its anti-malarial activity by generation of organic free radicals through cleavage of the endoperoxide bridge within its active moiety [24, 25]. The radical molecules can cause macromolecular damage by alkylating and poisoning essential malarial proteins [25]. Our data indicate that ART causes a loss of ΔΨm and induces cell death by generation of ROS. This conclusion is supported by the finding that the ROS scavenger NAC blocked ROS generation and, consequently, inhibited ART-induced cell death.
While apoptosis is increasingly well defined at the molecular level, the biochemical determinants of necrotic/oncotic cell death remain less well defined. Recent research suggests that oncosis is associated with intracellular events such as enhanced generation of ROS, swelling of mitochondria, ATP depletion, failure of Ca2+ homeostasis, activation of some proteases (e.g., calpains and cathepsins), lysosomal rupture, and ultimately plasma membrane rupture [26]. Although each of these events alone is not specifically responsible for oncosis, it is possible that a collection of these events in an organized, programmed cascade of self-destruction defines oncosis. Apoptosis and oncosis have some features in common, such as the mitochondrial permeability transition and loss of ΔΨm; the ultimate fate of cells is mostly due to the intensity and duration of the death signal and the cell’s genetic and metabolic status [27]. Cells in an aerobic environment are constantly generating ROS. While physiologic levels of ROS can serve as signaling molecules to regulate transcription, excessive production of ROS leads to oxidative stress, damage of intracellular molecules and organelles, and ultimately necrosis [28]. Accordingly, the triggering of ROS mediated oncosis-like cell death induced by ART may become an alternative approach to eradicate tumor cells, particularly tumor cells with defects in apoptosis pathway.
Recently, Youns et al. [29] showed that ART inhibited growth and induced apoptosis of human pancreatic cancer cells and the effect was more prominent with the poorly differentiated MiaPaCa-2 cells. Also, ART was identified as a novel topoisomerase II alpha inhibitor that inhibits pancreatic cancer cell growth through modulation of multiple signaling pathways. It is likely that different types of pancreatic cancer cells have particular molecular actions peculiar to their cell death process. Further studies examining the biochemical and molecular mechanisms of ART-induced cell death should provide a deeper understanding of the nature and characteristics of the cytotoxic effect of ART.
In summary, the present results show that ART exhibits potent antitumor activity against human pancreatic cancer cells in vitro and tumor xenografts in vivo. It displays a distinctive mode of action in which it induces ROS-mediated oncosis-like cell death in pancreatic cancer cells. The findings in this study may underline the potential utility of induction of ROS-mediated oncosis as a novel therapeutic strategy for the treatment of cancer. More studies are needed to fully characterize the anti-tumor action of ART, including the identification of its primary target(s) and the basis for its preferential effect on tumor cells.
References
Kalthoff H, Westphal S (2003) Apoptosis: targets in pancreatic cancer. Mol Cancer 2:6
Eckel F, Schneider G, Schmid RM (2006) Pancreatic cancer: a review of recent advances. Expert Opin Investig Drugs 15:1395–1410
Schneider G, Siveke JT, Eckel F et al (2005) Pancreatic cancer: basic and clinical aspects. Gastroenterology 128:1606–1625
Rocha-Lima CM (2008) New directions in the management of advanced pancreatic cancer: a review. Anticancer Drugs 19:435–446
Nosten F, Price RN (1995) New antimalarials. A risk-benefit analysis. Drug Saf 12:264–273
Efferth T, Dunstan H, Sauerbrey A et al (2001) The anti-malarial artesunate is also active against cancer. Int J Oncol 18:767–773
Hou J, Wang D, Zhang R et al (2008) Experimental therapy of hepatoma with artemisinin and its derivatives: in vitro and in vivo activity, chemosensitization, and mechanisms of action. Clin Cancer Res 14(17):5519–5530
Chen T, Li M, Zhang R, et al (2008) Dihydroartemisinin induces apoptosis and sensitizes human ovarian cancer cells to carboplatin therapy. J Cell Mol Med, May 2. Epub ahead of print
Efferth T, Giaisi M, Merling A et al (2007) Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS ONE 2:e693
Dell’Eva R, Pfeffer U, Vene R et al (2004) Inhibition of angiogenesis in vivo and growth of Kaposi’s sarcoma xenograft tumors by the anti-malarial artesunate. Biochem Pharmacol 68:2359–2366
Huang XJ, Ma ZQ, Zhang WP et al (2007) Dihydroartemisinin exerts cytotoxic effects and inhibits hypoxia inducible factor-1alpha activation in C6 glioma cells. J Pharm Pharmacol 59:849–856
Li PC, Lam E, Roos WP et al (2008) Artesunate derived from traditional Chinese medicine induces DNA damage and repair. Cancer Res 68:4347–4351
Nicoletti I, Migliorati G, Pagliacci MC et al (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 139:271–279
Cichorek M, Kozłowska K, Bryl E (2007) Mitochondrial transmembrane potential in spontaneous and camptothecin-induced apoptosis of melanotic and amelanotic melanoma cells. Neoplasma 54:29–36
Gao F, Yi J, Yuan JQ et al (2004) The cell cycle related apoptotic susceptibility to arsenic trioxide is associated with the level of reactive oxygen species. Cell Res 14:81–85
Zong WX, Thompson CB (2006) Necrotic death as a cell fate. Genes Dev 20:1–15
Trump BF, Berezesky IK, Chang SH, Phelps PC (1997) The pathways of cell death: oncosis, apoptosis, and necrosis. Toxicol Pathol 25:82–88
Majno G, Joris I (1995) Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146:3–15
Van Cruchten S, Van den Broeck W (2002) Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat Histol Embryol 31:214–223
Kaufmann SH, Earnshaw WC (2000) Induction of apoptosis by cancer chemotherapy. Exp Cell Res 256:42–49
Efferth T, Sauerbrey A, Olbrich A et al (2003) Molecular modes of action of artesunate in tumor cell lines. Mol Pharmacol 64:382–394
Efferth T, Briehl MM, Tome ME (2003) Role of antioxidant genes for the activity of artesunate against tumor cells. Int J Oncol 23:1231–1235
Efferth T, Rücker G, Falkenberg M et al (1996) Detection of apoptosis in KG-1a leukemic cells treated with investigational drugs. Arzneimittelforschung 46:196–200
Meshnick SR (2002) Artemisinin: mechanisms of action, resistance and toxicity. Int J Parasitol 32:1655–1660
Meshnick SR, Taylor TE, Kamchonwongpaisan S (1996) Artemisinin and the antimalarial endoperoxides: from herbal remedy to targeted chemotherapy. Microbiol Rev 60:301–315
Golstein P, Kroemer G (2007) Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 32:37–43
Lemasters JJ, Nieminen AL, Qian T et al (1998) The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1366:177–196
Ricci MS, Zong WX (2006) Chemotherapeutic approaches for targeting cell death pathways. Oncologist 11:342–357
Youns M, Efferth T, Reichling J et al (2009) Gene expression profiling identifies novel key players involved in the cytotoxic effect of Artesunate on pancreatic cancer cells. Biochem Pharmacol 78:273–283
Acknowledgments
This work was supported by grants from the Science & Technology Foundation of Guangdong Province (No. 2006B35501009) and the Science & Technology Research Grants of Shenzhen (No. 200702114). The authors thank Dr. Ann Power Smith of Write Science Right for editorial revision of this manuscript.
Conflicts of interest
We declare that we have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Du, JH., Zhang, HD., Ma, ZJ. et al. Artesunate induces oncosis-like cell death in vitro and has antitumor activity against pancreatic cancer xenografts in vivo. Cancer Chemother Pharmacol 65, 895–902 (2010). https://doi.org/10.1007/s00280-009-1095-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-009-1095-5