
Abstract
Aims/hypothesis
Although the substitution of saturated fatty acids with oleate has been recommended in the management of type 2 diabetes mellitus, the mechanisms by which oleate improves insulin resistance in skeletal muscle cells are not completely known. Here, we examined whether oleate, through activation of AMP-activated protein kinase (AMPK), prevented palmitate-induced endoplasmic reticulum (ER) stress, which is involved in the link between lipid-induced inflammation and insulin resistance.
Methods
Studies were conducted in mouse C2C12 myotubes and in the human myogenic cell line LHCN-M2. To analyse the involvement of AMPK, activators and inhibitors of this kinase and overexpression of a dominant negative AMPK construct (K45R) were used.
Results
Palmitate increased the levels of ER stress markers, whereas oleate did not. In palmitate-exposed cells incubated with a lower concentration of oleate, the effects of palmitate were prevented. The induction of ER stress markers by palmitate was prevented by the presence of the AMPK activators AICAR and A-769662. Moreover, the ability of oleate to prevent palmitate-induced ER stress and inflammation (nuclear factor-kappa B [NF-κB] DNA-binding activity and expression and secretion of IL6) as well as insulin-stimulated Akt phosphorylation and 2-deoxyglucose uptake was reversed in the presence of the AMPK inhibitor compound C or by overexpression of a dominant negative AMPK construct. Finally, palmitate reduced phospho-AMPK levels, whereas this was not observed in oleate-exposed cells or in palmitate-exposed cells supplemented with oleate.
Conclusions/interpretation
Overall, these findings indicate that oleate prevents ER stress, inflammation and insulin resistance in palmitate-exposed skeletal muscle cells by activating AMPK.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It has long been recognised that elevated plasma NEFA cause insulin resistance in humans [1]. However, saturated and monounsaturated NEFA differ significantly in their contribution to insulin resistance [2]. It is generally accepted that saturated NEFA induce insulin resistance [2, 3], whereas monounsaturated NEFA increase insulin sensitivity in diabetic patients [4, 5] and healthy individuals [2]. The mechanisms underlying the association between elevated NEFA and insulin resistance are currently unclear but accumulating evidence points to a link between enhanced NEFA levels and activation of a chronic low-level inflammatory process [6]. Elevated saturated NEFA can induce inflammation and, thus, insulin resistance, through several mechanisms including diacylglycerol (DAG)-mediated activation of protein kinase Cθ (PKCθ) [7] and activation of Toll-like receptors [8]. Both mechanisms lead to activation of the pro-inflammatory transcription factor nuclear factor-kappaB (NF-κB), which has been linked to fatty acid-induced impairment of insulin action in skeletal muscle [9]. Once activated, NF-κB regulates the expression of multiple inflammatory mediators, including IL-6. This cytokine correlates strongly with insulin resistance and type 2 diabetes [10] and its plasma levels are increased two- to threefold in patients with obesity and type 2 diabetes compared with lean control individuals [10].
Recently, endoplasmic reticulum (ER) stress has become a new potential mechanism involved in the association between saturated NEFA-induced inflammation and insulin resistance [11, 12], and it is now well accepted that limitation of the former will affect the latter [12]. In fact, patients with type 2 diabetes [13] and diet-induced and genetic ob/ob obese mice [14] have elevated levels of key ER stress markers, and elevated levels of NEFA have been proposed to induce insulin resistance by causing ER stress [15].
In conjunction with its central role in lipid synthesis, protein folding and transportation, the ER serves as a major signal transduction organelle that integrates cellular responses to stress. The accumulation of misfolded proteins and other stresses lead to the activation of an adaptive programme by the ER, known as the unfolded protein response (UPR), to re-establish equilibrium [11]. Initiation of the canonical UPR involves activation of three key signalling proteins: inositol-requiring 1 transmembrane kinase/endonuclease-1 (IRE-1α), activating transcription factor 6 (ATF6), and eukaryotic translation initiation factor-2α kinase 3 (PERK). The endoribonuclease activity of IRE-1α cleaves a 26-base-pair segment from XBP1 mRNA, creating an alternative message that is translated into the active (or spliced) form of the transcription factor X-box binding protein 1 (XBP1) (sXBP1). ATF6 translocates to the nucleus in which it acts as a transcription factor and PERK phosphorylates and inhibits an essential initiator of translation, eukaryotic initiation factor 2 (eIF2). Together these pathways work to decrease translation and increase protein folding [16]. The three branches of the canonical UPR intersect with a variety of inflammatory and stress signalling systems, including the NF-κB pathway [11]. Thus, phosphorylation of eIF2α by PERK results in a general repression of mRNA translation. Since inhibitor of κB (IκB), which inhibits NF-κB, has a shorter half-life than NF-κB, UPR activation shifts the ratio of IκB to NF-κB, thereby releasing NF-κB, which translocates to the nucleus and increases the expression of its target genes, such as IL6 [12]. In addition, in response to ER stress, the cytoplasmatic domain of phosphorylated IRE1α can recruit TNF-α-receptor-associated factor 2 (TRAF2), forming a complex that interacts and activates IκB kinase (IKK), leading to NF-κB activation [12]. Of note, activation of AMP-activated protein kinase (AMPK) exhibits multiple protective effects, including inhibition of inflammation, oxidative stress and insulin resistance and reduces the risk for developing obesity and type 2 diabetes [17]. Recently it has been reported that AMPK activation protects against hypoxic injury [18], atherosclerosis [19, 20] and lipid-induced hepatic disorders [21] by reducing ER stress.
Saturated NEFA-induced insulin resistance affects mainly skeletal muscle since this tissue accounts for most insulin-stimulated glucose use [22]. The present study was designed to investigate, in skeletal muscle cells, the cellular mechanisms by which the two most common fatty acids, the saturated fatty acid palmitate and the monounsaturated oleate [23], exert their differential effects on ER stress and whether AMPK activation contributes to their differences in fatty acid-induced inflammation and impairment of insulin signalling. In addition, we aimed to discover whether oleate prevents the deleterious effects of palmitate. Our findings demonstrate that oleate prevents palmitate-induced ER stress in mouse and in human skeletal muscle cells through an AMPK-dependent mechanism, and contributes to the explanation of how the monounsaturated fatty acid prevents palmitate-induced inflammation and insulin resistance.
Methods
Cell culture and transfection studies
Mouse C2C12 myoblasts (ATCC) were maintained and cultured as previously described [7].
For in vitro overexpression studies, cells were transfected with Lipofectamine 2000 in OPTI-MEM reduced-serum medium, following the manufacturer’s recommendations (Invitrogen, Carlsbad, CA, USA). The constructs used were pcDNA3/pAMPKα2 K45R (Addgene plasmid 15992, Cambridge, MA, USA) and the corresponding LacZ-carrying plasmid as a control [24]. Transfection time and the DNA-to-Lipofectamine ratio for overexpression studies were set after optimisation with the corresponding LacZ-carrying plasmid and using a β-galactosidase reporter gene staining kit (Sigma-Aldrich Química, Madrid, Spain).
The human myogenic cell line LHCN-M2 [25] was grown as previously reported [26].
RNA preparation and quantitative RT-PCR
The relative levels of specific mRNAs were assessed by real-time RT-PCR, as previously described [27] (see electronic supplementary material [ESM] Table 1 for primers used). XBP1 splicing (sXBP1) was examined by gel electrophoresis as previously reported [28].
2-Deoxy-d-[14C]glucose uptake experiments
Determination of 2-deoxy-d-[14C]glucose (2-DG) uptake was performed as reported elsewhere [7].
Immunoblotting
Total proteins (30 μg) were separated by SDS-PAGE on 10% separation gels and transferred to Immobilon polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). For IRE-1α a phos-tag gel was used as previously described [29]. Western blot analysis was performed using antibodies against total (catalogue number 9272) and phospho-Akt (Ser473) (9271), total (2532) and phospho-AMPK (Thr172) (2531), total (3662) and phospho-ACC (Ser79) (3661), BiP/GRP78 (3183), IRE-1α (3294) (Cell Signaling Technology, Danvers, MA, USA), PP2A catalytic subunit (52F8) (sc-6110), IκBα (sc-371), liver kinase B1 (LKB1) (sc-5638) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and actin (A5441) (Sigma-Aldrich Química). Detection was achieved using the EZ-ECL chemiluminescence detection kit (Biological Industries, Beit Haemek, Israel). The equal loading of proteins was assessed by Ponceau S staining. The size of the proteins detected was estimated using protein molecular-mass standards (Invitrogen).
Isolation of nuclear extracts and electrophoretic mobility shift assay
The isolation of nuclear extracts and electrophoretic mobility shift assay (EMSA) were performed as described elsewhere [27].
High performance liquid chromatography measurement of AMP
Adenine nucleotides were separated by high performance liquid chromatography using an X-Bridge column with a 3.5 μm outer diameter (100 × 4.6 cm). Elution was done with 0.1 mmol/l potassium dihydrogen phosphate, pH 6, containing 4 mmmol/l tetrabutylammonium hydrogen sulfate and 15% (vol./vol.) methanol. The conditions were as follows: 20 μl sample injection, column at room temperature, flow rate of 0.6 ml/min and UV monitoring at 260 nm.
Statistical analyses
Results are expressed as means ± SD of six separate experiments. Significant differences were established by one-way ANOVA using the GraphPad Prism 4.03 program (GraphPad Software, San Diego, CA, USA). Differences were considered significant at p < 0.05.
Results
Oleate prevents palmitate-induced ER stress in mouse and human skeletal muscle cells
To evaluate the differential effects of palmitate and oleate on ER stress in skeletal muscle cells we examined their effects on the expression of the ER stress markers sXbp1, Atf3, Chop (also known as Ddit3), Hsp70 (also known as Hspa4) and Grp78/Bip (also known as Hspa5). Mouse C2C12 myotubes exposed to 0.5 mmol/l palmitate showed an increase in sXbp1 mRNA levels compared with cells exposed only to BSA (Fig. 1a). The increase in the sXbp1 mRNA levels caused by palmitate was of lower intensity than that induced by thapsigargin, a potent inducer of ER stress [30]. In contrast to palmitate, 0.5 mmol/l oleate did not increase sXbp1 mRNA levels (Fig. 1b). Interestingly, co-incubation of cells with palmitate (0.5 mmol/l) and oleate (0.3 mmol/l) completely abolished the increase in sXbp1 expression caused by the saturated fatty acid. Consistent with the changes in sXbp1, cells exposed to palmitate showed a marked increase in the expression of Atf3, Chop, Hsp70 and Bip (Fig. 1c–f). These changes were not observed in cells incubated with oleate and co-incubation of palmitate-exposed cells with oleate completely blocked the effects of the saturated fatty acid. In agreement with the changes in Bip mRNA levels, glucose-regulated protein 78 (GRP78/BIP) protein levels were only increased in cells exposed to palmitate (Fig. 1g). Activation of IRE1α promotes the splicing of Xbp1. Thus, we then evaluated whether fatty acids activated IRE1α, by using phos-tag reagent, which selectively binds to phosphorylated amino acid residues [29]. Palmitate induced an increase in the phosphorylated forms of IRE1α, detected by slower migration (Fig. 1h). In contrast, this increase was observed neither in cells exposed to oleate nor in those co-incubated with palmitate and oleate. The potent pharmacological ER-stress inducer tunicamycin elicited a large increase in IRE1α phosphorylation, which was reduced by co-incubation with oleate (Fig. 1i).

Oleate prevents palmitate-induced ER stress in mouse skeletal muscle cells. C2C12 myotubes were incubated for 16 h in the presence or absence (Ct, control) of different fatty acids: 0.5 mmol/l palmitate (Pal), 0.5 mmol/l oleate (Ole) or 0.5 mmol/l palmitate supplemented with 0.3 mmol/l oleate (Pal + Ole). Indicated cells were incubated with 1 μmol/l thapsigargin or 5 μg/ml tunicamycin (Tuni) for 16 h. sXbp1 (a, b), Atf3 (c), Chop (d), Hsp70 (e) and Bip (f) mRNA levels. The graphs represent the quantification of the Aprt-normalised mRNA levels, expressed as a percentage of control samples ± SD of six independent experiments. Cell lysates were analysed by western blot using antibodies against BIP (g) and IRE1α (h, i). Immunoblots from three separate experiments were quantified and presented in the corresponding bar graphs. ***p < 0.001 and **p < 0.01 vs Ct; ††† p < 0.001 and †† p < 0.01 vs Pal; ‡‡ p < 0.01 vs Ole
In the human myogenic cell line LHCN-M2 exposure to palmitate caused a marked increase in the expression levels of sXBP1, ATF3 and CHOP (Fig. 2). In contrast, oleate did not and supplementation of palmitate-exposed cells with oleate completely abolished the effect of the saturated fatty acid. Overall, these findings indicate that oleate also prevents palmitate-induced ER stress in human skeletal muscle cells.

Oleate prevents palmitate-induced ER stress in human skeletal muscle cells. LHCN-M2 myotubes were incubated for 16 h in the presence or absence (Ct, Control) of different fatty acids: 0.5 mmol/l palmitate (Pal), 0.5 mmol/l oleate (Ole) or 0.5 mmol/l palmitate supplemented with 0.3 mmol/l oleate (Pal + Ole). sXBP1 (a), ATF3 (b) and CHOP (c) mRNA levels. The graphs represent the quantification of the 18S-normalised mRNA levels, expressed as a percentage of control samples ± SD of six independent experiments. ***p < 0.001 vs Ct; ††† p < 0.001 vs Pal
The preventive effect of oleate on palmitate-induced ER stress is peroxisome proliferator-activated receptor-α -β/δ independent
Since we have previously reported that oleate prevents palmitate-mediated activation of the DAG-PKCθ–NF-κB pathway by activating peroxisome proliferator-activated receptor (PPAR)-α [7], we then assessed the potential involvement of PPARs in the effects of oleate on ER stress. Co-incubation of cells with palmitate and oleate and the PPAR antagonists did not reverse the effects of the monounsaturated fatty acid on the expression of the ER stress markers in C2C12 myotubes (ESM Fig. 1 a–f). In addition, since oleate activates protein kinase A (PKA) [31] and activation of this kinase can inhibit ER stress [32], we evaluated whether this mechanism was involved in the effects of oleate. In the presence of the PKA inhibitor H89, the effect of oleate on sXbp1 in palmitate-exposed cells was not modified (ESM Fig. 2a). Further, we have previously reported that inhibition of fatty-acid oxidation by etomoxir reversed the effects of oleate on palmitate-mediated activation of the DAG-PKCθ–NF-κB pathway [7]. However, etomoxir failed to reverse the effects of oleate on palmitate-induced ER stress (ESM Fig. 2b). Since depletion of ER Ca2+ may contribute to palmitate-induced ER stress [33], we next assessed the effects of calcimycin, a Ca2+ mobiliser, and BAPTA-AM, a [Ca2+]i chelator. Calcimycin increased sXbp1 and Chop mRNA levels (ESM Fig. 2c, d), confirming that Ca2+ mobilisation is involved in ER stress in mouse skeletal muscle cells, as previously described [19]. However, co-incubation of cells with palmitate plus BAPTA-AM reduced neither sXbp1 nor Chop expression, indicating that under our conditions Ca2+ mobilisation does not contribute to palmitate-induced ER stress. Finally, a recent study demonstrated that silent information regulator T1 (SIRT1) serves as a negative regulator of UPR in type 2 diabetes mellitus [34]. However, the SIRT1 inhibitor EX527 did not reverse the effect of oleate on palmitate-induced ER stress (ESM Fig. 2e, f), indicating that the effects of the monounsaturated fatty acid on palmitate-induced ER stress did not involve SIRT1.
Oleate prevents palmitate-induced ER stress through an AMPK-dependent mechanism
It is worth noting that activation of AMPK, which is considered a pharmacological target for insulin resistance and type 2 diabetes mellitus [35], inhibits ER stress [18, 19, 21] whereas reduction of this kinase promotes ER stress [20]. To confirm a role for AMPK in the preventive effects of oleate on palmitate-induced ER stress in C2C12 myotubes, we used the AMPK activators AICAR and A-769662 and the AMPK inhibitor compound C. The increase in the expression of sXbp1, Atf3, Chop and Bip caused by palmitate was reduced in cells co-incubated with palmitate plus either AICAR or A-769662 (Fig. 3), suggesting that AMPK activation prevents palmitate-induced ER stress. In addition, when we co-incubated C2C12 and LHCN-M2 cells exposed to palmitate plus oleate with the AMPK inhibitor compound C we observed that the effect of oleate on the expression of sXbp1/sXBP1, Atf3/AFT3 and Chop was abolished (Fig. 4). Given the association of ER stress with the activation of the inflammatory process and insulin resistance [14], next we tried to link the inhibitory effects of oleate on palmitate-induced ER stress with oleate’s reported ability to prevent palmitate-induced inflammation and insulin resistance [7]. We evaluated the effect of fatty acids on the expression of Il6 and its secretion, a cytokine under the transcriptional control of NF-κB, which is involved in insulin resistance [10, 36]. Palmitate caused a 5.5-fold induction (p < 0.01) in Il6 mRNA levels, which was of lower intensity to the induction observed with potent ER-stress inducer tunicamycin (Fig. 5a). In contrast to palmitate, oleate exposure did not affect Il6 expression (Fig. 5b), whereas co-incubation of palmitate-exposed cells with oleate prevented the increase in Il6 mRNA levels. Interestingly, in the presence of compound C, the effect of oleate in preventing the increase in Il6 mRNA in palmitate-exposed cells was partially reversed. A similar pattern was observed when we determined the secretion of IL-6 into the culture medium (Fig. 5c). Of note, human skeletal muscle cells showed a similar behaviour to that reported for C2C12 myotubes (Fig. 5d). Since palmitate induces IL6 mRNA expression and IL-6 secretion through NF-κB activation driven by the reduction in IκBα levels [7, 8], we next evaluated the effects of fatty acids on the protein levels of this NF-κB inhibitor. Consistent with the changes in Il6, palmitate exposure reduced IκBα protein levels in C2C12 myotubes (Fig. 5e). In contrast, oleate did not and co-incubation of palmitate-exposed cells with the monounsaturated fatty acid prevented this reduction. Moreover, co-incubation of palmitate-exposed cells with oleate plus compound C reversed the effect of oleate. Measurement of NF-κB DNA-binding activity by EMSA confirmed these results. NF-κB formed one main complex with nuclear proteins (Fig. 5f) and specificity of this DNA-binding complex was assessed in competition experiments by adding an excess of unlabelled NF-κB oligonucleotide. Palmitate-exposed cells showed increased NF-κB DNA-binding activity compared with control cells, whereas cells exposed to oleate did not; co-incubation of palmitate-exposed cells with oleate prevented this increase. Interestingly, in the presence of compound C the effect of oleate on palmitate-exposed cells was blunted (Fig. 5f).

AMPK activators prevent palmitate-induced ER stress in mouse skeletal muscle cells. C2C12 myotubes were incubated for 16 h in the presence or absence (Ct, Control) of 0.5 mmol/l palmitate (Pal) and the AMPK activators AICAR (2 mmol/l) or A-769662 (60 μmol/l). sXbp1 (a, b), Atf3 (c, d), Chop (e, f) and Bip (g) mRNA levels. The graphs represent the quantification of the aprt-normalised mRNA levels, expressed as a percentage of control samples ± SD of six independent experiments. ***p < 0.001 and **p < 0.01 vs Ct; ††† p < 0.001 and †† p < 0.01 vs Pal

Oleate prevents palmitate-induced ER stress in skeletal muscle cells through an AMPK-dependent mechanism. C2C12 or LHCN-M2 myotubes were incubated for 16 h in the presence or absence (Ct, Control) of different fatty acids: 0.5 mmol/l palmitate (Pal), 0.5 mmol/l oleate (Ole) or 0.5 mmol/l palmitate supplemented with 0.3 mmol/l oleate (Pal + Ole). Indicated cells were pre-incubated with 30 μmol/l of the AMPK inhibitor compound C (Cc) 8 h before the exposure to fatty acids. sXbp1 (a), Atf3 (b), Chop (c), human sXBP1 (d) and human ATF3 (e) mRNA levels. The graphs represent the quantification of the 18S- (LHCN-M2 human cells) or aprt-normalised mRNA levels, expressed as a percentage of control samples ± SD of six independent experiments. ***p < 0.001 and **p < 0.01 vs Ct; ††† p < 0.001 and † p < 0.05 vs Pal; ‡‡‡ p < 0.001, ‡‡ p < 0.01 and ‡ p < 0.05 vs Ole; §§§ p < 0.001 and §§ p < 0.01 vs Pal + Ole

Oleate prevents palmitate-induced inflammation in skeletal muscle cells through an AMPK-dependent mechanism. C2C12 or LHCN-M2 myotubes were incubated for 16 h in the presence or absence (Ct, Control) of different fatty acids: 0.5 mmol/l palmitate (Pal), 0.5 mmol/l oleate (Ole) or 0.5 mmol/l palmitate supplemented with 0.3 mmol/l oleate (Pal + Ole). Indicated cells were pre-incubated with 30 μmol/l of the AMPK inhibitor compound C (Cc) before the exposure to fatty acids. Relative quantification of mouse Il6 (a, b) and human IL6 (d) mRNA levels assessed by real-time RT-PCR. The graphs represent the quantification of the housekeeping-gene-normalised mRNA levels, expressed as a percentage of control samples ± SD of six independent experiments. Mouse IL-6 secretion levels (c) were analysed by ELISA. C2C12 cell lysates were analysed by western blot using antibodies against IκBα (e). Immunoblots from three separate experiments were quantified and presented in the corresponding bar graphs. (f) Autoradiograph of EMSA performed with 32P-labelled NF-κB nucleotide and nuclear protein extracts (NE). One main specific complex (I), based on competition with a molar excess of unlabelled probe, was formed. The autoradiograph is representative of three separate experiments. ***p < 0.001 and **p < 0.01 vs Ct; ††† p < 0.001, †† p < 0.01 and † p < 0.05 vs Pal; ‡‡‡ p < 0.001 and ‡‡ p < 0.01 vs Ole; §§§ p < 0.001 and §§ p < 0.01 vs Pal + Ole
Then we evaluated whether AMPK activation by oleate contributed to the prevention of the reduction in insulin-stimulated Akt phosphorylation caused by palmitate exposure in C2C12 myotubes. Palmitate reduced insulin-stimulated Akt phosphorylation and this effect was not observed in cells exposed to oleate or in palmitate-exposed cells supplemented with oleate (Fig. 6a). In addition, the AMPK activators AICAR and A-769662 prevented this effect of palmitate (Fig. 6a). Likewise, the ER stress inducer thapsigargin reduced insulin-stimulated Akt phosphorylation. Of note, the effect of oleate on palmitate-exposed cells was reversed in the presence of compound C, indicating that the ability of oleate to prevent palmitate-induced insulin resistance requires AMPK activation. To further underpin the importance of ER stress in palmitate-induced insulin resistance in our conditions we incubated cells with palmitate in the presence of phenylbutyric acid (PBA), a pharmacological chaperone that reduces cellular ER stress and improves insulin sensitivity [37]. PBA prevented the reduction in insulin-stimulated Akt phosphorylation caused by palmitate (Fig. 6b). A similar trend to that reported for Akt was observed when we assessed 2-DG uptake (Fig. 6c). Interestingly, the ER stress inducer tunicamycin caused a similar reduction in insulin-stimulated 2-DG to that attained by palmitate.

Oleate prevents palmitate-induced insulin resistance in mouse skeletal muscle cells through an AMPK-dependent mechanism. C2C12 myotubes were incubated for 16 h in the presence or absence (Ct, Control) of different fatty acids: 0.5 mmol/l palmitate (Pal), 0.5 mmol/l oleate (Ole) or 0.5 mmol/l palmitate supplemented with 0.3 mmol/l oleate (Pal + Ole). Indicated cells were pre-incubated with 30 μmol/l of the AMPK inhibitor compound C, 2 mmol/l of AICAR, 60 μmol/l of A-769662, 2 mmol/l of the ER stress inhibitor PBA, 1 μmol/l of thapsigargin (Tapsi) or 5 μg/ml tunicamycin (Tuni). (a, b) C2C12 cell lysates were analysed by western blot using antibodies against total and phospho-Akt (Ser473). Indicated cells were incubated with 100 nmol/l insulin (Ins) for the last 10 min. Immunoblots from three separate experiments were quantified and presented in the corresponding bar graphs. (c) 2-DG uptake was assessed without or with insulin. Values are means ± SD of six independent experiments. §§§ p < 0.001 and § p < 0.05 vs Ct cells not exposed to insulin (Ct-Ins); ***p < 0.001 and *p < 0.05 vs Ct cells exposed to insulin (Ct + Ins); ††† p < 0.001 and † p < 0.05 vs Pal; ‡‡ p < 0.01 and ‡ p < 0.05 vs Pal + Ole
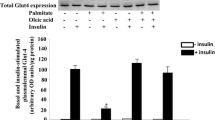
To clearly confirm the involvement of AMPK in the effects of oleate, we manipulated AMPK activity in C2C12 cells by a molecular approach, involving overproduction of an AMPKα subunit with a point mutation that causes the enzyme to function as a dominant negative suppressor of endogenous AMPK activity (K45R). Overproduction of this construct in C2C12 cells leads to the displacement of endogenous subunits, followed by degradation of free alpha subunit [24]. This resulted in a reduction in total protein AMPK levels (Fig. 7a) and in its activity, determined by the decrease in acetyl-CoA carboxylase (ACC) phosphorylation (Fig. 7b). ACC is a substrate for AMPK [38] and serves as an indicator of AMPK activity. Inhibition of AMPK activity attenuated the effect of oleate in palmitate-exposed cells on IRE1α activation (Fig. 7c), IκBα protein levels (Fig. 7d) and insulin-stimulated Akt phosphorylation (Fig. 7e), suggesting that oleate prevents palmitate-induced inflammation and insulin resistance through an AMPK-dependent mechanism.

The effects of oleate on ER stress, inflammation and insulin sensitivity in palmitate-exposed mouse skeletal muscle cells are AMPK dependent. C2C12 myotubes transfected with LacZ- or pAMPKα2 K45R-carrying plasmids were incubated for 16 h in the presence or absence of different fatty acids: 0.5 mmol/l palmitate (Pal), 0.5 mmol/l oleate (Ole) or 0.5 mmol/l palmitate supplemented with 0.3 mmol/l oleate (Pal + Ole). Cell lysates were analysed by western blot using antibodies against total AMPK and actin (a), total and phospho-ACC (Ser79) (b), IRE-1α (c) IκBα (d) and total and phospho-Akt (Ser473) (e). Indicated cells were incubated with 100 nmol/l insulin (Ins) for the last 10 min. Data are expressed as mean ± SD of four experiments. ¶¶ p < 0.01 and ¶ p < 0.05 vs LacZ−Ins; ***p < 0.001, **p < 0.01 and *p < 0.05 vs LacZ + Ins; ††† p < 0.001 and †† p < 0.01 vs Pal; ‡‡‡ p < 0.001 and ‡‡ p < 0.01 vs Ole; §§§ p < 0.001 and § p < 0.05 vs Pal + Ole; DN, dominant negative
Oleate prevents the reduction in phospho-AMPK induced by palmitate
Finally, we evaluated the potential mechanism responsible for the effect of fatty acids on AMPK activity. AMPK activation was monitored by western blot by incubating with a specific antibody against phosphorylated Thr172 of AMPK, which is essential for its activity [39]. When we examined the total and phospho-AMPK protein levels in C2C12 skeletal muscle cells exposed to fatty acids we observed that palmitate caused a reduction in phospho-AMPK levels compared with control cells (Fig. 8a). Unlike palmitate, oleate did not affect phospho-AMPK levels. Further, oleate supplementation prevented the reduction in phospho-AMPK levels caused by palmitate. Consistent with the phospho-AMPK levels, ACC-Ser79 was reduced in palmitate-exposed cells, whereas this reduction was not observed in cells exposed to oleate or in palmitate-exposed cells supplemented with oleate (Fig. 8b). The control of AMPK is complex and its phosphorylation status is regulated by both phosphatases and kinases [35]. Since a previous study reported that palmitate inhibited AMPK phosphorylation via ceramide-dependent phosphatase 2A (PP2A) activation in endothelial cells [40], we first examined the abundance of the PP2A/AC catalytic subunit. Fatty acids did not affect the protein levels of this phosphatase (Fig. 8c), making its contribution to the changes observed unlikely. Although in skeletal muscle cells LKB1 is the main upstream kinase regulating AMPK activity [41], we did not observe changes in the protein levels of LKB1 (Fig. 8d). Moreover, AMPK is activated allosterically by an increase in the intracellular AMP levels [35]. Even with minimal reduction in cellular ATP, changes in the concentration of AMP can activate AMPK [42]. Thus, we measured the AMP levels by HPLC to determine whether fatty acids affected its concentrations. Interestingly, palmitate reduced AMP levels compared with control cells or cells exposed to oleate, whereas in palmitate-exposed cells supplemented with oleate a significant increase was observed (Fig. 8e).

Oleate prevents the palmitate-mediated reduction in phospho-AMPK levels in mouse skeletal muscle cells. C2C12 myotubes were incubated for 16 h in the presence or absence (Ct, Control) of different fatty acids: 0.5 mmol/l palmitate (Pal), 0.5 mmol/l oleate (Ole) or 0.5 mmol/l palmitate supplemented with 0.3 mmol/l oleate (Pal + Ole). Cell lysates were analysed by western blot using antibodies against total and phospho-AMPK (Thr172) (a), total and phospho-ACC (Ser79) (b), PP2A (c) and LKB1 (d). (e) AMP levels in C2C12 myotubes exposed to fatty acids. Data are expressed as mean ± SD of four experiments. ***p < 0.001 and **p < 0.01 vs Ct; ††† p < 0.001 vs Pal; ‡‡‡ p < 0.001 vs Ole
Discussion
High-fat diets are known to cause insulin resistance and type 2 diabetes mellitus, mainly due to their fatty-acid content. However, whereas saturated fatty acids promote insulin resistance [2, 3], the monounsaturated oleic acid improves insulin sensitivity [4, 5]. This has led to the suggestion that dietary intake of oleic acid should be used as a substitute for saturated fatty acids in the management of type 2 diabetes mellitus [43]. There is increasing evidence that the Mediterranean diet has a protective effect on both obesity and diabetes. This diet is characterised by a specific fatty acid pattern; it is low in saturated fatty acids (7–8% of energy) and high in monounsaturated fatty acids (over 20% of total energy), because the fat source consists primarily of olive oil [44]. However, the mechanisms by which oleate may improve insulin resistance are not completely known.
This study provides the first evidence that the monounsaturated fatty acid oleate prevents saturated-fatty-acid-induced ER stress, inflammation and insulin resistance through AMPK activation. First, we show that oleate, in contrast to palmitate, does not increase the levels of ER stress markers. In addition, oleate supplementation at a low concentration provides a marked protection against ER stress and reduces the levels of these markers to those present in control cells. It is worth noting that the changes observed in the mouse cell line C2C12 were confirmed in human skeletal muscle cells, indicating that the effects of oleate are not species specific. Our findings discard the involvement of PPARs, Ca2+, SIRT1, PKA or increased fatty acid oxidation in the effects attained by oleate. Likewise, even though extracellular-signal-regulated kinase (ERK)1/2 has been involved in palmitate-induced NF-κB activation [45], no changes were observed in ER stress markers in cells co-incubated with palmitate plus U0126 (a MEK1/2 [mitogen-activated protein kinase]–ERK1/2 inhibitor) compared with cells incubated with only the saturated fatty acid, whereas IL6 mRNA levels were reduced (data not shown). Furthermore, oleate exposure did not affect palmitate uptake since we have previously reported that the total content of intracellular lipids is similar in palmitate- and palmitate-plus-oleate-exposed cells [7]. However, AMPK activation prevented the increase in ER stress markers in palmitate-exposed skeletal muscle cells. This is consistent with previous studies reporting that AMPK activation protects against hypoxic injury [18], atherosclerosis [19] and liver damage [21] by reducing ER stress. By using the AMPK inhibitor compound C and overproduction of a dominant negative AMPK construct we demonstrated that activation of this kinase was responsible for the reduction in ER stress attained by oleate in palmitate-exposed cells. Since ER stress is emerging as a potential site for the intersection of inflammation and insulin resistance [11], we assessed in our conditions the contribution of ER stress and AMPK activation to these processes. Interestingly, ER stress can activate NF-κB via translational suppression of IκΒα, resulting in the upregulation of mediators of inflammation and insulin resistance, such as IL-6 [46]. Consistent with this, the ER stress inducers tunicamycin and thapsigargin increased the expression of IL6 and reduced insulin-stimulated Akt phosphorylation, respectively. Likewise, PBA, which reduces cellular ER stress, prevented the reduction in insulin-stimulated Akt phosphorylation caused by palmitate. These findings are in agreement with those of a previous study [15] but contrast with those reported in a recent study indicating that ER stress does not mediate palmitate-induced insulin resistance [47]. We do not know the reasons for this discrepancy, but differences in the fatty acid–BSA conjugation could be involved.
Overall, these findings confirm the role of ER stress in inflammation and insulin resistance in skeletal muscle cells. Moreover, the ability of oleate to prevent the reduction in IκBα protein levels in palmitate-exposed cells was blocked by the AMPK inhibitor compound C and by overproduction of the dominant negative AMPK construct, suggesting that AMPK activation by oleate contributes to the prevention of palmitate-induced inflammation. In agreement with the changes in markers of inflammation, AICAR, A-769662 and oleate improved the reduction in insulin-stimulated Akt phosphorylation that had been caused by palmitate. The improvement achieved by oleate was prevented in cells supplemented with oleate plus compound C or cells in which the dominant negative AMPK was overproduced. These data confirm that oleate improves palmitate-induced insulin resistance through AMPK activation.
As a whole, the findings of this study show that AMPK activation by oleate contributes to the prevention of palmitate-induced inflammation and insulin resistance. In addition, our data also show that AMPK activation by oleate also prevents palmitate-induced NF-κB activation in human skeletal muscle cells. This is an interesting point, since NF-κB is activated in myocytes from obese individuals with type 2 diabetes compared with non-obese control individuals, whereas AMPK activation attenuates NF-κB activation [48].
Interestingly, it has been reported that reduction in AMPK levels promotes ER stress, suggesting that AMPK functions as a physiological suppressor of ER stress [20]. When we examined the potential mechanisms responsible for the increase in ER stress following palmitate exposure and the protective effects of oleate, we observed that the saturated fatty acid reduced phospho-AMPK levels. In contrast, neither oleate- nor palmitate-exposed cells supplemented with oleate showed changes in the levels of this kinase. Since it has been reported that high-fat-diet feeding significantly decreases phospho-AMPK in the liver and muscles of rodents [49], these findings suggest that the saturated fatty acids in these diets contribute to this reduction.
The regulation of AMPK is complex, involving allosteric control by an increase in the cellular content of AMP and covalent regulation through phosphorylation of Thr172 within the catalytic subunit by upstream kinase LKB1 and dephosphorylation by PP2A. Our data show that palmitate reduces AMP levels, whereas no changes were observed in cells exposed to oleate; in those exposed to palmitate and supplemented with oleate, AMP levels were higher than in control cells. These findings indicate that palmitate reduces AMPK activity through a reduction in AMP levels, whereas oleate supplementation can prevent the reduction caused by palmitate in the concentration of this nucleotide.
In summary, on the basis of our findings we propose that oleate prevents palmitate-induced ER stress, inflammation and insulin resistance in skeletal muscle cells through AMPK activation. These findings offer a new mechanistic approach to the beneficial effects of oleate vs the saturated fatty acid palmitate in skeletal muscle insulin resistance.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- AMPK:
-
AMP-activated protein kinase
- ATF3:
-
Activating transcription factor 3
- ATF6:
-
Activating transcription factor 6
- CHOP:
-
CCAAT/enhancer-binding protein homologous protein
- DAG:
-
Diacylglycerol
- eIF2:
-
Eukaryotic initiation factor 2
- EMSA:
-
Electrophoretic mobility shift assay
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular-signal-regulated kinase
- GRP78:
-
Glucose-regulated protein 78 (BIP)
- HSP70:
-
Heat shock protein 70
- IκB:
-
Inhibitor of κB
- IRE-1α:
-
Inositol-requiring 1 transmembrane kinase/endonuclease-1
- LKB1:
-
Liver kinase B1
- NF-κB:
-
Nuclear factor-κB
- PBA:
-
Phenylbutyric acid
- PERK:
-
Eukaryotic translation initiation factor-2α kinase 3
- PKA:
-
Protein kinase A
- PKCθ:
-
Protein kinase Cθ
- PP2A:
-
Ceramide-dependent phosphatase 2A
- PPAR:
-
Peroxisome proliferator-activated receptor
- SIRT1:
-
Silent information regulator T1
- TRAF2:
-
TNF-α-receptor-associated factor 2
- UPR:
-
Unfolded protein response
References
Boden G (1997) Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 46:3–10
Vessby B, Uusitupa M, Hermansen K et al (2001) Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: the KANWU study. Diabetologia 44:312–319
Hu FB, van Dam RM, Liu S (2001) Diet and risk of type II diabetes: the role of types of fat and carbohydrate. Diabetologia 44:805–817
Parillo M, Rivellese AA, Ciardullo AV et al (1992) A high-monounsaturated-fat/low-carbohydrate diet improves peripheral insulin sensitivity in non-insulin-dependent diabetic patients. Metabolism 41:1373–1378
Ryan M, McInerney D, Owens D, Collins P, Johnson A, Tomkin GH (2000) Diabetes and the Mediterranean diet: a beneficial effect of oleic acid on insulin sensitivity, adipocyte glucose transport and endothelium-dependent vasoreactivity. QJM 93:85–91
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119
Coll T, Eyre E, Rodríguez-Calvo R et al (2008) Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J Biol Chem 283:11107–11116
Senn JJ (2006) Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem 281:26865–26875
Kim JK, Kim YJ, Fillmore JJ et al (2001) Prevention of fat-induced insulin resistance by salicylate. J Clin Invest 108:437–446
Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G (2001) Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 280:E745–E751
Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140:900–917
Zhang K, Kaufman RJ (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462
Marchetti P, Bugliani M, Lupi R et al (2007) The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 50:2486–2494
Ozcan U, Cao Q, Yilmaz E et al (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Peng G, Li L, Liu Y et al (2011) Oleate blocks palmitate-induced abnormal lipid distribution, endoplasmic reticulum expansion and stress, and insulin resistance in skeletal muscle. Endocrinology 152:2206–2218
Eizirik DL, Cardozo AK, Cnop M (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29:42–61
Shaw RJ, Lamia KA, Vasquez D et al (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310:1642–1646
Terai K, Hiramoto Y, Masaki M et al (2005) AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol 25:9554–9575
Dong Y, Zhang M, Wang S et al (2010) Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 59:1386–1396
Dong Y, Zhang M, Liang B et al (2010) Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 121:792–803
Wang Y, Wu Z, Li D et al (2011) Involvement of oxygen-regulated protein 150 in AMP-activated protein kinase-mediated alleviation of lipid-induced endoplasmic reticulum stress. J Biol Chem 286:11119–11131
Silveira LR, Fiamoncini J, Hirabara SM et al (2008) Updating the effects of fatty acids on skeletal muscle. J Cell Physiol 217:1–12
Gorski J, Nawrocki A, Murthy M (1998) Characterization of free and glyceride-esterified long chain fatty acids in different skeletal muscle types of the rat. Mol Cell Biochem 178:113–118
Mu J, Brozinick JT, Valladares O, Bucan M, Birnbaum MJ (2001) A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell 7:1085–1094
Zhu CH, Mouly V, Cooper RN et al (2007) Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell 6:515–523
Esk C, Chen CY, Johannes L, Brodsky FM (2010) The clathrin heavy chain isoform CHC22 functions in a novel endosomal sorting step. J Cell Biol 188:131–144
Palomer X, Alvarez-Guardia D, Rodríguez-Calvo R et al (2009) TNF-alpha reduces PGC-1alpha expression through NF-kappaB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc Res 81:703–712
Guo W, Wong S, Xie W, Lei T, Luo Z (2007) Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am J Physiol Endocrinol Metab 293:E576–E586
Martinon F, Chen X, Lee AH, Glimcher LH (2010) TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11:411–418
Srivastava RK, Sollott SJ, Khan L, Hansford R, Lakatta EG, Longo DL (1999) Bcl-2 and Bcl-X(L) block thapsigargin-induced nitric oxide generation, c-Jun NH(2)-terminal kinase activity, and apoptosis. Mol Cell Biol 19:5659–5674
Chang CH, Chey WY, Chang TM (2000) Cellular mechanism of sodium oleate-stimulated secretion of cholecystokinin and secretin. Am J Physiol Gastrointest Liver Physiol 279:G295–G303
Yusta B, Baggio LL, Estall JL et al (2006) GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab 4:391–406
Gwiazda KS, Yang TL, Lin Y, Johnson JD (2009) Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am J Physiol Endocrinol Metab 296:E690–E701
Li Y, Xu S, Giles A et al (2011) Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J 25:1664–1679
Zhang BB, Zhou G, Li C (2009) AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab 9:407–416
Pickup JC, Mattock MB, Chusney GD, Burt D (1997) NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40:1286–1292
Ozcan U, Yilmaz E, Ozcan L et al (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313:1137–1140
Ruderman NB, Saha AK, Vavvas D, Witters LA (1999) Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol 276:E1–E18
Hawley SA, Davison M, Woods A et al (1996) Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271:27879–27887
Wu Y, Song P, Xu J, Zhang M, Zou MH (2007) Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem 282:9777–9788
Watt MJ, Steinberg GR, Chen ZP, Kemp BE, Febbraio MA (2006) Fatty acids stimulate AMP-activated protein kinase and enhance fatty acid oxidation in L6 myotubes. J Physiol 574:139–147
Zhou M, Lin BZ, Coughlin S, Vallega G, Pilch PF (2000) UCP-3 expression in skeletal muscle: effects of exercise, hypoxia, and AMP-activated protein kinase. Am J Physiol Endocrinol Metab 279:E622–E629
Berry EM (1997) Dietary fatty acids in the management of diabetes mellitus. Am J Clin Nutr 66:991S–997S
Bergouignan A, Momken I, Schoeller DA, Simon C, Blanc S (2009) Metabolic fate of saturated and monounsaturated dietary fats: the Mediterranean diet revisited from epidemiological evidence to cellular mechanisms. Prog Lipid Res 48:128–147
Green CJ, McRae K, Fogarty S, Hardie DG, Sakamoto K, Hundal HS (2011) Counter-modulation of fatty acid-induced pro-inflammatory nuclear factor kB signalling in rat skeletal muscle cells by AMP-activated protein kinase. Biochem J 435:463–474
Deng J, Lu PD, Zhang Y et al (2004) Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 24:10161–10168
Hage Hassan R, Hainault I, Vilquin JT et al (2012) Endoplasmic reticulum stress does not mediate palmitate-induced insulin resistance in mouse and human muscle cells. Diabetologia 55:204–214
Green CJ, Pedersen M, Pedersen BK, Scheele C (2011) Elevated NF-{kappa}B activation is conserved in human myocytes cultured from obese type 2 diabetic patients and attenuated by AMP-activated protein kinase. Diabetes 60:2810–2819
Muse ED, Obici S, Bhanot S et al (2004) Role of resistin in diet-induced hepatic insulin resistance. J Clin Invest 114:232–239
Acknowledgements
We thank A. Orozco (Department of Biochemistry and Molecular Biology, University of Barcelona, Barcelona, Spain) for experimental assistance with human myotube cultures. We thank M. J. Birnbaum (Howard Hughes Medical Institute, University of Pennsylvania, Philadelphia, PA, USA) for the pcDNA3/pAMPKalpha2-K45R plasmid. We would like to thank the University of Barcelona’s Language Advisory Service for revising the manuscript. M. Vázquez-Carrera is the guarantor of this work, had full access to all the data, and takes full responsibility for the integrity of data and the accuracy of data analysis.
Funding
This study was partly supported by funds from the Spanish Ministerio de Economía y Competitividad (SAF2009-06939 and SAF2012-30708) and European Union ERDF funds. CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) is an Instituto de Salud Carlos III project. L. Salvadó was supported by an FPI grant from the Spanish Ministerio de Economía y Competitividad.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
LS, TC, ES, EB, AMG-F, XP and MV-C processed the samples, analysed and prepared the data and were involved in drafting the article. LS, ES, AMG-F and XP contributed to the interpretation of the data and revised the article. MV-C designed the experiments, analysed and interpreted the data and wrote the manuscript. All authors approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 5 kb)
ESM Fig. 1
Oleate prevents palmitate-induced ER stress in skeletal muscle cells through a PPARindependent mechanism. C2C12 myotubes were incubated for 16 h in the presence or absence of different fatty acids (0.5 mmol/l palmitate, 0.5 mmol/l oleate or 0.5 mmol/l palmitate supplemented with 0.3 mmol/l oleate). When indicated cells were pre-incubated with 10 μmol/l of the PPARα antagonist GW6471, 1 μmol/l of the PPARβ/δ antagonist GSK0660 or with 20 μmol/l of the PPARγ antagonist GW9662 1 h before the exposure to fatty acids. sXbp1 (A), Atf3 (B), Chop (C), sXbp1 (D), Atf3 (E and F) mRNA levels. The graphics represent the quantification of the aprt-normalized mRNA levels, expressed as a percentage of control samples ± SD of six independent experiments. *** P < 0.001 vs. control, ††† P < 0.001 vs. palmitate-exposed cells. (PDF 90 kb)
ESM Fig. 2
Oleate prevents palmitate-induced ER stress in skeletal muscle cells through mechanisms independent of PKA, mitochondrial β-oxidation, Ca2+ mobilization and Sirt1. C2C12 myotubes were incubated for 16 h in the presence or absence of different fatty acids (0.5 mM palmitate, 0.5 mM oleate or 0.5 mM palmitate supplemented with 0.3 mM oleate). When indicated cells were pre-incubated with 10 μM of the PKA inhibitor H89, 40 μM of the carnitine palmitoyltransferase 1 inhibitor etomoxir, 30 μM of the [Ca2+]i chelator BAPTA-AM, 5 μM of the Ca2+ mobilizer calcimycin and 10 μM of Sirt1 inhibitor EX527 30 min before the exposure to fatty acids. sXbp1 (A-C), Chop (D and F) and Atf3 (E) mRNA levels. The graphics represent the quantification of the aprt-normalized mRNA levels, expressed as a percentage of control samples ± SD of six independent experiments. *** P < 0.001 vs. control, ††† P < 0.001 vs. palmitate-exposed cells. ‡‡‡ P < 0.001 vs. oleate-exposed cells. §§§ P < 0.001 vs. palmitate-exposed cells supplemented with 0.3 mM oleate. ¶¶¶ P < 0.001 vs. palmitate-exposed cells supplemented with 0.3 mM oleate and EX527. (PDF 105 kb)
Rights and permissions
About this article
Cite this article
Salvadó, L., Coll, T., Gómez-Foix, A.M. et al. Oleate prevents saturated-fatty-acid-induced ER stress, inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 56, 1372–1382 (2013). https://doi.org/10.1007/s00125-013-2867-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2867-3