{kind=link}
{kind=link}
This package is aimed at providing a way of retrieving variant information using ANNOVAR and myvariant.info. In particular, it is suited for bioinformaticians interested in aggregating variant information into a single NoSQL database (MongoDB solely at the moment).
Documentation now live at: http://vapr.readthedocs.io/en/latest/
DOI: Efficient population-scale variant analysis and prioritization with VAPr
- Amanda Birmingham (abirmingham@ucsd.edu)
- Adam Mark, M.S. (a1mark@ucsd.edu)
- Carlo Mazzaferro
- Guorong Xu, Ph.D.
- Kathleen Fisch, Ph.D. (kfisch@ucsd.edu)
This project is licensed under the MIT License - see the LICENSE file for details
- Background
1.1. Data Models - Getting Started
- Tutorial
3.1. Workflow Overview
3.2. VaprAnnotator - Tips on usage
3.2.1 Arguments
3.3. Core Methods
3.3.1 Annovar
3.3.2 Annotation
3.3.3 Filtering
3.3.1 Output Files
VAPr was developed to simplify the steps required to get mutation data from a VCF file to a downstream analysis process. A query system was implemented allowing users to quickly slice the genomic variant (GV) data and select variants according to their characteristics, allowing researchers to focus their analysis only on the subset of data that contains meaningful information. Further, this query system allows the user to select the format in which the data can be retrieved. Most notably, CSV or VCF files can be retrieved from the database, allowing any researcher to quickly filter variants and retrieve them in commonly used formats. The package can also be installed and used without having to download ANNOVAR. In that case, variant data can be retrieved solely by MyVariant.info and rapidly parsed to the MongoDB instance.
The annotation process identifies every unique variant in the union of variants found for the input samples; it then submits batches (of a user-specifiable size) of variant ids to MyVariant.info and stores the resulting annotation information to the local MongoDB. Subsequent filtering and output of the resulting annotations is done against the MongoDB rather than via additional calls to MyVariant.info, allowing the user to investigate multiple different filtering strategies on a given annotation run without additional overhead. Note that, by design, each run of annotate() performs new annotation calls to MyVariant.info rather than attempting to find potentially relevant past annotations in the MongoDB; this is because MyVariant.info is continually updated live, and we anticipate that users will want to receive the latest annotations each time they choose to annotate, rather than potentially “stale” annotations from past runs.
Intuitively, variant data could be stored in SQL-like databases, since annotation files are usually produced in VCF or CSV formats. However, a different approach may be more fruitful. As explained on our paper (currently under review), the abundance and diversity of genomic variant data causes SQL schemas to perform poorly for variant storage and querying. As it can be the case for many variants, the number of different fields and sub-fields it can have can be over 500, with even more diverse nested sub-fields. Creating a pre-defined schema (as required by SQL-like engined) becomes rather impossible: representing such variant in a table format would thus result in a highly sparse and inefficient storage. Representing instead a variant atomically, that is, as a standalone JSON object having no pre-defied schema, it is possible to compress the rich data into a more manageable format. A sample entry in the Mongo Database will look like this. The variety of data that can be retrieved from the sources results from the richness of databases that can be accessed through MyVariant.info. However, not every variant will have such data readily available. In some cases, the data will be restricted to what can be inferred from the vcf file and the annotation carried out with Annovar. In that case, the entries that will be found in the document will be the following:
{'1000g2015aug_all': 0.00579073,
'_id': ObjectId('5a0d4c5b59f987f13d76aa17'),
'alt': 'A',
'cadd': {'1000g': {'af': 0.01, 'afr': 0.002, 'amr': 0.01, 'eur': 0.02},
'_license': 'http://goo.gl/bkpNhq',
'esp': {'af': 0.017, 'afr': 0.005, 'eur': 0.023},
'gerp': {'n': 3.47, 'rs': 350.8, 'rs_pval': 8.50723e-58, 's': 1.47},
'phred': 19.55,
'polyphen': {'cat': 'benign', 'val': 0.017},
'sift': {'cat': 'tolerated', 'val': 0.43}},
'chr': '1',
'clinvar': {'_license': 'https://goo.gl/OaHML9',
'rcv': [{'accession': 'RCV000017600',
'clinical_significance': 'risk factor',
'conditions': {'identifiers': {'medgen': 'C2751604'},
'name': 'Epilepsy, juvenile myoclonic 7 '
'(EJM7)',
'synonyms': ['EPILEPSY, JUVENILE '
'MYOCLONIC, SUSCEPTIBILITY '
'TO, 7',
'EPILEPSY, IDIOPATHIC '
'GENERALIZED, SUSCEPTIBILITY '
'TO, 10; EPILEPSY, JUVENILE '
'MYOCLONIC, SUSCEPTIBILITY '
'TO, 7']}},
{'accession': 'RCV000017599',
'clinical_significance': 'risk factor',
'conditions': {'identifiers': {'medgen': 'C3150401'},
'name': 'Generalized epilepsy with '
'febrile seizures plus type 5 '
'(GEFSP5)'}},
{'accession': 'RCV000022558',
'clinical_significance': 'risk factor',
'conditions': {'identifiers': {'medgen': 'C2751603',
'omim': '613060'},
'name': 'Epilepsy, idiopathic generalized '
'10 (EIG10)',
'synonyms': ['EPILEPSY, IDIOPATHIC '
'GENERALIZED, SUSCEPTIBILITY '
'TO, 10']}}]},
'dbsnp': {'_license': 'https://goo.gl/Ztr5rl', 'rsid': 'rs41307846'},
'end': 1959699,
'exonicfunc_knowngene': 'nonsynonymous SNV',
'func_knowngene': 'exonic',
'gene_knowngene': 'GABRD',
'hgvs_id': 'chr1:g.1959699G>A',
'ref': 'G',
'samples': [{'AD': [17, 20],
'genotype': '0/1',
'genotype_likelihoods': [400.0, 0.0, 314.0],
'genotype_subclass_by_class': {'heterozygous': 'reference'},
'sample_id': 'S1'}],
'start': 1959699,
'wellderly': {'_license': 'https://goo.gl/e8OO17',
'alleles': [{'allele': 'A', 'freq': 0.015},
{'allele': 'G', 'freq': 0.985}]}}
These instructions will get you a copy of the package up and running on your local machine, and will enable you to run annotation jobs on any number of vcf files while storing the data in MongoDB. See the workflow
- MongoDB Community Edition. Installation instructions
- Python (2.7 and 3.5 currently supported and tested)
- BCFtools
- Tabix
- Annovar scripts (optional)
VAPr is written in Python and stores variant annotations in NoSQL database, using a locally-installed instance of MongoDB. Installation instructions
BCFtools will be used for VCF file merging between samples. To download and install:
wget https://github.com/samtools/bcftools/releases/download/1.6/bcftools-1.6.tar.bz2
tar -vxjf bcftools-1.6.tar.bz2
cd bcftools-1.6
make
make install
export PATH=/where/to/install/bin:$PATH
Refer here for installation debugging.
Tabix and bgzip binaries are available through the HTSlib project:
wget https://github.com/samtools/htslib/releases/download/1.6/htslib-1.6.tar.bz2
tar -vxjf htslib-1.6.tar.bz2
cd htslib-1.6
make
make install
export PATH=/where/to/install/bin:$PATH
Refer here for installation debugging.
(It is possible to proceed without installing ANNOVAR. Variants will only be annotated with MyVariant.info. In that case, users can skip the next steps and go straight to the section Known Variant Annotation and Storage)
Users who wish to annotate novel variants will also need to have a local installation of the popular command-line software ANNOVAR(1), which VAPr wraps with a Python interface. If you use ANNOVAR's functionality through VAPr, please remember to cite the ANNOVAR publication (see #1 in Citations)!
The base ANNOVAR program must be installed by each user individually, since its license agreement does not permit
redistribution. Please visit the ANNOVAR download form here,
ensure that you meet the requirements for a free license, and fill out the required form. You will then receive an email
providing a link to the latest ANNOVAR release file. Download this file (which will usually have a name like
annovar.latest.tar.gz) and place it in the location on your machine in which you would like the ANNOVAR program and
its data to be installed--the entire disk size of the databases will be around 25 GB, so make sure you have such space
available!
VAPr is available from PyPi. Once the above requirements have been installed, VAPr itself can be installed by just running:
pip install VAPr
See this jupyter notebook to create your first annotation job
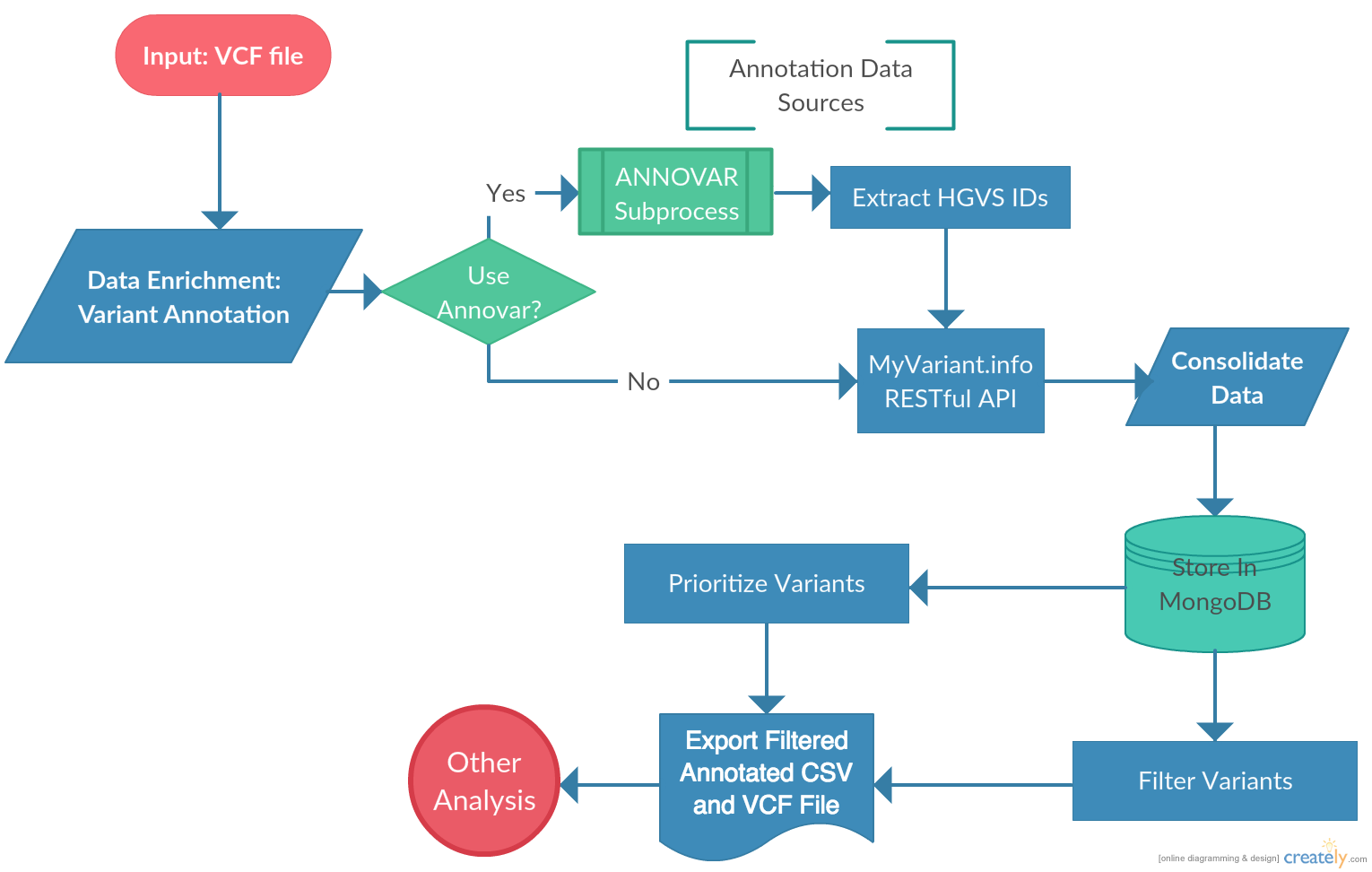
An annotation project can be started by providing the API with a small set of information and then running the core methods provided to spawn annotation jobs. This is done in the following manner:
# Import core module
from VAPr import vapr_core
import os
# Start by specifying the project information
IN_PATH = "/path/to/vcf"
OUT_PATH = "/path/to/out"
ANNOVAR_PATH = "/path/to/annovar"
MONGODB = 'VariantDatabase'
COLLECTION = 'Cancer'
annotator = vapr_core.VaprAnnotator(input_dir=IN_PATH,
output_dir=OUT_PATH,
mongo_db_name=MONGODB,
mongo_collection_name=COLLECTION,
build_ver='hg19',
vcfs_gzipped=False,
annovar_install_path=ANNOVAR_PATH)This will allow you to use any of core methods in the package.
If you plan to use Annovar, please make sure to download the necessary Annovar databases. When Annovar is first installed, it does not install Annovar databases by default. The vapr_core has a method download_annovar_databases() that will download the necessary annovar databases. Note: this command only needs to be run the first time you use VAPr.
annotator.download_annovar_databases()
dataset = annotator.annotate(num_processes=8)
The first four arguments are required to run the full annotation pipeline. These are:
-
input_dir: the path to the directory where all vcf files live. They may also be inside subdirectories, and VAPr will find them. VCF files may contain information for single or multiple samples. -
output_dir: the path to the directory where the annotated csv files will be written to. It will be used in two different instances: writing the file outputs from Annovar, and writing the file outputs from VAPr, in case these are needed -
mongo_db_name: Database used for variant storage as well as the output of annovar i.e. 'VariantDatabase'. -
mongo_collection_name: Collection name for this analysis i.e. 'cancer_analysis_01012018'. -
build_ver: Human genome build. VAPr currently supports the two human genome builds,hg19,hg38.
-
annovar_install_path: Path location of annovar installtion. NOTE: we can't provide the ANNOVAR package as it requires registration to be downloaded. It is, however, freely available here. Download, unzip and place it in whatever directory you'd like. Make sure you have enough space on disk (~15 GB for the datasets used for annotation). It is required that you specify the location to which you downloaded annovar. The folder where annovar lives looks like this:... /annovar/ annotate_variation.pl coding_change.pl convert2annovar.pl example/
humandb/ retrieve_seq_from_fasta.pl table_annovar.pl variants_reduction.pl -
vcfs_gzipped: Boolean. Only files with one vcf extension will be processed. If you are only analyzing one vcf, the file will not be bgzipped. If you are providing a directory or design file with multiple vcf files, they will be bgzipped and merged. If they are already bgzipped, please specifyvcfs_gzipped=Trueand the bgzip step will be skipped. -
design_file: Path to design file. The purpose of an optional design file is to accommodate VCF files scattered throughout a file system. The design file must be set up as a CSV file with the first field name as "Sample_Names", where the column should be populated with full file paths to each VCF you wish to include in the analysis. We anticipate in the future to be able to accommodate meta-data as successive columns which would be included as sample information in each variant document. A sample design file:Sample_Names /path/to/file1.vcf /path/to/file2.vcf /path/to/file3.vcf
The VaprAnnotator object has a variety of methods from it. These include:
- Annovar method:
download_annovar_databases - Annotation and storing methods:
annotateandannotate_lite
The differences and nuances of each will be discussed next.
download_annovar_databases(): this function downloads the databases required to run Annovar to the .../annovar/humandb/ directory.
It will download the databases according to the genome version specified. If your databases are out-of-date, re-running
this command will download the latest version of them. If you currently have the required databases, you may get an error.
Args:
Required:
- None
Optional:
- None
annotate(): this requires running Annovar beforehand, and will kick-start the main functionality
of this package. Namely, it will collect all the variant data from Annovar annotations, combine it with data coming
from MyVariant.info, and parse it to MongoDB, in the database and collection specified in project_data.
Args:
Required:
- None
Optional:
n_processes: An integer value that specifies the number of parallel processing jobs to be used. Larger vcf files will always benefit from a larger number of parallel processes, but that may not be the case for smaller vcf files. As a rule of thumb, use at mostnumber of CPU cores - 1, and for smaller vcf files (less than 50 thousand variants) 4-5 cores at most. Default: 4.verbose: An integer value from 0 to 3 that specifies the verbosity level. Default: 0.
annotate_lite(): Execution will skip annotating with Annovar. It will grab the HGVS ids from the
vcf files and query the variant data from MyVariant.info. It is subject to the issue of potentially having completely
empty data for some of the variants, and inability to run native VAPr queries on the data.
Args:
Required:
- None
Optional:
n_processes: An integer value that specifies the number of parallel processing jobs to be used. Use herenumber of CPU cores - 1.
Four different pre-made filters that allow for the retrieval of specific variants have been implemented. Refer to the [README.md](link to readme/filters) file for more more information about the fields and thresholds used.
In order to use the filters, proceed as follows:
rare_deleterious_variants = dataset.get_rare_deleterious_variants()This will return a list of dictionaries, where each dictionary is contains variant containing annotations.
- criteria 1: 1000 Genomes (ALL) allele frequency (Annovar) < 0.05 or info not available
- criteria 2: ESP6500 allele frequency (MyVariant.info - CADD) < 0.05 or info not available
- criteria 3: cosmic70 (MyVariant.info) information is present
- criteria 4: Func_knownGene (Annovar) is exonic, splicing, or both
- criteria 5: ExonicFunc_knownGene (Annovar) is not "synonymous SNV"
get_rare_deleterious_variants(): this will retrieve all the variants in your collection matching the thresholds specified in the
README.md file.
Args:
Required:
- None
Optional:
sample_names_list: A list of strings specifying the sample names from which you'd like to extract your variants. If this is not used, all variants are queried (that is, variants from all sample sin your collection). Default:None
- criteria: cosmic70 (MyVariant.info) information is present or ClinVar data is present and clinical significance is not Benign or Likely Benign
get_known_disease_variants(): this will retrieve all the variants in your collection matching the thresholds specified in the
README.md file.
Args:
Required:
- None
Optional:
sample_names_list: A list of strings specifying the sample names from which you'd like to extract your variants. If this is not used, all variants are queried (that is, variants from all sample sin your collection). Default:None
- criteria 1: genotype_subclass_by_class (VAPr) is compound heterozygous
- criteria 2: CADD phred score (MyVariant.info - CADD) > 10
get_deleterious_compound_heterozygous_variants(): this will retrieve all the variants in your collection matching the thresholds specified in the
README.md file.
Args:
Required:
- None
Optional:
sample_names_list: A list of strings specifying the sample names from which you'd like to extract your variants. If this is not used, all variants are queried (that is, variants from all sample sin your collection). Default:None
- criteria 1: Variant present in proband
- criteria 2: Variant not present in either ancestor-1 or ancestor-2
get_de_novo_variants(): this will retrieve all the variants in your collection matching the thresholds
specified in the README.md file.
Args:
Required:
proband: first sample name as string.ancestor1: second sample name as string.ancestor2: third sample name as string. De novo variants will be looked for in the proband sample, i.e. variants that occur in the first sample but not in either ancestor1 or ancestor2.
As long as you have a MongoDB instance running, filtering can be performed through pymongo as shown by the code below.
If a list is intended to be created from it, simply add: filter2 = list(filter2)
If you'd like to customize your filters, a good idea would be to look at the available fields to be filtered. Looking at the myvariant.info documentation, you can see what are all the fields available and can be used for filtering.
from pymongo import MongoClient
client = MongoClient()
db = getattr(client, mongodb_name)
collection = getattr(db, mongo_collection_name)
filtered = collection.find({"$and": [
{"$or": [{"func_knowngene": "exonic"},
{"func_knowngene": "splicing"}]},
{"cosmic70": {"$exists": True}},
{"1000g2015aug_all": {"$lt": 0.05}}
]})
filtered = list(filtered)write_unfiltered_annotated_csv(): All variants will be written to a CSV file.
Args:
Required:
output_fp: Name of output file path
write_filtered_annotated_csv(): List of filtered variants will be written to a CSV file.
Args:
Required:
-
filtered_variants: List of filtered variants retrieved by VAPr filters or custom filters. -
output_fp: Name of output file path.
write_unfiltered_annotated_vcf(): All variants will be written to a VCF file.
Args:
Required:
vcf_out_path: Name of output file path
Optional:
info_out: if set to true (Default), will write all annotation data to INFO column, else, it won't.
write_filtered_annotated_vcf(): List of filtered variants will be written to a VCF file.
Args:
Required:
-
filtered_variants: List of filtered variants retrieved by VAPr filters or custom filters. -
vcf_out_path: Name of output file path.
# List of rare deleterious variants
filtered_variants = dataset.get_rare_deleterious_variants()
# Write variants to vcf file
dataset.write_filtered_annotated_vcf(filtered_variants, output_dir + “/myfile.csv”)Citations
- Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data Nucleic Acids Research , 38:e164, 2010
- Xin J, Mark A, Afrasiabi C, Tsueng G, Juchler M, Gopal N, Stupp GS, Putman TE, Ainscough BJ, Griffith OL, Torkamani A, Whetzel PL, Mungall CJ, Mooney SD, Su AI, Wu C (2016) High-performance web services for querying gene and variant annotation. Genome Biology 17(1):1-7