{kind=link}
{kind=link}
{kind=link}
{kind=link}

CRISPResso is a software pipeline for the analysis of targeted CRISPR-Cas9 sequencing data. This algorithm allows for the quantification of both non-homologous end joining (NHEJ) and homologous directed repair (HDR) occurrences.
CRISPResso automatizes and performs the following steps summarized in the figure below:
- filters low quality reads,
- trims adapters,
- aligns the reads to a reference amplicon,
- quantifies the proportion of HDR and NHEJ outcomes,
- quantifies frameshift/inframe mutations (if applicable) and identifies affected splice sites,
- produces a graphical report to visualize and quantify the indels distribution and position.
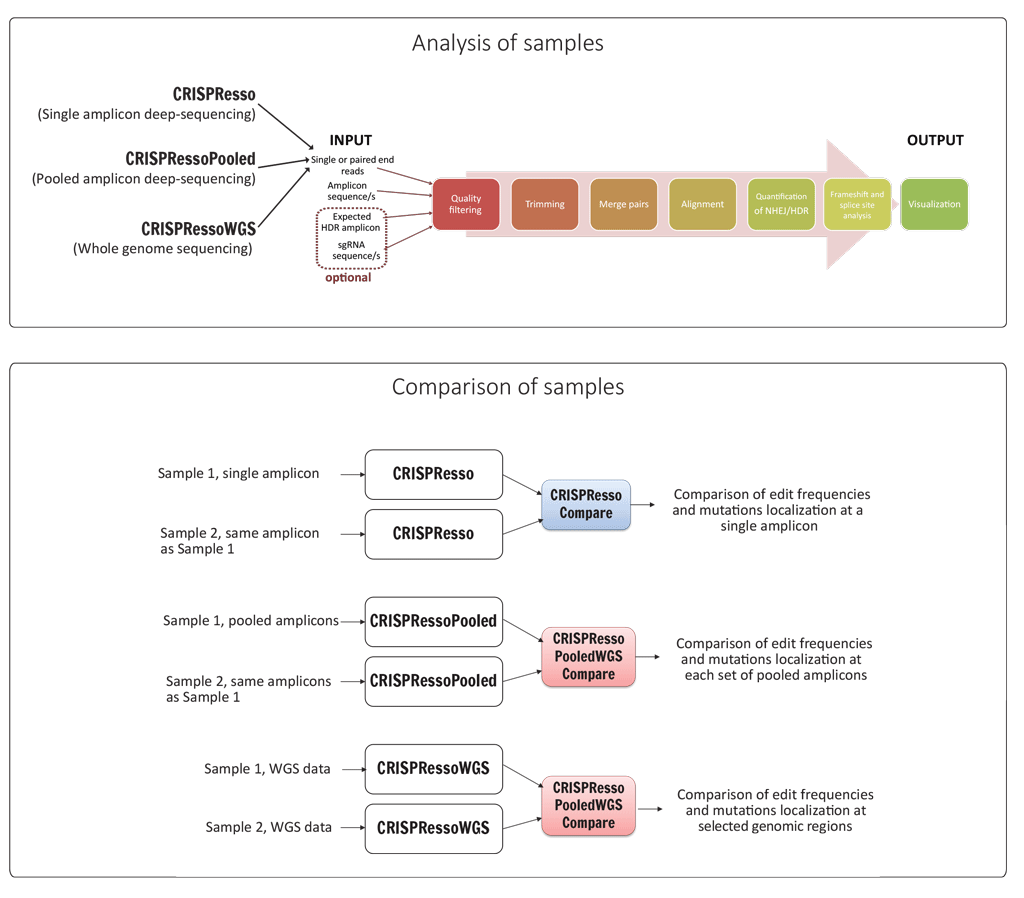
The CRISPResso suite accommodates single or pooled amplicon deep sequencing, WGS datasets and allows the direct comparison of individual experiments. In fact four additional utilities are provided:
- CRISPRessoPooled: a tool for the analysis of pooled amplicon experiments
- CRISPRessoWGS: a tool for the analysis of WGS data or prealigned reads in .bam format
- CRISPRessoCompare:a tool for the comparison of two CRISPResso analyses, useful for example to compare treated and untreated samples or to compare different experimental conditions
- CRISPRessoPooledCompare: a tool to compare experiments involving several regions analyzed by either CRISPRessoPooled or CRISPRessoWGS
If you don't like command line tools you can also use CRISPResso online here: http://crispresso.rocks
To install the command line version of CRISPResso, some dependencies must be installed before running the setup:
- Python 2.7 Anaconda: http://continuum.io/downloads
- Java: http://java.com/download
- C compiler / make. For Mac with OSX 10.7 or greater, open the terminal app and type and execute the command 'make', which will trigger the installation of OSX developer tools.Windows systems are not officially supported, although CRISPResso may work with Cygwin (https://www.cygwin.com/).
After checking that the required software is installed you can install CRISPResso from the official Python repository following these steps:
- Open a terminal window
- Type the command:
pip install CRISPResso --no-use-wheel --verbose- Close the terminal window
Alternatively if want to install the package without the PIP utility:
- Download the setup file: https://github.com/lucapinello/CRISPResso/archive/master.zip and decompress it
- Open a terminal window and go to the folder where you have decompressed the zip file
- Type the command: python setup.py install
- Close the terminal window and open a new one (this is important in order to setup correctly the PATH variable in your system).
The Setup will try to install these software for you:
- Trimmomatic(tested with v0.33): http://www.usadellab.org/cms/?page=trimmomatic
- Flash(tested with v1.2.11): http://ccb.jhu.edu/software/FLASH/
- Needle from the EMBOSS suite(tested with 6.6.0): ftp://emboss.open-bio.org/pub/EMBOSS/
If the setup fails on your machine you have to install them manually and put these utilities/binary files in your path!
To check that the installation worked, open a terminal window and execute CRISPResso --help, you should see the help page.
The setup will automatically create a folder in your home folder called CRISPResso_dependencies (if this folder is deleted, CRISPResso will not work!)! If you want to put the folder in a different location, you need to set the environment variable: CRISPRESSO_DEPENDENCIES_FOLDER. For example to put the folder in /home/lpinello/other_stuff you can write in the terminal BEFORE the installation:
export CRISPRESSO_DEPENDENCIES_FOLDER=/home/lpinello/other_stuffIf you like Docker, we provide a Docker image ready to use, so no installation is required!
https://hub.docker.com/r/lucapinello/crispresso/
To use the image first install Docker: https://docs.docker.com/engine/installation/
Then type the command:
docker pull lucapinello/crispresso
See an example on how to run CRISPResso from a Docker image in the section TESTING CRISPResso below.
The output of CRISPResso consists of a set of informative graphs that allow for the quantification and visualization of the position and type of outcomes within an amplicon sequence. An example is shown below:

CRISPResso requires two inputs: (1) paired-end reads (two files) or single-end reads (single file) in .fastq format (fastq.gz files are also accepted) from a deep sequencing experiment and (2) a reference amplicon sequence to assess and quantify the efficiency of the targeted mutagenesis. The amplicon sequence expected after HDR can be provided as an optional input to assess HDR frequency. One or more sgRNA sequences (without PAM sequences) can be provided to compare the predicted cleavage position/s to the position of the observed mutations. Coding sequence/s may be provided to quantify frameshift and potential splice site mutations.
The reads are first filtered based on the quality score (phred33) in order to remove potentially false positive indels. The filtering based on the phred33 quality score can be modulated by adjusting the optimal parameters (see additional notes below). The adapters are trimmed from the reads using Trimmomatic and then sequences are merged with FLASha (if using paired-end data).The remaining reads are then aligned with needle from the EMBOSS suite, an optimal global sequence aligner based on the Needleman-Wunsch algorithm that can easily accounts for gaps. Finally, after analyzing the aligned reads, a set of informative graphs are generated, allowing for the quantification and visualization of the position and type of outcomes within the amplicon sequence.
NHEJ events:
The required inputs are:
- Two files for paired-end reads or a single file for single-end reads in fastq format (fastq.gz files are also accepted). The reads are assumed to be already trimmed for adapters. If reads are not trimmed, please use the --trim_sequences option and the --trimmomatic_options_string if you are using an adapter different than Nextera.
- The reference amplicon sequence must also be provided.
Example:
CRISPResso -r1 reads1.fastq.gz -r2 reads2.fastq.gz -a AATGTCCCCCAATGGGAAGTTCATCTGGCACTGCCCACAGGTGAGGAGGTCATGATCCCCTTCTGGAGCTCCCAACGGGCCGTGGTCTGGTTCATCATCTGTAAGAATGGCTTCAAGAGGCTCGGCTGTGGTTHDR events: The required inputs are:
- Two files for paired-end reads or a single file for single-end reads in fastq format (fastq.gz files are also accepted). The reads are assumed to be already trimmed for adapters.
- The reference amplicon sequence.
- The expected amplicon sequence after HDR must also be provided.
Example:
CRISPResso -r1 reads1.fastq.gz -r2 reads2.fastq.gz -a GCTTACACTTGCTTCTGACACAACTGTGTTCACGAGCAACCTCAAACAGACACCATGGTGCATCTGACTCCTGAGGAGAAGAATGCCGTCACCACCCTGTGGGGCAAGGTGAACGTGGATGAAGTTGGTGGTGAGGCCCTGGGCAGGTTGGTATCAAGGTTACAAGA -e GCTTACACTTGCTTCTGACACAACTGTGTTCACGAGCAACCTCAAACAGACACCATGGTGCATCTGACTCCTGTGGAAAAAAACGCCGTCACGACGTTATGGGGCAAGGTGAACGTGGATGAAGTTGGTGGTGAGGCCCTGGGCAGGTTGGTATCAAGGTTACAAGAIMPORTANT: You must input the entire reference amplicon sequence (’Expected HDR Amplicon sequence’ is the reference for the sequenced amplicon, not simply the donor sequence). If only the donor sequence is provided, an error will result
- Required parameters
- To run CRISPResso, only 2 parameters are required for single end reads, or 3 for paired end reads:
-r1 or --fastq_r1: This parameter allows for the specification of the first fastq file.
-r2 or --fastq_r2 FASTQ_R2: This parameter allows for the specification of the second fastq file for paired end reads.
-a or --amplicon_seq: This parameter allows the user to enter the amplicon sequence used for the experiment.
- Optional parameters
- In addition to the required parameters explained in the previous section, several optional parameters can be adjusted to tweak your analysis, and to ensure CRISPResso analyzes your data in the best possible way.
-g or --guide_seq or: This parameter allows for the specification of the sgRNA sequence. If more than one sequence are included, please separate by comma/s. If the guide RNA sequence is entered, then the position of the guide RNA and the cleavage site will be indicated on the output analysis plots. Note that the sgRNA needs to be input as the guide RNA sequence (usually 20 nt) immediately 5' of the PAM sequence (usually NGG for SpCas9). If the PAM is found on the opposite strand with respect to the Amplicon Sequence, ensure the sgRNA sequence is also found on the opposite strand. The CRISPResso convention is to depict the expected cleavage position using the value of the parameter cleavage_offset nt 3' from the end of the guide. In addition, the use of alternate nucleases to SpCas9 is supported. For example, if using the Cpf1 system, enter the sequence (usually 20 nt) immediately 3' of the PAM sequence and explicitly set the cleavage_offset parameter to 1, since the default setting of -3 is suitable only for SpCas9. (default:None)
-e or --expected_hdr_amplicon_seq: This parameter allows for the specification of the amplicon sequence expected after HDR. If the data to be analyzed were derived from an experiment using a donor repair template for homology-directed repair (HDR for short), then you have the option to input the sequence of the expected HDR amplicon. This sequence is necessary for CRISPResso to be able to identify successful HDR events within the sequencing data.
--hdr_perfect_alignment_threshold: Sequence homology percentage for an HDR occurrence (default: 98.0). This parameter allows for the user to set a threshold for sequence homology for CRISPResso to count instances of successful HDR. This is useful to improve the analysis allowing some tolerance for technical artifacts present in the sequencing data such as sequencing errors or single nucleotide polymorphisms (SNPs) in the cells used in the experiment. Therefore, if you have a read that exhibits successful HDR but has a SNP or sequencing error within the amplicon, you can lower the sequence homology in order allow CRISPResso to count the read as a successful HDR event. If the data are completely free of sequencing errors or polymorphisms, then consider to set parameter to 100.
-d or -donor_seq:This parameter allows the user to highlight the critical subsequence of the expected HDR amplicon in plots. This parameter does not have any effect on the quantification of HDR events.
-c, --coding_seq:This parameter allows for the specification of the subsequence/s of the amplicon sequence covering one or more coding sequences for the frameshift analysis. If more than one (for example, split by intron/s), please separate by comma. (default: None)
-q, or --min_average_read_quality: This parameter allows for the specification of the minimum average quality score (phred33) to include a read for the analysis.(default: 0, minimum: 0, maximum: 40). This parameter is helpful to filter out low quality reads. If filtering based on average base quality is desired, a reasonable value for this parameter is greater than 30.
-s or --min_single_bp_quality: This parameter allows for the specification of the minimum single bp score (phred33) to include a read for the analysis (default: 0, minimum: 0, maximum: 40). This parameter is helpful to filter out low quality reads. This filtering is more aggressive, since any read with a single bp below the threshold will be discarded. If you want to filter your reads based on single base quality to have very high quality reads, a reasonable value for this parameter is greater than 20.
--min_identity_score: This parameter allows for the specification of the min identity score for the alignment (default: 60.0). In order for a read to be considered properly aligned, it should pass this threshold. We suggest to lower this threshold only if really large insertions or deletions are expected in the experiment (>40% of the amplicon length).
-n or --name: This parameter allows for the specification of the output name of the report (default: the names is obtained from the filename of the fastq file/s used in input).
-o or --output_folder: This parameter allows for the specification of the output folder to use for the analysis (default: current folder).
--split_paired_end: Splits a single fastq file contating paired end reads in two files before running CRISPResso (default: False). If you got your data from the MGH sequencing core in Boston (https://dnacore.mgh.harvard.edu/new-cgi-bin/site/pages/crispr_sequencing_main.jsp), you need this option!
--trim_sequences: This parameter enables the trimming of Illumina adapters with Trimmomatic (default: False)
--trimmomatic_options_string: This parameter allows the user the ability to override options for Trimmomatic (default: ILLUMINACLIP:/Users/luca/anaconda/lib/python2.7/site-packages/CRISPResso-0.8.0-py2.7.egg/CRISPResso/data/NexteraPE-PE.fa:0:90:10:0:true). This parameter is useful to specify different adaptor sequences used in the experiment if you need to trim them.
--min_paired_end_reads_overlap: This parameter allows for the specification of the minimum required overlap length between two reads to provide a confident overlap during the merging step. (default: 4, minimum: 1, max: read length)
--hide_mutations_outside_window_NHEJ: This parameter allows the user to visualize only the mutations overlapping the window around the cleavage site and used to classify a read as NHEJ. This parameter has no effect on the quantification of NHEJ. With the default setting (False), all mutations are visualized including those that do not overlap the window, even though these are not used to classify a read as NHEJ. It may be desirable in certain cases to hide pre-existing and known mutations or sequencing errors outside the window and hence not used for quantification of NHEJ events (default: False).
-w ,--window_around_sgrna: This parameter allows for the specification of a window(s) in bp around each sgRNA to quantify the indels. The window is centered on the predicted cleavage site specified by each sgRNA. Any indels not overlapping or substitutions not adjacent to the window are excluded. A value of 0 will disable this filter (default: 1). This parameter is important since sequencing artifacts and/or SNPs can lead to false positives or false negatives in the quantification of indels and HDR occurrences. Therefore, the user can choose to create a window around the predicted double strand break site of the nuclease used in the experiment. This can help limit sequencing or amplification errors or non-editing polymorphisms from being inappropriately quantified in CRISPResso analysis. Note: any indels that fully or partially overlap the window will be quantified.
--cleavage_offset: This parameter allows for the specification of the cleavage offset to use with respect to the provided sgRNA sequence. Remember that the sgRNA sequence must be entered without the PAM. The default is -3 and is suitable for the SpCas9 system. For alternate nucleases, other cleavage offsets may be appropriate, for example, if using Cpf1 set this parameter to 1. (default: -3, minimum:1, max: reference amplicon length). Note: any large indel that partially overlap the window will be also fully quantified.
--exclude_bp_from_left: Exclude bp from the left side of the amplicon sequence for the quantification of the indels (default: 15). This parameter is helpful to avoid artifacts due to imperfect trimming of the reads.
--exclude_bp_from_right: Exclude bp from the right side of the amplicon sequence for the quantification of the indels (default: 15). This parameter is helpful to avoid artifacts due to imperfect trimming of the reads.
--ignore_substitutions: Ignore substitutions events for the quantification and visualization (default: False).
--ignore_insertions: Ignore insertions events for the quantification and visualization (default: False).
--ignore_deletions: Ignore deletions events for the quantification and visualization (default: False).
--needle_options_string: This parameter allows the user to override options for the Needle aligner (default: -gapopen=10 -gapextend=0.5 -awidth3=5000). More information on the meaning of these parameters can be found in the needle documentation (http://embossgui.sourceforge.net/demo/manual/needle.html). We suggest that only experienced users modify these values.
--keep_intermediate: This parameter allows the user to keep all the intermediate files (default: False). We suggest keeping this parameter disabled for most applications, since the intermediate files (processed reads and alignments) can be really large.
--dump: This parameter allows to dump numpy arrays and pandas dataframes to file for debugging purposes (default: False).
--save_also_png: This parameter allows the user to also save.png images when creating the report., in addition to .pdf files.
-p, --n_processes Specify the number of processes to use for the quantification. This parameter is useful to speed up the quantification and generation of the mutation profiles when multiple CPUs are available. Please use with caution since increasing this parameter will increase significantly the memory required to run CRISPResso (default: 1).
- It is important to check if your reads are trimmed or not. CRISPResso assumes that the reads are already trimmed! If reads are not trimmed, use the option --trim_sequences. The default adapter file used is the Nextera. If you want to specify a custom adapter use the option --trimmomatic_options_string.
- It is possible to use CRISPResso with single end reads. In this case, just omit the option -r2 to specify the second fastq file.
- It is possible to filter based on read quality before aligning reads using the option -q. A reasonable value for this parameter (phred33) is 30.
- The command line CRISPResso tool for use on Mac computers requires OS 10.7 or greater. It also requires that command line tools are installed on your machine. After the installation of Anaconda, open the Terminal app and type make, this should prompt you to install command line tools (requires internet connection).
- Once installed, simply typing CRISPResso into any new terminal should load CRISPResso (you will be greeted by the CRISPResso cup)
- Paired end sequencing files are assumed to contain overlapping sequences (at least 1 bp), if not run CRISPResso on each single fastq file of the pair in single mode.
- Use the following command to get to your folder (directory) with sequencing files, assuming that is /home/lpinello/Desktop/CRISPResso_Folder/Sequencing_Files_Folder: cd /home/lpinello/Desktop/CRISPResso_Folder/Sequencing_Files_Folder
- CRISPResso’s default setting is to output analysis files into your directory, otherwise use the --output parameter.
- Download the two fastq files:
- http://bcb.dfci.harvard.edu/~lpinello/CRISPResso/reads1.fastq.gz
- http://bcb.dfci.harvard.edu/~lpinello/CRISPResso/reads2.fastq.gz
- Open a terminal and go to the folder where you have stored the files
- Type:
CRISPResso -r1 reads1.fastq.gz -r2 reads2.fastq.gz -a AATGTCCCCCAATGGGAAGTTCATCTGGCACTGCCCACAGGTGAGGAGGTCATGATCCCCTTCTGGAGCTCCCAACGGGCCGTGGTCTGGTTCATCATCTGTAAGAATGGCTTCAAGAGGCTCGGCTGTGGTT -g TGAACCAGACCACGGCCCGT- CRISPResso will create a folder with the processed data and the figures.
If you use a Docker image instead run with the following command:
docker run -v ${PWD}:/DATA -w /DATA -i lucapinello/crispresso CRISPResso -r1 reads1.fastq.gz -r2 reads2.fastq.gz -a AATGTCCCCCAATGGGAAGTTCATCTGGCACTGCCCACAGGTGAGGAGGTCATGATCCCCTTCTGGAGCTCCCAACGGGCCGTGGTCTGGTTCATCATCTGTAAGAATGGCTTCAAGAGGCTCGGCTGTGGTT -g TGAACCAGACCACGGCCCGTIf you run Docker on Window you have to specify the full path:
docker run -v //c/Users/luca/Downloads:/DATA -w /DATA lucapinello/crispresso CRISPResso -r1 reads1.fastq.gz -r2 reads2.fastq.gz -a AATGTCCCCCAATGGGAAGTTCATCTGGCACTGCCCACAGGTGAGGAGGTCATGATCCCCTTCTGGAGCTCCCAACGGGCCGTGGTCTGGTTCATCATCTGTAAGAATGGCTTCAAGAGGCTCGGCTGTGGTT -g TGAACCAGACCACGGCCCGT- The log of the external utilities called are stored in the file CRISPResso_RUNNING_LOG.txt
- You can specify the output folder with the option --output_folder
- You can inspect intermediate files with the option --keep_intermediate
- All the processed raw data used to generate the figures are available in the following plain text files:
- Mapping_statistics.txt: this file contains number of: reads in input, reads after preprocessing (merging or quality filtering) and reads properly aligned.
- Quantification_of_editing_frequency.txt: quantification of editing frequency: number of reads aligned, reads with NHEJ, reads with HDR, and reads with mixed HDR-NHEJ); In addition to each of these categories we also provide an overall report summarizing the total numbers of insertions, deletions and substitutions;
- Alleles_frequency_table.txt: number or reads and percentage for each allele discovered in the sequencing data.
- Frameshift_analysis.txt: number of modified reads with frameshift, in-frame and noncoding mutations;
- Splice_sites_analysis.txt: number of reads corresponding to potential affected splicing sites;
- effect_vector_combined.txt: location of mutations (including deletions, insertions, and substitutions) with respect to the reference amplicon;
- effect_vector_deletion.txt : location of deletions;
- effect_vector_insertion.txt: location of insertions;
- effect_vector_substitution.txt: location of substitutions.
- position_dependent_vector_avg_insertion_size.txt: average length of the insertions for each position.
- position_dependent_vector_avg_deletion_size.txt: average length of the deletions for each position.
- indel_histogram.txt: processed data used to generate figure 1 in the output report.
- insertion_histogram.txt: processed data used to generate the insertion histogram in figure 3 in the output report.
- deletion_histogram.txt: processed data used to generate the deletion histogram in figure 3 in the output report.
- substitution_histogram.txt: processed data used to generate the substitution histogram in figure 3 in the output report.
In order to help you to familiarize with the output of CRISPResso we provide several precomputed analyses, using the standard settings, for different simulated sequencing datasets with sequencing artifact modeled after the Illumina Miseq platform (using the ART simulation tool: http://www.niehs.nih.gov/research/resources/software/biostatistics/art/ ) and with known editing efficiency and mutagenesis profile:
- 1000 unmodified reads: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_unmodified_amplicon_MISEQ_ERROR_WINDOW_1bp.zip
- 1000 unmodified reads, 1000 reads with 1 substitution: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_amplicon_1_substitution_MISEQ_ERROR_WINDOW_1bp.zip
- 1000 unmodified reads, 1000 reads with 2 substitutions: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_amplicon_2_substitution_MISEQ_ERROR_WINDOW_1bp.zip
- 1000 unmodified reads, 1000 reads with 3 substitutions: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_amplicon_3_substitution_MISEQ_ERROR_WINDOW_1bp.zip
- 1000 unmodified reads, 1000 reads with an insertion of 5 bp: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_amplicon_5_ins_MISEQ_ERROR_WINDOW_1bp.zip
- 1000 unmodified reads, 1000 reads with an insertion of 10 bp: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_amplicon_10_ins_MISEQ_ERROR_WINDOW_1bp.zip
- 1000 unmodified reads, 1000 reads with an insertion of 50 bp: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_amplicon_50_ins_MISEQ_ERROR_WINDOW_1bp.zip
- 1000 unmodified reads, 1000 reads with a deletion of 5 bp: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_amplicon_5_del_MISEQ_ERROR_WINDOW_1bp.zip
- 1000 unmodified reads, 1000 reads with a deletion of 10 bp: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_amplicon_10_del_MISEQ_ERROR_WINDOW_1bp.zip
- 1000 unmodified reads, 1000 reads with a deletion of 50 bp: http://crispresso.rocks/static/examples/CRISPResso_on_SIMULATION_amplicon_50_del_MISEQ_ERROR_WINDOW_1bp.zip
CRISPRessoPooled is a utility to analyze and quantify targeted sequencing CRISPR/Cas9 experiments involving sequencing libraries with pooled amplicons. One common experimental strategy is to pool multiple amplicons (e.g. a single on-target site plus a set of potential off-target sites) into a single deep sequencing reaction (briefly, genomic DNA samples for pooled applications can be prepared by first amplifying the target regions for each gene/target of interest with regions of 150-400bp depending on the desired coverage. In a second round of PCR, with minimized cycle numbers, barcode and adaptors are added. With optimization, these two rounds of PCR can be merged into a single reaction. These reactions are then quantified, normalized, pooled, and undergo quality control before being sequenced). CRISPRessoPooled demultiplexes reads from multiple amplicons and runs the CRISPResso utility with appropriate reads for each amplicon separately.
Installation
CRISPRessoPooled is installed automatically during the installation of CRISPResso, but to use it two additional programs must be installed:
samtools: http://samtools.sourceforge.net/
bowtie2: http://bowtie-bio.sourceforge.net/bowtie2
To install these tools please refer to their documentation.
Usage
This tool can run in 3 different modes:
Amplicons mode: Given a set of amplicon sequences, in this mode the tool demultiplexes the reads, aligning each read to the amplicon with best alignment, and creates separate compressed FASTQ files, one for each amplicon. Reads that do not align to any amplicon are discarded. After this preprocessing, CRISPResso is run for each FASTQ file, and separated reports are generated, one for each amplicon.
To run the tool in this mode the user must provide:
- Paired-end reads (two files) or single-end reads (single file) in [FASTQ format ](http://en.wikipedia.org/wiki/FASTQ_format)(fastq.gz files are also accepted)
- A description file containing the amplicon sequences used to enrich regions in the genome and some additional information. In particular, this file, is a tab delimited text file with up to 5 columns (first 2 columns required):
- AMPLICON_NAME: an identifier for the amplicon (must be unique).
- AMPLICON_SEQUENCE: amplicon sequence used in the design of the experiment.
- sgRNA_SEQUENCE (OPTIONAL): sgRNA sequence used for this amplicon without the PAM sequence. If not available, enter NA.
- EXPECTED_AMPLICON_AFTER_HDR (OPTIONAL): expected amplicon sequence in case of HDR. If more than one, separate by commas and not spaces. If not available, enter NA.
- CODING_SEQUENCE (OPTIONAL): Subsequence(s) of the amplicon corresponding to coding sequences. If more than one, separate by commas and not spaces. If not available, enter NA.
A file in the right format should look like this:
Site1 CACACTGTGGCCCCTGTGCCCAGCCCTGGGCTCTCTGTACATGAAGCAAC CCCTGTGCCCAGCCC NA NA
Site2 GTCCTGGTTTTTGGTTTGGGAAATATAGTCATC NA GTCCTGGTTTTTGGTTTAAAAAAATATAGTCATC NA
Site 3 TTTCTGGTTTTTGGTTTGGGAAATATAGTCATC NA NA GGAAATATA
Note: no column titles should be entered. Also the colors here are used only for illustrative purposes and in a plain text file will be not be present and saved.
The user can easily create this file with any text editor or with spreadsheet software like Excel (Microsoft), Numbers (Apple) or Sheets (Google Docs) and then save it as tab delimited file.
Example:
CRISPRessoPooled -r1 SRR1046762\_1.fastq.gz -r2 SRR1046762\_2.fastq.gz -f AMPLICONS\_FILE.txt --name ONLY\_AMPLICONS\_SRR1046762 --gene\_annotations gencode\_v19.gzThe output of CRISPRessoPooled Amplicons mode consists of:
- REPORT_READS_ALIGNED_TO_AMPLICONS.txt: this file contains the
same information provided in the input description file, plus some
additional columns:
- Demultiplexed_fastq.gz_filename: name of the files containing the raw reads for each amplicon.
- n_reads: number of reads recovered for each amplicon.
- A set of fastq.gz files, one for each amplicon.
- A set of folders, one for each amplicon, containing a full CRISPResso report.
- SAMPLES_QUANTIFICATION_SUMMARY.txt: this file contains a summary of the quantification and the alignment statistics for each region analyzed (read counts and percentages for the various classes: Unmodified, NHEJ, point mutations, and HDR).
- CRISPRessoPooled_RUNNING_LOG.txt: execution log and messages for the external utilities called.
Genome mode: In this mode the tool aligns each read to the best location in the genome. Then potential amplicons are discovered looking for regions with enough reads (the default setting is to have at least 1000 reads, but the parameter can be adjusted with the option --min_reads_to_use_region). If a gene annotation file from UCSC is provided, the tool also reports the overlapping gene/s to the region. In this way it is possible to check if the amplified regions map to expected genomic locations and/or also to pseudogenes or other problematic regions. Finally CRISPResso is run in each region discovered.
To run the tool in this mode the user must provide:
- Paired-end reads (two files) or single-end reads (single file) in [FASTQ format ](http://en.wikipedia.org/wiki/FASTQ_format)(fastq.gz files are also accepted)
- The full path of the reference genome in bowtie2 format (e.g. /homes/luca/genomes/human_hg19/hg19). Instructions on how to build a custom index or precomputed index for human and mouse genome assembly can be downloaded from the bowtie2 website: http://bowtie-bio.sourceforge.net/bowtie2/index.shtml.
- Optionally the full path of a gene annotations file from UCSC. The user can download this file from the UCSC Genome Browser ( http://genome.ucsc.edu/cgi-bin/hgTables?command=start ) selecting as table "knowGene", as output format "all fields from selected table" and as file returned "gzip compressed". (e.g. like: homes/luca/genomes/human_hg19/gencode_v19.gz)
Example:
CRISPRessoPooled -r1 SRR1046762\_1.fastq.gz -r2 SRR1046762\_2.fastq.gz -x /gcdata/gcproj/Luca/GENOMES/hg19/hg19 --name ONLY\_GENOME\_SRR1046762 --gene\_annotations gencode\_v19.gzThe output of CRISPRessoPooled Genome mode consists of:
- REPORT_READS_ALIGNED_TO_GENOME_ONLY.txt: this file contains the list of all the regions discovered, one per line with the following information:
- chr_id: chromosome of the region in the reference genome.
- bpstart: start coordinate of the region in the reference genome.
- bpend: end coordinate of the region in the reference genome.
- fastq_file: location of the fastq.gz file containing the reads mapped to the region.
- n_reads: number of reads mapped to the region.
- sequence: the sequence, on the reference genome for the region.
MAPPED_REGIONS (folder): this folder contains all the fastq.gz files for the discovered regions.
A set of folders with the CRISPResso report on the regions with enough reads.
SAMPLES_QUANTIFICATION_SUMMARY.txt: this file contains a summary of the quantification and the alignment statistics for each region analyzed (read counts and percentages for the various classes: Unmodified, NHEJ, point mutations, and HDR).
CRISPRessoPooled_RUNNING_LOG.txt: execution log and messages for the external utilities called.
This running mode is particular useful to check if there are mapping artifacts or contaminations in the library. In an optimal experiment, the list of the regions discovered should contain only the regions for which amplicons were designed.
Mixed mode (Amplicons + Genome): in this mode, the tool first aligns reads to the genome and, as in the Genome mode, discovers aligning regions with reads exceeding a tunable threshold. Next it will align the amplicon sequences to the reference genome and will use only the reads that match both the amplicon locations and the discovered genomic locations, excluding spurious reads coming from other regions, or reads not properly trimmed. Finally CRISPResso is run using each of the surviving regions.
To run the tool in this mode the user must provide:
- Paired-end reads (two files) or single-end reads (single file) in [FASTQ format ](http://en.wikipedia.org/wiki/FASTQ_format)(fastq.gz files are also accepted)
- A description file containing the amplicon sequences used to enrich regions in the genome and some additional information (as described in the Amplicons mode section).
- The reference genome in bowtie2 format (as described in Genome mode section).
- Optionally the gene annotations from UCSC (as described in Genome mode section).
Example:
CRISPRessoPooled -r1 SRR1046762\_1.fastq.gz -r2 SRR1046762\_2.fastq.gz -f AMPLICONS\_FILE.txt -x /gcdata/gcproj/Luca/GENOMES /hg19/hg19 --name AMPLICONS\_AND\_GENOME\_SRR1046762 --gene\_annotations gencode\_v19.gzThe output of CRISPRessoPooled Mixed Amplicons + Genome mode consists of these files:
- REPORT_READS_ALIGNED_TO_GENOME_AND_AMPLICONS.txt: this file
contains the same information provided in the input description
file, plus some additional columns:
- Amplicon_Specific_fastq.gz_filename: name of the file containing the raw reads recovered for the amplicon.
- n_reads: number of reads recovered for the amplicon.
- Gene_overlapping: gene/s overlapping the amplicon region.
- chr_id: chromosome of the amplicon in the reference genome.
- bpstart: start coordinate of the amplicon in the reference genome.
- bpend: end coordinate of the amplicon in the reference genome.
- Reference_Sequence: sequence in the reference genome for the region mapped for the amplicon.
- MAPPED_REGIONS (folder): this folder contains all the fastq.gz files for the discovered regions.
- A set of folders with the CRISPResso report on the amplicons with enough reads.
- SAMPLES_QUANTIFICATION_SUMMARY.txt: this file contains a summary of the quantification and the alignment statistics for each region analyzed (read counts and percentages for the various classes: Unmodified, NHEJ, point mutations, and HDR).
- CRISPRessoPooled_RUNNING_LOG.txt: execution log and messages for the external utilities called.
The Mixed mode combines the benefits of the two previous running modes. In this mode it is possible to recover in an unbiased way all the genomic regions contained in the library, and hence discover contaminations or mapping artifacts. In addition, by knowing the location of the amplicon with respect to the reference genome, reads not properly trimmed or mapped to pseudogenes or other problematic regions will be automatically discarded, providing the cleanest set of reads to quantify the mutations in the target regions with CRISPResso.
If the focus of the analysis is to obtain the best quantification of editing efficiency for a set of amplicons, we suggest running the tool in the Mixed mode. The Genome mode is instead suggested to check problematic libraries, since a report is generated for each region discovered, even if the region is not mappable to any amplicon (however, his may be time consuming). Finally the Amplicon mode is the fastest, although the least reliable in terms of quantification accuracy.
CRISPRessoWGS is a utility for the analysis of genome editing experiment from whole genome sequencing (WGS) data. CRISPRessoWGS allows exploring any region of the genome to quantify targeted editing or potentially off-target effects.
Installation
CRISPRessoWGS is installed automatically during the installation of CRISPResso, but to use it two additional programs must be installed:
samtools: http://samtools.sourceforge.net/
bowtie2: http://bowtie-bio.sourceforge.net/bowtie2
To install these tools please refer to their documentation.
To run CRISPRessoWGS you must provide:
- A genome aligned BAM file. To align reads from a WGS experiment to the genome there are many options available, we suggest using either Bowtie2 (**<http://bowtie-bio.sourceforge.net/bowtie2/>) or **BWA (**<http://bio-bwa.sourceforge.net/>).**
- A FASTA file containing the reference sequence used to align the reads and create the BAM file (the reference files for the most common organism can be download from UCSC: http://hgdownload.soe.ucsc.edu/downloads.html. Download and uncompress only the file ending with .fa.gz, for example for the last version of the human genome download and uncompress the file hg38.fa.gz)
- Descriptions file containing the coordinates of the regions to
analyze and some additional information. In particular, this file is
a tab delimited text file with up to 7 columns (4 required):
- chr_id: chromosome of the region in the reference genome.
- bpstart: start coordinate of the region in the reference genome.
- bpend: end coordinate of the region in the reference genome.
- REGION_NAME: an identifier for the region (must be unique).
- sgRNA_SEQUENCE (OPTIONAL): sgRNA sequence used for this genomic segment without the PAM sequence. If not available, enter NA.
- EXPECTED_SEGMENT_AFTER_HDR (OPTIONAL): expected genomic segment sequence in case of HDR. If more than one, separate by commas and not spaces. If not available, enter NA.
- CODING_SEQUENCE (OPTIONAL): Subsequence(s) of the genomic segment corresponding to coding sequences. If more than one, separate by commas and not spaces. If not available, enter NA.
> A file in the right format should look like this:
chr1 65118211 65118261 R1 CTACAGAGCCCCAGTCCTGG NA NA
chr6 51002798 51002820 R2 NA NA NA
Note: no column titles should be entered. As you may have noticed this file is just a BED file with extra columns. For this reason a normal BED file with 4 columns, is also accepted by this utility.
- Optionally the full path of a gene annotations file from UCSC. You can download the this file from the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgTables?command=start) selecting as table "knowGene", as output format "all fields from selected table" and as file returned "gzip compressed". (something like: homes/luca/genomes/human_hg19/gencode_v19.gz)
Example:
CRISPRessoWGS -b WGS/50/50\_sorted\_rmdup\_fixed\_groups.bam -f WGS\_TEST.txt -r /gcdata/gcproj/Luca/GENOMES/mm9/mm9.fa --gene\_annotations ensemble\_mm9.txt.gz --name CRISPR\_WGS\_SRR1542350The output from these files will consist of:
REPORT_READS_ALIGNED_TO_SELECTED_REGIONS_WGS.txt: this file contains the same information provided in the input description file, plus some additional columns:
- sequence: sequence in the reference genome for the region specified.
- gene_overlapping: gene/s overlapping the region specified.
- n_reads: number of reads recovered for the region.
- bam_file_with_reads_in_region: file containing only the subset of the reads that overlap, also partially, with the region. This file is indexed and can be easily loaded for example on IGV for visualization of single reads or for the comparison of two conditions. For example, in the figure below (fig X) we show reads mapped to a region inside the coding sequence of the gene Crygc subjected to NHEJ (CRISPR_WGS_SRR1542350) vs reads from a control experiment (CONTROL_WGS_SRR1542349).
- fastq.gz_file_trimmed_reads_in_region: file containing only the subset of reads fully covering the specified regions, and trimmed to match the sequence in that region. These reads are used for the subsequent analysis with CRISPResso.
ANALYZED_REGIONS (folder): this folder contains all the BAM and FASTQ files, one for each region analyzed.
A set of folders with the CRISPResso report on the regions provided in input with enough reads (the default setting is to have at least 10 reads, but the parameter can be adjusted with the option
--min_reads_to_use_region).
CRISPRessoPooled_RUNNING_LOG.txt: execution log and messages for the external utilities called.
This utility is particular useful to investigate and quantify mutation frequency in a list of potential target or off-target sites, coming for example from prediction tools, or from other orthogonal assays.
CRISPRessoCompare is a utility for the comparison of a pair of CRISPResso analyses. CRISPRessoCompare produces a summary of differences between two conditions, for example a CRISPR treated and an untreated control sample (see figure below). Informative plots are generated showing the differences in editing rates and localization within the reference amplicon,
Installation
CRISPRessoCompare is installed automatically during the installation of CRISPResso
To run CRISPRessoCompare you must provide:
- Two output folders generated with CRISPResso using the same reference amplicon and settings but on different datasets.
- Optionally a name for each condition to use for the plots, and the name of the output folder
Example:
CRISPRessoCompare -n1 "VEGFA CRISPR" -n2 "VEGFA CONTROL" -n VEGFA_Site_1_SRR10467_VS_SRR1046787 CRISPResso_on_VEGFA_Site_1_SRR1046762/ CRISPResso_on_VEGFA_Site_1_SRR1046787/
The output will consist of:
- Comparison_Efficiency.pdf: a figure containing a comparison of the edit frequencies for each category (NHEJ, MIXED NHEJ-HDR and HDR) and as well the net effect subtracting the second sample (second folder in the command line) provided in the analysis from the first sample (first folder in the command line).
- Comparison_Combined_Insertion_Deletion_Substitution_Locations.pdf: a figure showing the average profile for the mutations for the two samples in the same scale and their difference with the same convention used in the previous figure (first sample – second sample).
- CRISPRessoCompare_RUNNING_LOG.txt: detailed execution log.
CRISPRessoPooledWGSCompare is an extension of the CRIPRessoCompare utility allowing the user to run and summarize multiple CRISPRessoCompare analyses where several regions are analyzed in two different conditions, as in the case of the CRISPRessoPooled or CRISPRessoWGS utilities.
Installation
CRISPRessoPooledWGSCompare is installed automatically during the installation of CRISPResso.
To run CRISPRessoPooledWGSCompare you must provide: 1. Two output folders generated with CRISPRessoPooled or CRISPRessoWGS using the same reference amplicon and settings but on different datasets. 2. Optionally a name for each condition to use for the plots, and the name of the output folder
Example:
CRISPRessoPooledWGSCompare CRISPRessoPooled_on_AMPLICONS_AND_GENOME_SRR1046762/ CRISPRessoPooled_on_AMPLICONS_AND_GENOME_SRR1046787/ -n1 SRR1046762 -n2 SRR1046787 -n AMPLICONS_AND_GENOME_SRR1046762_VS_SRR1046787The output from these files will consist of: 1. COMPARISON_SAMPLES_QUANTIFICATION_SUMMARIES.txt: this file contains a summary of the quantification for each of the two conditions for each region and their difference (read counts and percentages for the various classes: Unmodified, NHEJ, MIXED NHEJ-HDR and HDR). 2. A set of folders with CRISPRessoCompare reports on the common regions with enough reads in both conditions. 3. CRISPRessoPooledWGSCompare_RUNNING_LOG.txt: detailed execution log.
If you use CRISPResso in your work please cite:
Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, Yuan GC. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol. 2016 Jul 12;34(7):695-697. doi: 10.1038/nbt.3583. PubMed PMID: 27404874.
We are grateful to Feng Zhang and David Scott for useful feedback and suggestions; the FAS Research Computing Team, in particular Daniel Kelleher, for great support in hosting the web application of CRISPResso; and Sorel Fitz-Gibbon from UCLA for help in sharing data. Finally, we thank all members of the Guo-Cheng Yuan lab for testing the software.