
Abstract
The Wnt signaling pathway plays key roles in differentiation and development and alterations in this signaling pathway are causally associated with numerous human diseases. While several laboratories were examining roles for Wnt signaling in skeletal development during the 1990s, interest in the pathway rose exponentially when three key papers were published in 2001–2002. One report found that loss of the Wnt co-receptor, Low-density lipoprotein related protein-5 (LRP5), was the underlying genetic cause of the syndrome Osteoporosis pseudoglioma (OPPG). OPPG is characterized by early-onset osteoporosis causing increased susceptibility to debilitating fractures. Shortly thereafter, two groups reported that individuals carrying a specific point mutation in LRP5 (G171V) develop high-bone mass. Subsequent to this, the causative mechanisms for these observations heightened the need to understand the mechanisms by which Wnt signaling controlled bone development and homeostasis and encouraged significant investment from biotechnology and pharmaceutical companies to develop methods to activate Wnt signaling to increase bone mass to treat osteoporosis and other bone disease. In this review, we will briefly summarize the cellular mechanisms underlying Wnt signaling and discuss the observations related to OPPG and the high-bone mass disorders that heightened the appreciation of the role of Wnt signaling in normal bone development and homeostasis. We will then present a comprehensive overview of the core components of the pathway with an emphasis on the phenotypes associated with mice carrying genetically engineered mutations in these genes and clinical observations that further link alterations in the pathway to changes in human bone.
Similar content being viewed by others
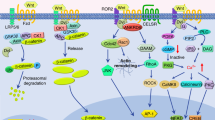


Overview of Wnt signal transduction
Wnts are a family of 19 mammalian proteins characterized by a conserved pattern of cysteine residues (1). Wnts can activate several signaling pathways after binding to their cognate receptors, however, the bulk of this review will focus on the best characterized of these pathways, the so-called canonical pathway (Figure 1). This pathway results in the stabilization of the β-catenin protein and subsequent transactivation of target genes (2). It is initiated by Wnt ligands binding to a receptor complex that includes a member of the Frizzled (Fzd) family of seven-transmembrane receptors and either Lrp5, or the related Lrp6 protein (3). Engagement of this complex results in the phosphorylation of the cytoplasmic domain of Lrp5 or Lrp6, creating a binding site for the Axin protein. In the absence of an upstreamsignal, Axin exists in a multiprotein complex that also includes Adenomatous polyposis coli (APC) and the serine/threonine protein kinase, Glycogen synthase kinase 3 α/β. This complex facilitates β-catenin for GSK3-dependent phosphorylation, targeting it for ubiquitin-dependent proteolysis. Binding of Axin to phosphorylated Lrp5/6 inhibits the ability of this complex to induce β-catenin degradation. The increased levels of cytoplasmic β-catenin can eventually enter the nucleus (4,5) and increase target gene transcription.

Overview of Wnt/β-catenin signaling. Production and secretion of Wnt ligands is dependent on the Porcupine-dependent lipid modification of Wnt and the presence of Wntless to facilitate the transport of lipid-modified Wnt to the plasma membrane (10). Once secreted, Wnt proteins bind to a receptor complex that includes either Lrp5 or Lrp6 and a member of the Frizzled family of seven-transmembrane receptors (3). In the absence of a Wnt ligand, a multiprotein complex, which includes GSK3, Axin, and APC, facilitates Casein kinase 1 (CKI)-primed and GSK3-dependent phosphorylation of β-catenin, targeting it for proteolytic degradation via the E3-ubiquitin ligase β-TrCP (11). Activation of the Wnt receptor complex leads to the activation of Dishevelled (Dvl) and the phosphorylation of the cytoplasmic domain of Lrp5/6 leading to the recruitment of Axin to the plasma membrane. This inhibits the processes, which induce the degradation of β-catenin, leading to increased accumulation in the cytoplasm. β-catenin can subsequently enter the nucleus where it can bind to members of the LEF/TCF family and activate target gene transcription via the recruitment of factors such as BCL9, Pygopus, and Parafibromin (a component of the PAF complex) to target gene promoters (12, 13). In the absence of β-catenin nuclear localization, TCF/LEF proteins can associate with members of the Groucho family to facilitate transcriptional repression. β-catenin also plays a key role in mediating cellular adhesion via its interactions with Cadherins. Finally, several extracellular inhibitors of this process, such as Dkks, Sost, and potentially Kremen, can negatively regulate the formation of the Wnt receptor complex.
Built onto this core pathway are a large number of proteins that modulate activation of the pathway. For example, several extracellular proteins such as Dkks (6), Sclerostin (7), and Secreted frizzled-related proteins (Sfrps) (8) inhibit formation of the Wnt-Lrp5/6-Fzd receptor complex by various mechanisms. In addition, cadherindependent cell adhesion, which links cell-cell contacts to the cytoskeleton (Figure 1), may also play key roles in mediating Wnt/β-catenin signaling due to the key role that β-catenin plays in this process (9).
Finally, the mechanisms necessary for the production and secretion of Wnt from cells have recently become better defined (14). This process is dependent on at least two genes, Porcupine (Porcn) and Wntless (Wls). Porcupine is an acyltransferase that mediates the addition of a lipid modification to all mammalian Wnts, while Wls binds to lipid-modified Wnts and facilitates their transport to the plasma membrane for secretion.
Several key observations emphasized the importance of Wnt signaling in skeletal homeostasis
While several groups had been studying the role of Wnt signaling in skeletal patterning and development during the 1990s, it was arguably the demonstration that mutations in human genes encoding components of the pathway that intensified the interest of the skeletal biology field in this pathway. Due to their conceptual importance, we will briefly overview these developments from the early part of the last decade.
Osteoporosis pseudoglioma and high-bone mass phenotypes linked to LRP5 mutations
Osteoporosis pseudoglioma syndrome (OPPG) (259770, OMIM 2012) is a rare, recessive disorder characterized by severe juvenile osteoporosis andearly-onset blindness related to hyperplastic primary vitreous (15). Additional abnormalities may include short stature, abnormal bone shape and frequent bone fracture. OPPG can be readily distinguished from other childhood bone-fragility disorders by normal collagen synthesis, hormone level, and calcium level (16). The OPPG gene was originally mapped to chromosome 17q12-13 (16), followed by the identification of the Wnt co-receptor, LRP5, as the target of the causative mutation underlying OPPG (17). Many subsequent studies have identified additionalmissense, nonsense, and splice-site mutations in LRP5 that result in OPPG (18–24). The diverse causative mutations are consistent with OPPG being due to a loss of Lrp5 signaling, as opposed to a gain in function. Carriers of OPPG-related LRP5 alleles have lower bone mineral density and increased incidence of bone fracture (17,25).
Further support for the role of LRP5 in bone growth was provided in 2002 when two groups independently reported that a point mutation in LRP5 (G171V) was present in affected individuals in families displaying an autosomally dominant high bone mass trait (26, 27). In one of these reports, data was presented suggesting that disruptions in the ability of Dkk1 to inhibit LRP5induced canonical Wnt signaling may underlie the phenotypes (26).
Sclerosteosis
Sclerosteosis (269500, OMIM 2012) is a rare disorder most commonly seen in the Afrikaner population of South Africa. Sclerosteosis is characterized by osteopetrosis and syndactyly, with the latter being the diagnostic discriminating between sclerosteosis and other sclerosing bone dysplasias (28). Additional features of this disorder include gigantism, facial hypoplasia, resistance to bone fracture, and gradual deafness and facial paralysis due to bony encroachmenton the ear canal and cranial nerves with age (28). The first sclerosteosis gene was mapped to 17q12-21 (29), with subsequent identification of both nonsense and splice mutation alleles of a novel gene the authors named sclerostin (SOST); although the authors acknowledge a U.S. patent application, PCT/US99/27990, which had named this gene BEER (30). Additional nonsense (31), missense (32), and frameshift (33) mutations in SOST have been identified. While the original characterization of the SOST protein suggested that it could bind bone morphogenetic proteins (BMP), thereby functioning as a BMP antagonist (34,35), another study revealed no influence of SOST on classical BMP signaling in vitro (36). SOST was then shown to inhibit Wnt signaling by interacting with LRP5/6 (37,38) and potentially with LRP4 (39). The role of the SOST-LRP4 interaction in sclerosteosis may be particularly relevant because two LRP4 mutations were identified in unrelated sclerosteosis patients from the Mediterranean who had normal SOST gene structure (39–41) (614305, OMIM 2012). These mutations reduced the SOST-LRP4 interaction, and it was shown that LRP4 is necessary for SOST's inhibitory effect on bone formation (39).
van Buchem syndrome
van Buchem syndrome (VBCH), also known as hyperostosis corticalis generalisata (239100, OMIM), is an autosomal recessive disorder most commonly seen in the Dutch. Like sclerosteosis, VBCH is a progressive bone dysplasia producing abnormal bone thickening that can lead to facial nerve palsy and hearing loss from bony encroachment on the cranial nerves and ear canal. Visually, the most pronounced feature of VBCH is the enlargement of the skull and mandible (42). Clinically, the best way to distinguish it from sclerosteosis is the absence of gigantism and syndactyly in VBCH (28,43). Due to their clinical similarities, it is not surprising that VBCH and sclerosteosis were originally mapped to the same genomic location, 17q12-21 (42). However, unlike sclerosteosis, which results from mutations that disrupt the coding region of the SOST gene, the VBCH mutation is due to an Alu recombination-mediated, 52-kb deletion that is 35 kb downstream from SOST (44,45). This region has been shown to regulate the transcription of SOST in osteoblast-like cells (46). While carriers of this mutant allele show lower serum levels of SOST, there does not appear to be an effect on bone mineral density (BMD) (47). A recent study identified an ECR5 enhancer element in this region, and Mef2C is the primary transcription factor binding this site to enhance SOST transcription (48).
It is worth noting that gain-of-function mutations in LRP5 can result in sclerosing bone disorders that resemble VBCH and sclerosteosis (49). Patients may present with greatly elevated BMD, abnormal cranial bone growth, including enlarged jaw and torus palatinus, and bony encroachment on cranial nerves and ear canals. However, there is a clear difference in the pattern of inheritance between these patients and those with VBCH or sclerosteosis:while the SOST-related sclerosing bone disorders require two inactive alleles of SOST, excessive bone growth due to a gain-of-function mutation in LRP5 is phenotypically dominant (26,27,49).
Evidence for specific functions of Wnt signaling components in skeletal development or disease
In this section, we discuss the phenotypes observed in mouse models, and in human patients, that are associated with alterations in many of the core components of the Wnt signaling pathway. In addition, this information is also summarized in Table 2, which is an extension of the information presented in an excellent review recently written by Westendorf and colleagues (50). In addition, because of the widespread use of conditional deletions within the osteochondral lineage to assess Wnt pathway function in skeletal disease, a summary of several key transgenic mouse strains is provided in Table 1.
Wnts
Wnts are secreted, cysteine-rich glycoproteins that are highly insoluble due to a conserved palmitoylation site. While glycosylation may be dispensable for efficient Wnt secretion, palmitoylation by the acyl-transferase porcupine is essential for both Wnt secretion and function (51,52). There are 19 WNTproteins in humans; they signal through binding of one of the frizzled class of receptors in combination with a co-receptor. The specific combination of Wnt + frizzled receptor + co-receptor determines whether the cellular response is β-catenin-dependent (canonical) or independent (non-canonical) (51). Because of their low solubility, Wnts diffuse poorly and are likely to have a limited range of signaling. Effective diffusion likely occurs by association of Wnts with lipoproteins (160) or exosomes (161), but whether either of these secretory modes influences receptor association and signaling is not known. The expression of all 19 Wnts in the limbs of developing mouse embryos has been documented, and it revealed highly regulated expression of each Wnt temporally and spatially (162). This information, combined with the abnormal axis phenotypes seen in numerous Wnt mouse models, highlights the critical importance of Wnt proteins in controlling body patterning during development.
Mouse models support crucial and non-redundant roles of most of the Wnt proteins during embryogenesis. For example, germline deletion of Wnts 1, 3, 3a, 4, 5a, 7b, 9a, 9b, and 11 are all lethal, and deletion of Wnt2 results in decreased viability (66,68,71,163–166). Because Wnts were initially characterized for their importance in body patterning in Drosophila and Xenopus (167, 168), it is not surprising that mutation of individual Wnt proteinsin mice results in either failure of axial body patterning (Wnt1, 3, 3a, 5a, and 8a) (66,71,76,163,165) and/or abnormal limb development (Wnt3, 5a, 7a, 9a) (64,69,71,74). However, most Wnts appear to have at best a redundant rolein body patterning in the mouse; they may have more critical roles in the development of specific organs or tissues (Wnt2, 2b, 4, 5b, 6, 7b, 8b, 9b, 10b, 11, and 16) (78–81,83,166,169–176). While many mice have been created carrying floxed alleles of individual Wnt genes for conditional deletion in specific tissues, most of these have not yet been crossed with mice carrying bone-specific Cre recombinases (64,69,75,78,79,169,170,177–182).
While not all Wnt models have been assessed for specific changes in bone structural qualities, several are clearly important for bone mechanical properties. For example, Wnt3a+/− and Wnt5a+/− mice have reduced BMD, bone volume to tissue volume (BV/TV) ratio, and trabecular number (Tb N) and increased trabecular spacing (Tb Sp) (65), which was traced back to abnormal chondrocyte and osteoblast function in Wnt5a mutants (72). Interestingly, overexpression of a Wnt5a or Wnt5b transgene in chondrocytes via the Col2a1 promoter resulted in a skeletal phenotype similar to that of Wnt5a-null mice: shortened skeletal elements and delayed ossification (72). Although they have not been fully assessed, hypomorphic Wnt3a-vt mice have abnormal development and ossification of vertebrae (67). Interestingly, mice expressing a Col2a1-Wnt4 transgene in cartilage have abnormal chondrocyte zoning and dwarfism but normal BMD (70).
When some Wnts are specifically deleted from bone tissue, there are often decreases in bone quality. For example, removal of Wnt7b in bone by Dermo1-Cre results in decreased skeletal ossification and smaller bones during embryonic development (75). The loss of Wnt9a results in decreased size and mineralization of appendicular long bones, as well as fusion of joints. Whereas Wnt5a seems to have the most profound effect on distal skeletal elements (72), Wnt9a seems important for the development of proximal long bones. Global knock-out of Wnt4 results in no abnormal joint phenotype, but concomitant loss of Wnt4 and Wnt9a exacerbates the joint fusion phenotype seen in Wnt9a knock-outs (69). The role of Wnt9a in joint formation was further supported by the finding of loss of chondrocytes and reduced cartilage in mice when a Wnt9a transgene was overexpressed in chondrocytes via a Col2a1 promoter (77).
Overexpression of a Wnt10b transgene with the Fabp4 adipocyte-specific promoter enhanced trabecular bone formation and the mechanical properties of long bones. This is likely a consequence of expression from the bone marrow compartment (81). Further evidence to support Wnt10b's role in bone development comes from knock-out mice, which have a 30% reduction in bone volume and BMD at 8 weeks of age. This was coupled to a reduction in serum osteocalcin (OC) but no change in TRAP5b, which suggests a loss of osteoblasts and no change in osteoclasts (81). Wnt16-null mice, while appearing to have normal skeletal development and structure, have significantly reduced cortical bone thickness and strength at 24 weeks of age (83).
Several human disorders have been directly linked to mutations in various WNTproteins. Many of these reflect the abnormal skeletal development in the Wnt mouse models. A Gln83X mutation in WNT3 has been identified in tetraamelia, autosomal recessive (OMIM: 273395). This disorder produces malformed heads, failure to develop limbs, cleft palate, and pelvic hypoplasia (183). Two autosomal dominant mutations affecting WNT5a (Cys182Arg and Cys83Ser) have been identified in Robinow syndrome (OMIM: 180700). Robinow patients have hypertelorism, craniofacial dysmorphism, short stature, and mesomelic shortening of the limbs (184). Two mutations in WNT7a result in similar, but clinically distinct disorders. The first is Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome (Arg292Cys or Gly204Ser) (OMIM: 276820), which results in absence of the ulna and fibula and severe limb deficiencies including truncated limbs and loss of digits (185,186). The second is Fuhrmann syndrome (Ala109Thr) (OMIM: 228930), which is less severe and causes bowing of the femurs, aplasia or hypoplasia of the fibula, and poly-, syn-, or oligo-dactyly (185).
While no mouse model currently exists for WNT10a, several mutations have been identified that result in odontoonychodermal dysplasia (OMIM: 257980) (187–190). These patients have several distinguishable malformations including dystrophic nails, malformed or undeveloped teeth, and hyperhidrosis of hands and feet. Less severe mutations of WNT10a (Asp217Asn and Gly95Lys) lead to tooth agenesis, selective 4 (OMIM: 150400), which produces loss or truncation of the lateral incisors (189,190). Mutations in WNT10b result in splithand/root malformation 6 (OMIM: 225300). Both missense (Arg332-Trp) and frameshift (4-BP DUP, 458AGCA) mutations have been identified in this disorder, which presents with syndactyly, median clefts of the hands or feet, and aplasia or hypoplasia of the phalanges, metacarpals, and metatarsals. Additional abnormalities include mental retardation, ectodermal, craniofacial, and orofacial clefting (191,192). While no specific disorder has been linked to WNT16, individual SNPs have been identified in the WNT16 gene that are associated with cortical bone thickness, BMD, and fracture risk (83).
Porcupine and Wntless
The mechanism of Wnt secretion has been a hot topic in the literature, and two proteins have been identified that are essential for the process. One is porcupine (Porcn), an ER-resident O-acyltransferase that is specifically involved in the palmitoylation of Wnts (193). The second is Wntless (Wls; aka Evi, Sprinter, or Gpr177) which serves as a Wnt chaperone (194). Proper function of both of these proteins is necessary for Wnts to be secreted; the absence of either Porcn or Wls results in the intracellular accumulation of Wnt proteins (51,52,193). Both Porcn and Wls mouse models produce severe phenotypes that are reminiscent of several Wnt knock-out mice, further supporting the concept that both Porcn and Wls have crucial and non-redundant roles in Wnt signaling (84–87,195–197).
Porcn is a member of the MBOAT family, having a low sequence similarity (<35%) to lysophospholipid acyltransferases (198). The gene lies on the X-chromosome, and mutations that disrupt the function of PORCN are male embryonic lethal and cause the female X-linked dominant disorder known as focal dermal hypoplasia (305600, OMIM 2012) (199). The structure of Wls is not conclusively defined, but it is predicted to be a multi-pass transmembrane protein (194). Wls is found in both the Golgi and the ER, and it is recycled via endosomes (200). Recognition of Wnts by Wls depends upon palmitoylation of Wnt by Porcn (200). The palmitoleate-binding domain of PORCN is believed to reside within amino acids 43–200, because loss of these residues abolishes WNT-WLS interactions (201). The mechanism of how Wnts dissociate from Wls upon reaching the cell surface has not been determined, but one study suggests that changes in pH may play a role (201). Crystal structures would greatly assist in answering these questions.
The loss of one functional copy of Porcn during embryogenesis is highly lethal to female mice, but is completely lethal in male embryos by day E15.5. Females that do survive have a spectrum of abnormal phenotypes with varying degrees of penetrance. These results are not surprising due to stochastic epigenetic silencing of one copy of the X-chromosome during female development. Skeletal abnormalities that are frequently seen in these mice include loss or fusions of digits, shortened or truncated limbs and body axis, and cleft palate. Crossing Porcn-floxed mice with mice carrying Prx1-Cre or Msx2-Cre removes Porcn from the developing limb ectoderm, leading to viable male offspring that have severely shortened and malformed limbs (84,85). No studies yet have used bone-specific Cre promoters to examine the role of Porcn in the maintenance of bone quality throughout life.
Homozygous deletion of Wls by homologous recombination, or by using Cre expressed early in embryogenesis to remove a floxed portion of the gene, is embryonic lethal before E10.5, with failure to develop mesoderm by E8.5 (195–197). However, heterozygotes develop without appreciable abnormalities. Interestingly, limb ectoderm-specific loss of Wls driven by Prx1-Cre is more severe than the related Prx1-Cre-Porcn–floxed model: these mice die during weaning due to such severe limb malformations that the mice are unable to acquire food or water. Their limbs are hypoplastic, shortened, and had truncated autopods. Further investigation revealed delayed cartilage hypertrophy and osteogenesis. The use of Msx2-Cre resulted in truncated autopods in all limbs, but also truncated zeugopods in hind limbs. Due to some expression of Msx2-Cre during craniofacial development, there was also a failure of fusion of the skull sutures (87).
Crossing the Wls-floxed mice with those carrying an OC-Cre allele resulted in osteoblast-specific loss of Wls. These mice have normal skeletal development and BMD at birth, but they are prone to spontaneous fractures at a young age. This likely has to do with deficits in bone repair, as there was a significant reduction in BMD by 20 days that became more significant with age. Less than 20% of these mice survive past two months, and only a handful make it to 4 months of age. There was a 90% decrease in BV/TVratio by 7 weeks and a greater than 50% decrease in cortical bone of the femur. The mice showed a progressive loss of trabecular bone, calvarial bone, and cartilage. There was also evidence of decreased osteoblastic activity (fewer osteoblasts and decreased serum OC) and increased osteoclastic activity (increased TRAP staining of femur sections). There was an apparent increase in cartilage degradation, because serum levels of CTX-II were markedly elevated (86).
Recently, mutations in PORCN have been identified as driving the X-linked dominant syndrome known as focal dermal hypoplasia (FDH) or Goltz syndrome (OMIM: 305600). These include both nonsense and missense mutations (13-BP DUP, NT1059; Gly60Arg; Arg124X; Trp74X; and Arg365Gln) (199,202,203). FDH is only seen in heterozygous females, which suggests that a complete loss of PORCN is embryonic lethal in humans. Stochastic X-linked inactivation leads to asymmetrical skeletal deformities such as syndactyly, absence or underdevelopment of digits or extremities, ectrodactyly, and polydactyly in various combinations. Additional abnormalities include osteopathia striata of long bones, scoliosis, and clavicular dysplasia (204). No human disease has yet been directly associated with WLS, but SNPs have been identified within an intron of WLS that are associated with BMD in Asian and European populations (205,206).
Frizzled (Fzd)
The Frizzled receptors form a family of 10 human genes. The proteins are characterized by seven transmembrane domains, an extracellular N-terminal domain that is N-glycosylated, a large extracellular cysteine-rich domain that is responsible for Wnt binding, three intracellular loops, and a cytoplasmic C-terminal tail that contains multiple phosphorylation sites. Fzds have a strong similarity to other G-protein-coupled receptors, and Wnt-mediated G-protein signaling through frizzleds may be important for some non-canonical Wnt signaling pathways (207). Crystallization of Xenopus Wnt8 with Fzd8 revealed a specific hydrophobic binding interaction between the conserved N-terminal lipid “thumb†domain on Wnt8 and a conserved site on Fzd8. The data suggested how specific Wnts may interact with specific Fzds due to interactions between the conserved hydrophobic C-terminal “index finger†domain and a less conserved binding groove on Fzd8 (1).
Although mouse germline deletions exist for Fzd1-6, 8, and 9 (90,201,208–216), only mouse models of Fzd9 deficiency have been thoroughly characterized for skeletal abnormalities (90). The individual Fzd receptor knockouts do not produce profound malformations in body patterning. However, most of the receptors are crucial for organ functioning: several knockouts result in late embryonic (Fzd5 (211)) or post-natal lethality (Fzd2, 3, 4 (201,208,209)). Two of the models do produce skeletal abnormalities; for example, craniofacial cleft palate and hypognathia in Fzd2-null mice (201), and curly tail in Fzd3-null mice (208). Fzd1 and Fzd8 germline deletions produce no overt phenotype (201,215), but further probing is likely to reveal subtle phenotypic consequences, such as the loss of Fzd6 resulting in abnormal hair patterning (214).
Deletion of Fzd9 results in no abnormal phenotype of skeletal development, nor is there a difference in bone volume by 6 weeks, but there is a 35% decrease in trabecular bone volume of the vertebrae by 24 weeks. This was due to lower trabecular number, not a change in trabecular thickness. No difference in osteoclast activity was apparent by TRAP staining, but calcein labeling revealed a significant decrease of bone anabolism and osteoblast surface. Both homozygous-null and heterozygous mutant animals had reduced cortical thickness, lower trabecular bone volume, fewer osteoblasts, decreased bone formation rates, and decreased mechanical resistance (90). As the study authors remarked, just because there is not an overt phenotype does not mean there is no consequence of Fzd loss on bone quality; further investigation into bone phenotypes of the other Fzd models is warranted. No human skeletal disorders have been directly linked to any of the frizzled receptors.
Low-density lipoprotein-related proteins 5 and 6 (Lrp5/6)
Lrp5 and Lrp6 are part of the β-catenin-dependent (canonical) Wnt signaling pathway that is primarily involved in cell differentiation and proliferation (217). Intracellular PPP(S/T)P repeats in the cytomplasmic tail of LRP5 and LRP6 are phosphorylated during Wnt signaling (218,219). These sites can be phosphorylated by numerous kinases including GSK3 (218), GPCR kinase 5 and 6 (220), protein kinase A (PKA) (221), mitogen-activated protein kinase (MAPK) (222), and cyclin-dependent kinase 14 (CDK14) (223). Phosphorylation of these sites then promotes further phosphorylation of neighboring serine residues in PPSPXS sites by members of the casein kinase I (CKI) family (224). This allows the binding of axin and recruitment of the β-catenin destruction complex to the cell membrane (217). Inhibition of canonical Wnt signaling by sclerostin (Sost) and Dickkopf-1 (Dkk1) is mediated by binding to Lrp5/6 (37,225).
Lrp4 does not have an overlapping function with Lrp5/6, and its role in modulating canonical Wnt signaling has only recently begun to be appreciated. Lrp4 binds both Sost and Dkk1 in vitro (92). In addition, Lrp4 enhances the ability of Sost to inhibit canonical Wnt signaling, but it appears to have no influence on Dkk1 activity. This is further supported by the identification of LRP4 mutations in patients with sclerosteosis who have normal SOST function (39). Lrp4 has also been shown to play an important role in bone development, and Lrp4 knock-out models have similar phenotypes as other Wnt pathway knock-outs (93,226).
Several Lrp4 mutant mice that display similar phenotypes have been identified from mutational screens. These mice all display brachydactyly and syndactyly on all limbs, along with shortening of distal segments and multiple fusions. The most interesting observation is that the abnormal digit duplications occur in the dorsoventral axis, which means they are out of plane. While producing similar gross anatomical consequences, digitation anormale (dan) and malformed digits (mdig) mice are viable as homozygotes, but mitten (mitt) and mitaine (mte) mice die at birth (93,94). Mice that carry a mutation that results in the secretion of only the Lrp4 extracellular domain have lower bodyweight, polysyndactyly, and abnormal tooth development with full penetrance. In-depth analyses of bone phenotypes revealed no change in bone shape, but femurs were shorter and have a narrower midshaft. There was a decrease in total BMD, but no change in cortical bone measurements. Vertebrae had reduced trabecular bone mass, and serum markers of osteoblast activity (OC and ALP) and osteoclast activity (DPD) were all elevated, suggesting increased bone turnover (92,226). Interestingly, this model must have retained some Lrp4 activity, because deletion of exon 1 causes perinatal lethality (91). A floxed mouse exists but has not been used to assess skeletal phenotype (227).
Mice carrying a germline homozygous deletion in Lrp5 are viable and fertile. Skeletal development is normal, but low bone mass is seen in juveniles when mice begin developing spontaneous fractures. Heterozygotes have lower bone mass than wild types, but the decrease is not as severe as in the homozygous mutants. Bone formation rate and the number of osteoblasts are decreased, but there is generally no change inthe number of osteoclasts or their activity (99,100). Studies using floxed alleles of Lrp5 that are conditionally deleted in osteoblasts have yielded conflicting results. Deletion of Lrp5 by Col1a1-Cre failed to mimic the low-bone-mass phenotype of Lrp5 knock-out mice. The authors then went on to show that they could reproduce the low-bone-mass phenotype when Lrp5 was conditionally deleted in the intestines via Villin-Cre (98); this was attributed to an increase in circulating serotonin. Another group removed Lrp5 from osteocytes via Dmp1-Cre, which also resulted in a low-bone-mass phenotype. This study also removed Lrp5 from the intestines using Villin Cre, but there was no consequence on bone mass, nor was there any change in circulating serotonin following the use of either Cre (101). One potential difference between these two studies that may be influencing the results is the location of the loxP sites in the Lrp5 locus. In the first study, Yadav et al used an Lrp5 allele that contained loxP sites around exon 6; previously this group had generated and characterized a germline mutant mouse lacking exon 6 and found that deletion of this exon still resulted in the production of a truncated protein product (97). The Lrp5-floxed mouse of Cui et al resulted in the loss of exon 2 as well as a frame shift after Cre recombination (101). It is possible that the truncated protein product produced after Cre recombination in the Yadav et al model has some confounding function. It is also possible that the different versions of the cre transgenic mice may account for some of the observed differences. Elucidation of the underlying reasons for these differences will be a key area of emphasis in the immediate future.
Knock-in models for gain-of-function mutations in Lrp5 have also been produced. Mice containing A213V or G170V mutations mimic two high-bone-mass alleles seen in the human population (A214V and G171V, respectively). To avoid confusion with the literature, we will refer to the two lines by the names of their human homologs. Either mutation results in a global increase in cortical bone mass, BV/TV ratio, and trabecular number and thickness, with a decrease in trabecular spacing. However, while A214V promoted the greatest increase in cortical bone properties, G171V appeared to have the greatest effect on trabecular bone growth. The consequence of this was most apparent in mechanical tests, where the G171V mutation showed enhanced stiffness only in response to axial loading, but not to three-point compression; these characteristics are most influenced by changes to periosteal and endosteal bone structure, respectively (102). Furthermore, the A214V mutation favored increased periosteal bone formation, while the G171V mutation favored endosteal bone formation in response to mechanical loading (228). In further support of the local effect of Lrp5 on bone mass, the use ofPrx1-Cre to drive expression in the appendicular skeleton specifically resulted in high-bone-mass traits in limbs (101). Similar to the Lrp5 conditional knock-out models, there are two conflicting results in the literature: one group found that the high-bone-mass trait is a consequence of intestine-specific expression of mutant Lrp5 and changes in serotonin (98), while the other found that it was a result of mutant Lrp5 expression in bone and that intestinal expression of mutant Lrp5 had no influence on serotonin (101,102).
There is also a mouse generated in an ENU mutation screen that produced a frameshift mutation in exon 23 of Lrp5, which resulted in the replacement of the last 39 amino acids of the C-terminal tail by 20 new amino acids. There was a loss of the last three PPP(S/T)P motifs, which are crucial for Lrp5-mediated canonical Wnt signaling. These mice have been exclusively studied in exudative vitreoretinopathy (229), and they may be interesting in bone studies due to the retention of a fully functioning extracellular domain, which might compete for ligand binding with Lrp6.
Lrp6-null mice die between E14.5 and birth with numerous defects that mimic mutations in other Wnt signaling molecules, such as truncation of the axial skeleton, limb defects, and urogenital malformation (99,100,103). Lrp6+/− mice have normal skeletogenesis and BMD but a reduced BV/TV ratio. Lrp6+/−Lrp5+/− compound heterozygotes have limb abnormalities not seen in either single heterozygotic background, including loss of limbs, synostosis, or reduction of postaxial digits. Lrp6+/−Lrp5−/− mice showed even more extreme limb abnormalities (99). Conditional deletion of Lrp6 in the mesenchyme via Dermo1-Cre revealed mostly normal skeletogenesis with only a slight delay in ossification of the skull at E17.5. A more severe skeletal phenotype became apparent when Lrp5 was concomitantly deleted in the mesenchyme (Lrp5flox/flox Lrp6flox/flox ), which resulted in perinatal mortality. These embryos had reduced body size, cranial malformation, global decreases in ossification, and failure of fusion in the sternum. There were also numerous areas of ectopic cartilage formation. The major skeletal defects were attributed to a lack of osteoblastogenesis characterized by a lack of osterix and osteocalcin expression. Growth plates revealed a loss of hypertrophic chondrocytes (100).
Three Lrp6 mutant mice have been identified during mutational screens. Crooked tail (Cd) is a G494D mutation within the second YWTD-EGF repeat domain that is predicted to disrupt protein-protein interactions. It was shown to prevent Dkk-1-mediated inhibition of canonical Wnt signaling, but not Dkk-1's ability to bind Lrp6. The mutation also appeared to result in decreased interaction with Mesd, which might reduce the ability of the mutant receptor to be displayed on the cell surface. The mutation results in a crooked tail phenotype and exencephaly with incomplete penetrance. Those mice that complete neurulation have severe malformations of caudal vertebrae, including misshapen and fused vertebrae. Evidence suggests a delay in the ossification of digits, but no evidence of increased polysyndactyly (104).
Like Cd mice, ringelschwanz (rs) mutant mice are the result of a spontaneous mutation in Lrp6 (R886W) located in the second YWTD-EGF repeat. As this lies in the third beta propeller region and this residue is highly conserved, this mutation is also predicted to disrupt Dkk interactions with Lrp6. Approximately one-third of rs homozygotes die within a week after birth. Like Cd mice, the pups show malformations of the vertebral column and neural tube, but instead of exencephaly, spina bifida is the predominant disorder. Oligodactyly is also common, with loss of the fifth digit in one or more limbs. rs mice also show evidence of delayed ossification of metatarsal and phalangeal bones. Bone mass in the tibia is reduced, with a 16% decrease in BMD and 29% reduction in cortical bone thickness (107). Another study found a reduction in trabecular bone volume and increased bone resorption along with normal osteoblast number and mineralization.
The rs mutation, like the Cd mutation, decreased the ability of Lrp6 to interact with Mesd (108). A third identified mutation, known as Gwazi (Gw), results in heterozygous mice with crooked tails due to formation of a sesamoid bone between caudal vertebrae (105). The mutation is a D549G amino acid substitution in the last YWTD-EGF repeat of the second propeller domain of Lrp6. The tail kink phenotype is found in 65% of heterozygotes and 85.5% of homozygotes of the Gw allele. The phenotype is fully penetrant in Dkk1+/−Lrp6+/Gw compound heterozygotes but drops to 21.5% in Wnt3+/− Lrp6+/Gw compound heterozygotes, suggesting that the Gw mutation results in a gain of function (106).
As mentioned above, LRP5 function was linked to human skeletal disorders by three seminal papers (17,26,27). Subsequently, families with these and other mutations in LRP5 were identified. The originally identified families with high-bone mass carried Gly171Val mutations (26,27). Another mutation at this codon, Gly171Arg, may be more detrimental, as the family reported severe headaches, which may be indicative of cranial nerve compression (49). Ala242Thr, Ala214Thr, Ala214Val, and Thr253Ile all lead to increased bone density with varied phenotypic consequences (49). Besides OPPG, inactivating mutations in LRP5 are also associated with Familial exudative vitroretinopathy (FEVR) (230), a disease caused by incomplete development of the retinal vasculature. Some FEVR-associated mutations in LRP5, but perhaps not all, are also associated with low bone mass (231–233).
A missense mutation in LRP6 (Arg611Cys) has been identified in LRP6 that causes the complex, metabolic disorder coronary artery disease, autosomal dominant 2 (Arg611Cys) (OMIM: 610947). All individuals that carried a copy of the mutant allele had hyperlipidemia, hyper-tension, and lower bone density. The mutated residue is highly conserved across species and lies within one of the LRP6 EGF domains. In vitro studies have shown that this mutation results in a 48% reduction in canonical Wnt signaling in response to WNT3a (234).
Numerous individual LRP4 mutations have been found in patients with Cenani-Lenz syndactyly syndrome (CLSS) (228). This disorder is characterized by mild face dysmorphisms including a prominent forehead, hypertelorism, and micrognathia. Limb malformations are more severe, including syndactylyl of hands and feet and malformation (and frequent fusion) of the radius, ulna, metacarpals, and metatarsals. LRP4 mutations have also been shown to lead to sclerosteosis 2 (OMIM: 614305) (39). Mutation in the third propeller domain of LRP4 favors sclerosteosis 2, which is less severe than CLSS, presenting with polysyndactyly and thickening of the cortical layer of the skull and long bones.
Dickkopfs (Dkks)
Four unique proteins of the Dickkopf family are encoded in the human genome. All contain an N-terminal signal peptide and two conserved cysteine-rich domains. The greatest conservation between Dkks is in the second CRD. Dkk1 has a single N-glycosylation site near the C-terminus. Dkk4 contains no N-glycans (235). Dkk1 can potentially form a complex on the cell surface between Lrp6 and Kremen, which results in the internalization of Lrp6 and subsequently inhibits canonical Wnt signaling (225). Dkk3 has been shown to have conflicting roles in regulating Wnt signaling depending upon cell type (236–238), and it does not bind or influence the level of Lrp5/6 as do the rest of the Dkk family. Dkk3 may exert its effects through Kremen interactions (239), but this interaction also seems inconsistent (225). For these reasons, Dkk3 is not usually considered a direct modulator of Wnt signaling. Dkk4 expression is a direct target of canonical Wnt signaling and may serve as a negative feedback mechanism (240). Dkk4 may inhibit the canonical WNT pathway, but may favor non-canonical Wnt signaling through JNK (241).
Dkk1-null mice die at birth and have some striking developmental abnormalities. Most malformations become apparent after E9.5, as these mice fail to develop eyes, olfactory placodes, the frontonasal mass, or mandible. All craniofacial features anterior to the ears fail to develop by late gestation. These mice also show fore- and hind-limb malformations, including fusion of digits and ectopic digits (110). Dkk1 heterozygous mutant mice are viable, fertile, and are the same size as Dkk1 wild-type littermates. However, the BV/TV ratio is increased, with an increase in trabecular number and size and a decrease in trabecular spacing. Histomorphometry has revealed that the mineralizing surface and matrix apposition rate are elevated two-fold in Dkk1 heterozygous mice; the number of osteoblasts is increased with no change in osteoclasts. Axial compression tests on the ulna show increased mechanical strength in Dkk1 heterozygous mice, with a higher maximum load and energy (109). Dkk1-floxed mice do exist but have not yet been used to assess skeletal phenoltypes with bone-specific expression of Cre (242). In opposition to what was seen in the Dkk1 mouse models, deletion of Dkk2 results in osteopenia, with major defects in mineral apposition rates. This has been attributed to an increase in the number of osteoclasts (115). No Dkk4 mouse models have been described in the literature. No human skeletal disorders have been linked to any of the DKK family members.
Sclerostin
Sclerostin (SOST) is a secreted WNT inhibitor that prevents Wnt signaling by direct interaction with the LRP4/5/6 co-receptors (37–39). It is 190 amino acids long, glycosylated, and contains a cysteine-knot motif. SOST also contains a heparin binding site, and both the N-terminal (1–55) and C-terminal (145–189) ends are unstructured. It contains three loop domains, the second of which is bound by the SOST-inhibiting antibody ScI-AbI (243). This is the same site responsible for binding to LRP6 (244). Serum levels of SOST increase during fracture healing (245). ScI-AbI speeds fracture healing in mice (246) and non-human primates (116). SOST is expressed by osteocytes (36,247) and articular chondrocytes (248). Polymorphisms in SOST are associated with BMD and fracture risk (249, 250). An ECR5 enhancer element downstream of the SOST coding region is critical for SOST expression and is lost in van Buchem syndrome (44–46,48). This enhancer region is bound by the Mef2 family of transcription factors (48,251).
Because SOST was originally discovered based on its role in sclerosteosis, it is not surprising that Sost mouse models have been well characterized for their bone phenotypes. Sost knock-out mice have accelerated fracture healing with faster callus maturation, mineralization, and enhanced stress resistance (116). There is also increased cortical bone area in long bones (102,117). When compared with high-bone-mass—promoting alleles of Lrp5,Sost-null mice were found to have bone quality changes similar to those of the Lrp5A214V mice, but not to the Lrp5G171V mutation. Similar to what is seen in sclerosteosis, Sost-null mice have a 60–80% increase in cranial parietal thickness and reduced foramen size (102). Mice have also been made that mimic van Buchem syndrome by inserting loxP sites around the ECR5 regulatory domain downstream of Sost. Conditional deletion of this enhancer in osteoblasts by crossing these mice to Col1a1-Cre-expressing mice resulted in a 41% increase in the BV/TV ratio by enhancing trabecular bone growth. These mice have an increased osteoblast surface with no change in osteoclasts. The high-bone-mass phenotype is not as extreme as that of Sost-null mice, supporting the idea that deletion of the regulatory element produces a hypomorphic allele (48). Mice have also been generated that overexpress Sost in osteoblasts due to the expression of a transgene behind an osteocalcin promoter. These mice were osteopenic with disorganized bone architecture, thin cortices, reduced trabecular bone, impaired lamellar bone formation, and chondrodysplasia. Not surprisingly, these bones were more fragile and less resistant to fracture by compression and four-point bending. Calcein labeling revealed a smaller osteoblast surface and decreased bone formation rate, without a change in markers of resorption (34).
As discussed above, numerous mutations have been identified in SOST that lead to sclerosteosis (Gln24X; IVS1DS, A-T, +3 and/or IVS1AS, A-C, −67; Trp124X; and Arg126X) (30,31), and van Buchem syndrome results from the deletion of an enhancer sequence approximately 35kb downstream of SOST (44). In addition to these well characterized but rare disorders, SOST has been implicated in the exceedingly rare craniodiaphyseal dysplasia, autosomal dominant disorder (OMIM: 122860). Mutation of Val21 in the signal peptide of SOST to either Leu or Met significantly diminished SOST secretion. These mutations were documented in two unrelated individuals with this disorder but were not found in samples from healthy patients (252).
Due to the profound effect SOST has on bone mass and quality, this protein has garnished significant attention from scientists and clinicians as a potential therapeutic target for osteoporosis. The anti-SOST antibody (Scl-Ab) has shown very promising results in both human and animal models of low bone mass disorders and fracture healing. To avoid redundancy and to give full attention to the exciting results from Scl-Ab studies, we encourage you to read the recently published and highly thorough review by Ke et al (253).
Secreted frizzled-related proteins (Sfrps)
Secreted frizzled-related proteins (sFRPs), like Dkk proteins, are secreted inhibitors of Wnt signaling. The first discovered was FRZB/SFRP-3, from the cartilage of mammalian long bones during embryonic and fetal development (254), and it was subsequently identified as a Wnt binding protein and an inhibitor of Wnt signaling (255,256). In total, five sFRPs have been identified in mice and humans (257); all five are expressed during mouse limb development (162). The sFRPs and frizzled receptors both have a cysteine-rich domain (CRD). Additionally, sFRPs contain netrin (NTR) domains, in common with netrin, complement proteins, and type I pro-collagen C-proteinase enhancer proteins (258). sFRPs and Wnts are generally expressed in polar opposite regions, which likely helps to establish a Wnt gradient (257). While sFRPs bind Wnts and can inhibit Wnt signaling, they may also be involved in enhancing the range of Wnt diffusion in the ECM when expressed together (257,259). Further, specific sFRP-Wnt interactions control Wnt signaling (260). For example, while Sfrp-1 and −2 can inhibit Wnt3A-stimulated canonical Wnt signaling, Sfrp-3 cannot (261). FRZBb-2/SFRP-4 is highly expressed in bone cells, and its mRNA is highly up-regulated in osteoarthritis cartilage samples (262). SFRP-1 negatively regulates osteoblastogenesis and trabecular bone volume (119), and it also directly binds RANKL and can inhibit osteoclastogenesis (263).
Germline deletion of the Sfrp1gene in mice results in an interesting bone phenotype. Initial inspection revealed no overt gross anatomical phenotype, and the mice were fertile. Additionally, there was no difference at 52 weeks in overall BMD, femoral diaphyseal BMD, thickness, or periosteal and endosteal circumference; femur length was not affected. Most serum biomarkers of bone turnover were normal, except that phosphate was elevated at 20 weeks in males only (but returned to normal in older mice). However, histology of the proximal femur revealed a 149% increase in trabecular bone volume in females and 62% in males compared with wild-type littermates at 35 weeks, whereas there had been no difference at 20 weeks; this suggested a prevention of age-related bone loss. Mineral apposition rate was only significantly increased in females (32%), further supporting influence of sex on phenotype. There was a noticeable decrease in apoptosis rates of osteoblasts and osteocytes in calvaria as determined by TUNEL, with no increase in proliferation (119). Sfrp2-deficient mice were viable, fertile, and had a normal lifespan and no overt physical phenotype. Close inspection revealed shortened metacarpals/metatarsals and phalangeal bones, resulting in significantly shorter digits. Ossification of the phalanges was also delayed. This was determined to be a consequence of reduced hypertrophic chondrocytes, not influenced by changes in apoptosis (120).
Deletion of Sfrp3 similarly resulted in no overt phenotype or changes in serum markers. Mice had no difference in subchondral bone mass, but both male and female mice had increased BMD, thickness, and stiffness in femurs and tibias; neither sex showed changes in measurements of trabecular bone. The most interesting consequence of Sfrp3 deletion was an enhanced anabolic response to ulna loading, even at loads below the threshold of response for wild-type mice (121).
While no skeletal assessment has been made on Sfrp4 knock-out mice, increased expression of Sfrp4 leads to decreases in bone mass. With a Sap-Sfrp4 transgene, mice had smaller increases in BMD and BMC from 5 to 15 weeks relative to wild-type development. This resulted in lower areal BMD and BMC at 15 weeks. Unexpectedly, calcein injection did not show a difference in mineral apposition rates between the transgenic mice and wildtype. There was also no difference in serum OC or CTX levels (123). Similar results were seen with a Col1a1-Sfrp4 transgene (124). The senescence-accelerated mouse P6 (SAMP6), which has lower peak bone mass at 4 months than other SAMPs, has mutations in the Sfrp4 promoter that create a hypermorphic allele of Sfrp4 with 40-fold increased expression. These mice have lower numbers of osteoblasts, and osteoblasts from these mice are less response to Wnt3A induction of canonical Wnt signaling ex vivo (264). No skeletal phenotypes have been assessed in Sfrp5 knock-out mice (265).
No human skeletal disorders have been directly linked to any of the sFRP family members, but SNPs in SFRP3/FRZB have been associated with the risk of osteoarthritis (OA) and osteoporotic fracture (266–268), as well as with hip shape (122). However, the association of these SNPs with OA or osteoporotic fracture is not consistently observed in all studies (269–271). Serum levels of FRZB may also be a marker of rheumatoid arthritis and response to therapy (272). Similarly, SNPs in SFRP4 were found to correlate with hip fracture risk, height, and body composition in Danish males, but the findings were not replicated in a Belgian population (273). Also the rs16879765 polymorphism was found to increase the risk of developing Dupuytren disorder nearly twofold in a Dutch population (274).
Dishevelled
Dishevelled (Dvl) proteins were originally discovered through its crucial role in cell polarity and identity. Loss of the Dvl gene phenocopies loss of the Wnt-1 homolog, Wingless, in Drosophila, supporting a critical role in down stream Wnt signaling (275). In mammals, three dishevelled homologs have been identified: Dvl-1, Dvl-2, and Dvl-3 (276–279). In canonical Wnt signaling, the three share mostly redundant roles (280), but they are involved in both β-catenin-dependent and independent Wnt signaling (281). All mammalian Dvl proteins contain three highly conserved domains. There is an N-terminal DIX domain that allows both homotypic and heterotypic interactions with other DIX-containing proteins such as axin1/2. This domain is necessary for Dvl's promotion of β-catenin accumulation (282). Dvls also contain a PDZ/DLG domain that may promote interaction with APC and frizzled (283,284), and a Dvl/egl-10/pleckstrin (DEP) domain found in several proteins that associate with G-proteins (285) and is critical for Dvl's promotion of JNK activation (286) and membrane localization (287). All three conserved domains, DIX, PDZ, and DEP, are necessary for β-catenin signaling through LEF1 (286).
The C-terminal of the PDZ domain is a conserved stretch of basic residues that functions as a nuclear localization signal in many proteins, but dishevelled has been found to be predominantly cytoplasmic (288). The N-terminal of PDZ contains a proline-rich, putative Src homology 3 (SH3) binding domain (289), which may recruit and promote Tyr654 phosphorylation of β-catenin by Src (290). Wnt/Wingless signaling results in the hyperphosphorylation of Dvl as well as its membrane localization (288,291). The PDZ domain interacts with and is phosphorylated by casein kinase 2 (CK2) during Wnt signaling (292). Dvl promotes dephosphorylation of axin by recruiting protein phosphatase 2C alpha (PP2C) via its PDZ domain to the Dvl-axin-β-catenin complex, which promotes Wnt signaling (293). Dvl is also phosphorylated by casein kinase 1 (CK1) during Wnt signaling (294, 295), which enhances Dvl association with Frat-1 and activation of the Wnt pathway (296). Dvl is necessary for the recruitment of axin to the membrane during Wnt signaling (297). Several proteins inhibit Wnt signaling by interacting with Dvl, including Idax (298), Daple (299), and Dab2 (300). Dvl proteins are phosphorylated within the DEP domain by Abl kinases, which is critical for planar cell polarity function, but has no known role in canonical Wnt signaling (301). Dvl is ubiquitinylated and targeted for proteasomal destruction by the HECTcontaining, Nedd4-like, ubiquitin E3 ligase (ITCH), and this interaction is dependent upon Dvl's DEP domain (302).
Dvl mouse models have not been thoroughly assessed for skeletal phenotypes. Dvl1 knock-out mice display no overt physical phenotype, but have abnormal behaviors including lower social interaction, not barbering, subordinance, not sleeping huddled, and not building nests (303). Dvl2 deletion results in 50% perinatal lethality due to cardiac anomalies. Interestingly, the surviving homozygous knock-outs are predominantly female and these mice have a normal lifespan. Some physical malformations in Dvl2 mutant mice were documented, including kinked tails, scoliosis, and vestigial tail, and 90% had mild abnormalities of vertebral bodies and ribs. The number of ribswere normal, but they were often forked and fused. The physical abnormalities were exacerbated if Dvl2 mutants were crossed with Dvl1-null mice (125). The majority of Dvl3 knock-out mice die before weaning, likely due to cardiac abnormalities, but no skeletal defects have been noted in these mice (304). None of the DVL family members have been directly linked to any human skeletal disorders.
Axin
The axin-1 protein is 832 or 868 amino acids in length, depending upon splicing, and is crucial for axis development (305). Axin negatively regulates Wnt signaling by interacting with GSK3 and β-catenin and by enhancing GSK3 phosphorylation of β-catenin. Axin contains RGS (regulator of G-protein signaling) and Dvl (DIX) domains. Axin is phosphorylated by GSK3 (306,307), which may positively promote the association between β-catenin and axin (307). As a small-molecule inhibitor, axin appears critical in restraining β-catenin's function and preventing it from activating Wnt signaling and promoting osteoblastogenesis from mesenchymal stem cells (308). Axin interacts with APC through the RGS domain (309) and with protein phosphatase 2A via an N-terminal domain (310) that may regulate APC phosphorylation (311). Axin can dimerize and interact with Dvl via the DIX domain, which appears to be important for inhibiting Wnt signaling (282,312), the heterotypic interaction by Dvl likely acting in a dominant negative fashion (313).
Axin forms a complex with β-catenin and casein kinase I to promote Ser-45 phosphorylation of β-catenin, thus priming β-catenin for further phosphorylation by GSK3 (314). Sumoylation on the C-terminal end appears to be critical for axin promotion of JNK activation (315) and enhances axin's stability (316). Lys505 of axin is ubiquitinylated by the E3 ubiquitin ligase Smurf2, which promotes axin's proteasomal degradation (317). The stability of axin is increased by protein arginine methyltransferase (PRMT1) via decreased ubiquitinylation and enhanced interaction with GSK3 (318). Axins are parsylated by tankyrase 1 and 2, which promotes axin polyubiquitinylation (319) by the RING-domain E3 ubiquitin ligase RNF146 (320, 321). Axin contains six RXL cyclin/CDK2 binding motifs spread throughout its structure, as well as numerous S/TP consensus target sequences for phosphorylation by CDKs (322). Most notably, CDK2 (322) and GSK3 (307) can both phosphorylate Thr609, Ser614, and Ser621 of axin, and phosphorylation of these sites enhances the axin-β-catenin interaction. Ser80, Ser82, and Ser222 can be phosphorylated by CKI, which enhances the axin-GSK3 interaction, and these residues can be dephosphorylated by protein phosphatase I (PPI) (323). Axins may mediate cross-talk between TGFβ and Wnt signaling (324). Activation of Wnt signaling results in the recruitment of axin to the cytoplasmic tail of LRP5/6 (325,326). Axin may also be involved in the nucleocytoplasmic shuffling of β-catenin (327, 328).
Research suggests that axin-1 and axin-2/conductin are functionally redundant (329,330), albeit, with different expression patterns during development (330,331), and axin-2 functions in a negative feedback loop during Wnt signaling (331,332). Axin-2 is 840 amino acids in length, shares 44% homology with axin-1, and retains the RGS and DIX domains, as well as binding sites for β-catenin, GSK3, and APC (329).
Axin was originally discovered as being the causative gene producing the “fused†phenotype in early mouse studies. Three alleles were identified that produced identical phenotypes with different levels of penetrance: Fused, Knobbly, and Kinky. Homozygosity for these mutations typically results in embryonic lethality by E9.5, with forebrain and neural tube defects as well as axis duplications. Heterozygotes survive, but often have rib fusions and tail bifurcation. Fused and Knobbly both result from IAP retrotransposon integrations, into intron 6 and exon 7 of Axin1, respectively. Kinky has been mapped to the Fused locus, but the causative mutation has not been identified (126, 127). To support that concept these mutations result in null alleles, two separate models with germline deletions of Axin1 produced identical phenotypes to the fused mice with full penetrance (126, 333). Some very interesting knock-in models have also been developed. Knock-in of Axin2 behind the Axin1 promoter demonstrated that the functions of these two proteins are highly redundant, as the mice had no overt phenotypes and were fertile (330). However, it would not be surprising if subtle differences remain to be discovered. Such studies could potentially reveal unique roles of axin-1/-2.
Mouse models have also been used to assess the importance of the RGS and KVEKVD domains in axin-1 function. Removal of either of these domains results in phenotypes identical to the complete knock-out of axin-1. However, in the ΔKVEKVD model, it is difficult to determine whether the embryonic lethal phenotype is a really a consequence specific to the loss of this domain, because this mutation resulted in a 3to 4-fold decrease in axin-1 protein (334). A very interesting mouse model that allows both temporal and tissue-specific overexpression of Axin1 is the TRE2-Axin1-GFP transgene. Axin1 can be overexpressed in specific tissues via tissue-specific promoter control of the reverse tetracycline transactivator (tTA), and temporally by the use of doxycycline to promote transcription via the tetracycline response element promoter (TRE). For example, crossing TRE2Axin1-GFP mice with mice carrying the MMTV—tTA transgene resulted in abnormal mammary gland development with decreased Wnt signaling in the glands (335). TRE2-Axin1-GFP mice have also been crossed with Axin2-tTA mice to assess the consequence of overexpressing axin-1 during canonical Wnt signaling. This resulted in abnormal neural development, but there were no in-depth assessments of skeletal phenotypes (336).
Fewer models have been developed for Axin2, but there have been some interesting studies published. Homozygous deletion of Axin2 results in skull defects at early postnatal periods, including craniosynostosis (premature fusing of cranial sutures) due to alterations in intramembraneous ossification. The mice show evidence of aberrant activation of Wnt signaling, which likely accounts for the increased osteoblast proliferation and differentiation seen ex vivo, and ultimately for the enhanced bone mineralization and ossification in vivo (130). Further assessment of these mice revealed an important role of axin-2 in mature bone: significant differences in bone volume and BMD are seen at 6 and 12 months when compared with wild-type mice, but no differences in bone volume or BMD are observed at 2 months of age (129). These mice also show an abnormal chondrocyte phenotype due to expedited maturation, resulting in a shorter hypertrophic zone in the growth plate of long bones (128). An Axin2 mutation was also identified in the canopus (canp) mouse: mice that were homozygous for the V26D missense mutation in Axin2 were arrested at midgestation with abnormal heart development and shortened or doubled tails. In vitro analyses revealed that this mutation greatly enhances the stability of the axin-2 protein, tripling its half-life (131).
Axin1 and Axin2 have both been implicated in cases of developmental disorders. Axin1 was found to be methylated in a pair of discordant monozygotic twins with caudal duplication anomaly (OMIM: 607864) (337). Supporting the critical role of Axin2 in craniofacial development, multiple studies in humans have identified polymorphisms/mutations in Axin2 related to tooth agenesis (338–341) and oral clefts (132,340). Mutations in Axin2 are also associated with oligodontia-colorectal cancer syndrome (Arg656X) (OMIM: 608615) (338).
Adenomatous polyposis coli (APC)
The name of the protein APC, adenomatous polyposis coli, originates from an autosomal dominant condition known as familial adenomatous polyposis (FAP), which is characterized by the presence of numerous pre-cancerous polyps of the colon and other regions of the gastrointestinal tract (MIM:175100). Affected individuals may develop hundreds to thousands of adenomatous polyps within the colon and rectum, which progress to carcinomas. FAP is also associated with several skeletal defects including dental abnormalities (342,343), osteosarcoma (344), osteoma of the jaw (345), and desmoids tumors (346).
The human APC gene is found on chromosome 5q22 and has 15 exons encoding a 2843-amino acid protein (347) containing two N-terminal coil-coiled regions, seven internal armadillo repeats, and several other functional domains (348). APC interacts with a diverse set of proteins and other cellular components to regulate cell migration and adhesion, cytoskeleton regulation, and chromosome segregation. Within the β-catenin destruction complex, APC joins the scaffolding protein axin by binding β-catenin through its armadillo repeats in order to assist glycogen synthase kinase 3 (GSK3) in phosphorylating β-catenin. Phosphorylated β-catenin is rapidly degraded through recognition by the F-box/WD40-repeat-containing protein β-TrCP, targeting β-catenin to the proteosome (349). APC may also be a nucleocytoplasmic shuttle protein (350), which has been speculated to chaperone β-catenin out of the nucleus (351,352) and thus preventing β-catenin from binding to TCF/LEF (350,353,354). As a negative regulator of βcatenin, defects in or loss of APC results in increased bone mass: FAP patients with APC mutations within the β-catenin binding domains have increased bone mineral density (355).
Consistent with APC being a negative regulator of β-catenin, Ocn-Cre-mediated deletion of Apc within mature osteoblasts and osteocytes (Apc CKO) produces early-onset, severe osteopetrosis leading to animal death at an early age (134). Osteoclasts were entirely absent in bones of these mice by 2 weeks of age. Ocn-Cre- mediated deletion of Apc is associated with elevated expression of the osteoclast inhibitory factor osteoprotegerin (Opg) within serum and in calvarial osteoblast cultures. Interestingly, no quantifiable differences were observed in proliferation or density during in vitro osteoblast differentiation. Deletion of APC via the Col2aI driven Cre promoter resulted in impaired skeletogenesis due to the inability of chondrocytes and osteoblasts to terminally differentiate (133). From these data, it is clear that the role of APC in regulating β-catenin activity is crucial for both cell autonomous and intercellular regulation of skeletal cell types.
Glycogen synthase kinase 3
Glycogen synthase kinase 3 (GSK3), originally found to phosphorylate and inactivate the enzyme glycogen synthase, is a negative regulator of several signaling pathways involved in osteogenesis, such as insulin/IGF, hedgehog, and Wnt/β-catenin. Within mammals, two GSK3 genes encode the α and β isoforms that differ mainly due to the presence of an N-terminal glycine-rich extension within the α isoform; the isoforms share 98% sequence identity within the kinase domain, which makes up the majority of the protein (356).
GSK3β has several substrates within the Wnt pathway, including Lrp5/6, β-catenin, axin, and APC, each of which play distinct roles in regulating the Wnt signal (357). GSK3 kinases typically prefer prephosphorylated or primed substrates by recognizing the consensus sequence Ser/Thr-X-X-X-Ser/Thr-P. Within the β-catenin destruction complex, casein kinase I (CKI) primes β-catenin by catalyzing Ser45 phosphorylation, in turn, facilitating sequential phosphorylation of three adjacent GSK3 consensus sites (Ser41, Ser37, Ser33) and promoting rapid degradation of β-catenin (314,358–361). Additionally, GSK3β-mediated phosphorylation of APC and axin increases the stability of the complex and its affinity to β-catenin (282,306,307). GSK3 phosphorylation of Lrp5/6 facilitates Wnt receptor complex internalization to form multiunit signalosomes, sequestering GSK3 and promoting β-catenin stabilization and Wnt target gene transcription (2,362,363).
There is a plethora of data indicating that Gsk3β is an inhibitor of bone formation in several contexts and biological pathways. Germline deletion of Gsk3β results in embryonic lethality due to high hepatocyte apoptosis (135). However, Gsk3β heterozygote mice develop without gross anatomical abnormalities and have a higher trabecular bone volume density, an increased number of osteoblasts, and an accelerated rate of bone formation (136). This phenotype was attributed to the loss of Gsk3β-mediated phosphorylation and inhibition of Runx2, an important transcriptional regulator throughout osteoblast differentiation. However, inhibition of Gsk3β via oral lithium chloride treatment of Lrp5−/− mice results in increased bone mass formation and decreased bone marrow adiposity, indicating that β-catenin stabilization favors osteoblast differentiation independent of Runx2 activity (136). Lithium chloride treatment of Lrp5−/− primary calvarial osteoblast cultures restores β-catenin TCF reporter activity to wild-type levels (96). Lithium chloride treatment via oral gavage also increases bone mass in C57BL/6 and SAMP6 mice. Other specific dual Gsk3 (α & β) inhibitrs induce expression of osteoblast markers such as osteocalcin, collagen I and V, runx2, and TCF reporter activity in vitro, as well as increased bone mass, strength, and other markers of osteoblastic differentiation and function within ovariectomized rats (364). Deletion of Gsk3β within chondrocytes via Col2a1-Cre results in a compensatory up-regulation of the Gsk3β isoform with a minimal effect on skeletal growth or development, indicating the importance of cell type specificity (140).
Lithium chloride treatment, which has been used for decades to treat bi-polar disorders (365–367), increases bone formation in the distal femurs of mice regardless of physical activity (368). While some studies have shown a reduced fracture risk (369), others were unable to observe this protective effect (370). Chronic treatment with lithium results in prolonged β-catenin stabilization, yet doesn't result increased susceptibility to several cancer types, in which elevated Wnt signaling is implicated, such as colon, breast or prostate (371–373). The importance of Gsk3 in bone formation is evident, but its mechanisms appear to be numerous and complex. Future cell-based and in vivo approaches are needed to elucidate cell autonomous and intercellular effects that GSK3 has on bone turnover.
β-catenin
β-catenin, encoded by the CTTNB1 gene, is the component of the Wnt pathway that enters the nucleus to drive Wnt target gene expression. Upon binding of a Wnt to the co-receptors Lrp5/6 and frizzled, the β-catenin destruction complex is inhibited, leading to cytoplasmic accumulation and nuclear translocation of β-catenin. Target genes including AXIN2 and DKK1 (Dickkopf-related protein 1), which are antagonists of the Wnt pathway, act as negative feedback regulators (331,374). This process is regulated through a cascade of post translational modifications of β-catenin and the molecules that regulate it. Destruction of β-catenin occurs via the GSK3-dependent process described above. Nuclear β-catenin binds to TCF/LEF DNA binding proteins to elicit the transcription of Wnt target genes. The deletion of exon 3 of β-catenin removes key residues and results in a non-degradable, stable form of β-catenin (375). Use of the knowledge of these mechanisms in concert with the Cre-lox system has generated several genetic studies into the role of β-catenin, Wnt signaling, and their role in bone formation.
β-catenin controls the fate of mesenchymal cells, from which chondrocytes, osteoblasts, and osteocytes originate. Mouse embryos lacking Cnntb1 within mesenchymal progenitors (via Dermo-Cre or Prx1-Cre) have severely diminished osteogenesis and form a truncated cartilaginous skeleton (59,144,149,376). This loss of β-catenin within skeletal precursors also causes ectopic cartilage formation, indicating that β-catenin is a positive regulator of the osteoblast lineage and an inhibitor of chondrogenesis (377). The importance of β-catenin in maintaining the osteoblast identity was further supported by removing β-catenin in pre-osteoblasts using the Dox-inhibitable Osx-Cre. Pre-natal deletion of Cnntb1 resulted in a lack of skeletal ossification and mineralization with formation of ectopic chondrocytes from osteoblast precursors (59). Intriguingly, if Dox was given until 2 or 4 months post-natally, there was a dramatic loss of trabecular bone with a concomitant increase in bone marrow adiposity. Additionally, ex vivo analyses revealed enhanced adipocyte differentiation of adult BMSCs in culture, suggesting that β-catenin suppresses adipogenesis (150). Genetic deletion of Cnntb1 within mature osteoblasts (using Ocn-Cre) results in dramatic reductions in trabecular and cortical bone mass due to defective osteoblast differentiation (62,134). This phenotype was attributed to a decrease in osteoprotegrin, resulting in elevated RANK ligand-stimulated osteoclast activation and increased bone resorption (134). Cnntb1 deletion via Collagen I-Cre results in a low-bone-mass phenotype, despite having normal numbers of osteoblasts. Bones from these mice display more TRAP-positive osteoclasts, suggesting that this phenotype is due to increase resorption and that β-catenin signaling in osteoblasts regulates osteoclast differentiation (143). Osteocytespecific deletion of Cnntb1 via Dmp1-Cre causes progressive bone loss and premature lethality. This lowbone-mass phenotype is associated with an increased number of osteoclasts and higher osteoclast activity due to disruption of the OPG/RANK axis, although osteoblast function and the osteocyte density is normal (151). These results indicate that β-catenin is a master regulator of the osteochondral lineage, directing embryonic bone formation and postnatal bone homeostasis.
Overactivation of Wnt signaling within limb buds (Prx1-Cre-mediated) results in abnormalities of the appendicular skeleton, with only undifferentiated, Sox9-expressing cell types remaining. This is a further indication that β-catenin activity must be spatially and temporally controlled to direct bone formation (144). Stabilization of β-catenin within osteoblast precursors (via osterix-Cre mediated excision of the third exon of β-catenin) results in premature mineralization during endochondral ossification but a loss in osteocalcin-expressing osteoblasts, signifying an inability to terminally differentiate (59,143). Although the number of osteoblasts was not affected in this model, the blockade of osteoblast differentiation reduced the osteoclast population, perhaps due to alteration of the RANK/RANKL regulatory axis. From these results, the cessation of β-catenin signaling appears to be required for terminal differentiation of osteoblasts and for initiating subsequent programs of bone formation such as osteoclast and chondrocyte differentiation and activity.
The word “cateninâ€â€”from the Latin “catenaâ€, which means chain, was based on the original identification of α-, β-, and γ-catenin (plakoglobin) as molecules that linked cytoplasmic domains of cadherins at cell-cell junctions with the actin cytoskeleton (378–380). This Wnt-independent role for β-catenin may be important in regulating bone mass accrual in response to mechanical stress. Mechanical deformation within calvarial osteoblasts induces β-catenin nuclear localization and activation of the Tcf/Lef reporter, independent of Lrp5 (381,382). This phenotype is enhanced with the addition of lithium chloride treatment, which further inhibits GSK3β (383). However, this mechanism is exclusively cell autonomous, as mechanical loading in vivo reduces SOST expression by osteocytes, resulting in elevated Lrp5/6-Δ-catenin signaling within osteoblasts. Conversely, mechanical unloading via tail suspension produces the opposite effect (384). From these data, it is apparent that there are multiple overlapping systems—from developmental cues to the response to physical stimuli for regulating β-catenin activity within osteoprogenitor cells in order to control cell fate and the deposition of bone.
Human skeletal diseases are more commonly caused by mutations in genes that regulate β-catenin and the Wnt pathway (Section I) than by mutations within CTNNB1 itself. However, a meta-analysis of five human genome-wide association studies of the femoral neck and lumbar spine bone mineral density found an association between CTNNB1 polymorphisms and altered BMD (205). Other developmental diseases, including cancer, have been attributed to defective regulation of β-catenin signaling (385),but β-catenin mutations alone do not appear sufficient for their pathogenesis. The regulation of β-catenin is dynamic via the integration of multiple points of regulation, from cell surface receptors and cytoplasmic modifiers to its nuclear translocation and transcription.
TCF/LEF
The TCF/LEF HMG box-containing family of transcription factors originated from the discovery of T-cell factor 1 (TCF1) and lymphoid enhancer factor (LEF-1) through investigations of the transcriptional regulation of the CD3E and T cell receptor (TCR) genes, respectively (386–390). TCF family members bind to the DNA consensus sequence YCTTTGWW (391) via their C-terminus and contact β-catenin through their N-terminus (392–394). There are four genes in this family: LEF1, TCF1, TCF3, and TCF4. TCF1 and LEF1 have overlapping expression patterns throughout development. Tcf3 is ubiquitously expressed during early murine embryogenesis (E6.5) and gradually declines by day E10. TCF4 is expressed later during development (E13.5) and is restricted to the diencephelon, mesencephelon, and the intestinal epithelium (154). Of the three TCF genes, only TCF1 and TCF4 are reported to be expressed within differentiating osteoblasts (143,395).
Tcf1−/− mice develop without any gross anatomical abnormalities other than a severe defect in T lymphogenesis (396), but they have slightly lower BMD at one month of age (143). Although osteoblast numbers were not affected within this model, the low-bone-mass phenotype was attributed to reduced expression of the target gene and osteoclast activating factor, Opg. Mice carrying heterozygous muations in Ctnnb1 and Tcf1 also have decreased bone mass with a decreased expression of Opg. Through chromatin immunoprecipitation, both Tcf1 and Tcf4 were found to co-occupy with β-catenin on the Opg promoter in D10 calvarial osteoblasts, indicating that Opg is a direct transcriptional target of β-catenin/TCF activity within the osteoblasts (143).
Lef1−/− mice die perinatally and lack hair follicles, teeth, mammary glands, and portions of the trigeminal nerve (158). Mice lacking both Tcf1 and Lef1 die at birth and have an arrest of cell differentiation within a and β T cells at the CD8+ single positive stage (397). These mice also have abnormal neural tube formation and defects in differentiation of the paraxial mesoderm, closely resembling the phenotype of Wnt3a-deficient mice (156). Mice heterozygous for an inactivating mutation in Lef-1 may show a gender and age-dependent role for Lef-1 during bone mass regulation. Three to four month-old Lef1+/− females have reduced bone mass, whereas males do not. This Lef1 haploinsufficient low-bone-mass female phenotype can be rescued by crossing with Gsk3β+/− mice. Male Lef1+/− mice have normal bone mass, but show a reduction in bone mass upon androgen receptor inactivation (157). Loss of the β-catenin binding domain within Lef-1 results in a similar overall phenotype, having severely reduced trabecular bone volume not seen within heterozygotes (50).
Mammalian Tcf1 and Lef1 genes harbor alternative promoters which produce truncated isoforms lacking the β-catenin binding domains to act as dominate negative co-repressors (398). Additionally, the Tcf1 gene contains two promoters, four alternative exons, and three splice acceptors within the last exon encoding an extended CTD, yielding up to 96 different mRNA transcripts (399). Although full-length Lef-1 expression was not detected within differentiating osteoblasts (143), Lef-1ΔN is driven by Bmp2 within mature osteoblasts to act as a negative regulator of Wnt transcription (400). Overexpression of Lef1 inhibited osteoblast differentiation (401), yet expression of Lef-1ΔN allowed maturation (402). This dominant negative form of Lef-1 is known to inhibit β-catenin transcription (398,403). Lef-1ΔN retains low affinity to β-catenin through a second domain, and expression within mature osteoblasts (2.3Col1a1) results in slightly higher bone formation rates, trabecular bone, and an increase in serum osteocalcin (159). Future work is needed to tease out the temporal expression of different Tcf/Lef transcription factors in order to determine their roles in gene expression within osteoblasts during the formation of bone and how other “dominant negative†transcription factors such as dnTcf1 regulate bone formation.
Other components of the nuclear β-catenin transcriptional complex
In the absence of nuclear β-catenin, Tcf/Lef proteins bind the transcriptional repressor Groucho/TLE on Wnt responsive elements (WREs) in order to facilitate HDAC-mediated repression of Wnt target genes (404–406). Upon entry of β-catenin to the nucleus, it binds to Lef or Tcf through its central armadillo repeats and initiates the formation of nuclear transcription complexes tethered to WREs via DNA-bound Tcf/Lef proteins. The N-terminus of β-catenin binds the transcriptional co-activators BCL-9 and pygopus, which associate with activated chromatin (marked by methylation of H3 [H3K4me]) to further decode histone tails via its PHD fingers and facilitate Wnt-induced transcription (407,408). The C-terminal domain (CTD) of β-catenin binds several chromatin remodeling complexes, such as MLL1/2, TIP60, and CBP/p300. In particular, a core member of the polymerase associated factor (PAF) complex, parafibromin (CDC73), binds to the β-catenin CTD in order to recruit the PAF complex and RNA polymerase II (Pol II) to WREs (409–411). Binding of β-catenin/parafibromin may be mediated by protein tyrosine phosphatase, PTPN11 (Shp2), which dephosphoylates parafibromin and thus allows it to bind β-catenin, recruit the PAF complex, and activate transcription (13). Interestingly, loss-of-function mutations within the coding sequence of parafibromin lead to parathyroid adenomas and ossifying fibromas of the jaw (412–414). Further work using cell-specific knock-outs are needed to help determine how these regulators of β-catenin/TCF-mediated transcription govern bone formation.
β-catenin target genes
Ultimately, activation of the Wnt pathway leads to the expression of distinct sets of genes that drive progenitor cell proliferation, differentiation, or survival. Several Wnt/β-catenin target genes have been identified and validated; they are enumerated on the Wnt home page (http://www.stanford.edu/group/nusselab/cgi-bin/wnt/). However, validating Wnt target genes has proven difficult due to the diverse set of cell types, states, and stimuli that may differentially activate the transcription of specific target genes through different Tcf/Lef transcription factors. For example, Lef1 expression is mainly restricted to developing lymphoid tissues and absent in osteoblasts, where Tcf1 and Tcf4 are expressed within differentiating osteoblasts. Within the pluripotent mesenchymal cell line C3H10T1/2, Wnt3a treatment upregulates genes involved in adipocyte and osteoblast differentiation. An Affymetrix oligonucleotide genomewide expression analysis performed on these cells identified 447 Wnt3a-regulated genes (415). Several of these are genes involved in the regulation of the Wnt pathway, such as an up-regulation of Axin2 and Fzd7, or a down-regulation of Tcf3, Sfrp2, and Fzd4. Wnt3a induces expression of 13 osteoblast-regulated genes including Bmp4, Opg, Col1a1, Hspg2, and the transcription factors Atf4 and Twist, but fails to elevate Runx2 expression (415). From gene expression profiling studies and the evaluation of osteoblast-specific knockouts of β-catenin, it is clear that Wnt target genes are required for osteochondral fate decisions and the expression of lineage-specific transcription factors such as Atf4 and Twist. Yet, down-regulation of Wnt signaling is required for osteoblast maturation, mineralization, and regulation of osteoclastogenesis. Further work is needed to identify the temporal expression of lineage-specific Wnt target genes to better understand the role of Wnt signaling in bone formation.
Summary
The demonstration that alterations in components of the Wnt/β-catenin signaling pathway were causally associated with dramatic changes in bone mass in humans has stimulated extensive interest in developing methods to activate this pathway to treat bone diseases such as osteoporosis (reviewed in (253)). Several therapeutics based on the concept that activating this pathway can increase bone mass are currently in clinical development and preliminary results appear promising (253). Pharmaceutical companies are also evaluating therapies to inhibit Wnt signaling to treat several tumor types in which activation of the pathway is associated with tumorigenesis (for examples, see (416,417)). In this context, it will be important to carefully monitor patients for deleterious effects on bone health. Overall, the possibility that millions of patients may be treated with compounds designed to modulate this pathway makes a continued emphasis on understanding the precise mechanisms associated with Wnt-mediated control of normal bone health a critical research focus.
References
Bazan JF, Janda CY, Garcia KC . Structural architecture and functional evolution of Wnts. Dev Cell. 2012;23:227–232.
MacDonald BT, Tamai K, He X . Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26.
He X, Semenov M, Tamai K, Zeng X . LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development. 2004;131:1663–1677.
Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F . Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell. 2008;133:340–353.
Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW, Jones DA . A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623–634.
Niehrs C . Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25:7469–7481.
Mason JJ, Williams BO . SOST and DKK: Antagonists of LRP family signaling as targets for treating bone disease. J Osteoporos. 2010;2010:460120.
Kawano Y, Kypta R . Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116:2627–2634.
Nelson WJ, Nusse R . Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487.
Hausmann G, Banziger C, Basler K . Helping Wingless take flight: how WNT proteins are secreted. Nat Rev Mol Cell Biol. 2007;8:331–336.
Stamos JL, Weis WI . The beta-Catenin Destruction Complex. Cold Spring Harb Perspect Biol. 2012;5:a007898.
Valenta T, Hausmann G, Basler K . The many faces and functions of beta-catenin. EMBO J. 2012;31:2714–2736.
Takahashi A, Tsutsumi R, Kikuchi I, Obuse C, Saito Y, Seidi A, Karisch R, Fernandez M, Cho T, Ohnishi N, Rozenblatt-Rosen O, Meyerson M, Neel BG, Hatakeyama M . SHP2 tyrosine phosphatase converts parafibromin/Cdc73 from a tumor suppressor to an oncogenic driver. Mol Cell. 2011;43:45–56.
Herr P, Hausmann G, Basler K . WNT secretion and signalling in human disease. Trends Mol Med. 2012;18:483–493.
Steichen-Gersdorf E, Gassner I, Unsinn K, Sperl W . Persistent hyperplastic primary vitreous in a family with osteoporosis-pseudoglioma syndrome. Clin Dysmorphol. 1997;6:171–176.
Gong Y, Vikkula M, Boon L, Liu J, Beighton P, Ramesar R, Peltonen L, Somer H, Hirose T, Dallapiccola B, De Paepe A, Swoboda W, Zabel B, Superti-Furga A, Steinmann B, Brunner HG, Jans A, Boles RG, Adkins W, van den Boogaard MJ, Olsen BR, Warman ML . Osteoporosis-pseudoglioma syndrome, a disorder affecting skeletal strength and vision, is assigned to chromosome region 11q12–13. Am J Hum Genet. 1996;59:146–151.
Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML . LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523.
Ai M, Heeger S, Bartels CF, Schelling DK . Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am J Hum Genet. 2005;77:741–753.
Cheung WM, Jin LY, Smith DK, Cheung PT, Kwan EY, Low L, Kung AW . A family with osteoporosis pseudoglioma syndrome due to compound heterozygosity of two novel mutations in the LRP5 gene. Bone. 2006;39:470–476.
Barros ER, Dias da Silva MR, Kunii IS, Hauache OM, Lazaretti-Castro M . A novel mutation in the LRP5 gene is associated with osteoporosis-pseudoglioma syndrome. Osteoporos Int. 2007;18:1017–1018.
Narumi S, Numakura C, Shiihara T, Seiwa C, Nozaki Y, Yamagata T, Momoi MY, Watanabe Y, Yoshino M, Matsuishi T, Nishi E, Kawame H, Akahane T, Nishimura G, Emi M, Hasegawa T . Various types of LRP5 mutations in four patients with osteoporosis-pseudoglioma syndrome: identification of a 7.2-kb microdeletion using oligonucleotide tiling microarray. Am J Med Genet A. 2010;152A:133–140.
Marques-Pinheiro A, Levasseur R, Cormier C, Bonneau J, Boileau C, Varret M, Abifadel M, Allanore Y . Novel LRP5 gene mutation in a patient with osteoporosis-pseudoglioma syndrome. Joint Bone Spine. 2010;77:151–153.
Laine CM, Chung BD, Susic M, Prescott T, Semler O, Fiskerstrand T, D'Eufemia P, Castori M, Pekkinen M, Sochett E, Cole WG, Netzer C, Makitie O . Novel mutations affecting LRP5 splicing in patients with osteoporosis-pseudoglioma syndrome (OPPG). Eur J Hum Genet. 2011;19:875–881.
Tuysuz B, Bursali A, Alp Z, Suyugul N, Laine CM, Makitie O . Osteoporosis-pseudoglioma syndrome: three novel mutations in the LRP5 gene and response to bisphosphonate treatment. Horm Res Paediatr. 2012;77:115–120.
Lev D, Binson I, Foldes AJ, Watemberg N, Lerman-Sagie T . Decreased bone density in carriers and patients of an Israeli family with the osteoporosis-pseudoglioma syndrome. Isr Med Assoc J. 2003;5:419–421.
Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP . High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–1521.
Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, Lappe MM, Spitzer L, Zweier S, Braunschweiger K, Benchekroun Y, Hu X, Adair R, Chee L, FitzGerald MG, Tulig C, Caruso A, Tzellas N, Bawa A, Franklin B, McGuire S, Nogues X, Gong G, Allen KM, Anisowicz A, Morales AJ, Lomedico PT, Recker SM, van Eerdewegh P, Recker RR, Johnson ML . A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–19.
Beighton P . Sclerosteosis. J Med Genet. 1988;25:200–203.
Balemans W, van den Ende J, Freire Paes-Alves A, Dikkers FG, Willems PJ, Vanhoenacker F, de Almeida-Melo N, Alves CF, Stratakis CA, Hill SC, van Hul W . Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am J Hum Genet. 1999;64:1661–1669.
Brunkow ME, Gardner JC, van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J . Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–589.
Balemans W, Ebeling M, Patel N, van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, van den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, van Hul W . Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10:537–543.
Piters E, Culha C, Moester M, van Bezooijen R, Adriaensen D, Mueller T, Weidauer S, Jennes K, De Freitas F, Lowik C, Timmermans JP, van Hul W, Papapoulos S . First missense mutation in the SOST gene causing sclerosteosis by loss of sclerostin function. Hum Mutat. 2010;31:E1526–E1543.
Bhadada SK, Rastogi A, Steenackers E, Boudin E, Arya A, Dhiman V, van Hul W . Novel SOST gene mutation in a sclerosteosis patient and her parents. Bone. 2013;52:707–710.
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA . Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–6276.
Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, Thesleff I, Itoh N . Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem. 2003;278:24113–24117.
van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, De Wilt E, Karperien M, Hamersma H, Papapoulos SE, ten Dijke P, Lowik CW . Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–814.
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D . Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–19887.
Semenov M, Tamai K, He X . SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–26775.
Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, Bouwmeester T, Schirle M, Bueno-Lozano M, Fuentes FJ, Itin PH, Boudin E, De Freitas F, Jennes K, Brannetti B, Charara N, Ebersbach H, Geisse S, Lu CX, Bauer A, van Hul W, Kneissel M . Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286:19489–19500.
Bueno M, Olivan G, Jimenez A, Garagorri JM, Sarria A, Bueno AL, Bueno M, Jr., Ramos FJ . Sclerosteosis in a Spanish male: first report in a person of Mediterranean origin. J Med Genet. 1994;31:976–977.
Itin PH, Keseru B, Hauser V . Syndactyly/brachyphalangy and nail dysplasias as marker lesions for sclerosteosis. Dermatology. 2001;202:259–260.
Van Hul W, Balemans W, van Hul E, Dikkers FG, Obee H, Stokroos RJ, Hildering P, Vanhoenacker F, van Camp G, Willems PJ . Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am J Hum Genet. 1998;62:391–399.
Beighton P, Barnard A, Hamersma H, van der Wouden A . The syndromic status of sclerosteosis and van Buchem disease. Clin Genet. 1984;25:175–181.
Balemans W, Patel N, Ebeling M, van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, van Hul W . Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–97.
Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P, Papapoulos S, Hamersma H, Brunkow ME . A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–152.
Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, Ovcharenko D, Plajzer-Frick I, Rubin EM . Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15:928–935.
van Lierop A, Hamdy N, van Egmond M, Bakker E, Dikkers F, Papapoulos S . Van Buchem disease: Clinical, biochemical and densitometric features of patients and disease carriers. J Bone Miner Res. 2013;28:848–854.
Collette NM, Genetos DC, Economides AN, Xie L, Shahnazari M, Yao W, Lane NE, Harland RM, Loots GG . Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci U S A. 2012;109:14092–14097.
Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Benichou O, Scopelliti D, Key L, Renton T, Bartels C, Gong Y, Warman ML, De Vernejoul MC, Bollerslev J, van Hul W . Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet. 2003;72:763–771.
Monroe DG, McGee-Lawrence ME, Oursler MJ, Westendorf JJ . Update on Wnt signaling in bone cell biology and bone disease. Gene. 2012;492:1–18.
Najdi R, Proffitt K, Sprowl S, Kaur S, Yu J, Covey TM, Virshup DM, Waterman ML . A uniform human Wnt expression library reveals a shared secretory pathway and unique signaling activities. Differentiation. 2012;84:203–213.
Tang X, Wu Y, Belenkaya TY, Huang Q, Ray L, Qu J, Lin X . Roles of N-glycosylation and lipidation in Wg secretion and signaling. Dev Biol. 2012;364:32–41.
Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ . Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80.
Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM . Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074.
Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, Zhang Z, Martin JF, Behringer RR, Nakamura T, De Crombrugghe B . Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A. 2005;102:14665–14670.
Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR . Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–146.
Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS . Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2011;15:2865–2876.
Yang G, Cui F, Hou N, Cheng X, Zhang J, Wang Y, Jiang N, Gao X, Yang X . Transgenic mice that express Cre recombinase in hypertrophic chondrocytes. Genesis. 2005;42:33–36.
Rodda SJ, McMahon AP . Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133:3231–3244.
Rossert J, Eberspaecher H, De Crombrugghe B . Separate cis-acting DNA elements of the mouse pro-alpha 1(I) collagen promoter direct expression of reporter genes to different type I collagen-producing cells in transgenic mice. J Cell Biol. 1995;129:1421–1432.
Dacquin R, Starbuck M, Schinke T, Karsenty G . Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002;224:245–251.
Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A, Clemens TL . Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–44012.
Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, Feng JQ . DMP1-targeted Cre expression in odontoblasts and osteocytes. J Dent Res. 2007;86:320–325.
Barrow JR, Thomas KR, Boussadia-Zahui O, Moore R, Kemler R, Capecchi MR, McMahon AP . Ectodermal Wnt3/beta-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev. 2003;17:394–409.
Takada I, Mihara M, Suzawa M, Ohtake F, Kobayashi S, Igarashi M, Youn MY, Takeyama K, Nakamura T, Mezaki Y, Takezawa S, Yogiashi Y, Kitagawa H, Yamada G, Takada S, Minami Y, Shibuya H, Matsumoto K, Kato S . A histone lysine methyl-transferase activated by non-canonical Wnt signalling suppresses PPAR-gamma transactivation. Nat Cell Biol. 2007;9:1273–1285.
Takada S, Stark KL, Shea MJ, Vassileva G, McMahon JA, McMahon AP . Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes Dev. 1994;8:174–189.
Greco TL, Takada S, Newhouse MM, McMahon JA, McMahon AP, Camper SA . Analysis of the vestigial tail mutation demonstrates that Wnt-3a gene dosage regulates mouse axial development. Genes Dev. 1996;10:313–324.
Stark K, Vainio S, Vassileva G, McMahon AP . Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1994;372:679–683.
Spater D, Hill TP, O'Sullivan R J, Gruber M, Conner DA, Hartmann C . Wnt9a signaling is required for joint integrity and regulation of Ihh during chondrogenesis. Development. 2006;133:3039–3049.
Lee HH, Behringer RR . Conditional expression of Wnt4 during chondrogenesis leads to dwarfism in mice. PLoS One. 2007;2:e450.
Yamaguchi TP, Bradley A, McMahon AP, Jones S . A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223.
Yang Y, Topol L, Lee H, Wu J . Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development. 2003;130:1003–1015.
Parr BA, McMahon AP . Dorsalizing signal Wnt-7a required for normal polarity of D-V and A-P axes of mouse limb. Nature. 1995;374:350–353.
Parr BA, Avery EJ, Cygan JA, McMahon AP . The classical mouse mutant postaxial hemimelia results from a mutation in the Wnt 7a gene. Dev Biol. 1998;202:228–234.
Tu X, Joeng KS, Nakayama KI, Nakayama K, Rajagopal J, Carroll TJ, McMahon AP, Long F . Noncanonical Wnt signaling through G protein-linked PKCdelta activation promotes bone formation. Dev Cell. 2007;12:113–127.
Popperl H, Schmidt C, Wilson V, Hume CR, Dodd J, Krumlauf R, Beddington RS . Misexpression of Cwnt8C in the mouse induces an ectopic embryonic axis and causes a truncation of the anterior neuroectoderm. Development. 1997;124:2997–3005.
Guo X, Day TF, Jiang X, Garrett-Beal L, Topol L, Yang Y . Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004;18:2404–2417.
Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP . Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell. 2005;9:283–292.
Jin YR, Han XH, Taketo MM, Yoon JK . Wnt9b-dependent FGF signaling is crucial for outgrowth of the nasal and maxillary processes during upper jaw and lip development. Development. 2012;139:1821–1830.
Juriloff DM, Harris MJ, McMahon AP, Carroll TJ, Lidral AC . Wnt9b is the mutated gene involved in multifactorial nonsyndromic cleft lip with or without cleft palate in A/WySn mice, as confirmed by a genetic complementation test. Birth Defects Res A Clin Mol Teratol. 2006;76:574–579.
Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, MacDougald OA . Regulation of osteoblasto-genesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102:3324–3329.
Stevens JR, Miranda-Carboni GA, Singer MA, Brugger SM, Lyons KM, Lane TF . Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J Bone Miner Res. 2010;25:2138–2147.
Zheng HF, Tobias JH, Duncan E, Evans DM, Eriksson J, Paternoster L, Yerges-Armstrong LM, Lehtimaki T, Bergstrom U, Kahonen M, Leo PJ, Raitakari O, Laaksonen M, Nicholson GC, Viikari J, Ladouceur M, Lyytikainen LP, Medina-Gomez C, Rivadeneira F, Prince RL, Sievanen H, Leslie WD, Mellstrom D, Eisman JA, Moverare-Skrtic S, Goltzman D, Hanley DA, Jones G, St Pourcain B, Xiao Y, Timpson NJ, Smith GD, Reid IR, Ring SM, Sambrook PN, Karlsson M, Dennison EM, Kemp JP, Danoy P, Sayers A, Wilson SG, Nethander M, McCloskey E, Vandenput L, Eastell R, Liu J, Spector T, Mitchell BD, Streeten EA, Brommage R, Pettersson-Kymmer U, Brown MA, Ohlsson C, Richards JB, Lorentzon M . WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. 2012;8:e1002745.
Barrott JJ, Cash GM, Smith AP, Barrow JR, Murtaugh LC . Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc Natl Acad Sci U S A. 2011;108:12752–12757.
Liu W, Shaver TM, Balasa A, Ljungberg MC, Wang X, Wen S, Nguyen H, van den Veyver IB . Deletion of Porcn in mice leads to multiple developmental defects and models human focal dermal hypoplasia (Goltz syndrome). PLoS One. 2012;7:e32331.
Zhong Z, Zylstra-Diegel CR, Schumacher CA, Baker JJ, Carpenter AC, Rao S, Yao W, Guan M, Helms JA, Lane NE, Lang RA, Williams BO . Wntless functions in mature osteoblasts to regulate bone mass. Proc Natl Acad Sci U S A. 2012;109:E2197–E2204.
Zhu X, Zhu H, Zhang L, Huang S, Cao J, Ma G, Feng G, He L, Yang Y, Guo X . Wls-mediated Wnts differentially regulate distal limb patterning and tissue morphogenesis. Dev Biol. 2012;365:328–338.
Maruyama T, Jiang M, Hsu W . Gpr177, a novel locus for bone-mineral-density and osteoporosis, regulates osteogenesis and chondrogenesis in skeletal development. J Bone Miner Res. 2012. [Epub ahead of print].
Yu H, Smallwood PM, Wang Y, Vidaltamayo R, Reed R, Nathans J . Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development. 2010;137:3707–3717.
Albers J, Schulze J, Beil FT, Gebauer M, Baranowsky A, Keller J, Marshall RP, Wintges K, Friedrich FW, Priemel M, Schilling AF, Rueger JM, Cornils K, Fehse B, Streichert T, Sauter G, Jakob F, Insogna KL, Pober B, Knobeloch KP, Francke U, Amling M, Schinke T . Control of bone formation by the serpentine receptor Frizzled-9. J Cell Biol. 2011;192:1057–1072.
Karner CM, Dietrich MF, Johnson EB, Kappesser N, Tennert C, Percin F, Wollnik B, Carroll TJ, Herz J . Lrp4 regulates initiation of ureteric budding and is crucial for kidney formation—a mouse model for Cenani-Lenz syndrome. PLoS One. 2010;5:e10418.
Choi HY, Dieckmann M, Herz J, Niemeier A . Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo . PLoS One. 2009;4:e7930.
Simon-Chazottes D, Tutois S, Kuehn M, Evans M, Bourgade F, Cook S, Davisson MT, Guenet JL . Mutations in the gene encoding the low-density lipoprotein receptor LRP4 cause abnormal limb development in the mouse. Genomics. 2006;87:673–677.
Weatherbee SD, Anderson KV, Niswander LA . LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000.
Iwaniec UT, Wronski TJ, Liu J, Rivera MF, Arzaga RR, Hansen G, Brommage R . PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. 2007;22:394–402.
Clement-Lacroix P, Ai M, Morvan F, Roman-Roman S, Vayssiere B, Belleville C, Estrera K, Warman ML, Baron R, Rawadi G . Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci U S A. 2005;102:17406–17411.
Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA, 2nd, Hartmann C, Li L, Hwang TH, Brayton CF, Lang RA, Karsenty G, Chan L . Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157:303–314.
Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, Mann JJ, Hen R, Ducy P, Karsenty G . Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825–837.
Holmen SL, Giambernardi TA, Zylstra CR, Buckner-Berghuis BD, Resau JH, Hess JF, Glatt V, Bouxsein ML, Ai M, Warman ML, Williams BO . Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. J Bone Miner Res. 2004;19:2033–2040.
Joeng KS, Schumacher CA, Zylstra-Diegel CR, Long F, Williams BO . Lrp5 and Lrp6 redundantly control skeletal development in the mouse embryo. Dev Biol. 2011;359:222–229.
Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR, Zhong Z, Matthes S, Jacobsen CM, Conlon RA, Brommage R, Liu Q, Mseeh F, Powell DR, Yang QM, Zambrowicz B, Gerrits H, Gossen JA, He X, Bader M, Williams BO, Warman ML, Robling AG . Lrp5 functions in bone to regulate bone mass. Nat Med. 2011;17:684–691.
Niziolek PJ, Farmer TL, Cui Y, Turner CH, Warman ML, Robling AG . High-bone-mass-producing mutations in the Wnt signaling pathway result in distinct skeletal phenotypes. Bone. 2011;49:1010–1019.
Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC . An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538.
Carter M, Chen X, Slowinska B, Minnerath S, Glickstein S, Shi L, Campagne F, Weinstein H, Ross ME . Crooked tail (Cd) model of human folate-responsive neural tube defects is mutated in Wnt coreceptor lipoprotein receptor-related protein 6. Proc Natl Acad Sci U S A. 2005;102:12843–12848.
Bogani D, Warr N, Elms P, Davies J, Tymowska-Lalanne Z, Goldsworthy M, Cox RD, Keays DA, Flint J, Wilson V, Nolan P, Arkell R . New semidominant mutations that affect mouse development. Genesis. 2004;40:109–117.
Fossat N, Jones V, Khoo PL, Bogani D, Hardy A, Steiner K, Mukhopadhyay M, Westphal H, Nolan PM, Arkell R, Tam PP . Stringent requirement of a proper level of canonical WNT signalling activity for head formation in mouse embryo. Development. 2011;138:667–676.
Kokubu C, Heinzmann U, Kokubu T, Sakai N, Kubota T, Kawai M, Wahl MB, Galceran J, Grosschedl R, Ozono K, Imai K . Skeletal defects in ringelschwanz mutant mice reveal that Lrp6 is required for proper somitogenesis and osteogenesis. Development. 2004;131:5469–5480.
Kubota T, Michigami T, Sakaguchi N, Kokubu C, Suzuki A, Namba N, Sakai N, Nakajima S, Imai K, Ozono K . Lrp6 hypomorphic mutation affects bone mass through bone resorption in mice and impairs interaction with Mesd. J Bone Miner Res. 2008;23:1661–1671.
Morvan F, Boulukos K, Clement-Lacroix P, Roman-Roman S, Suc-Royer I, Vayssiere B, Ammann P, Martin P, Pinho S, Pognonec P, Mollat P, Niehrs C, Baron R, Rawadi G . Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21:934–945.
Mukhopadhyay M, Shtrom S, Rodriguez-Esteban C, Chen L, Tsukui T, Gomer L, Dorward DW, Glinka A, Grinberg A, Huang SP, Niehrs C, Izpisua Belmonte JC, Westphal H . Dickkopf1 is required for embryonic head induction and limb morphogenesis in the mouse. Dev Cell. 2001;1:423–434.
De Rooy DP, Yeremenko NG, Wilson AG, Knevel R, Lindqvist E, Saxne T, Krabben A, Leijsma MK, Daha NA, Tsonaka S, Zhernakova A, Houwing-Duistermaat JJ, Huizinga TW, Toes RE, Baeten DL, Brouwer E, van der Helm-van Mil AH . Genetic studies on components of the Wnt signalling pathway and the severity of joint destruction in rheumatoid arthritis. Ann Rheum Dis. 2012. [Epub ahead of print].
Itoh S, Saito T, Hirata M, Ushita M, Ikeda T, Woodgett JR, Algul H, Schmid RM, Chung UI, Kawaguchi H . GSK-3alpha and GSK-3beta proteins are involved in early stages of chondrocyte differentiation with functional redundancy through RelA protein phosphorylation. J Biol Chem. 2012;287:29227–29236.
Oh H, Ryu JH, Jeon J, Yang S, Chun CH, Park H, Kim HJ, Kim WS, Kim HH, Kwon YG, Chun JS . Misexpression of Dickkopf-1 in endothelial cells, but not in chondrocytes or hypertrophic chondrocytes, causes defects in endochondral ossification. J Bone Miner Res. 2012;27:1335–1344.
Yao GQ, Wu JJ, Troiano N, Insogna K . Targeted overexpression of Dkk1 in osteoblasts reduces bone mass but does not impair the anabolic response to intermittent PTH treatment in mice. J Bone Miner Metab. 2011;29:141–148.
Li X, Liu P, Liu W, Maye P, Zhang J, Zhang Y, Hurley M, Guo C, Boskey A, Sun L, Harris SE, Rowe DW, Ke HZ, Wu D . Dkk2 has a role in terminal osteoblast differentiation and mineralized matrix formation. Nat Genet. 2005;37:945–952.
Ominsky MS, Li C, Li X, Tan HL, Lee E, Barrero M, Asuncion FJ, Dwyer D, Han CY, Vlasseros F, Samadfam R, Jolette J, Smith SY, Stolina M, Lacey DL, Simonet WS, Paszty C, Li G, Ke HZ . Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. J Bone Miner Res. 2011;26:1012–1021.
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C . Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–869.
Yerges LM, Klei L, Cauley JA, Roeder K, Kammerer CM, Moffett SP, Ensrud KE, Nestlerode CS, Marshall LM, Hoffman AR, Lewis C, Lang TF, Barrett-Connor E, Ferrell RE, Orwoll ES, Zmuda JM . High-density association study of 383 candidate genes for volumetric BMD at the femoral neck and lumbar spine among older men. J Bone Miner Res. 2009;24:2039–2049.
Bodine PV, Zhao W, Kharode YP, Bex FJ, Lambert AJ, Goad MB, Gaur T, Stein GS, Lian JB, Komm BS . The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol Endocrinol. 2004;18:1222–1237.
Morello R, Bertin TK, Schlaubitz S, Shaw CA, Kakuru S, Munivez E, Hermanns P, Chen Y, Zabel B, Lee B . Brachy-syndactyly caused by loss of Sfrp2 function. J Cell Physiol. 2008;217:127–137.
Lories RJ, Peeters J, Bakker A, Tylzanowski P, Derese I, Schrooten J, Thomas JT, Luyten FP . Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum. 2007;56:4095–4103.
Baker-Lepain JC, Lynch JA, Parimi N, McCulloch CE, Nevitt MC, Corr M, Lane NE . Variant alleles of the Wnt antagonist FRZB are determinants of hip shape and modify the relationship between hip shape and osteoarthritis. Arthritis Rheum. 2012;64:1457–1465.
Cho HY, Choi HJ, Sun HJ, Yang JY, An JH, Cho SW, Kim SW, Kim SY, Kim JE, Shin CS . Transgenic mice overexpressing secreted frizzled-related proteins (sFRP)4 under the control of serum amyloid P promoter exhibit low bone mass but did not result in disturbed phosphate homeostasis. Bone. 2010;47:263–271.
Nakanishi R, Akiyama H, Kimura H, Otsuki B, Shimizu M, Tsuboyama T, Nakamura T . Osteoblast-targeted expression of Sfrp4 in mice results in low bone mass. J Bone Miner Res. 2008;23:271–277.
Hamblet NS, Lijam N, Ruiz-Lozano P, Wang J, Yang Y, Luo Z, Mei L, Chien KR, Sussman DJ, Wynshaw-Boris A . Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002;129:5827–5838.
Perry WL, 3rd, Vasicek TJ, Lee JJ, Rossi JM, Zeng L, Zhang T, Tilghman SM, Costantini F . Phenotypic and molecular analysis of a transgenic insertional allele of the mouse Fused locus. Genetics. 1995;141:321–332.
Vasicek TJ, Zeng L, Guan XJ, Zhang T, Costantini F, Tilghman SM . Two dominant mutations in the mouse fused gene are the result of transposon insertions. Genetics. 1997;147:777–786.
Dao DY, Yang X, Flick LM, Chen D, Hilton MJ, O'Keefe RJ . Axin2 regulates chondrocyte maturation and axial skeletal development. J Orthop Res. 2010;28:89–95.
Yan Y, Tang D, Chen M, Huang J, Xie R, Jonason JH, Tan X, Hou W, Reynolds D, Hsu W, Harris SE, Puzas JE, Awad H, O'Keefe RJ, Boyce BF, Chen D . Axin2 controls bone remodeling through the beta-catenin-BMP signaling pathway in adult mice. J Cell Sci. 2009;122:3566–3578.
Yu HM, Jerchow B, Sheu TJ, Liu B, Costantini F, Puzas JE, Birchmeier W, Hsu W . The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development. 2005;132:1995–2005.
Qian L, Mahaffey JP, Alcorn HL, Anderson KV . Tissue-specific roles of Axin2 in the inhibition and activation of Wnt signaling in the mouse embryo. Proc Natl Acad Sci U S A. 2011;108:8692–8697.
Letra A, Bjork B, Cooper ME, Szabo-Rogers H, Deleyiannis FW, Field LL, Czeizel AE, Ma L, Garlet GP, Poletta FA, Mereb JC, Lopez-Camelo JS, Castilla EE, Orioli IM, Wendell S, Blanton SH, Liu K, Hecht JT, Marazita ML, Vieira AR, Silva RM . Association of AXIN2 with non-syndromic oral clefts in multiple populations. J Dent Res. 2012;91:473–478.
Miclea RL, Karperien M, Bosch CA, van der Horst G, van der Valk MA, Kobayashi T, Kronenberg HM, Rawadi G, Akcakaya P, Lowik CW, Fodde R, Wit JM, Robanus-Maandag EC . Adenomatous polyposis coli-mediated control of beta-catenin is essential for both chondrogenic and osteogenic differentiation of skeletal precursors. BMC Dev Biol. 2009;9:26.
Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, Williams BO . Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280:21162–21168.
Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR . Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90.
Kugimiya F, Kawaguchi H, Ohba S, Kawamura N, Hirata M, Chikuda H, Azuma Y, Woodgett JR, Nakamura K, Chung UI . GSK-3beta controls osteogenesis through regulating Runx2 activity. PLoS One. 2007;2:e837.
Nelson ER, Levi B, Sorkin M, James AW, Liu KJ, Quarto N, Longaker MT . Role of GSK-3beta in the osteogenic differentiation of palatal mesenchyme. PLoS One. 2011;6:e25847.
He F, Popkie AP, Xiong W, Li L, Wang Y, Phiel CJ, Chen Y . Gsk3beta is required in the epithelium for palatal elevation in mice. Dev Dyn. 2010;239:3235–3246.
Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR . Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28:6314–6328.
Gillespie JR, Ulici V, Dupuis H, Higgs A, Dimattia A, Patel S, Woodgett JR, Beier F . Deletion of glycogen synthase kinase-3beta in cartilage results in up-regulation of glycogen synthase kinase-3alpha protein expression. Endocrinology. 2011;152:1755–1766.
Soshnikova N, Zechner D, Huelsken J, Mishina Y, Behringer RR, Taketo MM, Crenshaw EB, 3rd, Birchmeier W . Genetic interaction between Wnt/beta-catenin and BMP receptor signaling during formation of the AER and the dorsal-ventral axis in the limb. Genes Dev. 2003;17:1963–1968.
Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, McCrea PD, De Crombrugghe B . Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;18:1072–1087.
Glass DA, 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G . Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–764.
Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C . Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–738.
Zhu M, Tang D, Wu Q, Hao S, Chen M, Xie C, Rosier RN, O'Keefe RJ, Zuscik M, Chen D . Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J Bone Miner Res. 2009;24:12–21.
Dao DY, Jonason JH, Zhang Y, Hsu W, Chen D, Hilton MJ, O'Keefe RJ . Cartilage-specific beta-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J Bone Miner Res. 2012;27:1680–1694.
Mirando AJ, Maruyama T, Fu J, Yu HM, Hsu W . beta-catenin/cyclin D1 mediated development of suture mesenchyme in calvarial morphogenesis. BMC Dev Biol. 2010;10:116.
Wei W, Zeve D, Suh JM, Wang X, Du Y, Zerwekh JE, Dechow PC, Graff JM, Wan Y . Biphasic and dosage-dependent regulation of osteoclastogenesis by beta-catenin. Mol Cell Biol. 2011;31:4706–4719.
Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F . Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132:49–60.
Song L, Liu M, Ono N, Bringhurst FR, Kronenberg HM, Guo J . Loss of wnt/beta-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J Bone Miner Res. 2012;27:2344–2358.
Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M . Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30:3071–3085.
Chen J, Long F . beta-catenin promotes bone formation and suppresses bone resorption in postnatal growing mice. J Bone Miner Res. 2012. [Epub ahead of print].
Mikasa M, Rokutanda S, Komori H, Ito K, Tsang YS, Date Y, Yoshida CA, Komori T . Regulation of Tcf7 by Runx2 in chondrocyte maturation and proliferation. J Bone Miner Metab. 2011;29:291–299.
Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H . Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19:379–383.
Brugmann SA, Goodnough LH, Gregorieff A, Leucht P, ten Berge D, Fuerer C, Clevers H, Nusse R, Helms JA . Wnt signaling mediates regional specification in the vertebrate face. Development. 2007;134:3283–3295.
Galceran J, Farinas I, Depew MJ, Clevers H, Grosschedl R . Wnt3a-/—like phenotype and limb deficiency in Lef1(-/-)Tcf1(-/-) mice. Genes Dev. 1999;13:709–717.
Noh T, Gabet Y, Cogan J, Shi Y, Tank A, Sasaki T, Criswell B, Dixon A, Lee C, Tam J, Kohler T, Segev E, Kockeritz L, Woodgett J, Muller R, Chai Y, Smith E, Bab I, Frenkel B . Lef1 haploinsufficient mice display a low turnover and low bone mass phenotype in a gender- and age-specific manner. PLoS One. 2009;4:e5438.
van Genderen C, Okamura RM, Farinas I, Quo RG, Parslow TG, Bruhn L, Grosschedl R . Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 1994;8:2691–2703.
Hoeppner LH, Secreto FJ, Razidlo DF, Whitney TJ, Westendorf JJ . Lef1DeltaN binds beta-catenin and increases osteoblast activity and trabecular bone mass. J Biol Chem. 2011;286:10950–10959.
Panakova D, Sprong H, Marois E, Thiele C, Eaton S . Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature. 2005;435:58–65.
Gross JC, Chaudhary V, Bartscherer K, Boutros M 2012 Active Wnt proteins are secreted on exosomes. Nat Cell Biol 14(10):1036–1045.
Witte F, Dokas J, Neuendorf F, Mundlos S, Stricker S . Comprehensive expression analysis of all Wnt genes and their major secreted antagonists during mouse limb development and cartilage differentiation. Gene Expr Patterns. 2009;9:215–223.
McMahon AP, Bradley A . The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–1085.
Monkley SJ, Delaney SJ, Pennisi DJ, Christiansen JH, Wainwright BJ . Targeted disruption of the Wnt2 gene results in placentation defects. Development. 1996;122:3343–3353.
Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A . Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22:361–365.
Parr BA, Cornish VA, Cybulsky MI, McMahon AP . Wnt7b regulates placental development in mice. Dev Biol. 2001;237:324–332.
Nusslein-Volhard C, Wieschaus E . Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801.
McMahon AP, Moon RT . Ectopic expression of the proto-oncogene int-1 in Xenopus embryos leads to duplication of the embryonic axis. Cell. 1989;58:1075–1084.
Goss AM, Tian Y, Tsukiyama T, Cohen ED, Zhou D, Lu MM, Yamaguchi TP, Morrisey EE . Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev Cell. 2009;17:290–298.
Tsukiyama T, Yamaguchi TP . Mice lacking Wnt2b are viable and display a postnatal olfactory bulb phenotype. Neurosci Lett. 2012;512:48–52.
Vainio S, Heikkila M, Kispert A, Chin N, McMahon AP . Female development in mammals is regulated by Wnt-4 signalling. Nature. 1999;397:405–409.
Agalliu D, Takada S, Agalliu I, McMahon AP, Jessell TM . Motor neurons with axial muscle projections specified by Wnt4/5 signaling. Neuron. 2009;61:708–720.
Potok MA, Cha KB, Hunt A, Brinkmeier ML, Leitges M, Kispert A, Camper SA . WNT signaling affects gene expression in the ventral diencephalon and pituitary gland growth. Dev Dyn. 2008;237:1006–1020.
Shu W, Jiang YQ, Lu MM, Morrisey EE . Wnt7b regulates mesenchymal proliferation and vascular development in the lung. Development. 2002;129:4831–4842.
Fotaki V, Larralde O, Zeng S, McLaughlin D, Nichols J, Price DJ, Theil T, Mason JO . Loss of Wnt8b has no overt effect on hippocampus development but leads to altered Wnt gene expression levels in dorsomedial telencephalon. Dev Dyn. 2010;239:284–296.
Majumdar A, Vainio S, Kispert A, McMahon J, McMahon AP . Wnt11 and Ret/Gdnf pathways cooperate in regulating ureteric branching during metanephric kidney development. Development. 2003;130:3175–3185.
Shan J, Jokela T, Peltoketo H, Vainio S . Generation of an allele to inactivate Wnt4 gene function conditionally in the mouse. Genesis. 2009;47:782–788.
Boyer A, Lapointe E, Zheng X, Cowan RG, Li H, Quirk SM, DeMayo FJ, Richards JS, Boerboom D . WNT4 is required for normal ovarian follicle development and female fertility. FASEB J. 2010;24:3010–3025.
Kobayashi A, Stewart CA, Wang Y, Fujioka K, Thomas NC, Jamin SP, Behringer RR . beta-Catenin is essential for Mullerian duct regression during male sexual differentiation. Development. 2011;138:1967–1975.
Dunlap KA, Filant J, Hayashi K, Rucker EB, 3rd, Song G, Deng JM, Behringer RR, DeMayo FJ, Lydon J, Jeong JW, Spencer TE . Postnatal deletion of Wnt7a inhibits uterine gland morphogenesis and compromises adult fertility in mice. Biol Reprod. 2011;85:386–396.
Lobov IB, Rao S, Carroll TJ, Vallance JE, Ito M, Ondr JK, Kurup S, Glass DA, Patel MS, Shu W, Morrisey EE, McMahon AP, Karsenty G, Lang RA . WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature. 2005;437:417–421.
Hayashi K, Yoshioka S, Reardon SN, Rucker EB, 3rd, Spencer TE, DeMayo FJ, Lydon JP, MacLean JA, 2nd . WNTs in the neonatal mouse uterus: potential regulation of endometrial gland development. Biol Reprod. 2011;84:308–319.
Niemann S, Zhao C, Pascu F, Stahl U, Aulepp U, Niswander L, Weber JL, Muller U . Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am J Hum Genet. 2004;74:558–563.
Person AD, Beiraghi S, Sieben CM, Hermanson S, Neumann AN, Robu ME, Schleiffarth JR, Billington CJ, Jr., van Bokhoven H, Hoogeboom JM, Mazzeu JF, Petryk A, Schimmenti LA, Brunner HG, Ekker SC, Lohr JL . WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev Dyn. 2010;239:327–337.
Woods CG, Stricker S, Seemann P, Stern R, Cox J, Sherridan E, Roberts E, Springell K, Scott S, Karbani G, Sharif SM, Toomes C, Bond J, Kumar D, Al-Gazali L, Mundlos S . Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am J Hum Genet. 2006;79:402–408.
Eyaid W, Al-Qattan MM, Al Abdulkareem I, Fetaini N, Al Balwi M . A novel homozygous missense mutation (c.610G>A, p.Gly204Ser) in the WNT7A gene causes tetra-amelia in two Saudi families. Am J Med Genet A. 2011;155A:599–604.
Adaimy L, Chouery E, Megarbane H, Mroueh S, Delague V, Nicolas E, Belguith H, De Mazancourt P, Megarbane A . Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am J Hum Genet. 2007;81:821–828.
Bohring A, Stamm T, Spaich C, Haase C, Spree K, Hehr U, Hoffmann M, Ledig S, Sel S, Wieacker P, Ropke A . WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet. 2009;85:97–105.
van den Boogaard MJ, Creton M, Bronkhorst Y, van der Hout A, Hennekam E, Lindhout D, Cune M, Ploos van Amstel HK . Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet. 2012;49:327–331.
Kantaputra P, Sripathomsawat W . WNT10A and isolated hypodontia. Am J Med Genet A. 2011;155A:1119–1122.
Ugur SA, Tolun A . Homozygous WNT10b mutation and complex inheritance in Split-Hand/Foot Malformation. Hum Mol Genet. 2008;17:2644–2653.
Blattner A, Huber AR, Rothlisberger B . Homozygous nonsense mutation in WNT10B and sporadic split-hand/foot malformation (SHFM) with autosomal recessive inheritance. Am J Med Genet A. 2010;152A:2053–2056.
Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H, Takao T, Takada S . Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell. 2006;11:791–801.
Banziger C, Soldini D, Schutt C, Zipperlen P, Hausmann G, Basler K . Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell. 2006;125:509–522.
Fu J, Jiang M, Mirando AJ, Yu HM, Hsu W . Reciprocal regulation of Wnt and Gpr177/mouse Wntless is required for embryonic axis formation. Proc Natl Acad Sci U S A. 2009;106:18598–18603.
Fu J, Ivy Yu HM, Maruyama T, Mirando AJ, Hsu W . Gpr177/mouse Wntless is essential for Wnt-mediated craniofacial and brain development. Dev Dyn. 2011;240:365–371.
Carpenter AC, Rao S, Wells JM, Campbell K, Lang RA . Generation of mice with a conditional null allele for Wntless. Genesis. 2010;48:554–558.
Proffitt KD, Virshup DM . Precise regulation of porcupine activity is required for physiological wnt signaling. J Biol Chem. 2012;287:34167–34178.
Wang X, Reid Sutton V, Omar Peraza-Llanes J, Yu Z, Rosetta R, Kou YC, Eble TN, Patel A, Thaller C, Fang P, van den Veyver IB . Mutations in X-linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat Genet. 2007;39:836–838.
Herr P, Basler K . Porcupine-mediated lipidation is required for Wnt recognition by Wls. Dev Biol. 2012;361:392–402.
Coombs GS, Yu J, Canning CA, Veltri CA, Covey TM, Cheong JK, Utomo V, Banerjee N, Zhang ZH, Jadulco RC, Concepcion GP, Bugni TS, Harper MK, Mihalek I, Jones CM, Ireland CM, Virshup DM . WLS-dependent secretion of WNT3A requires Ser209 acylation and vacuolar acidification. J Cell Sci. 2010;123:3357–3367.
Grzeschik KH, Bornholdt D, Oeffner F, Konig A, del Carmen Boente M, Enders H, Fritz B, Hertl M, Grasshoff U, Hofling K, Oji V, Paradisi M, Schuchardt C, Szalai Z, Tadini G, Traupe H, Happle R . Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet. 2007;39:833–835.
Leoyklang P, Suphapeetiporn K, Wananukul S, Shotelersuk V . Three novel mutations in the PORCN gene underlying focal dermal hypoplasia. Clin Genet. 2008;73:373–379.
Temple IK, MacDowall P, Baraitser M, Atherton DJ . Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180–187.
Rivadeneira F, Styrkarsdottir U, Estrada K, Halldorsson BV, Hsu YH, Richards JB, Zillikens MC, Kavvoura FK, Amin N, Aulchenko YS, Cupples LA, Deloukas P, Demissie S, Grundberg E, Hofman A, Kong A, Karasik D, van Meurs JB, Oostra B, Pastinen T, Pols HA, Sigurdsson G, Soranzo N, Thorleifsson G, Thorsteinsdottir U, Williams FM, Wilson SG, Zhou Y, Ralston SH, van Duijn CM, Spector T, Kiel DP, Stefansson K, Ioannidis JP, Uitterlinden AG . Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat Genet. 2009;41:1199–1206.
Styrkarsdottir U, Halldorsson BV, Gudbjartsson DF, Tang NL, Koh JM, Xiao SM, Kwok TC, Kim GS, Chan JC, Cherny S, Lee SH, Kwok A, Ho S, Gretarsdottir S, Kostic JP, Palsson ST, Sigurdsson G, Sham PC, Kim BJ, Kung AW, Kim SY, Woo J, Leung PC, Kong A, Thorsteinsdottir U, Stefansson K . European bone mineral density loci are also associated with BMD in East-Asian populations. PLoS One. 2010;5:e13217.
Wang HY, Liu T, Malbon CC . Structure-function analysis of Frizzleds. Cell Signal. 2006;18:934–941.
Wang Y, Thekdi N, Smallwood PM, Macke JP, Nathans J . Frizzled-3 is required for the development of major fiber tracts in the rostral CNS. J Neurosci. 2002;22:8563–8573.
Wang Y, Huso D, Cahill H, Ryugo D, Nathans J . Progressive cerebellar, auditory, and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J Neurosci. 2001;21:4761–4771.
Ye X, Wang Y, Cahill H, Yu M, Badea TC, Smallwood PM, Peachey NS, Nathans J . Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139:285–298.
Ishikawa T, Tamai Y, Zorn AM, Yoshida H, Seldin MF, Nishikawa S, Taketo MM . Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. Development. 2001;128:25–33.
Liu C, Wang Y, Smallwood PM, Nathans J . An essential role for Frizzled5 in neuronal survival in the parafascicular nucleus of the thalamus. J Neurosci. 2008;28:5641–5653.
van Es JH, Jay P, Gregorieff A, van Gijn ME, Jonkheer S, Hatzis P, Thiele A, van den Born M, Begthel H, Brabletz T, Taketo MM, Clevers H . Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol. 2005;7:381–386.
Guo N, Hawkins C, Nathans J . Frizzled6 controls hair patterning in mice. Proc Natl Acad Sci U S A. 2004;101:9277–9281.
Ye X, Wang Y, Rattner A, Nathans J . Genetic mosaic analysis reveals a major role for frizzled 4 and frizzled 8 in controlling ureteric growth in the developing kidney. Development. 2011;138:1161–1172.
Ranheim EA, Kwan HC, Reya T, Wang YK, Weissman IL, Francke U . Frizzled 9 knock-out mice have abnormal B-cell development. Blood. 2005;105:2487–2494.
Niehrs C . The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2010;13:767–779.
Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, Okamura H, Woodgett J, He X . A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–877.
Tamai K, Zeng X, Liu C, Zhang X, Harada Y, Chang Z, He X . A mechanism for Wnt coreceptor activation. Mol Cell. 2004;13:149–156.
Chen M, Philipp M, Wang J, Premont RT, Garrison TR, Caron MG, Lefkowitz RJ, Chen W . G Protein-coupled receptor kinases phosphorylate LRP6 in the Wnt pathway. J Biol Chem. 2009;284:35040–35048.
Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X . Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 2008;22:2968–2979.
Cervenka I, Wolf J, Masek J, Krejci P, Wilcox WR, Kozubik A, Schulte G, Gutkind JS, Bryja V . Mitogen-activated protein kinases promote WNT/beta-catenin signaling via phosphorylation of LRP6. Mol Cell Biol. 2011;31:179–189.
Davidson G, Shen J, Huang YL, Su Y, Karaulanov E, Bartscherer K, Hassler C, Stannek P, Boutros M, Niehrs C . Cell cycle control of wnt receptor activation. Dev Cell. 2009;17:788–799.
Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P, Glinka A, Niehrs C . Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867–872.
Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C . Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 2002;417:664–667.
Johnson EB, Hammer RE, Herz J . Abnormal development of the apical ectodermal ridge and polysyndactyly in Megf7-deficient mice. Hum Mol Genet. 2005;14:3523–3538.
Wu H, Lu Y, Shen C, Patel N, Gan L, Xiong WC, Mei L . Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron. 2012;75:94–107.
Niziolek PJ, Warman ML, Robling AG . Mechanotransduction in bone tissue: The A214V and G171V mutations in Lrp5 enhance load-induced osteogenesis in a surface-selective manner. Bone. 2012;51:459–465.
Xia CH, Liu H, Cheung D, Wang M, Cheng C, Du X, Chang B, Beutler B, Gong X . A model for familial exudative vitreoretinopathy caused by LPR5 mutations. Hum Mol Genet. 2008;17:1605–1612.
Warden SM, Andreoli CM, Mukai S . The Wnt signaling pathway in familial exudative vitreoretinopathy and Norrie disease. Semin Ophthalmol. 2007;22:211–217.
Toomes C, Bottomley HM, Jackson RM, Towns KV, Scott S, Mackey DA, Craig JE, Jiang L, Yang Z, Trembath R, Woodruff G, Gregory-Evans CY, Gregory-Evans K, Parker MJ, Black GC, Downey LM, Zhang K, Inglehearn CF . Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am J Hum Genet. 2004;74:721–730.
Qin M, Kondo H, Tahira T, Hayashi K . Moderate reduction of Norrin signaling activity associated with the causative missense mutations identified in patients with familial exudative vitreoretinopathy. Hum Genet. 2008;122:615–623.
Jiao X, Ventruto V, Trese MT, Shastry BS, Hejtmancik JF . Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am J Hum Genet. 2004;75:878–884.
Mani A, Radhakrishnan J, Wang H, Mani MA, Nelson-Williams C, Carew KS, Mane S, Najmabadi H, Wu D, Lifton RP . LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278–1282.
Krupnik VE, Sharp JD, Jiang C, Robison K, Chickering TW, Amaravadi L, Brown DE, Guyot D, Mays G, Leiby K, Chang B, Duong T, Goodearl AD, Gearing DP, Sokol SY, McCarthy SA . Functional and structural diversity of the human Dickkopf gene family. Gene. 1999;238:301–313.
Nakamura RE, Hunter DD, Yi H, Brunken WJ, Hackam AS . Identification of two novel activities of the Wnt signaling regulator Dickkopf 3 and characterization of its expression in the mouse retina. BMC Cell Biol. 2007;8:52.
Caricasole A, Ferraro T, Iacovelli L, Barletta E, Caruso A, Melchiorri D, Terstappen GC, Nicoletti F . Functional characterization of WNT7A signaling in PC12 cells: interaction with A FZD5 × LRP6 receptor complex and modulation by Dickkopf proteins. J Biol Chem. 2003;278:37024–37031.
Hoang BH, Kubo T, Healey JH, Yang R, Nathan SS, Kolb EA, Mazza B, Meyers PA, Gorlick R . Dickkopf 3 inhibits invasion and motility of Saos-2 osteosarcoma cells by modulating the Wnt-beta-catenin pathway. Cancer Res. 2004;64:2734–2739.
Nakamura RE, Hackam AS . Analysis of Dickkopf3 interactions with Wnt signaling receptors. Growth Factors. 2010;28:232–242.
Bazzi H, Fantauzzo KA, Richardson GD, Jahoda CA, Christiano AM . The Wnt inhibitor, Dickkopf 4, is induced by canonical Wnt signaling during ectodermal appendage morphogenesis. Dev Biol. 2007;305:498–507.
Hirata H, Hinoda Y, Majid S, Chen Y, Zaman MS, Ueno K, Nakajima K, Tabatabai ZL, Ishii N, Dahiya R . DICKKOPF-4 activates the noncanonical c-Jun-NH2 kinase signaling pathway while inhibiting the Wnt-canonical pathway in human renal cell carcinoma. Cancer. 2011;117:1649–1660.
Pietila I, Ellwanger K, Railo A, Jokela T, Barrantes Idel B, Shan J, Niehrs C, Vainio SJ . Secreted Wnt antagonist Dickkopf-1 controls kidney papilla development coordinated by Wnt-7b signalling. Dev Biol. 2011;353:50–60.
Veverka V, Henry AJ, Slocombe PM, Ventom A, Mulloy B, Muskett FW, Muzylak M, Greenslade K, Moore A, Zhang L, Gong J, Qian X, Paszty C, Taylor RJ, Robinson MK, Carr MD . Characterization of the structural features and interactions of sclerostin: molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. 2009;84:10890–10900.
Bourhis E, Wang W, Tam C, Hwang J, Zhang Y, Spittler D, Huang OW, Gong Y, Estevez A, Zilberleyb I, Rouge L, Chiu C, Wu Y, Costa M, Hannoush RN, Franke Y, Cochran AG . Wnt antagonists bind through a short peptide to the first beta-propeller domain of LRP5/6. Structure. 2011;19:1433–1442.
Sarahrudi K, Thomas A, Albrecht C, Aharinejad S . Strongly enhanced levels of sclerostin during human fracture healing. J Orthop Res. 2012;30:1549–1555.
McDonald MM, Morse A, Mikulec K, Peacock L, Yu N, Baldock PA, Birke O, Liu M, Ke HZ, Little DG . Inhibition of sclerostin by systemic treatment with sclerostin antibody enhances healing of proximal tibial defects in ovariectomized rats. J Orthop Res. 2012;30:1541–1548.
Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J . Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–1844.
Chan BY, Fuller ES, Russell AK, Smith SM, Smith MM, Jackson MT, Cake MA, Read RA, Bateman JF, Sambrook PN, Little CB . Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartilage. 2011;19:874–885.
Valero C, Zarrabeitia MT, Hernandez JL, Pineda B, Cano A, Garcia-Perez MA, Riancho JA . Relationship of sclerostin and secreted frizzled protein polymorphisms with bone mineral density: an association study with replication in postmenopausal women. Menopause. 2011;18:802–807.
Richards JB, Kavvoura FK, Rivadeneira F, Styrkarsdottir U, Estrada K, Halldorsson BV, Hsu YH, Zillikens MC, Wilson SG, Mullin BH, Amin N, Aulchenko YS, Cupples LA, Deloukas P, Demissie S, Hofman A, Kong A, Karasik D, van Meurs JB, Oostra BA, Pols HA, Sigurdsson G, Thorsteinsdottir U, Soranzo N, Williams FM, Zhou Y, Ralston SH, Thorleifsson G, van Duijn CM, Kiel DP, Stefansson K, Uitterlinden AG, Ioannidis JP, Spector TD . Collaborative meta-analysis: associations of 150 candidate genes with osteoporosis and osteoporotic fracture. Ann Intern Med. 2009;151:528–537.
Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, Keller H . Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res. 2007;22:1957–1967.
Kim SJ, Bieganski T, Sohn YB, Kozlowski K, Semenov M, Okamoto N, Kim CH, Ko AR, Ahn GH, Choi YL, Park SW, Ki CS, Kim OH, Nishimura G, Unger S, Superti-Furga A, Jin DK . Identification of signal peptide domain SOST mutations in autosomal dominant craniodiaphyseal dysplasia. Hum Genet. 2011;129:497–502.
Ke HZ, Richards WG, Li X, Ominsky MS . Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr Rev. 2012;33:747–783.
Hoang B, Moos M, Jr., Vukicevic S, Luyten FP . Primary structure and tissue distribution of FRZB, a novel protein related to Drosophila frizzled, suggest a role in skeletal morphogenesis. J Biol Chem. 1996;271:26131–26137.
Leyns L, Bouwmeester T, Kim SH, Piccolo S, De Robertis EM . Frzb-1 is a secreted antagonist of Wnt signaling expressed in the Spemann organizer. Cell. 1997;88:747–756.
Wang S, Krinks M, Lin K, Luyten FP, Moos M, Jr. . Frzb, a secreted protein expressed in the Spemann organizer, binds and inhibits Wnt-8. Cell. 1997;88:757–766.
Mii Y, Taira M . Secreted Wnt “inhibitors†are not just inhibitors: regulation of extracellular Wnt by secreted Frizzled-related proteins. Dev Growth Differ. 2011;53:911–923.
Banyai L, Patthy L . The NTR module: domains of netrins, secreted frizzled related proteins, and type I procollagen C-proteinase enhancer protein are homologous with tissue inhibitors of metalloproteases. Protein Sci. 1999;8:1636–1642.
Mii Y, Taira M . Secreted Frizzled-related proteins enhance the diffusion of Wnt ligands and expand their signalling range. Development. 2009;136:4083–4088.
Cho SW, Her SJ, Sun HJ, Choi OK, Yang JY, Kim SW, Kim SY, Shin CS . Differential effects of secreted frizzled-related proteins (sFRPs) on osteoblastic differentiation of mouse mesenchymal cells and apoptosis of osteoblasts. Biochem Biophys Res Commun. 2008;367:399–405.
Galli LM, Barnes T, Cheng T, Acosta L, Anglade A, Willert K, Nusse R, Burrus LW . Differential inhibition of Wnt-3a by Sfrp-1, Sfrp-2, and Sfrp-3. Dev Dyn. 2006;235:681–690.
James IE, Kumar S, Barnes MR, Gress CJ, Hand AT, Dodds RA, Connor JR, Bradley BR, Campbell DA, Grabill SE, Williams K, Blake SM, Gowen M, Lark MW . FrzB-2: a human secreted frizzled-related protein with a potential role in chondrocyte apoptosis. Osteoarthritis Cartilage. 2000;8:452–463.
Hausler KD, Horwood NJ, Chuman Y, Fisher JL, Ellis J, Martin TJ, Rubin JS, Gillespie MT . Secreted frizzled-related protein-1 inhibits RANKL-dependent osteoclast formation. J Bone Miner Res. 2004;19:1873–1881.
Nakanishi R, Shimizu M, Mori M, Akiyama H, Okudaira S, Otsuki B, Hashimoto M, Higuchi K, Hosokawa M, Tsuboyama T, Nakamura T . Secreted frizzled-related protein 4 is a negative regulator of peak BMD in SAMP6 mice. J Bone Miner Res. 2006;21:1713–1721.
Leaf I, Tennessen J, Mukhopadhyay M, Westphal H, Shawlot W . Sfrp5 is not essential for axis formation in the mouse. Genesis. 2006;44:573–578.
Rodriguez-Lopez J, Pombo-Suarez M, Liz M, Gomez-Reino JJ, Gonzalez A . Further evidence of the role of frizzled-related protein gene polymorphisms in osteoarthritis. Ann Rheum Dis. 2007;66:1052–1055.
Valdes AM, Loughlin J, Oene MV, Chapman K, Surdulescu GL, Doherty M, Spector TD . Sex and ethnic differences in the association of ASPN, CALM1, COL2A1, COMP, and FRZB with genetic susceptibility to osteoarthritis of the knee. Arthritis Rheum. 2007;56:137–146.
Lories RJ, Boonen S, Peeters J, De Vlam K, Luyten FP . Evidence for a differential association of the Arg200Trp single-nucleotide polymorphism in FRZB with hip osteoarthritis and osteoporosis. Rheumatology (Oxford). 2006;45:113–114.
Evangelou E, Chapman K, Meulenbelt I, Karassa FB, Loughlin J, Carr A, Doherty M, Doherty S, Gomez-Reino JJ, Gonzalez A, Halldorsson BV, Hauksson VB, Hofman A, Hart DJ, Ikegawa S, Ingvarsson T, Jiang Q, Jonsdottir I, Jonsson H, Kerkhof HJ, Kloppenburg M, Lane NE, Li J, Lories RJ, van Meurs JB, Nakki A, Nevitt MC, Rodriguez-Lopez J, Shi D, Slagboom PE, Stefansson K, Tsezou A, Wallis GA, Watson CM, Spector TD, Uitterlinden AG, Valdes AM, Ioannidis JP . Large-scale analysis of association between GDF5 and FRZB variants and osteoarthritis of the hip, knee, and hand. Arthritis Rheum. 2009;60:1710–1721.
Kerkhof JM, Uitterlinden AG, Valdes AM, Hart DJ, Rivadeneira F, Jhamai M, Hofman A, Pols HA, Bierma-Zeinstra SM, Spector TD, van Meurs JB . Radiographic osteoarthritis at three joint sites and FRZB, LRP5, and LRP6 polymorphisms in two population-based cohorts. Osteoarthritis Cartilage. 2008;16:1141–1149.
Min JL, Meulenbelt I, Kloppenburg M, van Duijn CM, Slagboom PE . Mutation analysis of candidate genes within the 2q33.3 linkage area for familial early-onset generalised osteoarthritis. Eur J Hum Genet. 2007;15:791–799.
Corallini F, Secchiero P, Castellino G, Montecucco M, Trotta F, Zauli G . Circulating levels of frizzled-related protein (FRZB) are increased in patients with early rheumatoid arthritis and decrease in response to disease-modifying antirheumatic drugs. Ann Rheum Dis. 2010;69:1733–1734.
Boudin E, Piters E, Nielsen TL, Andersen M, Roef G, Taes Y, Brixen K, van Hul W . Single nucleotide polymorphisms in sFRP4 are associated with bone and body composition related parameters in Danish but not in Belgian men. Mol Genet Metab. 2012;106:366–374.
Dolmans GH, Werker PM, Hennies HC, Furniss D, Festen EA, Franke L, Becker K, van der Vlies P, Wolffenbuttel BH, Tinschert S, Toliat MR, Nothnagel M, Franke A, Klopp N, Wichmann HE, Nurnberg P, Giele H, Ophoff RA, Wijmenga C . Wnt signaling and Dupuytren's disease. N Engl J Med. 2011;365:307–317.
Theisen H, Purcell J, Bennett M, Kansagara D, Syed A, Marsh JL . dishevelled is required during wingless signaling to establish both cell polarity and cell identity. Development. 1994;120:347–360.
Klingensmith J, Yang Y, Axelrod JD, Beier DR, Perrimon N, Sussman DJ . Conservation of dishevelled structure and function between flies and mice: isolation and characterization of Dvl2. Mech Dev. 1996;58:15–26.
Pizzuti A, Amati F, Calabrese G, Mari A, Colosimo A, Silani V, Giardino L, R Calza L, Palka G, Scarlato G, Novelli G, Dallapiccola B . cDNA characterization and chromosomal mapping of two human homologues of the Drosophila dishevelled polarity gene. Hum Mol Genet. 1996;5:953–958.
Sussman DJ, Klingensmith J, Salinas P, Adams PS, Nusse R, Perrimon N . Isolation and characterization of a mouse homolog of the Drosophila segment polarity gene dishevelled. Dev Biol. 1994;166:73–86.
Tsang M, Lijam N, Yang Y, Beier DR, Wynshaw-Boris A, Sussman DJ . Isolation and characterization of mouse dishevelled-3. Dev Dyn. 1996;207:253–262.
Lee YN, Gao Y, Wang HY . Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, −2, and −3. Cell Signal. 2008;20:443–452.
Gao C, Chen YG . Dishevelled: The hub of Wnt signaling. Cell Signal. 2010;22:717–727.
Kishida S, Yamamoto H, Hino S, Ikeda S, Kishida M, Kikuchi A . DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability. Mol Cell Biol. 1999;19:4414–4422.
Matsumine A, Ogai A, Senda T, Okumura N, Satoh K, Baeg GH, Kawahara T, Kobayashi S, Okada M, Toyoshima K, Akiyama T . Binding of APC to the human homolog of the Drosophila discs large tumor suppressor protein. Science. 1996;272:1020–1023.
Wong HC, Bourdelas A, Krauss A, Lee HJ, Shao Y, Wu D, Mlodzik M, Shi DL, Zheng J . Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol Cell. 2003;12:1251–1260.
Ponting CP, Bork P . Pleckstrin's repeat performance: a novel domain in G-protein signaling? Trends Biochem Sci. 1996;21:245–246.
Li L, Yuan H, Xie W, Mao J, Caruso AM, McMahon A, Sussman DJ, Wu D . Dishevelled proteins lead to two signaling pathways. Regulation of LEF-1 and c-Jun N-terminal kinase in mammalian cells. J Biol Chem. 1999;274:129–134.
Pan WJ, Pang SZ, Huang T, Guo HY, Wu D, Li L . Characterization of function of three domains in dishevelled-1: DEP domain is responsible for membrane translocation of dishevelled-1. Cell Res. 2004;14:324–330.
Yanagawa S, van Leeuwen F, Wodarz A, Klingensmith J, Nusse R . The dishevelled protein is modified by wingless signaling in Drosophila. Genes Dev. 1995;9:1087–1097.
Penton A, Wodarz A, Nusse R . A mutational analysis of dishevelled in Drosophila defines novel domains in the dishevelled protein as well as novel suppressing alleles of axin. Genetics. 2002;161:747–762.
Yokoyama N, Malbon CC . Dishevelled-2 docks and activates Src in a Wnt-dependent manner. J Cell Sci. 2009;122:4439–4451.
Lee JS, Ishimoto A, Yanagawa S . Characterization of mouse dishevelled (Dvl) proteins in Wnt/Wingless signaling pathway. J Biol Chem. 1999;274:21464–21470.
Willert K, Brink M, Wodarz A, Varmus H, Nusse R . Casein kinase 2 associates with and phosphorylates dishevelled. EMBO J. 1997;16:3089–3096.
Strovel ET, Wu D, Sussman DJ . Protein phosphatase 2Calpha dephosphorylates axin and activates LEF-1-dependent transcription. J Biol Chem. 2000;275:2399–2403.
McKay RM, Peters JM, Graff JM . The casein kinase I family in Wnt signaling. Dev Biol. 2001;235:388–396.
Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM . Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc Natl Acad Sci U S A. 2002;99:1182–1187.
Hino S, Michiue T, Asashima M, Kikuchi A . Casein kinase I epsilon enhances the binding of Dvl-1 to Frat-1 and is essential for Wnt-3a-induced accumulation of beta-catenin. J Biol Chem. 2003;278:14066–14073.
Cliffe A, Hamada F, Bienz M . A role of Dishevelled in relocating Axin to the plasma membrane during wingless signaling. Curr Biol. 2003;13:960–966.
Hino S, Kishida S, Michiue T, Fukui A, Sakamoto I, Takada S, Asashima M, Kikuchi A . Inhibition of the Wnt signaling pathway by Idax, a novel Dvl-binding protein. Mol Cell Biol. 2001;21:330–342.
Oshita A, Kishida S, Kobayashi H, Michiue T, Asahara T, Asashima M, Kikuchi A . Identification and characterization of a novel Dvl-binding protein that suppresses Wnt signalling pathway. Genes Cells. 2003;8:1005–1017.
Hocevar BA, Mou F, Rennolds JL, Morris SM, Cooper JA, Howe PH . Regulation of the Wnt signaling pathway by disabled-2 (Dab2). EMBO J. 2003;22:3084–3094.
Singh J, Yanfeng WA, Grumolato L, Aaronson SA, Mlodzik M . Abelson family kinases regulate Frizzled planar cell polarity signaling via Dsh phosphorylation. Genes Dev. 2010;24:2157–2168.
Wei W, Li M, Wang J, Nie F, Li L . The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol Cell Biol. 2012;32:3903–3912.
Lijam N, Paylor R, McDonald MP, Crawley JN, Deng CX, Herrup K, Stevens KE, Maccaferri G, McBain CJ, Sussman DJ, Wynshaw-Boris A . Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell. 1997;90:895–905.
Etheridge SL, Ray S, Li S, Hamblet NS, Lijam N, Tsang M, Greer J, Kardos N, Wang J, Sussman DJ, Chen P, Wynshaw-Boris A . Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008;4:e1000259.
Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL, 3rd, Lee JJ, Tilghman SM, Gumbiner BM, Costantini F . The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell. 1997;90:181–192.
Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A . Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17:1371–1384.
Jho E, Lomvardas S, Costantini F . A GSK3beta phosphorylation site in axin modulates interaction with beta-catenin and Tcf-mediated gene expression. Biochem Biophys Res Commun. 1999;266:28–35.
Gwak J, Hwang SG, Park HS, Choi SR, Park SH, Kim H, Ha NC, Bae SJ, Han JK, Kim DE, Cho JW, Oh S . Small molecule-based disruption of the Axin/beta-catenin protein complex regulates mesenchymal stem cell differentiation. Cell Res. 2012;2:237–247.
Nakamura T, Hamada F, Ishidate T, Anai K, Kawahara K, Toyoshima K, Akiyama T . Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells. 1998;3:395–403.
Hsu W, Zeng L, Costantini F . Identification of a domain of Axin that binds to the serine/threonine protein phosphatase 2A and a self-binding domain. J Biol Chem. 1999;274:3439–3445.
Ikeda S, Kishida M, Matsuura Y, Usui H, Kikuchi A . GSK-3beta-dependent phosphorylation of adenomatous poly-posis coli gene product can be modulated by beta-catenin and protein phosphatase 2A complexed with Axin. Oncogene. 2000;19:537–545.
Sakanaka C, Williams LT . Functional domains of axin. Importance of the C terminus as an oligomerization domain. J Biol Chem. 1999;274:14090–14093.
Fiedler M, Mendoza-Topaz C, Rutherford TJ, Mieszczanek J, Bienz M . Dishevelled interacts with the DIX domain polymerization interface of Axin to interfere with its function in down-regulating beta-catenin. Proc Natl Acad Sci U S A. 2011;108:1937–1942.
Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, Ben-Neriah Y, Alkalay I . Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002;16:1066–1076.
Rui HL, Fan E, Zhou HM, Xu Z, Zhang Y, Lin SC . SUMO-1 modification of the C-terminal KVEKVD of Axin is required for JNK activation but has no effect on Wnt signaling. J Biol Chem. 2002;277:42981–42986.
Kim MJ, Chia IV, Costantini F . SUMOylation target sites at the C terminus protect Axin from ubiquitination and confer protein stability. FASEB J. 2002;22:3785–3794.
Kim S, Jho EH . The protein stability of Axin, a negative regulator of Wnt signaling, is regulated by Smad ubiquitination regulatory factor 2 (Smurf2). J Biol Chem. 2010;285:36420–36426.
Cha B, Kim W, Kim YK, Hwang BN, Park SY, Yoon JW, Park WS, Cho JW, Bedford MT, Jho EH . Methylation by protein arginine methyltransferase 1 increases stability of Axin, a negative regulator of Wnt signaling. Oncogene. 2011;30:2379–2389.
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F . Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620.
Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A, Myer VE, Finan PM, Porter JA, Huang SM, Cong F . RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol. 2011;13:623–629.
Callow MG, Tran H, Phu L, Lau T, Lee J, Sandoval WN, Liu PS, Bheddah S, Tao J, Lill JR, Hongo JA, Davis D, Kirkpatrick DS, Polakis P, Costa M . Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS One. 2011;6:e22595.
Kim SI, Park CS, Lee MS, Kwon MS, Jho EH, Song WK . Cyclin-dependent kinase 2 regulates the interaction of Axin with beta-catenin. Biochem Biophys Res Commun. 2004;317:478–483.
Luo W, Peterson A, Garcia BA, Coombs G, Kofahl B, Heinrich R, Shabanowitz J, Hunt DF, Yost HJ, Virshup DM . Protein phosphatase 1 regulates assembly and function of the beta-catenin degradation complex. EMBO J. 2007;26:1511–1521.
Dao DY, Yang X, Chen D, Zuscik M, O'Keefe RJ . Axin1 and Axin2 are regulated by TGF- and mediate cross-talk between TGF- and Wnt signaling pathways. Ann N Y Acad Sci. 2007;1116:82–99.
Mao J, Wang J, Liu B, Pan W, Farr GH, 3rd, Flynn C, Yuan H, Takada S, Kimelman D, Li L, Wu D . Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell. 2001;7:801–809.
Liu G, Bafico A, Harris VK, Aaronson SA . A novel mechanism for Wnt activation of canonical signaling through the LRP6 receptor. Mol Cell Biol. 2003;23:5825–5835.
Cong F, Varmus H . Nuclear-cytoplasmic shuttling of Axin regulates subcellular localization of beta-catenin. Proc Natl Acad Sci U S A. 2004;101:2882–2887.
Wiechens N, Heinle K, Englmeier L, Schohl A, Fagotto F . Nucleo-cytoplasmic shuttling of Axin, a negative regulator of the Wnt-beta-catenin Pathway. J Biol Chem. 2004;279:5263–5267.
Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W . Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–599.
Chia IV, Costantini F . Mouse axin and axin2/conductin proteins are functionally equivalent in vivo . Mol Cell Biol. 2005;25:4371–4376.
Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F . Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183.
Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER . Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 2002;277:21657–21665.
Xie R, Jiang R, Chen D . Generation of Axin1 conditional mutant mice. Genesis. 2011;49:98–102.
Chia IV, Kim MJ, Itoh K, Sokol SY, Costantini F . Both the RGS domain and the six C-terminal amino acids of mouse Axin are required for normal embryogenesis. Genetics. 2009;181:1359–1368.
Hsu W, Shakya R, Costantini F . Impaired mammary gland and lymphoid development caused by inducible expression of Axin in transgenic mice. J Cell Biol. 2001;155:1055–1064.
Yu HM, Liu B, Costantini F, Hsu W . Impaired neural development caused by inducible expression of Axin in transgenic mice. Mech Dev. 2007;124:146–156.
Oates NA, van Vliet J, Duffy DL, Kroes HY, Martin NG, Boomsma DI, Campbell M, Coulthard MG, Whitelaw E, Chong S . Increased DNA methylation at the AXIN1 gene in a monozygotic twin from a pair discordant for a caudal duplication anomaly. Am J Hum Genet. 2006;79:155–162.
Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P . Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74:1043–1050.
Mostowska A, Biedziak B, Jagodzinski PP . Axis inhibition protein 2 (AXIN2) polymorphisms may be a risk factor for selective tooth agenesis. J Hum Genet. 2006;51:262–266.
Letra A, Menezes R, Granjeiro JM, Vieira AR . AXIN2 and CDH1 polymorphisms, tooth agenesis, and oral clefts. Birth Defects Res A Clin Mol Teratol. 2009;85:169–173.
Bergendal B, Klar J, Stecksen-Blicks C, Norderyd J, Dahl N . Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am J Med Genet. 2011;A 155A:1616–1622.
Carlsson G, Petrelli NJ, Nava H, Herrera L, Mittelman A . The value of colonoscopic surveillance after curative resection for colorectal cancer or synchronous adenomatous polyps. Arch Surg. 1987;122:1261–1263.
Jarvinen HJ, Peltokallio P, Landtman M, Wolf J . Gardner's stigmas in patients with familial adenomatosis coli. Br J Surg. 1982;69:718–721.
Hoffmann DC, Brooke BN . Familial sarcoma of bone in a polyposis coli family. Dis Colon Rectum. 1970;13:119–120.
Utsunomiya J, Nakamura T . The occult osteomatous changes in the mandible in patients with familial polyposis coli. Br J Surg. 1975;62:45–51.
Clark SK, Neale KF, Landgrebe JC, Phillips RK . Desmoid tumours complicating familial adenomatous polyposis. Br J Surg. 1999;6:1185–1189.
Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M . Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600.
Fodde R . The multiple functions of tumour suppressors: it's all in APC. Nat Cell Biol. 2003;5:190–192.
Aberle H, Bauer A, Stappert J, Kispert A, Kemler R . beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804.
Neufeld KL, Zhang F, Cullen BR, White RL . APC-mediated downregulation of beta-catenin activity involves nuclear sequestration and nuclear export. EMBO Rep. 2000;1:519–523.
Henderson BR, Fagotto F . The ins and outs of APC and beta-catenin nuclear transport. EMBO Rep. 2002;3:834–839.
Henderson BR . Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat Cell Biol. 2000;2:653–660.
Neufeld KL, Nix DA, Bogerd H, Kang Y, Beckerle MC, Cullen BR, White RL . Adenomatous polyposis coli protein contains two nuclear export signals and shuttles between the nucleus and cytoplasm. Proc Natl Acad Sci U S A. 2000;97:12085–12090.
Neufeld KL, White RL . Nuclear and cytoplasmic localizations of the adenomatous polyposis coli protein. Proc Natl Acad Sci U S A. 1997;94:3034–3039.
Miclea RL, Karperien M, Langers AM, Robanus-Maandag EC, van Lierop A, van der Hiel B, Stokkel MP, Ballieux BE, Oostdijk W, Wit JM, Vasen HF, Hamdy NA . APC mutations are associated with increased bone mineral density in patients with familial adenomatous polyposis. J Bone Miner Res. 2010;25:2624–2632.
Woodgett JR . Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438.
Westendorf JJ, Kahler RA, Schroeder TM . Wnt signaling in osteoblasts and bone diseases. Gene. 2004;1:19–39.
Hagen T, Vidal-Puig A . Characterisation of the phosphorylation of beta-catenin at the GSK-3 priming site Ser45. Biochem Biophys Res Commun. 2002;294:324–328.
Hagen T, Di Daniel E, Culbert AA, Reith AD . Expression and characterization of GSK-3 mutants and their effect on beta-catenin phosphorylation in intact cells. J Biol Chem. 2002;277:23330–23335.
Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X . Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847.
Sakanaka C . Phosphorylation and regulation of beta-catenin by casein kinase I epsilon. J Biochem. 2002;132:697–703.
Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C . Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science. 2007;316:1619–1622.
Chen G, Jiang Q, You Z, Yao J, Mou L, Lin X, Shen X, You T, Lin Q, Wen J, Lin L . Regulation of GSK-3 beta in the proliferation and apoptosis of human thyrocytes investigated using a GSK-3 beta-targeting RNAi adenovirus expression vector: involvement the Wnt/beta-catenin pathway. Mol Biol Rep. 2010;37:2773–2779.
Kulkarni NH, Onyia JE, Zeng Q, Tian X, Liu M, Halladay DL, Frolik CA, Engler T, Wei T, Kriauciunas A, Martin TJ, Sato M, Bryant HU, Ma YL . Orally bioavailable atti A, Penso D, GSK-3alpha/beta dual inhibitor increases markers of cellular differentiation in vitro and bone mass in vivo . J Bone Miner Res. 2006;21:910–920.
Detera-Wadleigh SD . Lithium-related genetics of bipolar disorder. Ann Med. 2001;33:272–285.
Lenox RH, Gould TD, Manji HK . Endophenotypes in bipolar disorder. Am J Med Genet. 2002;114:391–406.
Manji HK, Moore GJ, Chen G . Lithium at 50: have the neuroprotective effects of this unique cation been overlooked? Biol Psychiatry. 1999;46:929–940.
Haney EM, Warden SJ, Bliziotes MM . Effects of selective serotonin reuptake inhibitors on bone health in adults: time for recommendations about screening, prevention and management? Bone. 2010;46:13–17.
Vestergaard P, Rejnmark L, Mosekilde L . Reduced relative risk of fractures among users of lithium. Calcif Tissue Int. 2005;77:1–8.
Wilting I, De Vries F, Thio BM, Cooper C, Heerdink ER, Leufkens HG, Nolen WA, Egberts AC, van Staa TP . Lithium use and the risk of fractures. Bone. 2007;40:1252–1258.
Polakis P . Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4.
Robinson DR, Zylstra CR, Williams BO . Wnt signaling and prostate cancer. Curr Drug Targets. 2008;9:571–580.
Chen G, Shukeir N, Potti A, Sircar K, Aprikian A, Goltzman D, Rabbani SA . Up-regulation of Wnt-1 and beta-catenin production in patients with advanced metastatic prostate carcinoma: potential pathogenetic and prognostic implications. Cancer. 2004;101:1345–1356.
Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, Sugano S, Akiyama T . DKK1, a negative regulator of Wnt signaling, is a target of the beta-catenin/TCF pathway. Oncogene. 2004;23:8520–8526.
Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM . Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999;18:5931–5942.
Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R . Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264.
Day TF, Guo X, Garrett-Beal L, Yang Y . Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–750.
Ozawa M, Baribault H, Kemler R . The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J. 1989;8:1711–1717.
Abe K, Takeichi M . EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci U S A. 2008;105:13–19.
Pappas DJ, Rimm DL . Direct interaction of the C-terminal domain of alpha-catenin and F-actin is necessary for stabilized cell-cell adhesion. Cell Commun Adhes. 2006;13:151–170.
Case N, Ma M, Sen B, Xie Z, Gross TS, Rubin J . Beta-catenin levels influence rapid mechanical responses in osteoblasts. J Biol Chem. 2008;283:29196–29205.
Hens JR, Wilson KM, Dann P, Chen X, Horowitz MC, Wysolmerski JJ . TOPGAL mice show that the canonical Wnt signaling pathway is active during bone development and growth and is activated by mechanical loading in vitro . J Bone Miner Res. 2005;20:1103–1113.
Armstrong VJ, Muzylak M, Sunters A, Zaman G, Saxon LK, Price JS, Lanyon LE . Wnt/beta-catenin signaling is a component of osteoblastic bone cell early responses to load-bearing and requires estrogen receptor alpha. J Biol Chem. 2007;282:20715–20727.
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH . Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–5875.
Clevers H, Nusse R . Wnt/beta-catenin signaling and disease. Cell. 2012;49:1192–1205.
Oosterwegel MA, van de Wetering ML, Holstege FC, Prosser HM, Owen MJ, Clevers HC . TCF-1, a T cell-specific transcription factor of the HMG box family, interacts with sequence motifs in the TCR beta and TCR delta enhancers. Int Immunol. 1991;3:1189–1192.
Oosterwegel M, van de Wetering M, Dooijes D, Klomp L, Winoto A, Georgopoulos K, Meijlink F, Clevers H . Cloning of murine TCF-1, a T cell-specific transcription factor interacting with functional motifs in the CD3-epsilon and T cell receptor alpha enhancers. J Exp Med. 1991;173:1133–1142.
van de Wetering M, Oosterwegel M, Dooijes D, Clevers H . Identification and cloning of TCF-1, a T lymphocyte-specific transcription factor containing a sequence-specific HMG box. EMBO J. 1991;10:123–132.
Travis A, Amsterdam A, Belanger C, Grosschedl R . LEF-1, a gene encoding a lymphoid-specific protein with an HMG domain, regulates T-cell receptor alpha enhancer function [corrected]. Genes Dev. 1991;5:880–894.
Waterman ML, Fischer WH, Jones KA . A thymus-specific member of the HMG protein family regulates the human T cell receptor C alpha enhancer. Genes Dev. 1991;5:656–669.
Schilham MW, Clevers H . HMG box containing transcription factors in lymphocyte differentiation. Semin Immunol. 1998;10:127–132.
Clevers H, van de Wetering M . TCF/LEF factor earn their wings. Trends Genet. 1997;13:485–489.
Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W . Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642.
Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H . XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399.
Waterman ML . Lymphoid enhancer factor/T cell factor expression in colorectal cancer. Cancer Metastasis Rev. 2004;23:41–52.
Castrop J, Verbeek S, Hofhuis F, Clevers H . Circumvention of tolerance for the nuclear T cell protein TCF-1 by immunization of TCF-1 knock-out mice. Immunobiology. 1995;193:281–287.
Okamura RM, Sigvardsson M, Galceran J, Verbeek S, Clevers H, Grosschedl R . Redundant regulation of T cell differentiation and TCRalpha gene expression by the transcription factors LEF-1 and TCF-1. Immunity. 1998;8:11–20.
Vacik T, Lemke G . Dominant-negative isoforms of Tcf/Lef proteins in development and disease. Cell Cycle. 2011;10:4199–4200.
Van de Wetering M, Castrop J, Korinek V, Clevers H . Extensive alternative splicing and dual promoter usage generate Tcf-1 protein isoforms with differential transcription control properties. Mol Cell Biol. 1996;16:745–752.
Hoeppner LH, Secreto FJ, Westendorf JJ . Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets. 2009;13:485–496.
Kahler RA, Galindo M, Lian J, Stein GS, van Wijnen AJ, Westendorf JJ . Lymphocyte enhancer-binding factor 1 (Lef1) inhibits terminal differentiation of osteoblasts. J Cell Biochem. 2006;97:969–983.
Hoeppner LH, Secreto F, Jensen ED, Li X, Kahler RA, Westendorf JJ . Runx2 and bone morphogenic protein 2 regulate the expression of an alternative Lef1 transcript during osteoblast maturation. J Cell Physiol. 2009;221:480–489.
Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T, Lawrence Marsh J, Holcombe RF, Waterman ML . Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet. 2001;28:53–57.
Chen G, Fernandez J, Mische S, Courey AJ . A functional interaction between the histone deacetylase Rpd3 and the corepressor groucho in Drosophila development. Genes Dev. 1999;13:2218–2230.
Courey AJ, Jia S . Transcriptional repression: the long and the short of it. Genes Dev. 2001;15:2786–2796.
Turki-Judeh W, Courey AJ . Groucho: a corepressor with instructive roles in development. Curr Top Dev Biol. 2012;98:65–96.
Fiedler M, Sanchez-Barrena MJ, Nekrasov M, Mieszczanek J, Rybin V, Muller J, Evans P, Bienz M . Decoding of methylated histone H3 tail by the Pygo-BCL9 Wnt signaling complex. Mol Cell. 2008;30:507–518.
Mosimann C, Hausmann G, Basler K . Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol. 2009;10:276–286.
Mosimann C, Hausmann G, Basler K . Parafibromin/Hyrax activates Wnt/Wg target gene transcription by direct association with beta-catenin/Armadillo. Cell. 2006;125:327–341.
Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, Shanmugam KS, Copeland TD, Guszczynski T, Resau JH, Meyerson M . The parafibromin tumor suppressor protein is part of a human Paf1 complex. Mol Cell Biol. 2005;25:612–620.
Jaehning JA . The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta. 2010;1799:379–388.
Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD, Agarwal SK, Sood R, Jones MP, Moses TY, Haven C, Petillo D, Leotlela PD, Harding B, Cameron D, Pannett AA, Hoog A, Heath H, 3rd, James-Newton LA, Robinson B, Zarbo RJ, Cavaco BM, Wassif W, Perrier ND, Rosen IB, Kristoffersson U, Turnpenny PD, Farnebo LO, Besser GM, Jackson CE, Morreau H, Trent JM, Thakker RV, Marx SJ, Teh BT, Larsson C, Hobbs MR . HRPT2, encoding parafibromin, is mutated in hyperpara-thyroidism-jaw tumor syndrome. Nat Genet. 2002;32:676–680.
Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, Fleuren GJ, Robinson BG, Delbridge LW, Philips J, Nelson AE, Krause U, Hammje K, Dralle H, Hoang-Vu C, Gimm O, Marsh DJ, Morreau H, Teh BT . HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40:657–663.
Bradley KJ, Bowl MR, Williams SE, Ahmad BN, Partridge CJ, Patmanidi AL, Kennedy AM, Loh NY, Thakker RV . Parafibromin is a nuclear protein with a functional monopartite nuclear localization signal. Oncogene. 2007;26:1213–1221.
Jackson A, Vayssiere B, Garcia T, Newell W, Baron R, Roman-Roman S, Rawadi G . Gene array analysis of Wnt-regulated genes in C3H10T1/2 cells. Bone. 2005;36:585–598.
Lum L, Clevers H . Cell biology. The unusual case of Porcupine. Science. 2012;337:922–923.
Rey JP, Ellies DL . Wnt modulators in the biotech pipeline. Dev Dyn. 2010;239:102–114.
Acknowledgements
We thank David Nadziejka for extensive assistance with technical editing. The laboratory of BOW is supported by NIH grant AR053293 and the Van Andel Research Institute. KAM and CJD are supported by the Van Andel Institute Graduate School.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Maupin, K., Droscha, C. & Williams, B. A Comprehensive Overview of Skeletal Phenotypes Associated with Alterations in Wnt/β-catenin Signaling in Humans and Mice. Bone Res 1, 27–71 (2013). https://doi.org/10.4248/BR201301004
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.4248/BR201301004
