

Multi-Omics Approaches and Resources for Systems-Level Gene Function Prediction in the Plant Kingdom

 , , , ,
, , , , 
Abstract
:1. Introduction
2. The Omics-Platform
2.1. Genomics
2.1.1. Genomic-Assisted Gene Discovery for Crop Improvement
2.1.2. Single Cell Sequencing
2.1.3. Genome-Wide Association Study (GWAS)
2.1.4. Pan-Genomics
2.2. Transcriptomics
Transcriptome-Wide Association Studies: Prediction of Genes Governing Complex traits
2.3. Phenome
2.4. Epigenetic Modification
2.4.1. Interactomics
2.4.2. Resources for Plant Protein–Protein Interactions
2.4.3. Integrated Multi-Layer Omics Data for Functional Studies in Plant
- Genomics-transcriptomics
- Transcriptomics-proteomics
2.5. Candidate Gene Mining in the Context of Pathway Reconstruction
Challenges in Cellular Pathway Reconstruction
3. Guilt-by-Association (GBA), a Method for Gene Discovery
4. Predictive Modelling, Artificial Intelligence and Machine Learning Based Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parry, M.A.J.; Reynolds, M.; Salvucci, M.E.; Raines, C.; Andralojc, P.J.; Zhu, X.-G.; Price, D.G.; Condon, A.G.; Furbank, R.T. Raising yield potential of wheat. II. Increasing photosynthetic capacity and efficiency. J. Exp. Bot. 2011, 62, 453–467. [Google Scholar] [CrossRef]
- Pramanik, D.; Shelake, R.M.; Kim, M.J.; Kim, J.-Y. CRISPR-Mediated Engineering across the Central Dogma in Plant Biology for Basic Research and Crop Improvement. Mol. Plant 2021, 14, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, Y.; Chen, X.; Chen, Y. Plant Noncoding RNAs: Hidden Players in Development and Stress Responses. Annu. Rev. Cell Dev. Biol. 2019, 35, 407–431. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Huang, S.-S.C. Improving plant gene regulatory network inference by integrative analysis of multi-omics and high resolution data sets. Curr. Opin. Syst. Biol. 2022, 22, 8–15. [Google Scholar] [CrossRef]
- Herrgård, M.J.; Swainston, N.; Dobson, P.; Dunn, W.B.; Arga, K.Y.; Arvas, M.; Blüthgen, N.; Borger, S.; Costenoble, R.; Heinemann, M.; et al. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat. Biotechnol. 2008, 26, 1155–1160. [Google Scholar] [CrossRef]
- Raikhel, N.V.; Coruzzi, G.M. Achieving the in silico plant. Systems biology and the future of plant biological research. Plant Physiol. 2003, 132, 404–409. [Google Scholar]
- Santos, F.; Boele, J.; Teusink, B. A Practical Guide to Genome-Scale Metabolic Models and Their Analysis. Methods Enzymol. 2011, 500, 509–532. [Google Scholar] [CrossRef] [PubMed]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121. [Google Scholar] [CrossRef]
- McCloskey, D.; Palsson, B.; Feist, A.M. Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol. Syst. Biol. 2013, 9, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahood, E.H.; Kruse, L.H.; Moghe, G.D. Machine learning: A powerful tool for gene function prediction in plants. Appl. Plant Sci. 2020, 8, e11376. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural variant calling: The long and the short of it. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Weirauch, M.T.; Cote, A.; Norel, R.; Annala, M.; Zhao, Y.; Riley, T.R.; Saez-Rodriguez, J.; Cokelaer, T.; Vedenko, A.; Talukder, S.; et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 2013, 31, 126–134. [Google Scholar] [CrossRef]
- Vu, T.T.D.; Jung, J. Protein function prediction with gene ontology: From traditional to deep learning models. PeerJ 2021, 9, e12019. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Singh, S.; Rao, A.; Mishra, P.; Yadav, A.K.; Maurya, R.; Kaur, S.; Tandon, G. Bioinformatics in Next-Generation Genome Se-quencing. In Current Trends in Bioinformatics: An Insight; Wadhwa, G., Shanmughavel, P., Singh, A., Bellare, J., Eds.; Springer: Singapore, 2018; pp. 27–38. [Google Scholar]
- Kühner, S.; van Noort, V.; Betts, M.J.; Leo-Macias, A.; Batisse, C.; Rode, M.; Yamada, T.; Maier, T.; Bader, S.; Beltran-Alvarez, P.; et al. Proteome Organization in a Genome-Reduced Bacterium. Science 2009, 326, 1235–1240. [Google Scholar] [CrossRef]
- Edwards, D.; Batley, J. Plant bioinformatics: From genome to phenome. Trends Biotechnol. 2004, 22, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Bolger, M.E.; Weisshaar, B.; Scholz, U.; Stein, N.; Usadel, B.; Mayer, K.F. Plant genome sequencing—Applications for crop improvement. Curr. Opin. Biotechnol. 2014, 26, 31–37. [Google Scholar] [CrossRef]
- Cao, Y.; Fanning, S.; Proos, S.; Jordan, K.; Srikumar, S. A Review on the Applications of Next Generation Sequencing Tech-nologies as Applied to Food-Related Microbiome Studies. Front. Microbiol. 2017, 8, 1829. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Haynes, E.; Jimenez, E.; Pardo, M.A.; Helyar, S.J. The future of NGS (Next Generation Sequencing) analysis in testing food authenticity. Food Control 2019, 101, 134–143. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 28 July 2022).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Jiao, W.B.; Schneeberger, K. Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rear-rangements with altered evolutionary dynamics. Nat. Commun. 2020, 11, 989. [Google Scholar] [CrossRef] [Green Version]
- Abril, J.F.; Castellano Hereza, S. Genome Annotation. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Schönbach, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 195–209. [Google Scholar]
- Liu, Y.; Guo, J.; Hu, G.; Zhu, H. Gene prediction in metagenomic fragments based on the SVM algorithm. BMC Bioinform. 2013, 14, S12. [Google Scholar] [CrossRef] [Green Version]
- Scalzitti, N.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genom. 2020, 21, 293. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, Y.; Li, Y. A Brief Review of Computational Gene Prediction Methods. Genom. Proteom. Bioinform. 2004, 2, 216–221. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, S.-Y.; Deng, F. Well-characterized sequence features of eukaryote genomes and implications for ab initio gene prediction. Comput. Struct. Biotechnol. J. 2016, 14, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pati, A.; Ivanova, N.N.; Mikhailova, N.; Ovchinnikova, G.; Hooper, S.D.; Lykidis, A.; Kyrpides, N.C. GenePRIMP: A gene prediction improvement pipeline for prokaryotic genomes. Nat. Chem. Biol. 2010, 7, 455–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, I.; O’Toole, N.; Zabaneh, O.; Nourzadeh, R.; Dahdouli, M.; Abdellateef, M.; Gordon, P.M.; Soh, J.; Butler, G.; Sensen, C.W.; et al. SnowyOwl: Accurate prediction of fungal genes by using RNA-Seq and homology information to select among ab initio models. BMC Bioinform. 2014, 15, 229. [Google Scholar] [CrossRef] [Green Version]
- Testa, A.C.; Hane, J.K.; Ellwood, S.R.; Oliver, R.P. CodingQuarry: Highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genom. 2015, 16, 170. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef] [Green Version]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.S.; Law, M.; Holt, C.; Stein, J.C.; Moghe, G.; Hufnagel, D.; Lei, J.; Achawanantakun, R.; Jiao, D.; Lawrence, C.J.; et al. MAKER-P: A Tool Kit for the Rapid Creation, Management, and Quality Control of Plant Genome Annotations. Plant Physiol. 2013, 164, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.-L.; Rosli, R.; Tatarinova, T.V.; Hogan, M.; Firdaus-Raih, M.; Low, E.-T.L. Seqping: Gene prediction pipeline for plant genomes using self-training gene models and transcriptomic data. BMC Bioinform. 2017, 18, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Mao, L.; Ware, D.; Stein, L. Evidence-based gene predictions in plant genomes. Genome Res. 2009, 19, 1912–1923. [Google Scholar] [CrossRef] [Green Version]
- Flicek, P. Gene prediction: Compare and CONTRAST. Genome Biol. 2007, 8, 233. [Google Scholar] [CrossRef] [PubMed]
- Van Baren, M.J.; Koebbe, B.C.; Brent, M.R. Using N-SCAN or TWINSCAN to predict gene structures in genomic DNA se-quences. Curr. Protoc. Bioinform. 2007, 20, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T. Identification of complete gene structures in genomic DNA. Genome Biol. 2000, 1, reports222. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Henry, C.S.; Hanson, A.D. Frontiers in metabolic reconstruction and modeling of plant genomes. J. Exp. Bot. 2012, 63, 2247–2258. [Google Scholar] [CrossRef] [Green Version]
- Osterman, A.; Overbeek, R. Missing genes in metabolic pathways: A comparative genomics approach. Curr. Opin. Chem. Biol. 2003, 7, 238–251. [Google Scholar] [CrossRef]
- De Oliveira Dal’Molin, C.G.; Nielsen, L.K. Plant genome-scale metabolic reconstruction and modelling. Curr. Opin. Biotechnol. 2013, 24, 271–277. [Google Scholar] [CrossRef]
- Pont, C.; Wagner, S.; Kremer, A.; Orlando, L.; Plomion, C.; Salse, J. Paleogenomics: Reconstruction of plant evolutionary trajectories from modern and ancient DNA. Genome Biol. 2019, 20, 29. [Google Scholar] [CrossRef]
- Rai, A.; Saito, K. Omics data input for metabolic modeling. Curr. Opin. Biotechnol. 2016, 37, 127–134. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [Green Version]
- Bolser, D.; Staines, D.M.; Pritchard, E.; Kersey, P. Ensembl Plants: Integrating Tools for Visualizing, Mining, and Analyzing Plant Genomics Data. Methods Mol. Biol. 2016, 1374, 115–140. [Google Scholar] [CrossRef]
- Dong, Q.; Schlueter, S.D.; Brendel, V. PlantGDB, plant genome database and analysis tools. Nucleic Acids Res. 2004, 32, 354D–359D. [Google Scholar] [CrossRef] [PubMed]
- Proost, S.; Van Bel, M.; Vaneechoutte, D.; Van De Peer, Y.; Inzé, D.; Mueller-Roeber, B.; Vandepoele, K. PLAZA 3.0: An access point for plant comparative genomics. Nucleic Acids Res. 2015, 43, D974–D981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Jaiswal, P.; Hebbard, C.; Avraham, S.; Buckler, E.S.; Casstevens, T.; Hurwitz, B.; McCouch, S.; Ni, J.; Pujar, A.; et al. Gramene: A growing plant comparative genomics resource. Nucleic Acids Res. 2006, 36, D947–D953. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Sinha, P.; Singh, V.K.; Kumar, A.; Zhang, Q.; Bennetzen, J.L. 5Gs for crop genetic improvement. Curr. Opin. Plant Biol. 2020, 56, 190–196. [Google Scholar] [CrossRef]
- Varshney, R.K.; Pandey, M.K.; Bohra, A.; Singh, V.K.; Thudi, M.; Saxena, R.K. Toward the sequence-based breeding in legumes in the post-genome sequencing era. Theor. Appl. Genet. 2019, 132, 797–816. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Bohra, A.; Roorkiwal, M.; Barmukh, R.; Cowling, W.A.; Chitikineni, A.; Lam, H.-M.; Hickey, L.T.; Croser, J.S.; Bayer, P.E.; et al. Fast-forward breeding for a food-secure world. Trends Genet. 2021, 37, 1124–1136. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Minnoye, L.; Marinov, G.K.; Krausgruber, T.; Pan, L.; Marand, A.P.; Secchia, S.; Greenleaf, W.J.; Furlong, E.E.M.; Zhao, K.; Schmitz, R.J.; et al. Chromatin accessibility profiling methods. Nat. Rev. Methods Prim. 2021, 1, 58. [Google Scholar] [CrossRef]
- Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; Kaul, R.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Tu, X.; Marand, A.P.; Schmitz, R.J.; Zhong, S. A combinatorial indexing strategy for low-cost epigenomic profiling of plant single cells. Plant Comm. 2022, 3, 100308. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Villalon, A.; Brady, S.M. Single cell RNA sequencing and its promise in reconstructing plant vascular cell lineages. Curr. Opin. Plant Biol. 2019, 48, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Birnbaum, K.D.; Ehrhardt, D.W. Towards building a plant cell atlas. Trends Plant Sci. 2019, 24, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denyer, T.; Timmermans, M.C. Crafting a blueprint for single-cell RNA sequencing. Trends Plant Sci. 2022, 27, 92–103. [Google Scholar] [CrossRef]
- Giacomello, S. A new era for plant science: Spatial single-cell transcriptomics. Curr. Opin. Plant Biol. 2021, 60, 102041. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Gao, S.; Cui, F.; Chen, W.; Fan, L.; Qi, Y. Single-cell RNA sequencing reveals the landscape of maize root tips and assists in identification of cell type-specific nitrate-response genes. Crop J. 2022. [Google Scholar] [CrossRef]
- He, T.; Li, C. Harness the power of genomic selection and the potential of germplasm in crop breeding for global food security in the era with rapid climate change. Crop J. 2020, 8, 688–700. [Google Scholar] [CrossRef]
- Cuevas, J.; Crossa, J.; Montesinos-López, O.A.; Burgueño, J.; Pérez-Rodríguez, P.; de Los Campos, G. Bayesian Genomic Pre-diction with Genotype × Environment Interaction Kernel Models. G3 Genes Genomes Genet. 2017, 7, 41–53. [Google Scholar]
- Mulesa, T.H.; Westengen, O.T. Against the grain? A historical institutional analysis of access governance of plant genetic resources for food and agriculture in Ethiopia. J. World Intellect. Prop. 2020, 23, 82–120. [Google Scholar] [CrossRef] [Green Version]
- Yugander, A.; Sundaram, R.M.; Singh, K.; Ladhalakshmi, D.; Rao, L.V.S.; Madhav, M.S.; Badri, J.; Prasad, M.S.; Laha, G.S. Incorporation of the novel bacterial blight resistance gene Xa38 into the genetic background of elite rice variety Improved Samba Mahsuri. PLoS ONE 2018, 13, e0198260. [Google Scholar] [CrossRef] [PubMed]
- Ratna Madhavi, K.; Rambabu, R.; Abhilash Kumar, V.; Vijay Kumar, S.; Aruna, J.; Ramesh, S.; Sundaram, R.M.; Laha, G.S.; Sheshu Madhav, M.; Prasad, M.S. Marker assisted introgression of blast (Pi-2 and Pi-54) genes into the genetic background of elite, bacterial blight resistant indica rice variety, Improved Samba Mahsuri. Euphytica 2016, 212, 331–342. [Google Scholar] [CrossRef]
- Huang, C.; Chen, Z.; Liang, C. Oryza pan-genomics: A new foundation for future rice research and improvement. Crop J. 2021, 9, 622–632. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; Chai, S.; Yang, Z.; Zhang, Q.; Xin, H.; Xu, Y.; Lin, S.; Chen, X.; Yao, Z.; et al. Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 2022, 13, 682. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, J.; Zhang, H.; Liu, Z.; Wang, Y.; Xing, L.; He, Q.; Du, H. Plant pan-genomics: Recent advances, new challenges, and roads ahead. J. Genet. Genom. 2022. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, R.; Duraiyan, J.; Kaliyappan, K.; Palanisamy, M. Microarray and its applications. J. Pharm. Bioallied. Sci. 2012, 4, S310–S312. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.T.; Landry, J.-R. RNA-Seq—quantitative measurement of expression through massively parallel RNA-sequencing. Methods 2009, 48, 249–257. [Google Scholar] [CrossRef]
- Costa-Silva, J.; Domingues, D.; Martins Lopes, F. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.Y.; Medvedovic, M.; Bumgarner, R.E. From co-expression to co-regulation: How many microarray experiments do we need? Genome Biol. 2004, 5, R48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ederli, L.; Dawe, A.; Pasqualini, S.; Quaglia, M.; Xiong, L.; Gehring, C. Arabidopsis flower specific defense gene expression patterns affect resistance to pathogens. Front. Plant Sci. 2015, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Horimoto, K. Relationship between regulatory pattern of gene expression level and gene function. PLoS ONE 2017, 12, e0177430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Q.; Zhang, L.; Yi, X.; Zhang, K.; Yao, D.; Zhang, X.; Wang, Q.; Zhao, X.; Ling, Y.; Xu, W.; et al. Co-expression network analyses identify functional modules associated with development and stress response in Gossypium arboreum. Sci. Rep. 2016, 6, 38436. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.C.D.; Righetti, K.; Nijveen, H.; Yazdanpanah, F.; Ligterink, W.; Buitink, J.; Hilhorst, H.W.M. A gene co-expression network predicts functional genes controlling the re-establishment of desiccation tolerance in germinated Arabidopsis thaliana seeds. Planta 2015, 242, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, C.; Mutwil, M.; Saxe, F.; Eder, M.; Nikoloski, Z.; Persson, S. Large-Scale Co-Expression Approach to Dissect Secondary Cell Wall Formation Across Plant Species. Front. Plant Sci. 2011, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yin, Y.; Ma, Q.; Tang, X.; Hao, D.; Xu, Y. Genome-scale identification of cell-wall related genes in Arabidopsis based on co-expression network analysis. BMC Plant Biol. 2012, 12, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Edgar, R. Gene Expression Omnibus: Microarray Data Storage, Submission, Retrieval, and Analysis. Methods Enzymol. 2006, 411, 352–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huala, E.; Dickerman, A.W.; Garcia-Hernandez, M.; Weems, D.; Reiser, L.; LaFond, F.; Hanley, D.; Kiphart, D.; Zhuang, M.; Huang, W.; et al. The Arabidopsis Information Resource (TAIR): A comprehensive database and web-based information re-trieval, analysis, and visualization system for a model plant. Nucleic Acids Res. 2001, 29, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klepikova, A.V.; Kulakovskiy, I.V.; Kasianov, A.S.; Logacheva, M.D.; Penin, A.A. An update to database TraVA: Organ-specific cold stress response in Arabidopsis thaliana. BMC Plant Biol. 2019, 19, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Antonio, B.A.; Namiki, N.; Takehisa, H.; Minami, H.; Kamatsuki, K.; Sugimoto, K.; Shimizu, Y.; Hirochika, H.; Nagamura, Y. RiceXPro: A platform for monitoring gene expression in japonica rice grown under natural field conditions. Nucleic Acids Res. 2011, 39, D1141–D1148. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Oono, Y.; Wakimoto, H.; Ogata, J.; Kanamori, H.; Sasaki, H.; Mori, S.; Matsumoto, T.; Itoh, T. TENOR: Database for Comprehensive mRNA-Seq Experiments in Rice. Plant Cell Physiol. 2016, 57, e7. [Google Scholar] [CrossRef]
- Tanaka, T.; Sakai, H.; Fujii, N.; Kobayashi, F.; Nakamura, S.; Itoh, T.; Matsumoto, T.; Wu, J. bex-db: Bioinformatics workbench for comprehensive analysis of barley-expressed genes. Breed. Sci. 2013, 63, 430–434. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-R.; Liu, C.-C.; Sun, C.-H.; Chen, Y.-T. Plant stress RNA-seq Nexus: A stress-specific transcriptome database in plant cells. BMC Genom. 2018, 19, 966. [Google Scholar] [CrossRef] [Green Version]
- Saithong, T.; Rongsirikul, O.; Kalapanulak, S.; Chiewchankaset, P.; Siriwat, W.; Netrphan, S.; Suksangpanomrung, M.; Meechai, A.; Cheevadhanarak, S. Starch biosynthesis in cassava: A genome-based pathway reconstruction and its exploitation in data integration. BMC Syst. Biol. 2013, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Töpfer, N.; Caldana, C.; Grimbs, S.; Willmitzer, L.; Fernie, A.R.; Nikoloski, Z. Integration of Genome-Scale Modeling and Transcript Profiling Reveals Metabolic Pathways Underlying Light and Temperature Acclimation in Arabidopsis. Plant Cell 2013, 25, 1197–1211. [Google Scholar] [CrossRef] [Green Version]
- Dharmawardhana, P.; Ren, L.; Amarasinghe, V.; Monaco, M.; Thomason, J.; Ravenscroft, D.; McCouch, S.; Ware, D.; Jaiswal, P. A genome scale metabolic network for rice and accompanying analysis of tryptophan, auxin and serotonin biosynthesis regulation under biotic stress. Rice 2013, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Assefa, T.; Otyama, P.I.; Brown, A.V.; Kalberer, S.R.; Kulkarni, R.S.; Cannon, S.B. Genome-wide associations and epistatic interactions for internode number, plant height, seed weight and seed yield in soybean. BMC Genom. 2019, 20, 527. [Google Scholar] [CrossRef] [Green Version]
- Weiße, A.Y.; Oyarzún, D.A.; Danos, V.; Swain, P.S. Mechanistic links between cellular trade-offs, gene expression, and growth. Proc. Natl. Acad. Sci. USA 2015, 112, E1038–E1047. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, A.; Ichihara, H.; Asamizu, E.; Shirasawa, S.; Nakamura, Y.; Tabata, S.; Hirakawa, H. Plant Genome DataBase Japan (PGDBj). Methods Mol. Biol. 2017, 1533, 45–77. [Google Scholar] [CrossRef] [PubMed]
- Spannagl, M.; Nussbaumer, T.; Bader, K.C.; Martis, M.M.; Seidel, M.; Kugler, K.G.; Gundlach, H.; Mayer, K.F. PGSB PlantsDB: Updates to the database framework for comparative plant genome research. Nucleic Acids Res. 2016, 44, D1141–D1147. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Veeraraghavan, N.; Richter, A.; Wall, K.; Jansen, R.K.; Leebens-Mack, J.; Makalowska, I.; dePamphilis, C.W. Chloro-plastDB: The Chloroplast Genome Database. Nucleic Acids Res. 2006, 34, D692–D696. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.; Hamilton, J.; Childs, K.; Cepela, J.; Crisovan, E.; Vaillancourt, B.; Hirsch, C.N.; Habermann, M.; Neal, B.; Buell, C.R. Spud DB: A Resource for Mining Sequences, Genotypes, and Phenotypes to Accelerate Potato Breeding. Plant Genome 2014, 7, plantgenome2013-10. [Google Scholar] [CrossRef] [Green Version]
- Ruggieri, V.; Alexiou, K.; Morata, J.; Argyris, J.; Pujol, M.; Yano, R.; Nonaka, S.; Ezura, H.; Latrasse, D.; Boualem, A.; et al. An improved assembly and annotation of the melon (Cucumis melo L.) reference genome. Sci. Rep. 2018, 8, 8088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portwood, J.L., II; Woodhouse, M.R.; Cannon, E.K.; Gardiner, J.M.; Harper, L.C.; Schaeffer, M.L.; Walsh, J.R.; Sen, T.Z.; Cho, K.T.; Schott, D.A.; et al. MaizeGDB 2018: The maize multi-genome genetics and genomics database. Nucleic Acids Res. 2019, 47, D1146–D1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, H.; Lee, S.S.; Tanaka, T.; Numa, H.; Kim, J.; Kawahara, Y.; Wakimoto, H.; Yang, C.-C.; Iwamoto, M.; Abe, T.; et al. Rice Annotation Project Database (RAP-DB): An Integrative and Interactive Database for Rice Genomics. Plant Cell Physiol. 2013, 54, e6. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Yao, E.; Blake, V.C.; Cooper, L.; Wight, C.P.; Michel, S.; Cagirici, H.B.; Lazo, G.R.; Birkett, C.L.; Waring, D.J.; Jannink, J.-L.; et al. GrainGenes: A data-rich repository for small grains genetics and genomics. Database 2022, 2022, baac034. [Google Scholar] [CrossRef]
- Brown, A.V.; Conners, S.I.; Huang, W.; Wilkey, A.P.; Grant, D.; Weeks, N.T.; Cannon, S.B.; Graham, M.A.; Nelson, R.T. A new decade and new data at SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 2021, 49, D1496–D1501. [Google Scholar] [CrossRef]
- Jung, S.; Staton, M.; Lee, T.; Blenda, A.; Svancara, R.; Abbott, A.; Main, D. GDR (Genome Database for Rosaceae): Integrated web-database for Rosaceae genomics and genetics data. Nucleic Acids Res. 2008, 36, D1034–D1040. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, T.; He, X.; Cai, X.; Lin, R.; Liang, J.; Wu, J.; King, G.; Wang, X. BRAD V3.0: An upgraded Brassicaceae database. Nucleic Acids Res. 2022, 50, D1432–D1441. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Tamiru, M.; Salby, R.; Bolitho, C.; Williams, A.; Huggard, S.; Fisch, E.; Unsworth, K.; Whelan, J.; Lewsey, M.G. AgriSeqDB: An online RNA-Seq database for functional studies of agriculturally relevant plant species. BMC Plant Biol. 2018, 18, 200. [Google Scholar] [CrossRef] [PubMed]
- Waese, J.; Provart, N.J. The Bio-Analytic Resource for Plant Biology. In Plant Genomics Databases. Methods in Molecular Biology; Van Dijk, A., Ed.; Humana Press: New York, NJ, USA, 2017; Volume 1533, pp. 119–148. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, J.; Li, D.; Zhang, Z.; Liu, F.; Zhou, X.; Wang, T.; Ling, Y.; Su, Z. PMRD: Plant microRNA database. Nucleic Acids Res. 2010, 38, D806–D813. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. DIP: The database of interacting proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Wu, A.; Xu, X.-J.; Xiao, P.-P.; Lu, L.; Liu, J.; Cao, Y.; Chen, L.; Wu, J.; Zhao, X.-M. PPIM: A Protein-Protein Interaction Database for Maize. Plant Physiol. 2016, 170, 618–626. [Google Scholar] [CrossRef] [Green Version]
- del Toro, N.; Shrivastava, A.; Ragueneau, E.; Meldal, B.; Combe, C.; Barrera, E.; Perfetto, L.; How, K.; Ratan, P.; Shirodkar, G.; et al. The IntAct database: Efficient access to fine-grained molecular interaction data. Nucleic Acids Res. 2022, 50, D648–D653. [Google Scholar] [CrossRef]
- Gu, H.; Zhu, P.; Jiao, Y.; Meng, Y.; Chen, M. PRIN: A predicted rice interactome network. BMC Bioinform. 2011, 12, 161. [Google Scholar] [CrossRef] [Green Version]
- Bader, G.; Donaldson, I.; Wolting, C.; Ouellette, B.F.F.; Pawson, T.; Hogue, C.W.V. BIND--The Biomolecular Interaction Network Database. Nucleic Acids Res. 2003, 31, 248–250. [Google Scholar] [CrossRef] [Green Version]
- Chatr-Aryamontri, A.; Breitkreutz, B.-J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2014, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Brandão, M.M.; Dantas, L.L.; Silva-Filho, M.C. AtPIN: Arabidopsis thaliana Protein Interaction Network. BMC Bioinform. 2009, 10, 454. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, S.; Qi, H.; Wang, T.; Li, H.; Zhang, Z. PlaPPISite: A comprehensive resource for plant protein-protein interaction sites. BMC Plant Biol. 2020, 20, 61. [Google Scholar] [CrossRef]
- Stein, A.; Russell, R.B.; Aloy, P. 3did: Interacting protein domains of known three-dimensional structure. Nucleic Acids Res. 2005, 33, D413–D417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatr-Aryamontri, A.; Ceol, A.; Palazzi, L.M.; Nardelli, G.; Schneider, M.V.; Castagnoli, L.; Cesareni, G. MINT: The Molecular INTeraction database. Nucleic Acids Res. 2007, 35, D572–D574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, Y.; Okamura, Y.; Tadaka, S.; Kinoshita, K.; Obayashi, T. ATTED-II in 2016: A Plant Coexpression Database Towards Lineage-Specific Coexpression. Plant Cell Physiol. 2016, 57, e5. [Google Scholar] [CrossRef] [PubMed]
- Srinivasasainagendra, V.; Page, G.P.; Mehta, T.; Coulibaly, I.; Loraine, A.E. CressExpress: A Tool for Large-Scale Mining of Expression Data from Arabidopsis. Plant Physiol. 2008, 147, 1004–1016. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Yang, S.; Kim, E.; Ko, Y.; Hwang, S.; Shin, J.; Shim, J.E.; Shim, H.; Kim, H.; Kim, C.; et al. AraNet v2: An improved database of co-functional gene networks for the study of Arabidopsis thaliana and 27 other nonmodel plant species. Nucleic Acids Res. 2015, 43, D996–D1002. [Google Scholar] [CrossRef]
- Ogata, Y.; Suzuki, H.; Sakurai, N.; Shibata, D. CoP: A database for characterizing co-expressed gene modules with biological information in plants. Bioinformatics 2010, 26, 1267–1268. [Google Scholar] [CrossRef] [Green Version]
- Chien, C.-H.; Chow, C.-N.; Wu, N.-Y.; Chiang-Hsieh, Y.-F.; Hou, P.-F.; Chang, W.-C. EXPath: A database of comparative expression analysis inferring metabolic pathways for plants. BMC Genom. 2015, 16, S6. [Google Scholar] [CrossRef] [Green Version]
- Ohyanagi, H.; Takano, T.; Terashima, S.; Kobayashi, M.; Kanno, M.; Morimoto, K.; Kanegae, H.; Sasaki, Y.; Saito, M.; Asano, S.; et al. Plant Omics Data Center: An integrated web repository for interspecies gene expression networks with NLP-based cu-ration. Plant Cell Physiol. 2015, 56, e9. [Google Scholar] [CrossRef] [Green Version]
- Mutwil, M.; Klie, S.; Tohge, T.; Giorgi, F.; Wilkins, O.; Campbell, M.; Fernie, A.R.; Usadel, B.; Nikoloski, Z.; Persson, S. PlaNet: Combined Sequence and Expression Comparisons across Plant Networks Derived from Seven Species. Plant Cell 2011, 23, 895–910. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Hongo, K.; Suwabe, K.; Shimizu, A.; Nagayama, T.; Abe, R.; Kikuchi, S.; Yamamoto, N.; Fujii, T.; Yokoyama, K.; et al. OryzaExpress: An Integrated Database of Gene Expression Networks and Omics Annotations in Rice. Plant Cell Physiol. 2011, 52, 220–229. [Google Scholar] [CrossRef]
- Kudo, T.; Terashima, S.; Takaki, Y.; Tomita, K.; Saito, M.; Kanno, M.; Yokoyama, K.; Yano, K. PlantExpress: A Database Inte-grating OryzaExpress and ArthaExpress for Single-species and Cross-species Gene Expression Network Analyses with Mi-croarray-Based Transcriptome Data. Plant Cell Physiol. 2017, 58, e1. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Namiki, N.; Takehisa, H.; Kamatsuki, K.; Minami, H.; Ikawa, H.; Ohyanagi, H.; Sugimoto, K.; Itoh, J.-I.; Antonio, B.A.; et al. RiceFREND: A platform for retrieving coexpressed gene networks in rice. Nucleic Acids Res. 2013, 41, D1214–D1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.C.; Sweetman, C.; Drew, D.P.; Ford, C.M. VTCdb: A gene co-expression database for the crop species Vitis vinifera (grapevine). BMC Genom. 2013, 14, 882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [Green Version]
- Steinhauser, D.; Usadel, B.; Luedemann, A.; Thimm, O.; Kopka, J. CSB.DB: A comprehensive systems-biology database. Bioinformatics 2004, 20, 3647–3651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Jun, K.M.; Kim, J.S.; Chae, S.; Pahk, Y.-M.; Lee, T.-H.; Sohn, S.-I.; Lee, S.I.; Lim, M.-H.; Kim, C.-K.; et al. RapaNet: A Web Tool for the Co-Expression Analysis of Brassica rapa Genes. Evol. Bioinform. 2017, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Zou, D.; Sang, J.; Xu, X.; Yin, H.; Li, M.; Wu, S.; Hu, S.; Hao, L.; Zhang, Z. Rice Expression Database (RED): An integrated RNA-Seq-derived gene expression database for rice. J. Genet. Genom. 2017, 44, 235–241. [Google Scholar] [CrossRef]
- Ferrari, C.; Proost, S.; Ruprecht, C.; Mutwil, M. PhytoNet: Comparative co-expression network analyses across phytoplankton and land plants. Nucleic Acids Res. 2018, 46, W76–W83. [Google Scholar] [CrossRef] [PubMed]
- Proost, S.; Mutwil, M. CoNekT: An open-source framework for comparative genomic and transcriptomic network analyses. Nucleic Acids Res. 2018, 46, W133–W140. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Shah, M.; Ballouz, S.; Crow, M.; Gillis, J. CoCoCoNet: Conserved and comparative co-expression across a diverse set of species. Nucleic Acids Res. 2020, 48, W566–W571. [Google Scholar] [CrossRef]
- Tang, S.; Zhao, H.; Lu, S.; Yu, L.; Zhang, G.; Zhang, Y.; Yang, Q.Y.; Zhou, Y.; Wang, X.; Ma, W.; et al. Genome- and transcrip-tome-wide association studies provide insights into the genetic basis of natural variation of seed oil content in Brassica napus. Mol. Plant 2021, 14, 470–487. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; de Geus, E.J.C.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Kremling, K.A.G.; Diepenbrock, C.H.; Gore, M.A.; Buckler, E.S.; Bandillo, N.B. Transcriptome-Wide Association Supplements Genome-Wide Association in Zea mays. G3 Genes Genomes Genet. 2019, 9, 3023–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Li, X.; Tanaka, R.; Wood, J.C.; Tibbs-Cortes, L.E.; Magallanes-Lundback, M.; Bornowski, N.; Hamilton, J.P.; Vaillancourt, B.; Diepenbrock, C.H.; et al. Combining GWAS and TWAS to identify candidate causal genes for tocochromanol levels in maize grain. Genetics 2022, 221. [Google Scholar] [CrossRef]
- Perez-Sanz, F.; Navarro, P.J.; Egea-Cortines, M. Plant phenomics: An overview of image acquisition technologies and image data analysis algorithms. GigaScience 2017, 6, gix092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houle, D.; Govindaraju, D.R.; Omholt, S. Phenomics: The next challenge. Nat. Rev. Genet. 2010, 11, 855–866. [Google Scholar] [CrossRef]
- Lobos, G.A.; Camargo, A.V.; del Pozo, A.; Araus, J.L.; Ortiz, R.; Doonan, J.H. Editorial: Plant Phenotyping and Phenomics for Plant Breeding. Front. Plant Sci. 2017, 8, 2181. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Maurer, H.P.; Jenz, M.; Hahn, V.; Ruckelshausen, A.; Leiser, W.L.; Würschum, T. The performance of phenomic selection depends on the genetic architecture of the target trait. Theor. Appl. Genet. 2021, 135, 653–665. [Google Scholar] [CrossRef]
- Parmley, K.; Nagasubramanian, K.; Sarkar, S.; Ganapathysubramanian, B.; Singh, A.K. Development of Optimized Phenomic Predictors for Efficient Plant Breeding Decisions Using Phenomic-Assisted Selection in Soybean. Plant Phenomics 2019, 2019, 5809404. [Google Scholar] [CrossRef]
- Lane, H.M.; Murray, S.C.; Montesinos-Lopez, A.; Crossa, J.; Rooney, D.K.; Barrero-Farfan, I.D.; De la Fuente, G.N.; Morgan, C.L.S. Phenomic selection and prediction of maize grain yield from near-infrered reflectance spectroscopy of kernels. Plant Phenome. J. 2020, 3, e0117737. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, M.T.V.; Morota, G.; Costa, P.M.dA.; Vidigal, P.M.P.; Barbosa, M.H.P.; Peternelli, L.A. Near-infrared spec-troscopy outperforms genomics for predicting sugarcane feedstock quality traits. PLoS ONE 2021, 16, e0236853. [Google Scholar] [CrossRef]
- Li, Z.; Sillanpää, M.J. Dynamic Quantitative Trait Locus Analysis of Plant Phenomic Data. Trends Plant Sci. 2015, 20, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Epigenomics of Plant Responses to Environmental Stress. Epigenomes 2018, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Arneson, A.; Haghani, A.; Thompson, M.J.; Pellegrini, M.; Bin Kwon, S.; Vu, H.; Maciejewski, E.; Yao, M.; Li, C.Z.; Lu, A.T.; et al. A mammalian methylation array for profiling methylation levels at conserved sequences. Nat. Commun. 2022, 13, 783. [Google Scholar] [CrossRef]
- Grehl, C.; Wagner, M.; Lemnian, I.; Glaser, B.; Grosse, I. performance of mapping approaches for whole genome bisulfite sequencing data in crop plants. Front. Plant Sci. 2020, 11, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, K.; Muino, J.M.; Østerås, M.; Farinelli, L.; Krajewski, P.; Angenent, G.C. Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat. Protoc. 2010, 5, 457–472. [Google Scholar] [CrossRef]
- Tollefsbol, T.O. Advances in epigenetic technology. Methods Mol. Biol. 2011, 791, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajic, M.; Maher, K.A.; Deal, R.B. Identification of Open Chromatin Regions in Plant Genomes Using ATAC-Seq. Plant Chromatin Dyn. 2017, 1675, 183–201. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Carey, M.; Workman, J.L. The Role of Chromatin during Transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Li, R.; Hu, F.; Li, B.; Zhang, Y.; Chen, M.; Fan, T.; Wang, T. Whole genome bisulfite sequencing methylome analysis of mulberry (Morus alba) reveals epigenome modifications in response to drought stress. Sci. Rep. 2020, 10, 232. [Google Scholar] [CrossRef]
- Maher, K.A.; Bajic, M.; Kajala, K.; Reynoso, M.; Pauluzzi, G.; West, D.A.; Zumstein, K.; Woodhouse, M.; Bubb, K.L.; Dorrity, M.W.; et al. Profiling of Accessible Chromatin Regions across Multiple Plant Species and Cell Types Reveals Common Gene Regulatory Principles and New Control Modules. Plant Cell 2018, 30, 15–36. [Google Scholar] [CrossRef] [Green Version]
- Badad, O.; Lahssassi, N.; Zaid, N.; El-Baze, A.; Zaid, Y.; Meksem, J.; Lightfoot, D.A.; Tombuloglu, H.; Zaid, E.H.; Unver, T.; et al. Genome-wide MeDIP-Seq profiling of wild and cultivated olives trees suggests DNA methylation fin-gerprint on the sensory quality of olive oil. Plants 2021, 10, 1405. [Google Scholar] [CrossRef] [PubMed]
- Hawe, J.; Theis, F.J.; Heinig, M. Inferring Interaction Networks from Multi-Omics Data. Front. Genet. 2019, 10, 535. [Google Scholar] [CrossRef]
- Schaefer, R.J.; Michno, J.-M.; Myers, C.L. Unraveling gene function in agricultural species using gene co-expression networks. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provart, N.J. Correlation networks visualization. Front. Plant Sci. 2012, 3, 240. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yan, X.; Chen, S. Bioinformatic analysis of molecular network of glucosinolate biosynthesis. Comput. Biol. Chem. 2011, 35, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ye, C.-Y.; Bisaria, A.; Tuskan, G.A.; Kalluri, U.C. Identification of candidate genes in Arabidopsis and Populus cell wall biosynthesis using text-mining, co-expression network analysis and comparative genomics. Plant Sci. 2011, 181, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Van Verk, M.C.; Bol, J.F.; Linthorst, H.J. Prospecting for Genes involved in transcriptional regulation of plant defenses, a bioinformatics approach. BMC Plant Biol. 2011, 11, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashari, K.-S.; Abdullah-Zawawi, M.-R.; Harun, S.; Mohamed-Hussein, Z.-A. Reconstruction of the Transcriptional Regulatory Network in Arabidopsis thaliana Aliphatic Glucosinolate Biosynthetic Pathway. Sains Malays. 2018, 47, 2993–3002. [Google Scholar] [CrossRef]
- De Las Rivas, J.; Fontanillo, C. Protein–Protein Interactions Essentials: Key Concepts to Building and Analyzing Interactome Networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheth, B.P.; Thaker, V.S. Plant systems biology: Insights, advances and challenges. Planta 2014, 240, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Stagljar, I. Using the Yeast Two-Hybrid System to Identify Interacting Proteins. Methods Mol. Biol. 2004, 261, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Grefen, C.; Lalonde, S.; Obrdlik, P. Split-ubiquitin system for identifying protein-protein interactions in membrane and full-length proteins. Curr. Protoc. Neurosci. 2007, 41, 5–27. [Google Scholar] [CrossRef]
- Kerppola, T.K. Bimolecular Fluorescence Complementation (BiFC) Analysis as a Probe of Protein Interactions in Living Cells. Annu. Rev. Biophys. 2008, 37, 465–487. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.H.; Knudsen, G.M.; Verschueren, E.; Johnson, J.R.; Cimermancic, P.; Greninger, A.L.; Pico, A.R. Affinity purifica-tion-mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 2014, 9, 2539–2554. [Google Scholar] [CrossRef] [Green Version]
- Paul, C.; Rho, H.; Neiswinger, J.; Zhu, H. Characterization of Protein–Protein Interactions Using Protein Microarrays. Cold Spring Harb. Protoc. 2016, 2016, prot087965. [Google Scholar] [CrossRef]
- Harun, S.; Rohani, E.R.; Ohme-Takagi, M.; Goh, H.-H.; Mohamed-Hussein, Z.-A. ADAP is a possible negative regulator of glucosinolate biosynthesis in Arabidopsis thaliana based on clustering and gene expression analyses. J. Plant Res. 2021, 134, 327–339. [Google Scholar] [CrossRef]
- Liu, L.-Y.D.; Hsiao, Y.-C.; Chen, H.-C.; Yang, Y.-W.; Chang, M.-C. Construction of gene causal regulatory networks using microarray data with the coefficient of intrinsic dependence. Bot. Stud. 2019, 60, 22. [Google Scholar] [CrossRef] [PubMed]
- Mounet, F.; Moing, A.; Garcia, V.; Petit, J.; Maucourt, M.; Deborde, C.; Bernillon, S.; Le Gall, G.; Colquhoun, I.; Defernez, M.; et al. Gene and Metabolite Regulatory Network Analysis of Early Developing Fruit Tissues Highlights New Candidate Genes for the Control of Tomato Fruit Composition and Development. Plant Physiol. 2009, 149, 1505–1528. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. Is proteomics the new genomics? Cell 2007, 130, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gedeon, T.; Bokes, P. Delayed Protein Synthesis Reduces the Correlation between mRNA and Protein Fluctuations. Biophys. J. 2012, 103, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riba, A.; Di Nanni, N.; Mittal, N.; Arhné, E.; Schmidt, A.; Zavolan, M. Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. Proc. Natl. Acad. Sci. USA 2019, 116, 15023–15032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neymotin, B.; Ettorre, V.; Gresham, D. Multiple Transcript Properties Related to Translation Affect mRNA Degradation Rates in Saccharomyces cerevisiae. G3 Genes Genomes Genet. 2016, 6, 3475–3483. [Google Scholar] [CrossRef] [Green Version]
- Ponnala, L.; Wang, Y.; Sun, Q.; Van Wijk, K.J. Correlation of mRNA and protein abundance in the developing maize leaf. Plant J. 2014, 78, 424–440. [Google Scholar] [CrossRef]
- Osorio, S.; Alba, R.; Damasceno, C.M.; Lopez-Casado, G.; Lohse, M.; Zanor, M.I.; Tohge, T.; Usadel, B.; Rose, J.K.; Fei, Z.; et al. Systems biology of tomato fruit development: Combined transcript, protein, and metabolite analysis of tomato transcription factor (nor, rin) and ethylene receptor (Nr) mutants reveals novel regulatory interactions. Plant Physiol. 2011, 157, 405–425. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; He, S.; Gong, W.; Xu, F.; Pan, Z.; Jia, Y.; Geng, X.; Du, X. Integration of proteomic and transcriptomic profiles reveals multiple levels of genetic regulation of salt tolerance in cotton. BMC Plant Biol. 2018, 18, 128. [Google Scholar] [CrossRef] [Green Version]
- Syed, N.H.; Kalyna, M.; Marquez, Y.; Barta, A.; Brown, J.W. Alternative splicing in plants--coming of age. Trends Plant Sci. 2012, 17, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.Y.; Mutwil, M. Towards revealing the functions of all genes in plants. Trends Plant Sci. 2014, 19, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Lamesch, P.; Berardini, T.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef]
- Saito, K.; Hirai, M.Y.; Yonekura-Sakakibara, K. Decoding genes with coexpression networks and metabolomics—‘Majority report by precogs’. Trends Plant Sci. 2008, 13, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Provart, N.J.; Alonso, J.; Assmann, S.M.; Bergmann, D.; Brady, S.; Brkljacic, J.; Browse, J.; Chapple, C.; Colot, V.; Cutler, S.R.; et al. 50 years of Arabidopsis research: Highlights and future directions. New Phytol. 2015, 209, 921–944. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.-X.; Reiter, R.J. An evolutionary view of melatonin synthesis and metabolism related to its biological functions in plants. J. Exp. Bot. 2020, 71, 4677–4689. [Google Scholar] [CrossRef] [PubMed]
- Schnoes, A.M.; Brown, S.D.; Dodevski, I.; Babbitt, P.C. Annotation Error in Public Databases: Misannotation of Molecular Function in Enzyme Superfamilies. PLoS Comput. Biol. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Alvarez, R.V.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef]
- Lopez, R.; Silventoinen, V.; Robinson, S.; Kibria, A.; Gish, W. WU-Blast2 server at the European Bioinformatics Institute. Nucleic Acids Res. 2003, 31, 3795–3798. [Google Scholar] [CrossRef] [Green Version]
- Medema, M.H.; Osbourn, A. Computational genomic identification and functional reconstitution of plant natural product biosynthetic pathways. Nat. Prod. Rep. 2016, 33, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Oliver, S.G. Guilt-by-association goes global. Nature 2000, 403, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L. Guilt by Association: Contextual Information in Genome Analysis. Genome Res. 2000, 10, 1074–1077. [Google Scholar] [CrossRef]
- Hansen, B.O.; Vaid, N.; Musialak-Lange, M.; Janowski, M.; Mutwil, M. Elucidating gene function and function evolution through comparison of co-expression networks of plants. Front. Plant Sci. 2014, 5, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Yang, Y.; Zhong, J.; Gao, H.; Khan, L.; Thompson, D.K.; Zhou, J. Constructing gene co-expression networks and pre-dicting functions of unknown genes by random matrix theory. BMC Bioinform. 2007, 8, 299. [Google Scholar] [CrossRef] [Green Version]
- Allocco, D.J.; Kohane, I.S.; Butte, A.J. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinform. 2004, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassel, G.; Gaudinier, A.; Brady, S.; Hennig, L.; Rhee, S.Y.; De Smet, I. Systems Analysis of Plant Functional, Transcriptional, Physical Interaction, and Metabolic Networks. Plant Cell 2012, 24, 3859–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dam, S.; Võsa, U.; van der Graaf, A.; Franke, L.; de Magalhães, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- López-Kleine, L.; Leal, L.; López, C. Biostatistical approaches for the reconstruction of gene co-expression networks based on transcriptomic data. Briefings Funct. Genom. 2013, 12, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Mahanta, P.; Ahmed, H.A.; Bhattacharyya, D.K.; Kalita, J.K. An effective method for network module extraction from mi-croarray data. BMC Bioinform. 2012, 13, S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couto, C.M.V.; Comin, C.H.; Costa, L.D.F. Effects of threshold on the topology of gene co-expression networks. Mol. BioSyst. 2017, 13, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Borate, B.R.; Chesler, E.J.; Langston, M.A.; Saxton, A.M.; Voy, B.H. Comparison of threshold selection methods for microarray gene co-expression matrices. BMC Res. Notes 2009, 2, 240. [Google Scholar] [CrossRef] [Green Version]
- Perkins, A.D.; Langston, M.A. Threshold selection in gene co-expression networks using spectral graph theory techniques. BMC Bioinform. 2009, 10 (Suppl. S11), S4. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yin, L.; Xue, H. Co-expression Analysis Identifies CRC and AP1 the Regulator of Arabidopsis Fatty Acid Biosynthesis. J. Integr. Plant Biol. 2012, 54, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Bordych, C.; Eisenhut, M.; Pick, T.; Kuelahoglu, C.; Weber, A.P.M. Co-expression analysis as tool for the discovery of transport proteins in photorespiration. Plant Biol. 2013, 15, 686–693. [Google Scholar] [CrossRef]
- Leal, L.G.; Lopez, C.; López-Kleine, L. Construction and comparison of gene co-expression networks shows complex plant immune responses. PeerJ 2014, 2, e610. [Google Scholar] [CrossRef] [Green Version]
- Barah, P.; Jayavelu, N.D.; Sowdhamini, R.; Shameer, K.; Bones, A.M. Transcriptional regulatory networks in Ara-bidopsis thaliana during single and combined stresses. Nucleic Acids Res. 2016, 44, 3147–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barah, P.; Winge, P.; Kuśnierczyk, A.; Tran, D.H.; Bones, A.M. Molecular Signatures in Arabidopsis thaliana in Response to Insect Attack and Bacterial Infection. PLoS ONE 2013, 8, e58987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-M.; Lin, H.-H.; Liu, W.-Y.; Yu, C.-P.; Chen, H.-J.; Wartini, P.P.; Kao, Y.-Y.; Wu, Y.-H.; Lin, J.-J.; Lu, M.-Y.J.; et al. Comparative transcriptomics method to infer gene coexpression networks and its applications to maize and rice leaf transcriptomes. Proc. Natl. Acad. Sci. USA 2019, 116, 3091–3099. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, L.E.; Russo, P.S.T.; Gomes-Correia, B.; Araujo-Pereira, M.; Sepúlveda-Hermosilla, G.; Maracaja-Coutinho, V.; Nakaya, H.I. webCEMiTool: Co-expression Modular Analysis Made Easy. Front. Genet. 2019, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuva, D.D.; Cursons, J.; Smyth, G.K.; Davis, M.J. Differential co-expression-based detection of conditional relationships in transcriptional data: Comparative analysis and application to breast cancer. Genome Biol. 2019, 20, 236. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.A.; Ye, S.; Kendziorski, C. R/EBcoexpress: An empirical Bayesian framework for discovering differential co-expression. Bioinformatics 2012, 28, 1939–1940. [Google Scholar] [CrossRef]
- Li, D.; Brown, J.B.; Orsini, L.; Pan, Z.; Hu, G.; He, S. MODA: MOdule Differential Analysis for Weighted Gene Co-Expression Network. arXiv 2016, arXiv:1605.04739. [Google Scholar]
- Amar, D.; Safer, H.; Shamir, R. Dissection of Regulatory Networks that Are Altered in Disease via Differential Co-expression. PLoS Comput. Biol. 2013, 9, e1002955. [Google Scholar] [CrossRef] [Green Version]
- Tesson, B.M.; Breitling, R.; Jansen, R.C. DiffCoEx: A simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinform. 2010, 11, 497. [Google Scholar] [CrossRef] [Green Version]
- Henao, J.D. Coexnet: An R Package to Build CO-EXpression NETworks from Microarray Data; Version 1.8.0.; View on Bio-conductor; 2019. Available online: https://rdrr.io/bioc/coexnet/ (accessed on 15 April 2022).
- Yan, Q.; Wu, F.; Yan, Z.; Li, J.; Ma, T.; Zhang, Y.; Zhao, Y.; Wang, Y.; Zhang, J. Differential co-expression networks of long non-coding RNAs and mRNAs in Cleistogenes songorica under water stress and during recovery. BMC Plant Biol. 2019, 19, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Ding, Z.; Li, P. Maize network analysis revealed gene modules involved in development, nutrients utilization, metabolism, and stress response. BMC Plant Biol. 2017, 17, 131. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Cheng, F.; Zhong, Y.; Guo, X. Comparative transcriptome and co-expression network analysis of carpel quantitative variation in Paeonia rockii. BMC Genom. 2019, 20, 683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, F.; Augustine, R.C.; Marshall, R.S.; Li, F.; Kirkpatrick, L.D.; Otegui, M.S.; Vierstra, R.D. Maize multi-omics reveal roles for autophagic recycling in proteome remodelling and lipid turnover. Nat. Plants 2018, 4, 1056–1070. [Google Scholar] [CrossRef]
- Wang, H.; Wang, S.; Zhang, Y.; Bi, S.; Zhu, X. A brief review of machine learning methods for RNA methylation sites prediction. Methods 2022, 203, 399–421. [Google Scholar] [CrossRef]
- Banerjee, D.; Jindra, M.A.; Linot, A.J.; Pfleger, B.F.; Maranas, C.D. EnZymClass: Substrate specificity prediction tool of plant acyl-ACP thioesterases based on ensemble learning. Curr. Res. Biotechnol. 2022, 4, 1–9. [Google Scholar] [CrossRef]
- Sampaio, M.; Rocha, M.; Dias, O. Exploring synergies between plant metabolic modelling and machine learning. Comput. Struct. Biotechnol. J. 2022, 20, 1885–1900. [Google Scholar] [CrossRef] [PubMed]
- Campos, T.L.; Korhonen, P.K.; Hofmann, A.; Gasser, R.B.; Young, N.D. Harnessing model organism genomics to un-derpin the machine learning-based prediction of essential genes in eukaryotes-Biotechnological implications. Biotechnol. Adv. 2021, 54, 107822. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.D.J.; Kootstra, G.; Kruijer, W.; de Ridder, D. Machine learning in plant science and plant breeding. iScience 2021, 24, 101890. [Google Scholar] [CrossRef] [PubMed]







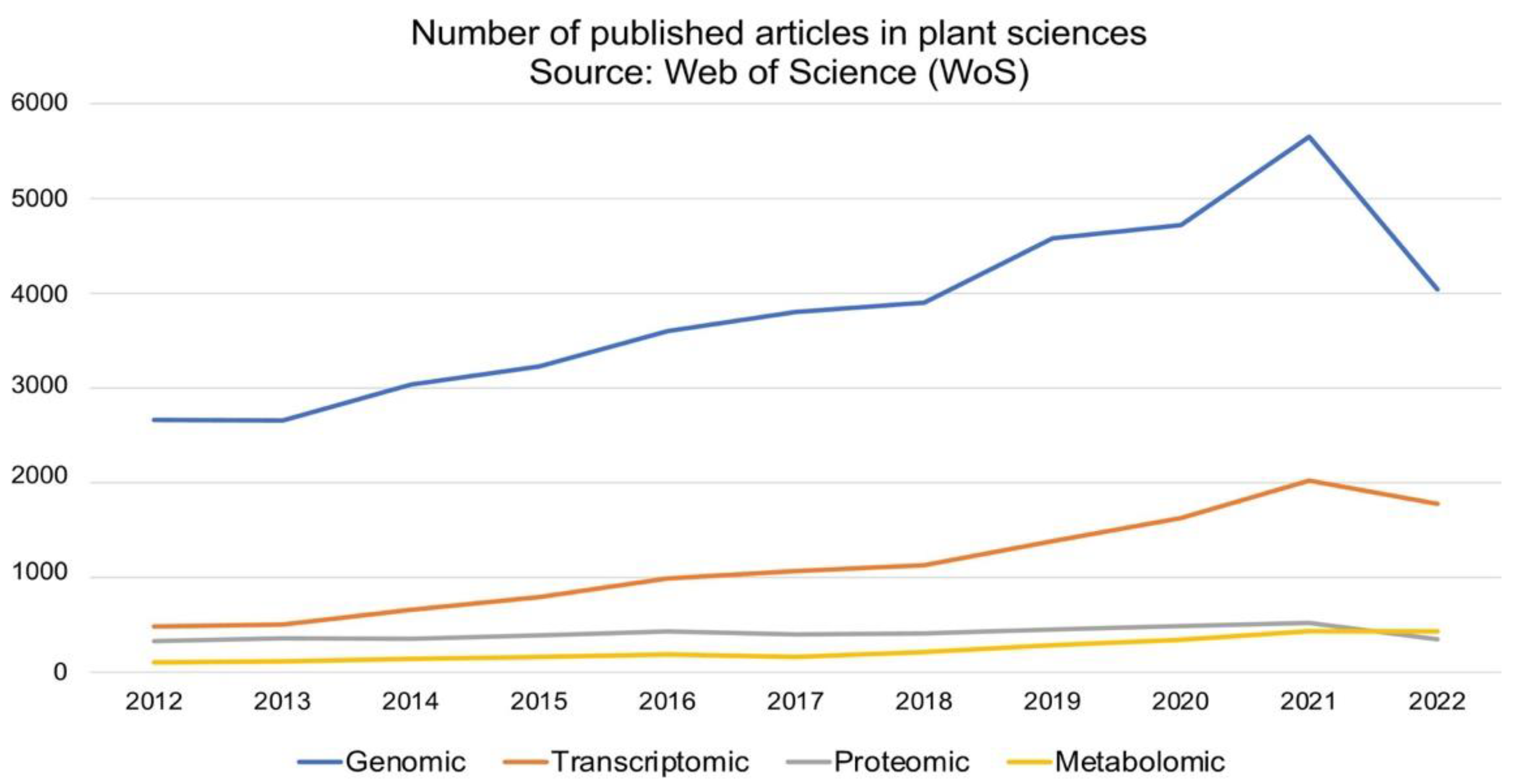{kind=link}
{kind=link}
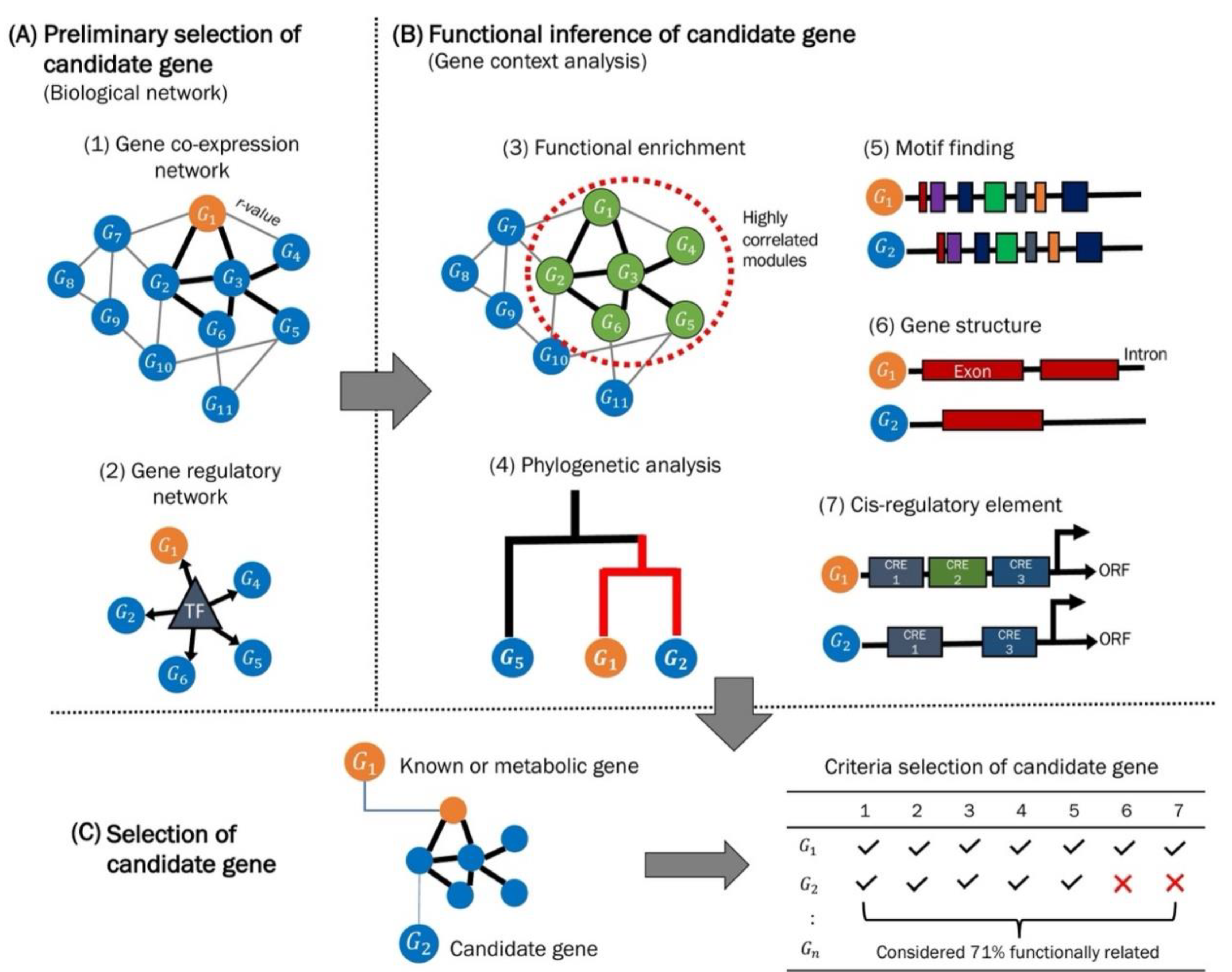{kind=link}
| Omics Type | Database | Organism | URL | References |
|---|---|---|---|---|
| Genomics | Plant Genome Database (PlantGDB) | Plants | http://www.plantgdb.org | [54] |
| Plant Genome DataBase Japan (PGDBj) | Plants | http://pgdbj.jp/?ln=en | [104] | |
| National Center for Biotechnology Information (NCBI) | Various | https://www.ncbi.nlm.nih.gov | [52] | |
| Ensembl Plants | Plants | http://plants.ensembl.org/ | [53] | |
| Phytozome | Plants | https://phytozome.jgi.doe.gov | [57] | |
| PLAZA | Plants | https://bioinformatics.psb.ugent.be/plaza/ | [55] | |
| Plant Genome and Systems Biology (PGSB PlantsDB) | Plants | http://pgsb.helmholtz-muenchen.de/plant/plantsdb.jsp | [105] | |
| Chloroplast Genome Database (ChloroplastDB) | Plants | http://chloroplast.cbio.psu.edu/ | [106] | |
| The Solanaceae Genomics Resource (Spud DB) | Potato | http://solanaceae.plantbiology.msu.edu | [107] | |
| Melon Genome Database (Melonomics) | Melon | https://www.melonomics.net/ | [108] | |
| Maize Genetics and Genomics Database (MaizeGDB) | Maize | https://www.maizegdb.org | [109] | |
| Rice Annotation Project Database (RAP-DB) | Rice | https://rapdb.dna.affrc.go.jp | [110] | |
| Rice Genome Annotation Project (RGAP) | Rice | http://rice.plantbiology.msu.edu | [111] | |
| GrainGenes | Wheat, Barley, rye, oat | http://wheat.pw.usda.gov/GG3/ | [112] | |
| SoyBase | Soy | Soybase.org | [113] | |
| Genome Database for Rosaceae (GDR) | Rosaceae plants | https://www.rosaceae.org/ | [114] | |
| Brassica Database (BRAD) | Brassica plants | http://brassicadb.org/brad/ | [115] | |
| Transcriptomics | Gene Expression Omnibus (GEO) | Various | https://www.ncbi.nlm.nih.gov/geo/ | [92] |
| AgriSeqDB | Plants | https://expression.latrobe.edu.au/agriseqdb | [116] | |
| The Bio-Analytic Resource for Plant Biology (BAR) | Plants | http://bar.utoronto.ca | [117] | |
| and | The Arabidopsis Information Resource (TAIR) | Arabidopsis | https://www.arabidopsis.org | [93] |
| Transcriptome Variation Analysis (TRAVA) | Arabidopsis | http://travadb.org | [94] | |
| The Rice Expression Profile Database (RiceXPro) | Rice | https://ricexpro.dna.affrc.go.jp | [95] | |
| Transcriptome Encycloperdia of Rice (TENOR) | Rice | http://tenor.dna.affrc.go.jp/ | [96] | |
| Barley Gene Expression Database (Bex-db) | Barley | http://barleyflc.dna.affrc.go.jp/hvdb/ | [97] | |
| Plant Stress RNA-seq Nexus (PSRN) | Plants | http://syslab5.nchu.edu.tw | [98] | |
| Plant microRNA database (PMRD) | Plants | http://bioinformatics.cau.edu.cn/PMRD/ | [118] | |
| Interactomics | STRING | Various | https://string-db.org | [119] |
| Database of Interacting Proteins (DIP) | Various | http://dip.doe-mbi.ucla.edu | [120] | |
| Protein–Protein Interaction Database for Maize (PPIM) | Maize | http://comp-sysbio.org/ppim | [121] | |
| IntAct | Various | https://www.ebi.ac.uk/intact/ | [122] | |
| Oryza sativa Protein–Protein Interaction Network (PRIN) | Rice | http://bis.zju.edu.cn/prin/ | [123] | |
| Biomolecular Interaction Network Database (BIND) | Various | http://bind.ca | [124] | |
| The Biological General Repository for Interaction Datasets (BioGRID) | Various | https://thebiogrid.org | [125] | |
| Arabidopsis thaliana Protein Interaction Network (AtPIN) | Arabidopsis | https://atpin.bioinfoguy.net | [126] | |
| PlaPPISite | Plants | http://zzdlab.com/plappisite/index.php | [127] | |
| 3D interacting domains (3did) | Various | https://3did.irbbarcelona.org | [128] | |
| Molecular INTeraction database (MINT) | Various | http://mint.bio.uniroma2.it/mint/ | [129] | |
| ATTED-II | Plants | http://atted.jp/ | [130] | |
| CressExpress | Arabidopsis | http://cressexpress.org/ | [131] | |
| Arabidopsis Network (AraNet) | Arabidopsis | http://www.inetbio.org/aranet/ | [132] | |
| Co-expressed Biological Processes (CoP) | Plants | http://webs2.kazusa.or.jp/kagiana/cop0911/ | [133] | |
| EXPath | Plants | http://expath.itps.ncku.edu.tw/ | [134] | |
| Plant Omics Data Center (PODC) | Plants | http://bioinf.mind.meiji.ac.jp/podc/ | [135] | |
| Plant Netwrok (PlaNet) | Plants | http://aranet.mpimp-golm.mpg.de/ | [136] | |
| OryzaExpress | Rice | http://plantomics.mind.meiji.ac.jp/OryzaExpress/ | [137] | |
| PlantExpress | Rice, Arabidopsis | http://plantomics.mind.meiji.ac.jp/PlantExpress/ | [138] | |
| Rice Functionally Related Gene Expression Network Database (RiceFREND) | Rice | http://ricefrend.dna.affrc.go.jp/ | [139] | |
| Vitis vinifera Co-expression Database (VTCdb) | Grape | http://vtcdb.adelaide.edu.au/ | [140] | |
| GeneMania | Various | http://genemania.org/ | [141] | |
| A Comprehensive Systems-Biology Database (CSB.DB) | Various | http://www.csbdb.de/csbdb/home/databases.html | [142] | |
| RapaNet | Brassica | http://bioinfo.mju.ac.kr/arraynet/brassica300k/query/ | [143] | |
| Rice Expression Database (RED) | Rice | http://expression.ic4r.org | [144] | |
| PhytoNet | Various | www.gene2function.de | [145] | |
| CoNekT | Plants | https://conekt.sbs.ntu.edu.sg | [146] | |
| CoCoCoNet | Plants | https://milton.cshl.edu/CoCoCoNet | [147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullah-Zawawi, M.-R.; Govender, N.; Harun, S.; Muhammad, N.A.N.; Zainal, Z.; Mohamed-Hussein, Z.-A. Multi-Omics Approaches and Resources for Systems-Level Gene Function Prediction in the Plant Kingdom. Plants 2022, 11, 2614. https://doi.org/10.3390/plants11192614
Abdullah-Zawawi M-R, Govender N, Harun S, Muhammad NAN, Zainal Z, Mohamed-Hussein Z-A. Multi-Omics Approaches and Resources for Systems-Level Gene Function Prediction in the Plant Kingdom. Plants. 2022; 11(19):2614. https://doi.org/10.3390/plants11192614
Chicago/Turabian StyleAbdullah-Zawawi, Muhammad-Redha, Nisha Govender, Sarahani Harun, Nor Azlan Nor Muhammad, Zamri Zainal, and Zeti-Azura Mohamed-Hussein. 2022. "Multi-Omics Approaches and Resources for Systems-Level Gene Function Prediction in the Plant Kingdom" Plants 11, no. 19: 2614. https://doi.org/10.3390/plants11192614