Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: Perpetrators or Bystanders?


Abstract
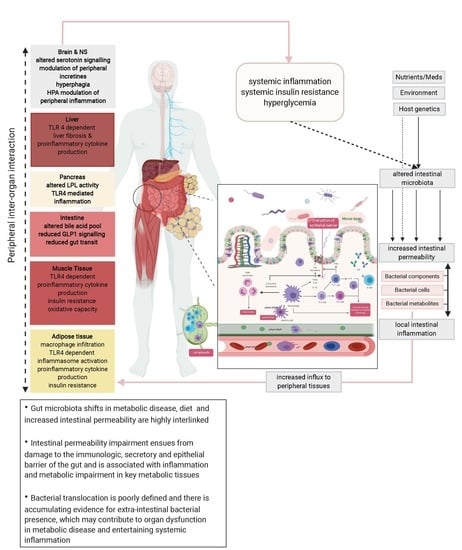
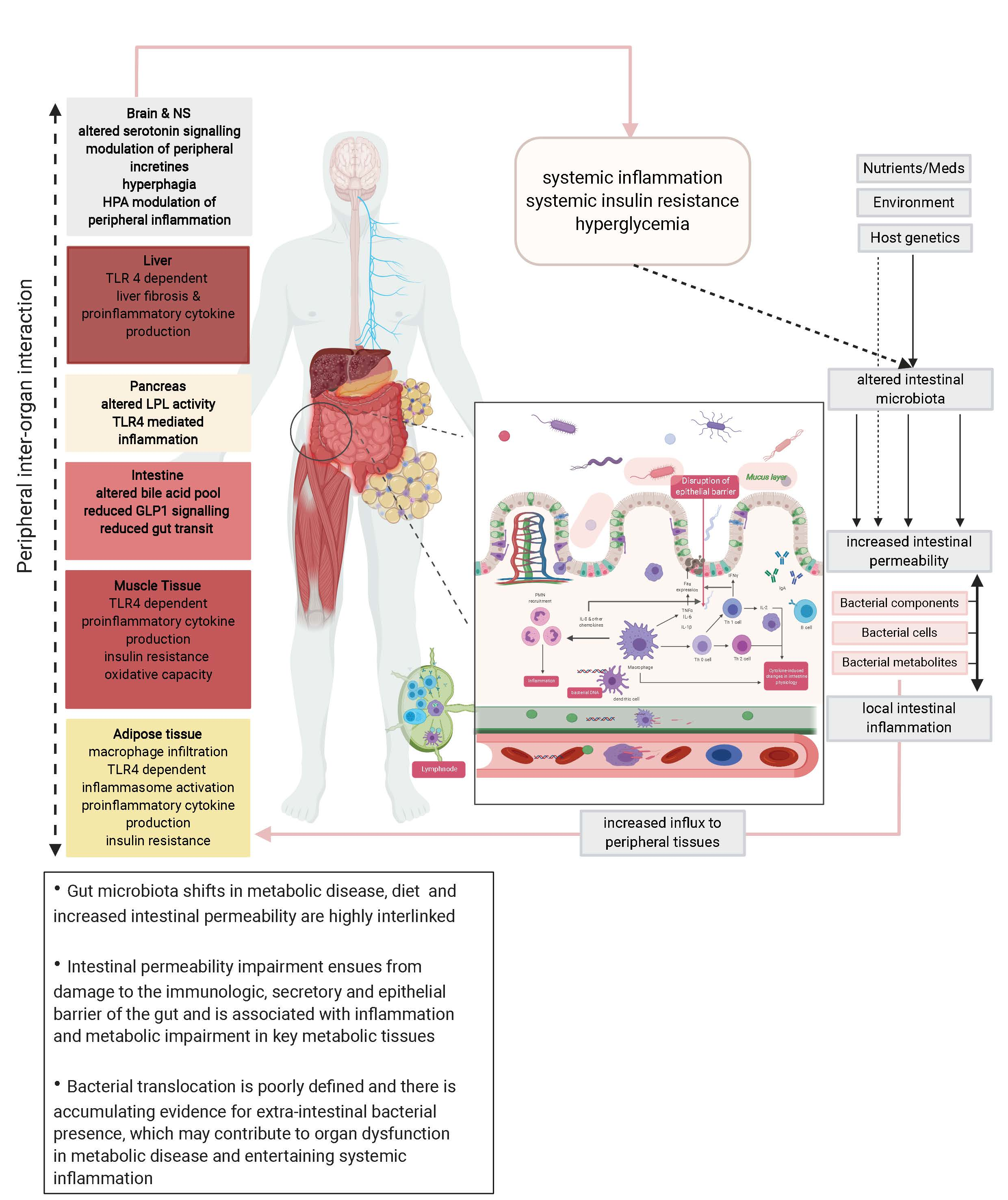
:
1. Introduction
2. Gut Microbiome Shifts, Diet, and Intestinal Permeability in Metabolic Disease
2.1. Compositional Gut Microbiota Shifts and Metabolic Disease Signatures
2.2. Quantitative Gut Microbiome Shifts in Metabolic Disease: When Numbers Matter
2.3. Dietary Signals in the Crosstalk between Gut Microbiome and Intestinal Permeability
3. Intestine’s Cerberus and the Leaky Gut
3.1. Lymph Nodes and Immune Cells
3.2. Secretory Compartment Including Mucus and IgA Antibodies
3.3. Intestinal Lining and Barrier Dysfunction
4. Breaking Down the Barriers: Markers of Bacterial Translocation
5. Bacterial Translocation and the Ominous T2D Octet
5.1. Adipose Tissue
5.2. Liver
5.3. Pancreas
5.4. Intestine
5.5. Muscle
5.6. Brain and Nervous System
6. Bacterial Presence in Remote Tissues
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.R.; Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G.; Hyland, N.P. Breaking down the barriers: The gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F. Programming of host metabolism by the gut microbiota. Ann. Nutr. Metab. 2011, 58 (Suppl. 2), 44–52. [Google Scholar]
- Cavalier-Smith, T.; Brasier, M.; Embley, T.M. Introduction: How and when did microbes change the world? Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Koppel, N.; Rekdal, V.M.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356. [Google Scholar] [CrossRef]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Berg, R.D.; Garlington, A.W. Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model. Infect. Immun. 1979, 23, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; Deal, C.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar]
- Albenberg, L.G.; Wu, G.D. Diet and the Intestinal Microbiome: Associations, Functions, and Implications for Health and Disease. Gastroenterology 2014, 146, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Duda-Chodak, A.; Tarko, T.; Satora, P.; Sroka, P. Interaction of dietary compounds, especially polyphenols, with the intestinal microbiota: A review. Eur. J. Nutr. 2015, 54, 325–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.; Barnes, S.; Demark-Wahnefried, W.; Morrow, C.; Salvador, C.; Skibola, C.; Tollefsbol, T.O. Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin. Epigenet. 2015, 7, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Gut Microbiome, Diet, and Links to Cardiometabolic and Chronic Disorders. Abstract. Europe PMC. Available online: https://europepmc.org/article/med/26616538 (accessed on 14 March 2020).
- Turnbaugh, P.J.; Gordon, J.I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009, 587, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, A.E.; Schwabkey, Z.; Wiesnoski, D.H.; Jenq, R.R. Clinical Evidence for the Microbiome in Inflammatory Diseases. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, T.; Bäckhed, F. The gut microbiota and metabolic disease: Current understanding and future perspectives. J. Intern. Med. 2016, 280, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids--at the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef]
- Deschasaux, M.; Bouter, K.E.; Prodan, A.; Levin, E.; Groen, A.K.; Herrema, H.; Tremaroli, V.; Bakker, G.J.; Attaye, I.; Pinto-Sietsma, S.-J.; et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 2018, 24, 1526–1531. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, D.; Fang, Z.; Jie, Z.; Qiu, X.; Zhang, C.; Chen, Y.; Ji, L. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS ONE 2013, 8, e71108. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef]
- Shin, N.-R.; Lee, J.-C.; Lee, H.-Y.; Kim, M.-S.; Whon, T.W.; Lee, M.-S.; Bae, J.-W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga–Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Kootte, R.S.; Levin, E.; Salojärvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; Hermes, G.; Bouter, K.E.; Koopen, A.M.; Holst, J.J.; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26, 611–619.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, K.; Yamori, Y.; Ooshima, A.; Okamoto, K. Effects of high or low sodium intake in spontaneously hypertensive rats. Jpn. Circ. J. 1972, 36, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Mell, B.; Jala, V.R.; Mathew, A.V.; Byun, J.; Waghulde, H.; Zhang, Y.; Haribabu, B.; Vijay-Kumar, M.; Pennathur, S.; Joe, B. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol. Genomics 2015, 47, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef]
- Sabaté, J.-M.; Jouët, P.; Harnois, F.; Mechler, C.; Msika, S.; Grossin, M.; Coffin, B. High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: A contributor to severe hepatic steatosis. Obes. Surg. 2008, 18, 371–377. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Lou, S.; Watthanasuntorn, K.; Kroner, P.T.; Cheungpasitporn, W.; Lukens, F.J.; Pungpapong, S.; Keaveny, A.P.; Ungprasert, P. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease: a systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2020, 32, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Wijarnpreecha, K.; Werlang, M.E.; Watthanasuntorn, K.; Panjawatanan, P.; Cheungpasitporn, W.; Gomez, V.; Lukens, F.J.; Ungprasert, P. Obesity and Risk of Small Intestine Bacterial Overgrowth: A Systematic Review and Meta-Analysis. Dig. Dis. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- El Kurdi, B.; Babar, S.; El Iskandarani, M.; Bataineh, A.; Lerch, M.M.; Young, M.; Singh, V.P. Factors That Affect Prevalence of Small Intestinal Bacterial Overgrowth in Chronic Pancreatitis: A Systematic Review, Meta-Analysis, and Meta-Regression. Clin. Transl. Gastroenterol. 2019, 10, e00072. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.; Morya, R.K.; Bhadada, S.K.; Rana, S. Type 1 diabetes mellitus: Complex interplay of oxidative stress, cytokines, gastrointestinal motility and small intestinal bacterial overgrowth. Eur. J. Clin. Invest. 2018, 48, e13021. [Google Scholar] [CrossRef] [PubMed]
- Zietz, B.; Lock, G.; Straub, R.H.; Braun, B.; Schölmerich, J.; Palitzsch, K.D. Small-bowel bacterial overgrowth in diabetic subjects is associated with cardiovascular autonomic neuropathy. Diabetes Care 2000, 23, 1200–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignata, C.; Budillon, G.; Monaco, G.; Nani, E.; Cuomo, R.; Parrilli, G.; Ciccimarra, F. Jejunal bacterial overgrowth and intestinal permeability in children with immunodeficiency syndromes. Gut 1990, 31, 879–882. [Google Scholar] [CrossRef] [Green Version]
- Vandeputte, D.; Kathagen, G.; D’hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017, 551, 507–511. [Google Scholar] [CrossRef]
- De Santis, S.; Cavalcanti, E.; Mastronardi, M.; Jirillo, E.; Chieppa, M. Nutritional Keys for Intestinal Barrier Modulation. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Mu, Q.; Kirby, J.; Reilly, C.M.; Luo, X.M. Leaky Gut as a Danger Signal for Autoimmune Diseases. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, M.; Lyle, B.J.; Madsen, K.L.; Sonnenburg, J.; Verbeke, K.; Wu, G.D. Role for diet in normal gut barrier function: Developing guidance within the framework of food-labeling regulations. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G17–G39. [Google Scholar] [CrossRef]
- Bisanz, J.E.; Upadhyay, V.; Turnbaugh, J.A.; Ly, K.; Turnbaugh, P. Diet Induces Reproducible Alterations in the Mouse and Human Gut Microbiome; Social Science Research Network: Rochester, NY, USA, 2019. [Google Scholar]
- Li, N.; Neu, J. Glutamine Deprivation Alters Intestinal Tight Junctions via a PI3-K/Akt Mediated Pathway in Caco-2 Cells. J. Nutr. 2009, 139, 710–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, J.; Fukumoto, K.; Fukushi, E.; Sonoyama, K.; Kawabata, J. Isolation of Tryptophan as an Inhibitor of Ovalbumin Permeation and Analysis of Its Suppressive Effect on Oral Sensitization. Biosci. Biotechnol. Biochem. 2004, 68, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Yan, H.; You, Z.; Wang, P.; Wang, S. Effects of enteral supplementation with glutamine granules on intestinal mucosal barrier function in severe burned patients. Burns J. Int. Soc. Burn Inj. 2004, 30, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Ang, Q.Y.; Turnbaugh, P.J. A diet-dependent enzyme from the human gut microbiome promotes Th17 accumulation and colitis. bioRxiv 2019, 766899. [Google Scholar]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.K.; Muir, J.G.; Gibson, P.R. Review article: Insights into colonic protein fermentation, its modulation and potential health implications. Aliment. Pharmacol. Ther. 2016, 43, 181–196. [Google Scholar] [CrossRef] [Green Version]
- Llewellyn, S.R.; Britton, G.J.; Contijoch, E.J.; Vennaro, O.H.; Mortha, A.; Colombel, J.-F.; Grinspan, A.; Clemente, J.C.; Merad, M.; Faith, J.J. Interactions Between Diet and the Intestinal Microbiota Alter Intestinal Permeability and Colitis Severity in Mice. Gastroenterology 2018, 154, 1037–1046.e2. [Google Scholar] [CrossRef]
- Chen, T.; Kim, C.Y.; Kaur, A.; Lamothe, L.; Shaikh, M.; Keshavarzian, A.; Hamaker, B.R. Dietary fibre-based SCFA mixtures promote both protection and repair of intestinal epithelial barrier function in a Caco-2 cell model. Food Funct. 2017, 8, 1166–1173. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Lerner, A.; Matthias, T. Changes in intestinal tight junction permeability associated with industrial food additives explain the rising incidence of autoimmune disease. Autoimmun. Rev. 2015, 14, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobin, K.; Stumpf, N.E.; Schwab, S.; Eichler, M.; Neubert, P.; Rauh, M.; Adamowski, M.; Babyak, O.; Hinze, D.; Sivalingam, S.; et al. A high-salt diet compromises antibacterial neutrophil responses through hormonal perturbation. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Fava, F.; Danese, S. Intestinal microbiota in inflammatory bowel disease: Friend of foe? World J. Gastroenterol. 2011, 17, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Thiennimitr, P.; Winter, S.E.; Winter, M.G.; Xavier, M.N.; Tolstikov, V.; Huseby, D.L.; Sterzenbach, T.; Tsolis, R.M.; Roth, J.R.; Bäumler, A.J. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. USA 2011, 108, 17480–17485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffen, E.K.; Berg, R.D.; Deitch, E.A. Comparison of translocation rates of various indigenous bacteria from the gastrointestinal tract to the mesenteric lymph node. J. Infect. Dis. 1988, 157, 1032–1038. [Google Scholar] [CrossRef]
- Wells, C.L. Relationship between intestinal microecology and the translocation of intestinal bacteria. Antonie Van Leeuwenhoek 1990, 58, 87–93. [Google Scholar] [CrossRef]
- Macutkiewicz, C.; Carlson, G.; Clark, E.; Dobrindt, U.; Roberts, I.; Warhurst, G. Characterisation of Escherichia coli strains involved in transcytosis across gut epithelial cells exposed to metabolic and inflammatory stress. Microbes Infect. 2008, 10, 424–431. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 2004, 303, 1662–1665. [Google Scholar] [CrossRef] [Green Version]
- Hapfelmeier, S.; Lawson, M.A.E.; Slack, E.; Kirundi, J.K.; Stoel, M.; Heikenwalder, M.; Cahenzli, J.; Velykoredko, Y.; Balmer, M.L.; Endt, K.; et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 2010, 328, 1705–1709. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, D.; Benson, A.; Mirpuri, J.; Pifer, R.; Hou, B.; DeFranco, A.L.; Yarovinsky, F. B cell-intrinsic MyD88 signaling prevents the lethal dissemination of commensal bacteria during colonic damage. Immunity 2012, 36, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, W.E.; Berg, R.D. Bacterial translocation from the gastrointestinal tract of athymic (nu/nu) mice. Infect. Immun. 1980, 27, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Choudhry, M.A.; Fazal, N.; Goto, M.; Gamelli, R.L.; Sayeed, M.M. Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G937–G947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboziev, I.; Karlsson, F.; Grisham, M.B. Gut-associated lymphoid tissue, T cell trafficking, and chronic intestinal inflammation. Ann. N. Y. Acad. Sci. 2010, 1207 Suppl 1, E86–E93. [Google Scholar] [CrossRef]
- Richard, C.; Wadowski, M.; Goruk, S.; Cameron, L.; Sharma, A.M.; Field, C.J. Individuals with obesity and type 2 diabetes have additional immune dysfunction compared with obese individuals who are metabolically healthy. BMJ Open Diabetes Res. Care 2017, 5, e000379. [Google Scholar] [CrossRef]
- Bertoni, A.G.; Saydah, S.; Brancati, F.L. Diabetes and the risk of infection-related mortality in the U.S. Diabetes Care 2001, 24, 1044–1049. [Google Scholar] [CrossRef] [Green Version]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef] [Green Version]
- Hodin, C.M.; Verdam, F.J.; Grootjans, J.; Rensen, S.S.; Verheyen, F.K.; Dejong, C.H.C.; Buurman, W.A.; Greve, J.W.; Lenaerts, K. Reduced Paneth cell antimicrobial protein levels correlate with activation of the unfolded protein response in the gut of obese individuals. J. Pathol. 2011, 225, 276–284. [Google Scholar] [CrossRef]
- Meyer-Hoffert, U.; Hornef, M.W.; Henriques-Normark, B.; Axelsson, L.-G.; Midtvedt, T.; Pütsep, K.; Andersson, M. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 2008, 57, 764–771. [Google Scholar] [CrossRef]
- Haeusler, R.A.; Astiarraga, B.; Camastra, S.; Accili, D.; Ferrannini, E. Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes 2013, 62, 4184–4191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlström, A.; Kovatcheva-Datchary, P.; Ståhlman, M.; Khan, M.-T.; Bäckhed, F.; Marschall, H.-U. Induction of farnesoid X receptor signaling in germ-free mice colonized with a human microbiota. J. Lipid Res. 2017, 58, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, R.W.; Clements, W.D.; Smye, M.G.; Pope, C.; Rowlands, B.J.; Diamond, T. Intestinal barrier dysfunction in clinical and experimental obstructive jaundice and its reversal by internal biliary drainage. Br. J. Surg. 1996, 83, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.V.; Murchan, P.; Leonard, N.; Clarke, P.; Keane, F.B.; Tanner, W.A. Gut barrier failure in experimental obstructive jaundice. J. Surg. Res. 1996, 62, 11–16. [Google Scholar] [CrossRef]
- Gutzeit, C.; Magri, G.; Cerutti, A. Intestinal IgA production and its role in host-microbe interaction. Immunol. Rev. 2014, 260, 76–85. [Google Scholar] [CrossRef]
- Luck, H.; Khan, S.; Kim, J.H.; Copeland, J.K.; Revelo, X.S.; Tsai, S.; Chakraborty, M.; Cheng, K.; Tao Chan, Y.; Nøhr, M.K.; et al. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat. Commun. 2019, 10, 3650. [Google Scholar] [CrossRef]
- Damms-Machado, A.; Louis, S.; Schnitzer, A.; Volynets, V.; Rings, A.; Basrai, M.; Bischoff, S.C. Gut permeability is related to body weight, fatty liver disease, and insulin resistance in obese individuals undergoing weight reduction. Am. J. Clin. Nutr. 2017, 105, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, T.F.S.; Souza, N.C.S.; Chiarello, P.G.; Franceschini, S.C.C.; Bressan, J.; Ferreira, C.L.L.F.; Maria do Carmo, G.P. Intestinal permeability parameters in obese patients are correlated with metabolic syndrome risk factors. Clin. Nutr. Edinb. Scotl. 2012, 31, 735–740. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Genser, L.; Aguanno, D.; Soula, H.A.; Dong, L.; Trystram, L.; Assmann, K.; Salem, J.-E.; Vaillant, J.-C.; Oppert, J.-M.; Laugerette, F.; et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J. Pathol. 2018, 246, 217–230. [Google Scholar] [CrossRef]
- Devriese, S.; Van den Bossche, L.; Van Welden, S.; Holvoet, T.; Pinheiro, I.; Hindryckx, P.; De Vos, M.; Laukens, D. T84 monolayers are superior to Caco-2 as a model system of colonocytes. Histochem. Cell Biol. 2017, 148, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Rah, B.; Bastola, D.; Dhawan, P.; Singh, A.B. Obesity-induces Organ and Tissue Specific Tight Junction Restructuring and Barrier Deregulation by Claudin Switching. Sci. Rep. 2017, 7, 5125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Thomas, T.C.; Storlien, L.H.; Huang, X.F. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int. J. Obes. 2000, 24, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Siegmund, B.; Sennello, J.A.; Jones-Carson, J.; Gamboni-Robertson, F.; Lehr, H.A.; Batra, A.; Fedke, I.; Zeitz, M.; Fantuzzi, G. Leptin receptor expression on T lymphocytes modulates chronic intestinal inflammation in mice. Gut 2004, 53, 965–972. [Google Scholar] [CrossRef]
- Sitaraman, S.; Liu, X.; Charrier, L.; Gu, L.H.; Ziegler, T.R.; Gewirtz, A.; Merlin, D. Colonic leptin: Source of a novel proinflammatory cytokine involved in IBD. FASEB J. 2004, 18, 696–698. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Böttcher, C.; Letizia, M.; Yerinde, C.; Wu, H.; Freise, I.; Rodriguez-Sillke, Y.; Stoyanova, A.K.; Kreis, M.E.; Asbach, P.; et al. Leptin induces TNFα-dependent inflammation in acquired generalized lipodystrophy and combined Crohn’s disease. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.-H. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Gerriets, V.A.; Danzaki, K.; Kishton, R.J.; Eisner, W.; Nichols, A.G.; Saucillo, D.C.; Shinohara, M.L.; MacIver, N.J. Leptin directly promotes T-cell glycolytic metabolism to drive effector T-cell differentiation in a mouse model of autoimmunity. Eur. J. Immunol. 2016, 46, 1970–1983. [Google Scholar] [CrossRef] [Green Version]
- White, R.H.; Frayn, K.N.; Little, R.A.; Threlfall, C.J.; Stoner, H.B.; Irving, M.H. Hormonal and metabolic responses to glucose infusion in sepsis studied by the hyperglycemic glucose clamp technique. JPEN J. Parenter. Enteral Nutr. 1987, 11, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Majdalawieh, A.; Ro, H.-S. LPS-induced suppression of macrophage cholesterol efflux is mediated by adipocyte enhancer-binding protein 1. Int. J. Biochem. Cell Biol. 2009, 41, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E.; et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G433–G441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thuy, S.; Ladurner, R.; Volynets, V.; Wagner, S.; Strahl, S.; Königsrainer, A.; Maier, K.-P.; Bischoff, S.C.; Bergheim, I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J. Nutr. 2008, 138, 1452–1455. [Google Scholar] [CrossRef] [Green Version]
- Lassenius, M.I.; Pietiläinen, K.H.; Kaartinen, K.; Pussinen, P.J.; Syrjänen, J.; Forsblom, C.; Pörsti, I.; Rissanen, A.; Kaprio, J.; Mustonen, J.; et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 2011, 34, 1809–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pussinen, P.J.; Havulinna, A.S.; Lehto, M.; Sundvall, J.; Salomaa, V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care 2011, 34, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.J.; Zhang, P.; Bowden, D.W.; Devereaux, B.; Davoren, P.M.; Cripps, A.W.; West, N.P. Increased intestinal permeability as a risk factor for type 2 diabetes. Diabetes Metab. 2017, 43, 163–166. [Google Scholar] [CrossRef]
- Ruiz, A.G.; Casafont, F.; Crespo, J.; Cayón, A.; Mayorga, M.; Estebanez, A.; Fernadez-Escalante, J.C.; Pons-Romero, F. Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression in obese patients: Evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes. Surg. 2007, 17, 1374–1380. [Google Scholar] [CrossRef]
- Ortiz, S.; Zapater, P.; Estrada, J.L.; Enriquez, P.; Rey, M.; Abad, A.; Such, J.; Lluis, F.; Francés, R. Bacterial DNA translocation holds increased insulin resistance and systemic inflammatory levels in morbid obese patients. J. Clin. Endocrinol. Metab. 2014, 99, 2575–2583. [Google Scholar] [CrossRef] [Green Version]
- Trøseid, M.; Nestvold, T.K.; Rudi, K.; Thoresen, H.; Nielsen, E.W.; Lappegård, K.T. Plasma lipopolysaccharide is closely associated with glycemic control and abdominal obesity: Evidence from bariatric surgery. Diabetes Care 2013, 36, 3627–3632. [Google Scholar] [CrossRef] [Green Version]
- Nádházi, Z.; Takáts, A.; Offenmüller, K.; Bertók, L. Plasma endotoxin level of healthy donors. Acta Microbiol. Immunol. Hung. 2002, 49, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Elsbach, P.; Weiss, J. The bactericidal/permeability-increasing protein (BPI), a potent element in host-defense Against gram-negative bacteria and lipopolysaccharide. Immunobiology 1993, 187, 417–429. [Google Scholar] [CrossRef]
- Hurley, J.C. Concordance of endotoxemia with gram-negative bacteremia in patients with gram-negative sepsis: A meta-analysis. J. Clin. Microbiol. 1994, 32, 2120–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yoshida, S.; Hara, H. Physiological concentrations of short-chain fatty acids immediately suppress colonic epithelial permeability. Br. J. Nutr. 2008, 100, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Plöger, S.; Stumpff, F.; Penner, G.B.; Schulzke, J.-D.; Gäbel, G.; Martens, H.; Shen, Z.; Günzel, D.; Aschenbach, J.R. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann. N. Y. Acad. Sci. 2012, 1258, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K.; Lutgendorff, F.; Phan, V.; Söderholm, J.D.; Sherman, P.M.; McKay, D.M. Enhanced translocation of bacteria across metabolically stressed epithelia is reduced by butyrate. Inflamm. Bowel Dis. 2010, 16, 1138–1148. [Google Scholar] [CrossRef]
- Ortega, F.J.; Sabater, M.; Moreno-Navarrete, J.M.; Pueyo, N.; Botas, P.; Delgado, E.; Ricart, W.; Frühbeck, G.; Fernández-Real, J.M. Serum and urinary concentrations of calprotectin as markers of insulin resistance and type 2 diabetes. Eur. J. Endocrinol. 2012, 167, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Tamboli, C.P.; Richard, F.; Colombel, J.-F. Fecal calprotectin in Crohn’s disease: New family ties. Gastroenterology 2003, 124, 1971–1974. [Google Scholar] [CrossRef]
- Kanda, T.; Nakatomi, Y.; Ishikawa, H.; Hitomi, M.; Matsubara, Y.; Ono, T.; Muto, T. Intestinal fatty acid-binding protein as a sensitive marker of intestinal ischemia. Dig. Dis. Sci. 1992, 37, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Fujii, H.; Tani, T.; Murakami, H.; Suda, T.; Sakai, Y.; Ono, T.; Hatakeyama, K. Intestinal fatty acid-binding protein is a useful diagnostic marker for mesenteric infarction in humans. Gastroenterology 1996, 110, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Thuijls, G.; van Wijck, K.; Grootjans, J.; Derikx, J.P.M.; van Bijnen, A.A.; Heineman, E.; Dejong, C.H.C.; Buurman, W.A.; Poeze, M. Early diagnosis of intestinal ischemia using urinary and plasma fatty acid binding proteins. Ann. Surg. 2011, 253, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Hawkesworth, S.; Moore, S.E.; Fulford, A.J.C.; Barclay, G.R.; Darboe, A.A.; Mark, H.; Nyan, O.A.; Prentice, A.M. Evidence for metabolic endotoxemia in obese and diabetic Gambian women. Nutr. Diabetes 2013, 3, e83. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Uzzau, S.; Goldblum, S.E.; Fasano, A. Human zonulin, a potential modulator of intestinal tight junctions. J. Cell Sci. 2000, 113 Pt 24, 4435–4440. [Google Scholar]
- Wang, L.; Llorente, C.; Hartmann, P.; Yang, A.-M.; Chen, P.; Schnabl, B. Methods to determine intestinal permeability and bacterial translocation during liver disease. J. Immunol. Methods 2015, 421, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, J.; Vogelsang, H.; Hübl, W.; Waldhöer, T.; Lochs, H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 1993, 341, 1437–1439. [Google Scholar] [CrossRef]
- Bosi, E.; Molteni, L.; Radaelli, M.G.; Folini, L.; Fermo, I.; Bazzigaluppi, E.; Piemonti, L.; Pastore, M.R.; Paroni, R. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia 2006, 49, 2824–2827. [Google Scholar] [CrossRef] [Green Version]
- Mooradian, A.D.; Morley, J.E.; Levine, A.S.; Prigge, W.F.; Gebhard, R.L. Abnormal intestinal permeability to sugars in diabetes mellitus. Diabetologia 1986, 29, 221–224. [Google Scholar] [CrossRef]
- Wilbrink, J.; Bernards, N.; Mujagic, Z.; van Avesaat, M.; Pijls, K.; Klaassen, T.; van Eijk, H.; Nienhuijs, S.; Stronkhorst, A.; Wilms, E.; et al. Intestinal barrier function in morbid obesity: Results of a prospective study on the effect of sleeve gastrectomy. Int. J. Obes. 2020, 44, 368–376. [Google Scholar] [CrossRef]
- Horton, F.; Wright, J.; Smith, L.; Hinton, P.J.; Robertson, M.D. Increased intestinal permeability to oral chromium (51 Cr) -EDTA in human Type 2 diabetes. Diabet. Med. J. Br. Diabet. Assoc. 2014, 31, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Navarrete, J.M.; Sabater, M.; Ortega, F.; Ricart, W.; Fernández-Real, J.M. Circulating zonulin, a marker of intestinal permeability, is increased in association with obesity-associated insulin resistance. PLoS ONE 2012, 7, e37160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zak-Gołąb, A.; Kocełak, P.; Aptekorz, M.; Zientara, M.; Juszczyk, L.; Martirosian, G.; Chudek, J.; Olszanecka-Glinianowicz, M. Gut microbiota, microinflammation, metabolic profile, and zonulin concentration in obese and normal weight subjects. Int. J. Endocrinol. 2013, 2013, 674106. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, L.; Nybo, M.; Poulsen, M.K.; Henriksen, J.E.; Dahl, J.; Rasmussen, L.M. Plasma calprotectin and its association with cardiovascular disease manifestations, obesity and the metabolic syndrome in type 2 diabetes mellitus patients. BMC Cardiovasc. Disord. 2014, 14, 196. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A. From the Triumvirate to the Ominous Octet: A New Paradigm for the Treatment of Type 2 Diabetes Mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Nakae, J.; Watanabe, N.; Kikuchi, T.; Tateya, S.; Tamori, Y.; Kaneko, M.; Abe, T.; Onodera, M.; Itoh, H. Colonic Pro-inflammatory Macrophages Cause Insulin Resistance in an Intestinal Ccl2/Ccr2-Dependent Manner. Cell Metab. 2016, 24, 295–310. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Bonecchi, R. Colonic Macrophages “Remote Control” Adipose Tissue Inflammation and Insulin Resistance. Cell Metab. 2016, 24, 196–198. [Google Scholar] [CrossRef] [Green Version]
- Vila, I.K.; Badin, P.-M.; Marques, M.-A.; Monbrun, L.; Lefort, C.; Mir, L.; Louche, K.; Bourlier, V.; Roussel, B.; Gui, P.; et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014, 7, 1116–1129. [Google Scholar] [CrossRef]
- Creely, S.J.; McTernan, P.G.; Kusminski, C.M.; Fisher, M.; Da Silva, N.F.; Khanolkar, M.; Evans, M.; Harte, A.L.; Kumar, S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E740–E747. [Google Scholar] [CrossRef] [Green Version]
- Vitseva, O.I.; Tanriverdi, K.; Tchkonia, T.T.; Kirkland, J.L.; McDonnell, M.E.; Apovian, C.M.; Freedman, J.; Gokce, N. Inducible Toll-like receptor and NF-kappaB regulatory pathway expression in human adipose tissue. Obesity 2008, 16, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Ilan, Y. Leaky gut and the liver: A role for bacterial translocation in nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, T.M.; Schwacha, H.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Aponte, J.J.; Pelz, K.; Berger, D.; Kist, M.; Blum, H.E. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am. J. Gastroenterol. 2002, 97, 2364–2370. [Google Scholar] [CrossRef]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Mizrahi, M.; Shabat, Y.; Ben Ya’acov, A.; Lalazar, G.; Adar, T.; Wong, V.; Muller, B.; Rawlin, G.; Ilan, Y. Alleviation of insulin resistance and liver damage by oral administration of Imm124-E is mediated by increased Tregs and associated with increased serum GLP-1 and adiponectin: Results of a phase I/II clinical trial in NASH. J. Inflamm. Res. 2012, 5, 141–150. [Google Scholar]
- Shen, J.; Sakaida, I.; Uchida, K.; Terai, S.; Okita, K. Leptin enhances TNF-alpha production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. Life Sci. 2005, 77, 1502–1515. [Google Scholar] [CrossRef]
- Balzan, S.; de Almeida Quadros, C.; de Cleva, R.; Zilberstein, B.; Cecconello, I. Bacterial translocation: Overview of mechanisms and clinical impact. J. Gastroenterol. Hepatol. 2007, 22, 464–471. [Google Scholar] [CrossRef]
- Mattijssen, F.; Alex, S.; Swarts, H.J.; Groen, A.K.; van Schothorst, E.M.; Kersten, S. Angptl4 serves as an endogenous inhibitor of intestinal lipid digestion. Mol. Metab. 2013, 3, 135–144. [Google Scholar] [CrossRef]
- Oresic, M.; Simell, S.; Sysi-Aho, M.; Näntö-Salonen, K.; Seppänen-Laakso, T.; Parikka, V.; Katajamaa, M.; Hekkala, A.; Mattila, I.; Keskinen, P.; et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J. Exp. Med. 2008, 205, 2975–2984. [Google Scholar] [CrossRef] [Green Version]
- King, C.; Sarvetnick, N. The incidence of type-1 diabetes in NOD mice is modulated by restricted flora not germ-free conditions. PLoS ONE 2011, 6, e17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiner, T.U.; Hyötyläinen, T.; Knip, M.; Bäckhed, F.; Orešič, M. The Gut Microbiota Modulates Glycaemic Control and Serum Metabolite Profiles in Non-Obese Diabetic Mice. PLoS ONE 2014, 9, e110359. [Google Scholar] [CrossRef] [PubMed]
- Amyot, J.; Semache, M.; Ferdaoussi, M.; Fontés, G.; Poitout, V. Lipopolysaccharides impair insulin gene expression in isolated islets of Langerhans via Toll-Like Receptor-4 and NF-κB signalling. PLoS ONE 2012, 7, e36200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Badman, M.K.; Flier, J.S. The gut and energy balance: Visceral allies in the obesity wars. Science 2005, 307, 1909–1914. [Google Scholar] [CrossRef] [Green Version]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Eissele, R.; Göke, R.; Willemer, S.; Harthus, H.P.; Vermeer, H.; Arnold, R.; Göke, B. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur. J. Clin. Invest. 1992, 22, 283–291. [Google Scholar] [CrossRef]
- Wichmann, A.; Allahyar, A.; Greiner, T.U.; Plovier, H.; Lundén, G.Ö.; Larsson, T.; Drucker, D.J.; Delzenne, N.M.; Cani, P.D.; Bäckhed, F. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 2013, 14, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef]
- Albaugh, V.L.; Banan, B.; Antoun, J.; Xiong, Y.; Guo, Y.; Ping, J.; Alikhan, M.; Clements, B.A.; Abumrad, N.N.; Flynn, C.R. Role of Bile Acids and GLP-1 in Mediating the Metabolic Improvements of Bariatric Surgery. Gastroenterology 2019, 156, 1041–1051.e4. [Google Scholar] [CrossRef] [Green Version]
- Grasset, E.; Puel, A.; Charpentier, J.; Collet, X.; Christensen, J.E.; Tercé, F.; Burcelin, R. A Specific Gut Microbiota Dysbiosis of Type 2 Diabetic Mice Induces GLP-1 Resistance through an Enteric NO-Dependent and Gut-Brain Axis Mechanism. Cell Metab. 2017, 25, 1075–1090.e5. [Google Scholar] [CrossRef] [Green Version]
- Chimerel, C.; Emery, E.; Summers, D.K.; Keyser, U.; Gribble, F.M.; Reimann, F. Bacterial Metabolite Indole Modulates Incretin Secretion from Intestinal Enteroendocrine L Cells. Cell Rep. 2014, 9, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J.; Erlich, P.; Asa, S.L.; Brubaker, P.L. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc. Natl. Acad. Sci. USA 1996, 93, 7911–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeppesen, P.B. Clinical significance of GLP-2 in short-bowel syndrome. J. Nutr. 2003, 133, 3721–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef] [Green Version]
- Warram, J.H.; Martin, B.C.; Krolewski, A.S.; Soeldner, J.S.; Kahn, C.R. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann. Intern. Med. 1990, 113, 909–915. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- van der Crabben, S.N.; Blümer, R.M.E.; Stegenga, M.E.; Ackermans, M.T.; Endert, E.; Tanck, M.W.T.; Serlie, M.J.; van der Poll, T.; Sauerwein, H.P. Early endotoxemia increases peripheral and hepatic insulin sensitivity in healthy humans. J. Clin. Endocrinol. Metab. 2009, 94, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Agwunobi, A.O.; Reid, C.; Maycock, P.; Little, R.A.; Carlson, G.L. Insulin resistance and substrate utilization in human endotoxemia. J. Clin. Endocrinol. Metab. 2000, 85, 3770–3778. [Google Scholar] [CrossRef]
- Van Cromphaut, S.J.; Vanhorebeek, I.; Van den Berghe, G. Glucose metabolism and insulin resistance in sepsis. Curr. Pharm. Des. 2008, 14, 1887–1899. [Google Scholar] [CrossRef]
- Virkamäki, A.; Yki-Järvinen, H. Mechanisms of insulin resistance during acute endotoxemia. Endocrinology 1994, 134, 2072–2078. [Google Scholar] [CrossRef] [PubMed]
- Fowelin, J.; Attvall, S.; Von Schenck, H.; Smith, U.; Lager, I. Combined effect of growth hormone and cortisol on late posthypoglycemic insulin resistance in humans. Diabetes 1989, 38, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Fowelin, J.; Attvall, S.; von Schenck, H.; Smith, U.; Lager, I. Characterization of the insulin-antagonistic effect of growth hormone in man. Diabetologia 1991, 34, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, R.A.; Nystrom, G.J.; Lang, C.H. Lipopolysaccharide regulates proinflammatory cytokine expression in mouse myoblasts and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R698–R709. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Hussey, S.E.; Sanchez-Avila, A.; Tantiwong, P.; Musi, N. Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PLoS ONE 2013, 8, e63983. [Google Scholar] [CrossRef]
- Frisard, M.I.; McMillan, R.P.; Marchand, J.; Wahlberg, K.A.; Wu, Y.; Voelker, K.A.; Heilbronn, L.; Haynie, K.; Muoio, B.; Li, L.; et al. Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E988–E998. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Mandard, S.; Dray, C.; Deckert, V.; Valet, P.; Besnard, P.; Drucker, D.J.; Lagrost, L.; Grober, J. Lipopolysaccharides-mediated increase in glucose-stimulated insulin secretion: Involvement of the GLP-1 pathway. Diabetes 2014, 63, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Manolakis, A.C.; Kapsoritakis, A.N.; Tiaka, E.K.; Sidiropoulos, A.; Gerovassili, A.; Satra, M.; Vamvakopoulou, D.; Tsiopoulos, F.; Papanas, N.; Skoularigis, I.; et al. TLR4 gene polymorphisms: Evidence for protection against type 2 diabetes but not for diabetes-associated ischaemic heart disease. Eur. J. Endocrinol. 2011, 165, 261–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, E.; Li, J.V.; Athanasiou, T.; Ashrafian, H.; Nicholson, J.K. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol. 2011, 19, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Savidge, T.; Shulman, R.J. Brain-gut microbiome interactions and functional bowel disorders. Gastroenterology 2014, 146, 1500–1512. [Google Scholar] [CrossRef] [Green Version]
- Foster, J.A.; McVey Neufeld, K.-A. Gut-brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef]
- Bliss, E.S.; Whiteside, E. The Gut-Brain Axis, the Human Gut Microbiota and Their Integration in the Development of Obesity. Front. Physiol. 2018, 9, 900. [Google Scholar] [CrossRef] [Green Version]
- Margolis, K.G.; Stevanovic, K.; Li, Z.; Yang, Q.M.; Oravecz, T.; Zambrowicz, B.; Jhaver, K.G.; Diacou, A.; Gershon, M.D. Pharmacological reduction of mucosal but not neuronal serotonin opposes inflammation in mouse intestine. Gut 2014, 63, 928–937. [Google Scholar] [CrossRef]
- Spohn, S.N.; Bianco, F.; Scott, R.B.; Keenan, C.M.; Linton, A.A.; O’Neill, C.H.; Bonora, E.; Dicay, M.; Lavoie, B.; Wilcox, R.L.; et al. Protective Actions of Epithelial 5-Hydroxytryptamine 4 Receptors in Normal and Inflamed Colon. Gastroenterology 2016, 151, 933–944. [Google Scholar] [CrossRef] [Green Version]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Zheng, P.; Zeng, B.; Zhou, C.; Liu, M.; Fang, Z.; Xu, X.; Zeng, L.; Chen, J.; Fan, S.; Du, X.; et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 2016, 21, 786–796. [Google Scholar] [CrossRef]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome–brain–β-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Suez, J.; Korem, T.; Zeevi, D.; Zilberman-Schapira, G.; Thaiss, C.A.; Maza, O.; Israeli, D.; Zmora, N.; Gilad, S.; Weinberger, A.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef]
- Swithers, S.E. Artificial sweeteners are not the answer to childhood obesity. Appetite 2015, 93, 85–90. [Google Scholar] [CrossRef]
- Cani, P.D.; Delzenne, N.M. The role of the gut microbiota in energy metabolism and metabolic disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charpentier, J.; Waget, A.; Klopp, P.; Magnan, C.; Cruciani-Guglielmacci, C.; Lee, S.J.; Burcelin, R.; Grasset, E. Lixisenatide requires a functional gut-vagus nerve-brain axis to trigger insulin secretion in controls and type 2 diabetic mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G671–G684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Punder, K.; Pruimboom, L. Stress Induces Endotoxemia and Low-Grade Inflammation by Increasing Barrier Permeability. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Castillo, D.J.; Rifkin, R.F.; Cowan, D.A.; Potgieter, M. The Healthy Human Blood Microbiome: Fact or Fiction? Front. Cell. Infect. Microbiol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, R.W.; Vali, H.; Lau, P.C.K.; Palfree, R.G.E.; De Ciccio, A.; Sirois, M.; Ahmad, D.; Villemur, R.; Desrosiers, M.; Chan, E.C.S. Are there naturally occurring pleomorphic bacteria in the blood of healthy humans? J. Clin. Microbiol. 2002, 40, 4771–4775. [Google Scholar] [CrossRef] [Green Version]
- Amar, J.; Serino, M.; Lange, C.; Chabo, C.; Iacovoni, J.; Mondot, S.; Lepage, P.; Klopp, C.; Mariette, J.; Bouchez, O.; et al. Involvement of tissue bacteria in the onset of diabetes in humans: Evidence for a concept. Diabetologia 2011, 54, 3055–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; Courtney, M.; Marre, M.; Charles, M.A.; Balkau, B.; et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: The D.E.S.I.R. study. PLoS ONE 2013, 8, e54461. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Kanazawa, A.; Ikeda, F.; Yoshihara, T.; Goto, H.; Abe, H.; Komiya, K.; Kawaguchi, M.; Shimizu, T.; Ogihara, T.; et al. Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care 2014, 37, 2343–2350. [Google Scholar] [CrossRef] [Green Version]
- Païssé, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Lelouvier, B.; Servant, F.; Païssé, S.; Brunet, A.-C.; Benyahya, S.; Serino, M.; Valle, C.; Ortiz, M.R.; Puig, J.; Courtney, M.; et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 2016, 64, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Schierwagen, R.; Alvarez-Silva, C.; Madsen, M.S.A.; Kolbe, C.C.; Meyer, C.; Thomas, D.; Uschner, F.E.; Magdaleno, F.; Jansen, C.; Pohlmann, A.; et al. Circulating microbiome in blood of different circulatory compartments. Gut 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaiser, R.A.; Halimi, A.; Alkharaan, H.; Lu, L.; Davanian, H.; Healy, K.; Hugerth, L.W.; Ateeb, Z.; Valente, R.; Moro, C.F.; et al. Enrichment of oral microbiota in early cystic precursors to invasive pancreatic cancer. Gut 2019, 68, 2186–2194. [Google Scholar] [CrossRef] [Green Version]
- Ben-Jonathan, N.; Hugo, E.R.; Brandebourg, T.D. Effects of bisphenol A on adipokine release from human adipose tissue: Implications for the metabolic syndrome. Mol. Cell. Endocrinol. 2009, 304, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-M.; Kim, K.-S.; Jacobs, D.R.; Lee, D.-H. Persistent organic pollutants in adipose tissue should be considered in obesity research. Obes. Rev. 2017, 18, 129–139. [Google Scholar] [CrossRef]
- Burcelin, R.; Serino, M.; Chabo, C.; Garidou, L.; Pomié, C.; Courtney, M.; Amar, J.; Bouloumié, A. Metagenome and metabolism: The tissue microbiota hypothesis. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 61–70. [Google Scholar] [CrossRef]
- Lluch, J.; Servant, F.; Païssé, S.; Valle, C.; Valière, S.; Kuchly, C.; Vilchez, G.; Donnadieu, C.; Courtney, M.; Burcelin, R.; et al. The Characterization of Novel Tissue Microbiota Using an Optimized 16S Metagenomic Sequencing Pipeline. PLoS ONE 2015, 10, e0142334. [Google Scholar] [CrossRef]
- Zulian, A.; Cancello, R.; Cesana, E.; Rizzi, E.; Consolandi, C.; Severgnini, M.; Panizzo, V.; Di Blasio, A.M.; Micheletto, G.; Invitti, C. Adipose tissue microbiota in humans: An open issue. Int. J. Obes. 2005 2016, 40, 1643–1648. [Google Scholar] [CrossRef]
- Udayappan, S.D.; Kovatcheva-Datchary, P.; Bakker, G.J.; Havik, S.R.; Herrema, H.; Cani, P.D.; Bouter, K.E.; Belzer, C.; Witjes, J.J.; Vrieze, A.; et al. Intestinal Ralstonia pickettii augments glucose intolerance in obesity. PLOS ONE 2017, 12, e0181693. [Google Scholar] [CrossRef] [Green Version]
- Pedicino, D.; Severino, A.; Ucci, S.; Bugli, F.; Flego, D.; Giglio, A.F.; Trotta, F.; Ruggio, A.; Lucci, C.; Iaconelli, A.; et al. Epicardial adipose tissue microbial colonization and inflammasome activation in acute coronary syndrome. Int. J. Cardiol. 2017, 236, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Jensen, B.A.H.; Varin, T.V.; Servant, F.; Blerk, S.V.; Richard, D.; Marceau, S.; Surette, M.; Biertho, L.; Lelouvier, B.; et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat. Metab. 2020, 2, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Massier, L.; Chakaroun, R.; Tabei, S.; Crane, A.; Didt, K.D.; Fallmann, J.; Von Bergen, M.; Haange, S.-B.; Heyne, H.O.; Stumvoll, M.; et al. Adipose Tissue Derived Bacteria are Associated with Inflammation in Obesity and Type 2 Diabetes. Gut. In press.
- Schierwagen, R.; Alvarez-Silva, C.; Servant, F.; Trebicka, J.; Lelouvier, B.; Arumugam, M. Trust is good, control is better: Technical considerations in blood microbiome analysis. Gut 2019. [Google Scholar] [CrossRef]
- Karstens, L.; Asquith, M.; Davin, S.; Fair, D.; Gregory, W.T.; Wolfe, A.J.; Braun, J.; McWeeney, S. Controlling for Contaminants in Low-Biomass 16S rRNA Gene Sequencing Experiments. mSystems 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Lauder, A.P.; Roche, A.M.; Sherrill-Mix, S.; Bailey, A.; Laughlin, A.L.; Bittinger, K.; Leite, R.; Elovitz, M.A.; Parry, S.; Bushman, F.D. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 2016, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Scarpellini, E.; Ianiro, G.; Attili, F.; Bassanelli, C.; De Santis, A.; Gasbarrini, A. The human gut microbiota and virome: Potential therapeutic implications. Dig. Liver Dis. 2015, 47, 1007–1012. [Google Scholar] [CrossRef] [Green Version]
- Nkamga, V.D.; Henrissat, B.; Drancourt, M. Archaea: Essential inhabitants of the human digestive microbiota. Hum. Microbiome J. 2017, 3, 1–8. [Google Scholar] [CrossRef]
- Manrique, P.; Dills, M.; Young, M.J. The Human Gut Phage Community and Its Implications for Health and Disease. Viruses 2017, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-Method Characterization of the Human Circulating Microbiome. Front. Microbiol. 2018, 9, 3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Permeability Marker | Tests | Direct/ Indirect | Sample Needed | Corresponding Literature |
|---|---|---|---|---|
| Functional tests | ||||
| Lactulose/Mannitol | Small intestinal permeability | direct | 24 h Urine | Bosi et al., 2006 [130] Teixeira et al., 2012 [90] Genser et al., 2018 [92] |
| lactulose/L-Rhamnoase | Small intestinal permeability | direct | 24 h Urine | Mooradian et al., 1986 [131] Wigg et al., 2001 [47] Wilbrink et al., 2019 [132] |
| Chrom-51-Ethylen diamine tetraacetic acid (51 Cr-EDTA) | Entire intestine permeability | direct | 24 h Urine | Horton et al., 2012 [133] |
| Circulating/fecal markers | ||||
| Zonulin | Tight junction dysfunction | indirect | Serum/Plasma/Feces | Wang et al., 2000 [127] Moreno-Navarrete et al., 2012 [134] Zak-Gołąb et al., 2013 [135] |
| Lipopolysaccharide (LPS) | Endotoxemia | indirect | Serum/Plasma | Cani et al., 2007 [136] Damms-Machado et al., 2017 [89] |
| Lipopolysaccharide Binding Protein (LBP) | Measurement via LPS Binding potential | indirect | Serum | Ruiz et al., 2007 [110] Ahmad et al., 2017 [94] Genser et al., 2018 [92] |
| Calprotectin | Gut inflammation | indirect | Serum Urin Plasma Feces | Ortega et al., 2012 [121] Pedersen et al., 2014 [137] |
| Endotoxin core antibodies | Endotoxemia | indirect | Plasma | Hawkesworth et al., 2013 [126] |
| intestinal fatty acid binding protein (iFABP) | Ischemia | indirect | Plasma/Serum | Cox et al., 2017 [109] |
| Ex Vivo | ||||
| Ussing chambers | Transepithelial electrical resistance (TEER) | direct | Intestinal biopsies | Genser et al., 2018 [92] |
| Reference | Study Population | Tissue | Detection Method | Findings | Limitations |
|---|---|---|---|---|---|
| Amar et al., 2011 [200] | 3280; 3149 without diabetes, 131 with incident diabetes | Blood | 16S rRNA gene concentration, pyrosequencing | 16S concentration slightly higher in diabetes (0.13 vs. 0.15, p = 0.04) Adj. OR of incident diabetes for 1 SD 16S: 1.35 [1.1–1.6], p = 0.002, Proteobacteria dominant phylum | Not matched for sex, age Group size with incident diabetes small, no negative controls reported, DNA was air-dried |
| Amar et al., 2013 [201] | 3936, with 3, 6, and 9 years follow-up (73 cardiovascular events) | Blood | 16S rRNA gene quantification | Concentration of Proteobacteria was positively correlated with onset of cardiovascular events (OR 1.56 [1.1–2.2], p = 0.007) | Quantification of all bacteria (Eubac) was lower compared with Proteobacteria (Probac), Tertiles not equally distributed, no negative controls reported, DNA was air-dried |
| Burcelin et al., 2013 [209] | Not reported, Patients grouped by body mass index (BMI) | Adipose tissue stromal vascular fraction | 16S rRNA gene pyrosequencing | Shift from Firmicutes to Proteobacteria with increasing BMI, Ralstonia was associated with BMI | Figure with previously unpublished data in Review article, no methods reported |
| Sato, Konazawa et al., 2014 [202] | 100, 50 with T2D, 50 control subjects | Blood, fecal samples | Targeted 16S rRNA gene amplification using Yakult Intestinal Flora-SCAN with group-, genus- and species-specific primers | Gut bacteria associated with T2D found in fecal samples (i.e., Lactobacillus) were detected at sig. Higher levels in blood of T2D subjects (28% vs. 4%, p < 0.01) | No sequencing data, bias due to selection of primers, no negative controls reported |
| Ortiz et al., 2014 [111] | 58 patients undergoing bariatric surgery and 3, 6, and 12 month follow-up | Blood | 16S rRNA gene quantification, LPS measurement (Limulus amoeboyte lysate (LAL)-test) | Translocation rate at baseline: 32.8% After follow-up: 13.8% (3 month), 1.8% (6 month), 5.2% (12 month) | Follow-up does not distinguish between surgery procedure (Roux-en-Y-gastric bypass (RYGB) or sleeve gastrectomy (SG)), no control group, no negative controls reported |
| Païssé et al., 2016 [203] | 30 healthy subjects | Whole blood, buffy coat, red blood cells, plasma | 16S rRNA gene quantification, and sequencing of V3-V4 region by MiSeq | Most blood bacteria located in buffy coat (93.7%), followed by red blood cells (6.2%) and plasma (0.1%) Dominant phyla are Proteobacteria (~80%), Actinobacteria, Firmicutes, Baceroidetes | Small cohort size, No negative controls reported |
| Lelouvier et al., 2016 [204] | Discovery cohort with 50 patients and validation cohort with 71 patients, all obese but with different stages of liver fibrosis | Blood | 16S rRNA gene quantification, and sequencing of V3-V4 region by MiSeq | Quantity of bacterial DNA increased in liver fibrosis, Actinobacteria decreased and Proteobacteria increased in liver fibrosis, Overall dominant phyla reported were Proteobacteria and Actinobacteria, Association between quantity and liver fibrosis but not bacterial taxa signature could be reproduced in validation cohort | 16S metagenomic sequencing of stool was performed using different region (V1–V3), and sequencing platform (454 FLX), no negative controls reported, tissue in cohorts differed (buffy coat vs. whole blood) + large differences in quantification (652.6 vs. 3.1 copies/µL) |
| Pedicino et al., 2017 [213] | 18 with acute coronary syndrome (ACS), 16 with stable angina (SA), and 13 controls from patients undergoing mitral insufficiency | Epicardial adipose tissue | 16S rRNA gene amplification (V1–V3) and sequencing (n = 3 per group) on GS junior platform | Predominant species in ACS: Cyanobacteria Streptophyta and Proteobacteria Rickettsiale, in SA Proteobacteria Moracellaceae and Pseudomonas | No technical negative controls, only few samples sequenced |
| Udayappanet al., 2017 [212] | 12 patients | Mesenteric-visceral adipose tissue | Denaturing gradient gel electrophoresis and Sanger sequencing | Bacteria were found in mesenteric tissue, Actinobacteria are dominant Gram-positive and Ralstonia Gram- negative bacteria. Fecal R. picetti increased in T2D | Small sample size, non-state-of-the-art method introduces bias in reported bacteria (cloning and Sanger sequencing instead of next-generation amplicon sequencing) |
| Schierwagen et al., 2018 [205] | 7 patients with decompensated liver cirrhosis | Central, hepatic, peripheral, and portal venous blood (buffy coat) | 16S rRNA sequencing | 4 Phyla reported, dominated by proteobacteria and Actinobacteria, composition did not differ between compartments, Pelomonas, Rahnella among other genera correlated positively with inflammatory markers, Esherchica and Salmonella negatively | Limited methods reported due to format (Letter), small sample size, no control group |
| Anhê, Jensen et al., 2020 [214] | 40 patients with obesity (20 without T2D, 20 with T2D) | Liver, blood, adipose tissue | 16S rRNA quantification and sequencing (V3-4) | Bacterial DNA is present in adipose tissue and liver, Highest amounts were observed in liver an omental adipose tissue, diversity was highest in mesenteric adipose tissue, dominant phyla were Proteobacteria and Firmicutes | Although a strong point is negative controls, it becomes not clear how they were analyzed, clinical data is reported but not included in analysis |
| Massier, Chakaroun et al., 2020 [215] | 75 patients with obesity (33 with T2D, 42 without T2D) | Omental, mesenteric, subcutaneous adipose tissue, blood | 16S rRNA quantification and sequencing (V4-5) catalyzed reporter deposition - fluorescence in situ hybridization (CARD-FISH) bacterial DNA challenge in immortalized human preadipocytes | Bacterial DNA is present in all tested adipose tissue depots as well we blood, with dissimilarities between tissues being influenced by overall host inflammation and insulin resistance. Highest amounts of bacterial DNA were detected in the blood. Bacterial quantity was associated with macrophages infiltration and expression of inflammatory markers in adipose tissue. Living bacterial cells were detected in adipose tissue via CARD-FISH. | No inclusion of lean subjects |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakaroun, R.M.; Massier, L.; Kovacs, P. Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: Perpetrators or Bystanders? Nutrients 2020, 12, 1082. https://doi.org/10.3390/nu12041082
Chakaroun RM, Massier L, Kovacs P. Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: Perpetrators or Bystanders? Nutrients. 2020; 12(4):1082. https://doi.org/10.3390/nu12041082
Chicago/Turabian StyleChakaroun, Rima M., Lucas Massier, and Peter Kovacs. 2020. "Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: Perpetrators or Bystanders?" Nutrients 12, no. 4: 1082. https://doi.org/10.3390/nu12041082