
Structural and Functional Changes and Possible Molecular Mechanisms in Aged Skin



Abstract
:1. Introduction
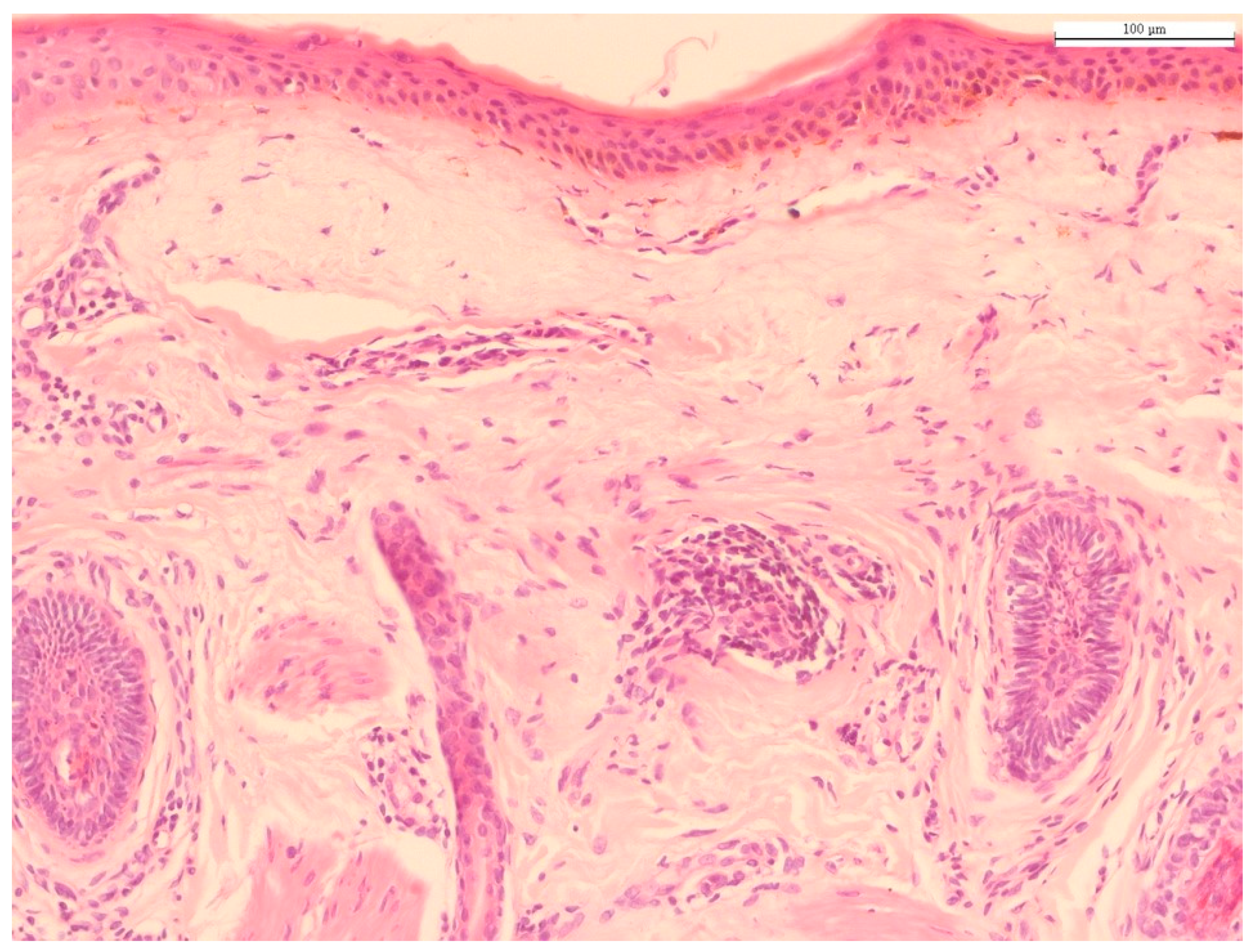
2. Histology of Chronological Aging and Photoaging
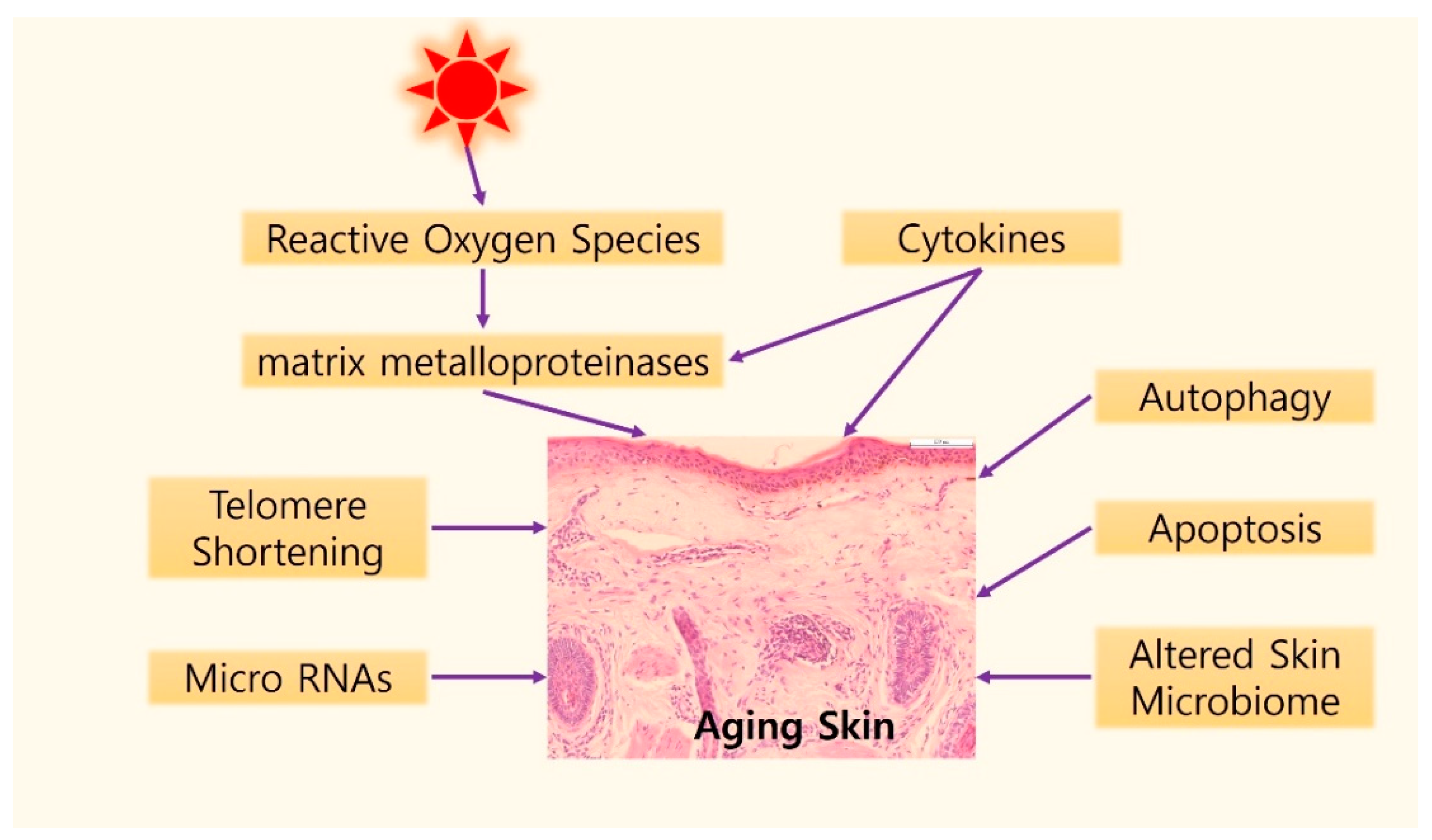
3. Molecular Mechanisms of Skin Aging
3.1. Telomere Shortening
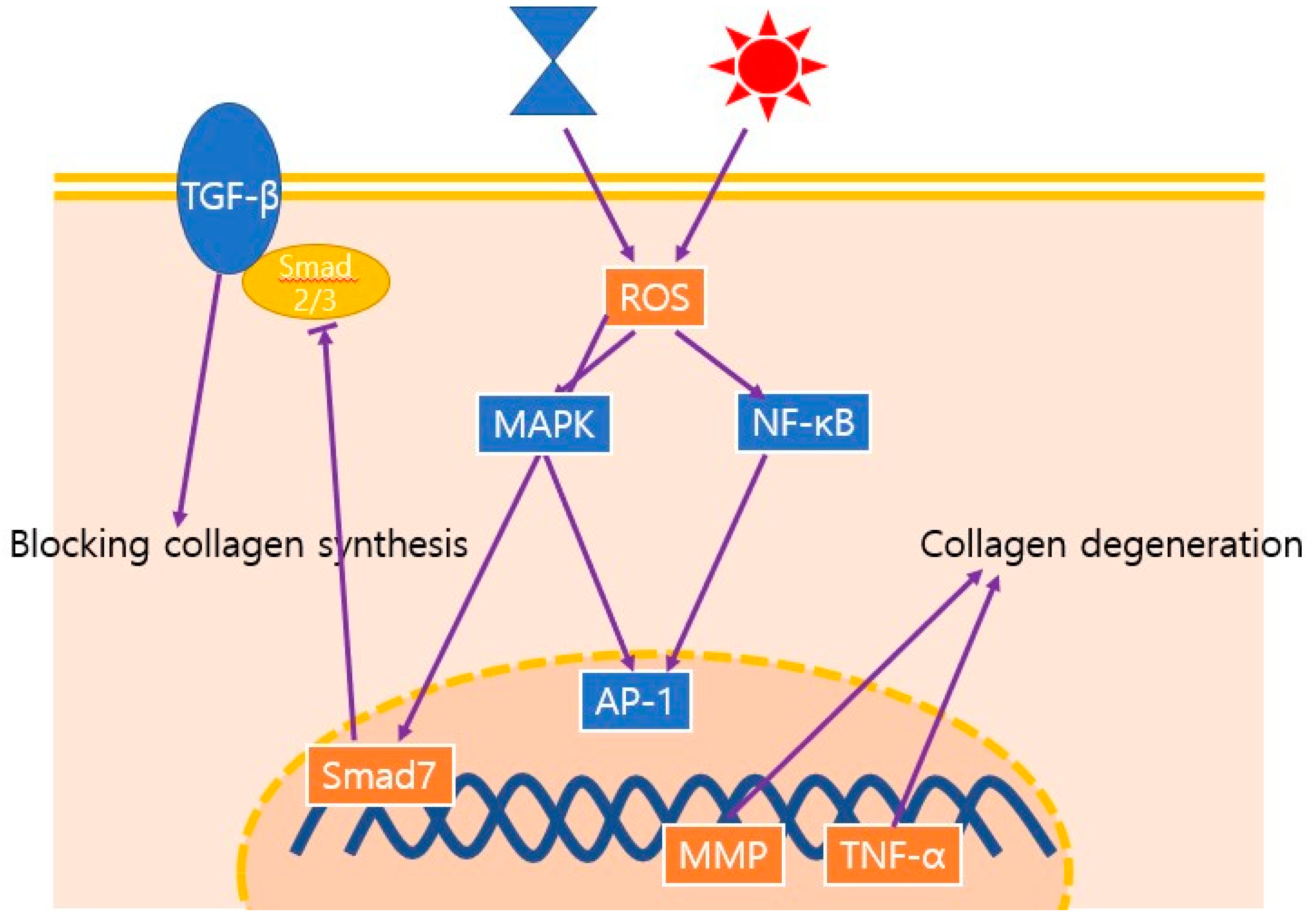
3.2. Oxidative Stress and MMPs
3.3. Cytokines in Aging Skin
3.4. Autophagic Control
3.5. Apoptosis in Skin Aging
3.6. Role of microRNAs in Skin Aging
3.7. Skin Microbiome
4. Other Environmental Stressors Associated with Skin Aging
4.1. Tobacco Smoke
4.2. Environmental Pollutants
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin barrier immunity and ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Koohgoli, R.; Hudson, L.; Naidoo, K.; Wilkinson, S.; Chavan, B.; Birch-Machin, M.A. Bad air gets under your skin. Exp. Dermatol. 2017, 26, 384–387. [Google Scholar] [CrossRef]
- Rittié, L.; Fisher, G.J. Natural and sun-induced aging of human skin. Cold Spring Harb. Perspect. Med. 2015, 5, a015370. [Google Scholar] [CrossRef] [PubMed]
- Laga, A.C.; Murphy, G.F. The Translational Basis of Human Cutaneous Photoaging: On Models, Methods, and Meaning. Am. J. Pathol. 2009, 174, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Stanley, J.R. Synergy of understanding dermatologic disease and epidermal biology. J. Clin. Investig. 2012, 122, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Biniek, K.; Kaczvinsky, J.; Matts, P.; Dauskardt, R.H. Understanding age-induced alterations to the biomechanical barrier function of human stratum corneum. J. Dermatol. Sci. 2015, 80, 94–101. [Google Scholar] [CrossRef]
- Choe, C.; Schleusener, J.; Lademann, J.; Darvin, M.E. Age related depth profiles of human Stratum Corneum barrier-related molecular parameters by confocal Raman microscopy in vivo. Mech. Ageing Dev. 2018, 172, 6–12. [Google Scholar] [CrossRef]
- Lavker, R.M. Structural alterations in exposed and unexposed aged skin. J. Investig. Dermatol. 1979, 73, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilchrest, B.A.; Blog, F.B.; Szabo, G. Effects of aging and chronic sun exposure on melanocytes in human skin. J. Investig. Dermatol. 1979, 73, 141–143. [Google Scholar] [CrossRef] [Green Version]
- Russell-Goldman, E.; Murphy, G.F. The Pathobiology of Skin Aging: New Insights into an Old Dilemma. Am. J. Pathol. 2020, 190, 1356–1369. [Google Scholar] [CrossRef]
- Sauermann, K.; Clemann, S.; Jaspers, S.; Gambichler, T.; Altmeyer, P.; Hoffmann, K.; Ennen, J. Age related changes of human skin investigated with histometric measurements by confocal laser scanning microscopy in vivo. Skin Res. Technol. 2002, 8, 52–56. [Google Scholar] [CrossRef]
- Wlaschek, M.; Maity, P.; Makrantonaki, E.; Scharffetter-Kochanek, K. Connective Tissue and Fibroblast Senescence in Skin Aging. J. Investig. Dermatol. 2021, 141, 985–992. [Google Scholar] [CrossRef]
- Ghosh, K.; Capell, B.C. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef] [Green Version]
- Varani, J.; Spearman, D.; Perone, P.; Fligiel, S.E.; Datta, S.C.; Wang, Z.Q.; Shao, Y.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Inhibition of type I procollagen synthesis by damaged collagen in photoaged skin and by collagenase-degraded collagen in vitro. Am. J. Pathol. 2001, 158, 931–942. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.J.; Quan, T.; Purohit, T.; Shao, Y.; Cho, M.K.; He, T.; Varani, J.; Kang, S.; Voorhees, J.J. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am. J. Pathol. 2009, 174, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varani, J.; Dame, M.K.; Rittie, L.; Fligiel, S.E.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Decreased collagen production in chronologically aged skin: Roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am. J. Pathol. 2006, 168, 1861–1868. [Google Scholar] [CrossRef] [Green Version]
- Makrantonaki, E.; Zouboulis, C.C. The skin as a mirror of the aging process in the human organism-state of the art and results of the aging research in the German National Genome Research Network 2 (NGFN-2). Exp. Gerontol. 2007, 42, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.H.; Eun, H.C. Angiogenesis in skin aging and photoaging. J. Dermatol. 2007, 34, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, H.; Davis, E.C.; Starcher, B.C.; Ouchi, T.; Yanagisawa, M.; Richardson, J.A.; Olson, E.N. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 2002, 415, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, R.; McGovern, A.; Ridley, C.; Cain, S.A.; Baldwin, A.; Wang, M.C.; Guo, C.; Mironov, A., Jr.; Drymoussi, Z.; Trump, D.; et al. Differential regulation of elastic fiber formation by fibulin-4 and -5. J. Biol. Chem. 2009, 284, 24553–24567. [Google Scholar] [CrossRef] [Green Version]
- Amano, S. Characterization and mechanisms of photoageing-related changes in skin. Damages of basement membrane and dermal structures. Exp. Dermatol. 2016, 25 (Suppl. S3), 14–19. [Google Scholar] [CrossRef] [PubMed]
- Langton, A.K.; Sherratt, M.J.; Griffiths, C.E.; Watson, R.E. Differential expression of elastic fibre components in intrinsically aged skin. Biogerontology 2012, 13, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Kwon, S.H.; Choi, J.Y.; Na, J.I.; Huh, C.H.; Choi, H.R.; Park, K.C. Molecular Mechanisms of Dermal Aging and Antiaging Approaches. Int. J. Mol. Sci. 2019, 20, 2126. [Google Scholar] [CrossRef] [Green Version]
- Kurban, R.S.; Bhawan, J. Histologic changes in skin associated with aging. J. Dermatol. Surg. Oncol. 1990, 16, 908–914. [Google Scholar] [CrossRef]
- Bhawan, J.; Andersen, W.; Lee, J.; Labadie, R.; Solares, G. Photoaging versus intrinsic aging: A morphologic assessment of facial skin. J. Cutan. Pathol. 1995, 22, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Han, A.; Chien, A.L.; Kang, S. Photoaging. Dermatol. Clin. 2014, 32, 291–299. [Google Scholar] [CrossRef]
- Eller, M.S.; Yaar, M.; Gilchrest, B.A. DNA damage and melanogenesis. Nature 1994, 372, 413–414. [Google Scholar] [CrossRef]
- Kaidbey, K.H.; Agin, P.P.; Sayre, R.M.; Kligman, A.M. Photoprotection by melanin—A comparison of black and Caucasian skin. J. Am. Acad. Dermatol. 1979, 1, 249–260. [Google Scholar] [CrossRef]
- Tsuji, T. Loss of dermal elastic tissue in solar elastosis. Arch. Dermatol. 1980, 116, 474–475. [Google Scholar] [CrossRef]
- Sellheyer, K. Pathogenesis of solar elastosis: Synthesis or degradation? J. Cutan. Pathol. 2003, 30, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Naylor, E.C.; Watson, R.E.; Sherratt, M.J. Molecular aspects of skin ageing. Maturitas 2011, 69, 249–256. [Google Scholar] [CrossRef]
- Fisher, G.J.; Wang, Z.Q.; Datta, S.C.; Varani, J.; Kang, S.; Voorhees, J.J. Pathophysiology of premature skin aging induced by ultraviolet light. N. Engl. J. Med. 1997, 337, 1419–1428. [Google Scholar] [CrossRef]
- Chakraborti, S.; Mandal, M.; Das, S.; Mandal, A.; Chakraborti, T. Regulation of matrix metalloproteinases: An overview. Mol. Cell. Biochem. 2003, 253, 269–285. [Google Scholar] [CrossRef]
- Ryu, J.; Park, S.J.; Kim, I.H.; Choi, Y.H.; Nam, T.J. Protective effect of porphyra-334 on UVA-induced photoaging in human skin fibroblasts. Int. J. Mol. Med. 2014, 34, 796–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.H.; Seo, J.Y.; Lee, M.K.; Eun, H.C.; Lee, J.H.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Ultraviolet modulation of human macrophage metalloelastase in human skin in vivo. J. Investig. Dermatol. 2002, 119, 507–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imokawa, G.; Ishida, K. Biological mechanisms underlying the ultraviolet radiation-induced formation of skin wrinkling and sagging I: Reduced skin elasticity, highly associated with enhanced dermal elastase activity, triggers wrinkling and sagging. Int. J. Mol. Sci. 2015, 16, 7753–7775. [Google Scholar] [CrossRef] [Green Version]
- Cenizo, V.; André, V.; Reymermier, C.; Sommer, P.; Damour, O.; Perrier, E. LOXL as a target to increase the elastin content in adult skin: A dill extract induces the LOXL gene expression. Exp. Dermatol. 2006, 15, 574–581. [Google Scholar] [CrossRef]
- Weihermann, A.C.; Lorencini, M.; Brohem, C.A.; de Carvalho, C.M. Elastin structure and its involvement in skin photoageing. Int. J. Cosmet. Sci. 2017, 39, 241–247. [Google Scholar] [CrossRef]
- Schwartz, E.; Feinberg, E.; Lebwohl, M.; Mariani, T.J.; Boyd, C.D. Ultraviolet radiation increases tropoelastin accumulation by a post-transcriptional mechanism in dermal fibroblasts. J. Investig. Dermatol. 1995, 105, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, T. Photoimmunosuppression. Photodermatol. Photoimmunol. Photomed. 2002, 18, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Bosset, S.; Bonnet-Duquennoy, M.; Barré, P.; Chalon, A.; Kurfurst, R.; Bonté, F.; Schnébert, S.; Le Varlet, B.; Nicolas, J.F. Photoageing shows histological features of chronic skin inflammation without clinical and molecular abnormalities. Br. J. Dermatol. 2003, 149, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Warner, R.L.; Gharaee-Kermani, M.; Phan, S.H.; Kang, S.; Chung, J.H.; Wang, Z.Q.; Datta, S.C.; Fisher, G.J.; Voorhees, J.J. Vitamin A antagonizes decreased cell growth and elevated collagen-degrading matrix metalloproteinases and stimulates collagen accumulation in naturally aged human skin. J. Investig. Dermatol. 2000, 114, 480–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zouboulis Ch, C. Intrinsic skin aging. A critical appraisal of the role of hormones. Der Hautarzt Z. Fur Dermatol. Venerol. Und Verwandte Geb. 2003, 54, 825–832. [Google Scholar]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef] [Green Version]
- D’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Klingelhutz, A.J. The role of telomeres in the ageing of human skin. Exp. Dermatol. 2011, 20, 297–302. [Google Scholar] [CrossRef]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, S.; Tada-Oikawa, S.; Kawanishi, S. Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry 2001, 40, 4763–4768. [Google Scholar] [CrossRef]
- Birch, J.; Barnes, P.J.; Passos, J.F. Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease. Pharmacol. Ther. 2018, 183, 34–49. [Google Scholar] [CrossRef] [Green Version]
- Krutmann, J.; Schroeder, P. Role of mitochondria in photoaging of human skin: The defective powerhouse model. J. Investig. Dermatol. Symp. Proc. 2009, 14, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, M.; Yamashita, R.; Ueda, M. Telomere length of the skin in association with chronological aging and photoaging. J. Dermatol. Sci. 2006, 43, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Poljšak, B.; Dahmane, R.G.; Godić, A. Intrinsic skin aging: The role of oxidative stress. Acta Dermatovenerol. Alp. Pannonica Adriat. 2012, 21, 33–36. [Google Scholar]
- Sajo, M.E.J.; Kim, C.S.; Kim, S.K.; Shim, K.Y.; Kang, T.Y.; Lee, K.J. Antioxidant and Anti-Inflammatory Effects of Shungite against Ultraviolet B Irradiation-Induced Skin Damage in Hairless Mice. Oxidative Med. Cell. Longev. 2017, 2017, 7340143. [Google Scholar] [CrossRef] [Green Version]
- Pandel, R.; Poljšak, B.; Godic, A.; Dahmane, R. Skin photoaging and the role of antioxidants in its prevention. ISRN Dermatol. 2013, 2013, 930164. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Han, J.; Jiang, C.; Zhang, Y. Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res. Rev. 2020, 59, 101036. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative stress in aging human skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Desprez, P.Y.; Campisi, J.; Velarde, M.C. Cell Autonomous and Non-Autonomous Effects of Senescent Cells in the Skin. J. Investig. Dermatol. 2015, 135, 1722–1726. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.J.; Talwar, H.S.; Lin, J.; Lin, P.; McPhillips, F.; Wang, Z.; Li, X.; Wan, Y.; Kang, S.; Voorhees, J.J. Retinoic acid inhibits induction of c-Jun protein by ultraviolet radiation that occurs subsequent to activation of mitogen-activated protein kinase pathways in human skin in vivo. J. Clin. Investig. 1998, 101, 1432–1440. [Google Scholar] [CrossRef]
- Fligiel, S.E.; Varani, J.; Datta, S.C.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Collagen degradation in aged/photodamaged skin in vivo and after exposure to matrix metalloproteinase-1 in vitro. J. Investig. Dermatol. 2003, 120, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.R.; Fingleton, B.; Rothenberg, M.L.; Matrisian, L.M. Matrix metalloproteinases: Biologic activity and clinical implications. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2000, 18, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, T.; He, T.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation induces Smad7 via induction of transcription factor AP-1 in human skin fibroblasts. J. Biol. Chem. 2005, 280, 8079–8085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipkiss, A.R. Accumulation of altered proteins and ageing: Causes and effects. Exp. Gerontol. 2006, 41, 464–473. [Google Scholar] [CrossRef]
- Borg, M.; Brincat, S.; Camilleri, G.; Schembri-Wismayer, P.; Brincat, M.; Calleja-Agius, J. The role of cytokines in skin aging. Climacteric J. Int. Menopause Soc. 2013, 16, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Ansary, T.M.; Hossain, M.R.; Kamiya, K.; Komine, M.; Ohtsuki, M. Inflammatory Molecules Associated with Ultraviolet Radiation-Mediated Skin Aging. Int. J. Mol. Sci. 2021, 22, 3974. [Google Scholar] [CrossRef] [PubMed]
- Ansel, J.; Perry, P.; Brown, J.; Damm, D.; Phan, T.; Hart, C.; Luger, T.; Hefeneider, S. Cytokine modulation of keratinocyte cytokines. J. Investig. Dermatol. 1990, 94, 101s–107s. [Google Scholar] [CrossRef] [Green Version]
- Pilkington, S.M.; Bulfone-Paus, S.; Griffiths, C.E.M.; Watson, R.E.B. Inflammaging and the Skin. J. Investig. Dermatol. 2021, 141, 1087–1095. [Google Scholar] [CrossRef]
- Youn, U.J.; Nam, K.W.; Kim, H.S.; Choi, G.; Jeong, W.S.; Lee, M.Y.; Chae, S. 3-Deoxysappanchalcone inhibits tumor necrosis factor-α-induced matrix metalloproteinase-9 expression in human keratinocytes through activated protein-1 inhibition and nuclear factor-kappa B DNA binding activity. Biol. Pharm. Bull. 2011, 34, 890–893. [Google Scholar] [CrossRef] [Green Version]
- Chou, D.H.; Lee, W.; McCulloch, C.A. TNF-alpha inactivation of collagen receptors: Implications for fibroblast function and fibrosis. J. Immunol. 1996, 156, 4354–4362. [Google Scholar]
- Crawford, H.C.; Matrisian, L.M. Mechanisms controlling the transcription of matrix metalloproteinase genes in normal and neoplastic cells. Enzym. Protein 1996, 49, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Hirao, T.; Aoki, H.; Yoshida, T.; Sato, Y.; Kamoda, H. Elevation of interleukin 1 receptor antagonist in the stratum corneum of sun-exposed and ultraviolet B-irradiated human skin. J. Investig. Dermatol. 1996, 106, 1102–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, L.A.; Raizner, K.; Wlaschek, M.; Brenneisen, P.; Gethöffer, K.; Scharffetter-Kochanek, K. UVA-1 exposure in vivo leads to an IL-6 surge within the skin. Exp. Dermatol. 2017, 26, 830–832. [Google Scholar] [CrossRef] [Green Version]
- Wlaschek, M.; Heinen, G.; Poswig, A.; Schwarz, A.; Krieg, T.; Scharffetter-Kochanek, K. UVA-induced autocrine stimulation of fibroblast-derived collagenase/MMP-1 by interrelated loops of interleukin-1 and interleukin-6. Photochem. Photobiol. 1994, 59, 550–556. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Cho, D.H.; Park, H.J. IL-18 and Cutaneous Inflammatory Diseases. Int. J. Mol. Sci. 2015, 16, 29357–29369. [Google Scholar] [CrossRef] [Green Version]
- Cohen, H.J.; Pieper, C.F.; Harris, T.; Rao, K.M.; Currie, M.S. The association of plasma IL-6 levels with functional disability in community-dwelling elderly. J. Gerontol. Ser. A Biol. Sci. Med Sci. 1997, 52, M201–M208. [Google Scholar] [CrossRef] [Green Version]
- Beyer, I.; Mets, T.; Bautmans, I. Chronic low-grade inflammation and age-related sarcopenia. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Fagot, D.; Asselineau, D.; Bernerd, F. Direct role of human dermal fibroblasts and indirect participation of epidermal keratinocytes in MMP-1 production after UV-B irradiation. Arch. Dermatol. Res. 2002, 293, 576–583. [Google Scholar] [CrossRef]
- Omoigui, S. The Interleukin-6 inflammation pathway from cholesterol to aging-role of statins, bisphosphonates and plant polyphenols in aging and age-related diseases. Immun. Ageing I A 2007, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Robichaud, P.; He, T.; Fisher, G.J.; Voorhees, J.J.; Quan, T. Oxidant exposure induces cysteine-rich protein 61 (CCN1) via c-Jun/AP-1 to reduce collagen expression in human dermal fibroblasts. PLoS ONE 2014, 9, e115402. [Google Scholar] [CrossRef] [Green Version]
- Quan, T.; Qin, Z.; Robichaud, P.; Voorhees, J.J.; Fisher, G.J. CCN1 contributes to skin connective tissue aging by inducing age-associated secretory phenotype in human skin dermal fibroblasts. J. Cell Commun. Signal. 2011, 5, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, T.; He, T.; Shao, Y.; Lin, L.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Elevated cysteine-rich 61 mediates aberrant collagen homeostasis in chronologically aged and photoaged human skin. Am. J. Pathol. 2006, 169, 482–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, M.A.; Quan, T.; Voorhees, J.J.; Fisher, G.J. Extracellular matrix regulation of fibroblast function: Redefining our perspective on skin aging. J. Cell Commun. Signal. 2018, 12, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dancourt, J.; Melia, T.J. Lipidation of the autophagy proteins LC3 and GABARAP is a membrane-curvature dependent process. Autophagy 2014, 10, 1470–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharaziha, P.; Panaretakis, T. Dynamics of Atg5-Atg12-Atg16L1 Aggregation and Deaggregation. Methods Enzymol. 2017, 587, 247–255. [Google Scholar]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Devkota, S. The autophagy process. Oncotarget 2017, 8, 18623. [Google Scholar] [CrossRef]
- Yamano, K.; Matsuda, N.; Tanaka, K. The ubiquitin signal and autophagy: An orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016, 17, 300–316. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pride, H.; Yu, Z.; Sunchu, B.; Mochnick, J.; Coles, A.; Zhang, Y.; Buffenstein, R.; Hornsby, P.J.; Austad, S.N.; Pérez, V.I. Long-lived species have improved proteostasis compared to phylogenetically-related shorter-lived species. Biochem. Biophys. Res. Commun. 2015, 457, 669–675. [Google Scholar] [CrossRef]
- Fujii, S.; Hara, H.; Araya, J.; Takasaka, N.; Kojima, J.; Ito, S.; Minagawa, S.; Yumino, Y.; Ishikawa, T.; Numata, T.; et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 2012, 1, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Gerland, L.M.; Peyrol, S.; Lallemand, C.; Branche, R.; Magaud, J.P.; Ffrench, M. Association of increased autophagic inclusions labeled for beta-galactosidase with fibroblastic aging. Exp. Gerontol. 2003, 38, 887–895. [Google Scholar] [CrossRef]
- Gosselin, K.; Deruy, E.; Martien, S.; Vercamer, C.; Bouali, F.; Dujardin, T.; Slomianny, C.; Houel-Renault, L.; Chelli, F.; De Launoit, Y.; et al. Senescent keratinocytes die by autophagic programmed cell death. Am. J. Pathol. 2009, 174, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Narzt, M.S.; Nagelreiter, I.M.; Hohensinner, P.; Terlecki-Zaniewicz, L.; Tschachler, E.; Grillari, J.; Gruber, F. Autophagy deficient keratinocytes display increased DNA damage, senescence and aberrant lipid composition after oxidative stress in vitro and in vivo. Redox Biol. 2017, 11, 219–230. [Google Scholar] [CrossRef]
- Gewirtz, D.A. Autophagy and senescence: A partnership in search of definition. Autophagy 2013, 9, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 2011, 6, e23367. [Google Scholar] [CrossRef] [Green Version]
- Cavinato, M.; Jansen-Dürr, P. Molecular mechanisms of UVB-induced senescence of dermal fibroblasts and its relevance for photoaging of the human skin. Exp. Gerontol. 2017, 94, 78–82. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kaarniranta, K. Apoptosis and aging: Increased resistance to apoptosis enhances the aging process. Cell. Mol. Life Sci. CMLS 2011, 68, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.L.; Holcakova, J.; Finlan, L.E.; Nekulova, M.; Hrstka, R.; Gueven, N.; DiRenzo, J.; Smith, G.; Hupp, T.R.; Vojtesek, B. DeltaNp63 transcriptionally regulates ATM to control p53 Serine-15 phosphorylation. Mol. Cancer 2010, 9, 195. [Google Scholar] [CrossRef] [Green Version]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K. ER stress and hormetic regulation of the aging process. Ageing Res. Rev. 2010, 9, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; Teresky, A.K.; Hernando, E.; Cordon-Cardo, C.; Levine, A.J. Declining p53 function in the aging process: A possible mechanism for the increased tumor incidence in older populations. Proc. Natl. Acad. Sci. USA 2007, 104, 16633–16638. [Google Scholar] [CrossRef] [Green Version]
- Keyes, W.M.; Wu, Y.; Vogel, H.; Guo, X.; Lowe, S.W.; Mills, A.A. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005, 19, 1986–1999. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K. Glycolysis links p53 function with NF-kappaB signaling: Impact on cancer and aging process. J. Cell. Physiol. 2010, 224, 1–6. [Google Scholar]
- Kinser, H.E.; Pincus, Z. MicroRNAs as modulators of longevity and the aging process. Hum. Genet. 2020, 139, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Grillari, J.; Hackl, M.; Grillari-Voglauer, R. miR-17-92 cluster: Ups and downs in cancer and aging. Biogerontology 2010, 11, 501–506. [Google Scholar] [CrossRef] [Green Version]
- Ratzinger, S.; Grässel, S.; Dowejko, A.; Reichert, T.E.; Bauer, R.J. Induction of type XVI collagen expression facilitates proliferation of oral cancer cells. Matrix Biol. J. Int. Soc. Matrix Biol. 2011, 30, 118–125. [Google Scholar] [CrossRef]
- Shin, K.H.; Pucar, A.; Kim, R.H.; Bae, S.D.; Chen, W.; Kang, M.K.; Park, N.H. Identification of senescence-inducing microRNAs in normal human keratinocytes. Int. J. Oncol. 2011, 39, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Mancini, M.; Lena, A.M.; Saintigny, G.; Mahé, C.; Di Daniele, N.; Melino, G.; Candi, E. MicroRNAs in human skin ageing. Ageing Res. Rev. 2014, 17, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Vartholomatos, G.; Tzavaras, T.; Hatziapostolou, M.; Fackelmayer, F.O.; Sandaltzopoulos, R.; Polytarchou, C.; Kolettas, E. Senescence-associated microRNAs target cell cycle regulatory genes in normal human lung fibroblasts. Exp. Gerontol. 2017, 96, 110–122. [Google Scholar] [CrossRef]
- Martinez, I.; Cazalla, D.; Almstead, L.L.; Steitz, J.A.; DiMaio, D. miR-29 and miR-30 regulate B-Myb expression during cellular senescence. Proc. Natl. Acad. Sci. USA 2011, 108, 522–527. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Chen, H.; Gao, J.; Liu, Y.; Li, J.; Wang, J. Bioactive Ingredients in Chinese Herbal Medicines That Target Non-coding RNAs: Promising New Choices for Disease Treatment. Front. Pharmacol. 2019, 10, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.P.; Qi, R.Q.; Chen, W.; Shi, Y.; Cui, Z.Z.; Gao, X.H.; Chen, H.D.; Zhou, L.; Mi, Q.S. Aging affects epidermal Langerhans cell development and function and alters their miRNA gene expression profile. Aging 2012, 4, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, C.; Yang, S.; Guo, Z. MiRNA-27a decreases ultraviolet B irradiation-induced cell damage. J. Cell. Biochem. 2020, 121, 1032–1038. [Google Scholar] [CrossRef]
- Li, W.; Wu, Y.F.; Xu, R.H.; Lu, H.; Hu, C.; Qian, H. miR-1246 releases RTKN2-dependent resistance to UVB-induced apoptosis in HaCaT cells. Mol. Cell. Biochem. 2014, 394, 299–306. [Google Scholar] [CrossRef]
- Srivastava, A.; Karlsson, M.; Marionnet, C.; Bernerd, F.; Gueniche, A.; Rawadi, C.E.L.; Ståhle, M.; Sonkoly, E.; Breton, L.; Pivarcsi, A. Identification of chronological and photoageing-associated microRNAs in human skin. Sci. Rep. 2018, 8, 12990. [Google Scholar] [CrossRef]
- Song, J.; Liu, P.; Yang, Z.; Li, L.; Su, H.; Lu, N.; Peng, Z. MiR-155 negatively regulates c-Jun expression at the post-transcriptional level in human dermal fibroblasts in vitro: Implications in UVA irradiation-induced photoaging. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2012, 29, 331–340. [Google Scholar] [CrossRef]
- Chen, Y.E.; Tsao, H. The skin microbiome: Current perspectives and future challenges. J. Am. Acad. Dermatol. 2013, 69, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Prescott, S.L.; Larcombe, D.L.; Logan, A.C.; West, C.; Burks, W.; Caraballo, L.; Levin, M.; Etten, E.V.; Horwitz, P.; Kozyrskyj, A.; et al. The skin microbiome: Impact of modern environments on skin ecology, barrier integrity, and systemic immune programming. World Allergy Organ. J. 2017, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bai, X.; Peng, T.; Yi, X.; Luo, L.; Yang, J.; Liu, J.; Wang, Y.; He, T.; Wang, X.; et al. New Insights Into the Skin Microbial Communities and Skin Aging. Front. Microbiol. 2020, 11, 565549. [Google Scholar] [CrossRef]
- Dréno, B.; Araviiskaia, E.; Berardesca, E.; Gontijo, G.; Sanchez Viera, M.; Xiang, L.F.; Martin, R.; Bieber, T. Microbiome in healthy skin, update for dermatologists. J. Eur. Acad. Dermatol. Venereol. JEADV 2016, 30, 2038–2047. [Google Scholar] [CrossRef]
- Shibagaki, N.; Suda, W.; Clavaud, C.; Bastien, P.; Takayasu, L.; Iioka, E.; Kurokawa, R.; Yamashita, N.; Hattori, Y.; Shindo, C.; et al. Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Sci. Rep. 2017, 7, 10567. [Google Scholar] [CrossRef] [PubMed]
- Wilantho, A.; Deekaew, P.; Srisuttiyakorn, C.; Tongsima, S.; Somboonna, N. Diversity of bacterial communities on the facial skin of different age-group Thai males. PeerJ 2017, 5, e4084. [Google Scholar] [CrossRef] [Green Version]
- Jugé, R.; Rouaud-Tinguely, P.; Breugnot, J.; Servaes, K.; Grimaldi, C.; Roth, M.P.; Coppin, H.; Closs, B. Shift in skin microbiota of Western European women across aging. J. Appl. Microbiol. 2018, 125, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Morita, A.; Torii, K.; Maeda, A.; Yamaguchi, Y. Molecular basis of tobacco smoke-induced premature skin aging. J. Investig. Dermatol. Symp. Proc. 2009, 14, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.; Maity, P.; Burkovski, A.; Schwab, J.; Müssel, C.; Singh, K.; Ferreira, F.F.; Krug, L.; Maier, H.J.; Wlaschek, M.; et al. A model of the onset of the senescence associated secretory phenotype after DNA damage induced senescence. PLoS Comput. Biol. 2017, 13, e1005741. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.C.; Young, D.A.; Waters, J.G.; Rowan, A.D.; Chantry, A.; Edwards, D.R.; Clark, I.M. The comparative role of activator protein 1 and Smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-beta 1. J. Biol. Chem. 2003, 278, 10304–10313. [Google Scholar] [CrossRef] [Green Version]
- Kim, O.Y.; Chae, J.S.; Paik, J.K.; Seo, H.S.; Jang, Y.; Cavaillon, J.M.; Lee, J.H. Effects of aging and menopause on serum interleukin-6 levels and peripheral blood mononuclear cell cytokine production in healthy nonobese women. Age 2012, 34, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Rivetti di Val Cervo, P.; Lena, A.M.; Nicoloso, M.; Rossi, S.; Mancini, M.; Zhou, H.; Saintigny, G.; Dellambra, E.; Odorisio, T.; Mahé, C.; et al. p63-microRNA feedback in keratinocyte senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Lahmann, C.; Bergemann, J.; Harrison, G.; Young, A.R. Matrix metalloproteinase-1 and skin ageing in smokers. Lancet 2001, 357, 935–936. [Google Scholar] [CrossRef]
- Yin, L.; Morita, A.; Tsuji, T. Alterations of extracellular matrix induced by tobacco smoke extract. Arch. Dermatol. Res. 2000, 292, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Morita, A.; Tsuji, T. Tobacco smoke extract induces age-related changes due to modulation of TGF-beta. Exp. Dermatol. 2003, 12 (Suppl. S2), 51–56. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Torii, K.; Fritsche, E.; Shintani, Y.; Nishida, E.; Nakamura, M.; Shirakata, Y.; Haarmann-Stemmann, T.; Abel, J.; Krutmann, J.; et al. Role of the aryl hydrocarbon receptor in tobacco smoke extract-induced matrix metalloproteinase-1 expression. Exp. Dermatol. 2013, 22, 349–353. [Google Scholar] [CrossRef]
- Schikowski, T.; Hüls, A. Air Pollution and Skin Aging. Curr. Environ. Health Rep. 2020, 7, 58–64. [Google Scholar] [CrossRef]
- Vierkötter, A.; Schikowski, T.; Ranft, U.; Sugiri, D.; Matsui, M.; Krämer, U.; Krutmann, J. Airborne particle exposure and extrinsic skin aging. J. Investig. Dermatol. 2010, 130, 2719–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.A.; Villano, C.M.; Dorn, R.; White, L.A. Interaction between the aryl hydrocarbon receptor and retinoic acid pathways increases matrix metalloproteinase-1 expression in keratinocytes. J. Biol. Chem. 2004, 279, 25284–25293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grether-Beck, S.; Felsner, I.; Brenden, H.; Marini, A.; Jaenicke, T.; Aue, N.; Welss, T.; Uthe, I.; Krutmann, J. Air pollution-induced tanning of human skin. Br. J. Dermatol. 2021. [Google Scholar] [CrossRef]
- Li, M.; Vierkötter, A.; Schikowski, T.; Hüls, A.; Ding, A.; Matsui, M.S.; Deng, B.; Ma, C.; Ren, A.; Zhang, J.; et al. Epidemiological evidence that indoor air pollution from cooking with solid fuels accelerates skin aging in Chinese women. J. Dermatol. Sci. 2015, 79, 148–154. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
| Aging Process | Histological Findings | References | |
|---|---|---|---|
| chronological aging | Epidermis | Stratum corneum | [6,7,8] |
| Epidermal atrophy | [9,10] | ||
| Langerhans cells | [11] | ||
| Dermis | Fibroblast senescence | [13,14,44] | |
| Collagen structure | [15,16,17,18,43,44] | ||
| Elastic fibers | [22,23] | ||
| Photoaging | Epidermal thickness | [25,26] | |
| Cell atypism | [27] | ||
| Solar elastosis | [32,34,36,38,40] | ||
| Molecular Mechanisms | Details | References |
|---|---|---|
| Telomere shortening | In intrinsic aging | [48] |
| In chronological aging | [49,52] | |
| Oxidative stress and matrix metalloproteinases | ROS and photoaging | [53,55,56] |
| Signal cascade activated by ROS | [58,59,61,63,132,133] | |
| Cytokines | Cytokines released by UV radiation | [65,66,67] |
| Tumor necrosis factor α | [69] | |
| Interleukins | [72,73,77,78,79,80,134] | |
| CCN1 | [82,83] | |
| Autophagic control | Association with aging process | [92,93,94,95,96,97,98] |
| Apoptosis | Association with aging process | [104,107,108,109] |
| MicroRNAs | Chronological aging | [113,114,115,116,117,118,119,135] |
| photoaging | [120,121,122,123] | |
| Skin microbiome | Association with aging process | [128,129,130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Hong, Y.; Kim, M. Structural and Functional Changes and Possible Molecular Mechanisms in Aged Skin. Int. J. Mol. Sci. 2021, 22, 12489. https://doi.org/10.3390/ijms222212489
Lee H, Hong Y, Kim M. Structural and Functional Changes and Possible Molecular Mechanisms in Aged Skin. International Journal of Molecular Sciences. 2021; 22(22):12489. https://doi.org/10.3390/ijms222212489
Chicago/Turabian StyleLee, Hyunji, Yongjun Hong, and Miri Kim. 2021. "Structural and Functional Changes and Possible Molecular Mechanisms in Aged Skin" International Journal of Molecular Sciences 22, no. 22: 12489. https://doi.org/10.3390/ijms222212489