An Overview of Drug Resistance in Protozoal Diseases


Abstract
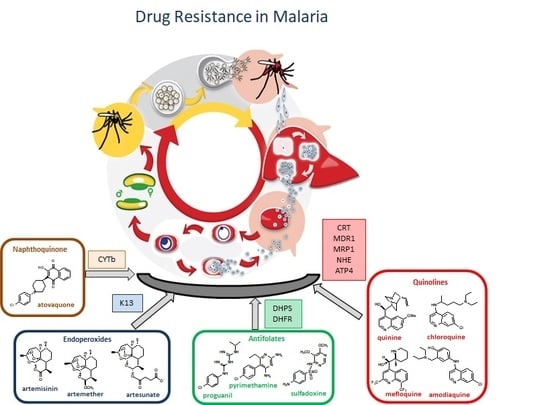
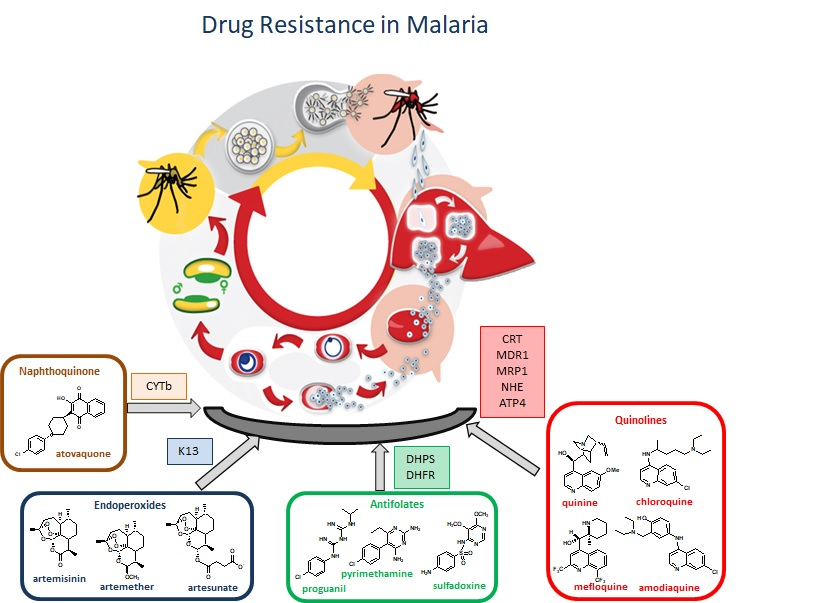
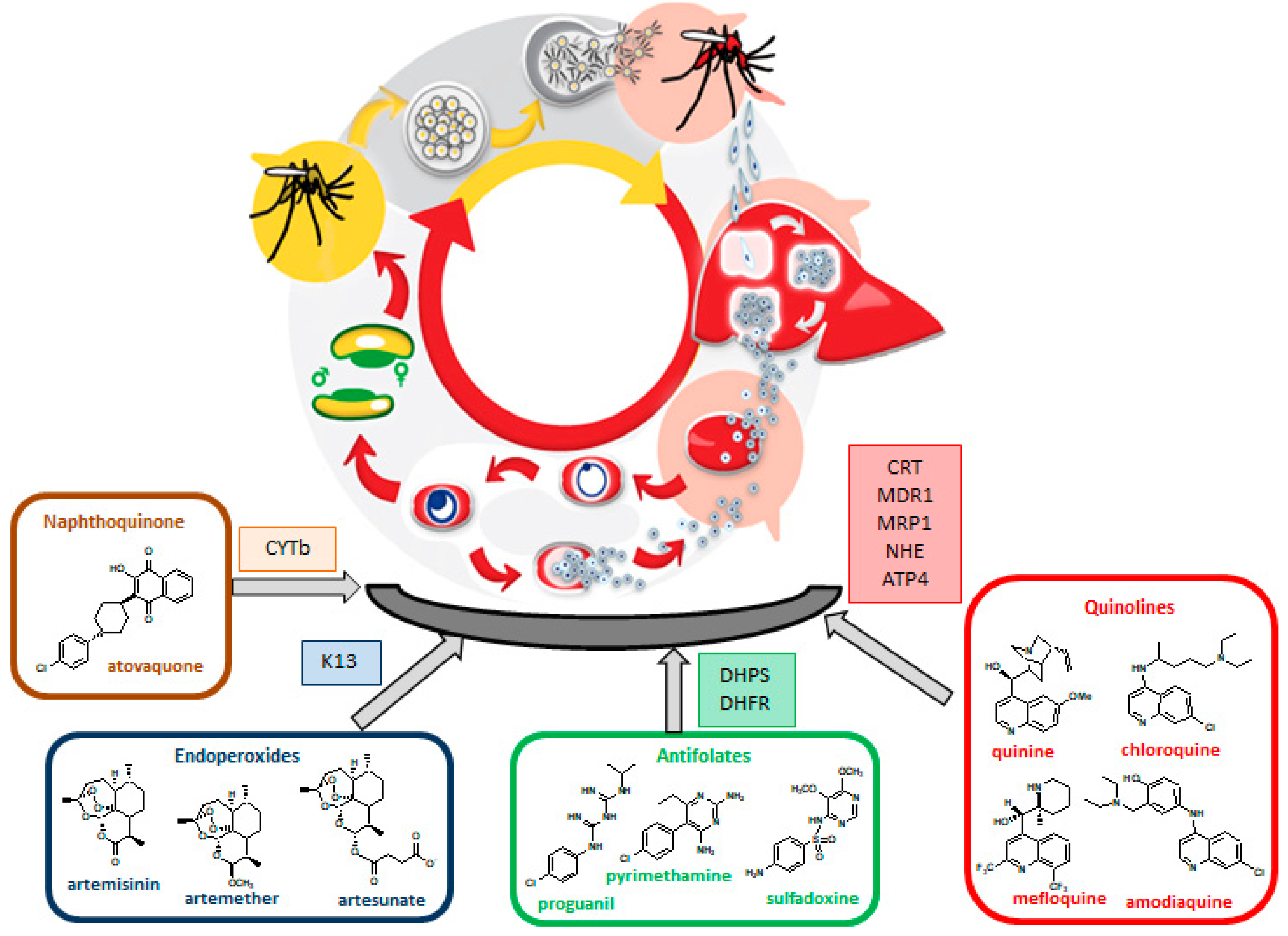
:
1. Introduction
2. The Triangle Relationship: Parasitic Protozoa, Host, and Drug Resistance
3. Antimalarial Drug Resistance
3.1. Resistance to Quinolines
Resistance-Associated Mutations
3.2. Resistance to Antifolates
Resistance-Associated Mutations
3.3. Resistance to Artemisinin
Resistance-Associated Mutations
3.4. Resistance to Atovaquone
Resistance-Associated Mutations
3.5. Global Surveillance on Malaria Resistance
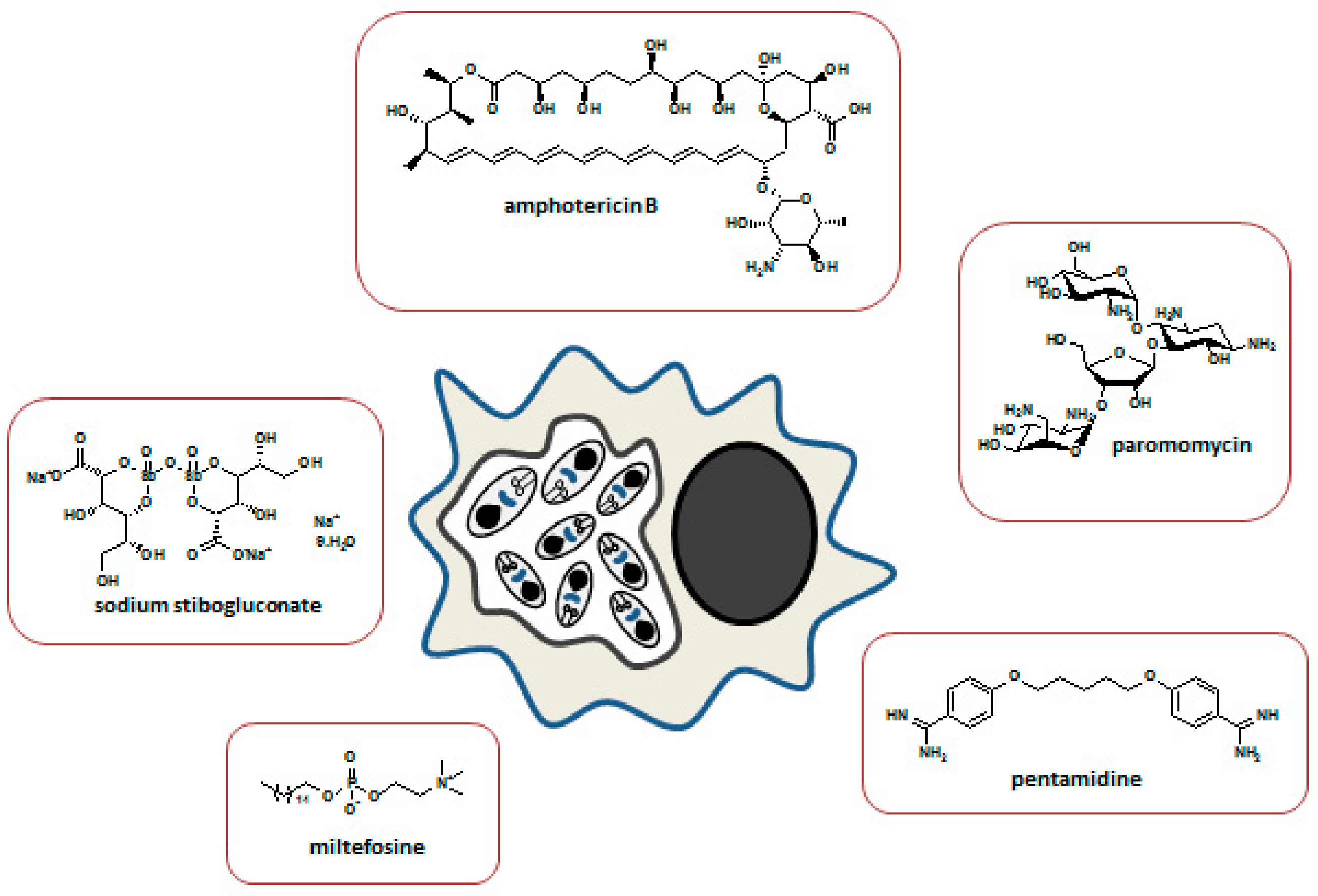
4. Antileishmanial Drug Resistance
4.1. Resistance to Antimonials
Resistance-Associated Mutations
4.2. Resistance to Pentamidine
Resistance-Associated Mutations
4.3. Resistance to Amphotericin B
Resistance-Associated Mutations
4.4. Resistance to Miltefosine
Resistance-Associated Mutations
4.5. Resistance to Paromomycin
Resistance-Associated Mutations
5. Antitrypanosomal Drug Resistance
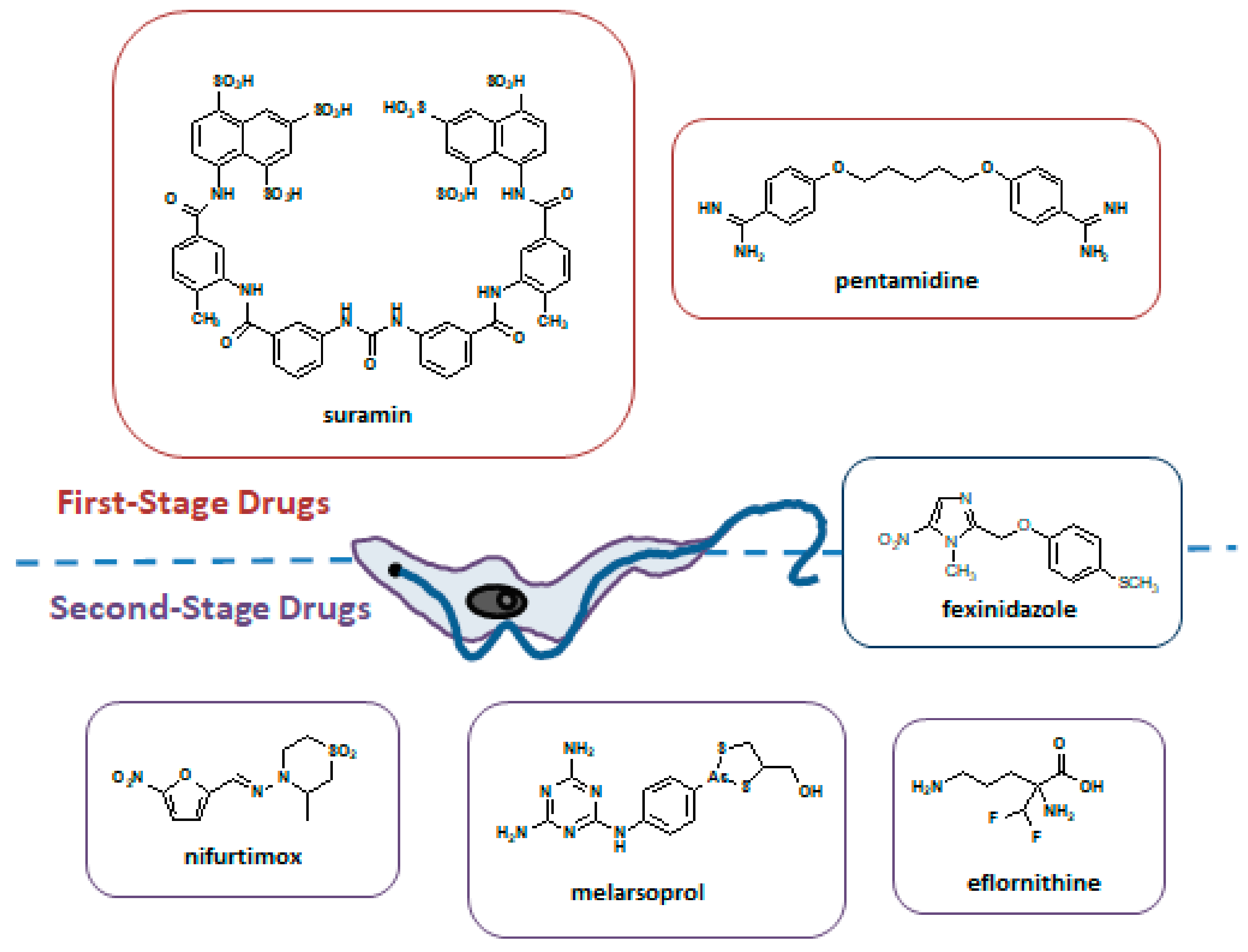
5.1. Resistance to Pentamidine
Resistance-Associated Mutations
5.2. Resistance to Suramin
5.3. Resistance to Melarsoprol
Resistance-Associated Mutations
5.4. Resistance to Eflornithine
Resistance-Associated Mutations
5.5. Resistance to Nifurtimox
Resistance-Associated Mutations
5.6. Resistance to Fexinidazole
6. Perspectives
Funding
Conflicts of Interest
References
- Fletcher, S.M.; Stark, D.; Harkness, J.; Ellisa, J. Enteric Protozoa in the Developed World: A Public Health Perspective. Clin. Microbiol. Rev. 2012, 25, 420–449. [Google Scholar] [CrossRef]
- Shanan, S.; Abd, H.; Bayoumi, M.; Saeed, A.; Sandstrom, G. Prevalence of Protozoa Species in Drinking and Environmental Water Sources in Sudan. Biomed. Res. Int. 2015, 5. [Google Scholar] [CrossRef]
- Imam, T.S. The Complexities in the Classification of Protozoa: A Challenge to Parasitologists. Bayero J. Pure Appl. Sci. 2009, 2, 159–164. [Google Scholar] [CrossRef]
- Antonovics, J.; Wilson, A.J.; Forbes, M.R.; Hauffe, H.C.; Kallio, E.R.; Leggett, H.C.; Longdon, B.; Okamura, B.; Sait, S.M.; Webster, J.P. The evolution of transmission mode. Philos. Trans. R. Soc. B-Biol. Sci. 2017, 372, 12. [Google Scholar] [CrossRef]
- Borst, P.; Ouellette, M. New mechanisms of drug-resistance in parasitic protozoa. Ann. Rev. Microbiol. 1995, 49, 427–460. [Google Scholar] [CrossRef]
- Monzote, L.; Siddiq, A. Drug development to protozoan diseases. Open Med. Chem. J. 2011, 5, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Aspinall, T.V.; Joynson, D.H.M.; Guy, E.; Hyde, J.E.; Sims, P.F.G. The molecular basis of sulfonamide resistance in Toxoplasma gondii and implications for the clinical management of toxoplasmosis. J. Infect. Dis. 2002, 185, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Malaria Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Tjitra, E.; Anstey, N.M.; Sugiarto, P.; Warikar, N.; Kenangalem, E.; Karyana, M.; Lampah, D.A.; Price, R.N. Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: A prospective study in Papua, Indonesia. PLoS Med. 2008, 5, 890–899. [Google Scholar] [CrossRef]
- Siciliano, G.; Alano, P. Enlightening the malaria parasite life cycle: Bioluminescent Plasmodium in fundamental and applied research. Front. Microbiol. 2015, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Shivapurkar, R.; Hingamire, T.; Kulkarni, A.S.; Rajamohanan, P.R.; Reddy, D.S.; Shanmugam, D. Evaluating antimalarial efficacy by tracking glycolysis in Plasmodium falciparum using NMR spectroscopy. Sci. Rep. 2018, 8, 10. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Camarda, G.; Jirawatcharadech, P.; Priestley, R.S.; Saif, A.; March, S.; Wong, M.H.L.; Leung, S.; Miller, A.B.; Baker, D.A.; Alano, P.; et al. Antimalarial activity of primaquine operates via a two-step biochemical relay. Nat. Commun. 2019, 10, 3226. [Google Scholar] [CrossRef]
- Wilson, D.W.; Langer, C.; Goodman, C.D.; McFadden, G.I.; Beeson, J.G. Defining the Timing of Action of Antimalarial Drugs against Plasmodium falciparum. Antimicrob. Agents Chemother. 2013, 57, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Capela, R.; Magalhaes, J.; Miranda, D.; Machado, M.; Sanches-Vaz, M.; Albuquerque, I.S.; Sharma, M.; Gut, J.; Rosenthal, P.J.; Frade, R.; et al. Endoperoxide-8-aminoquinoline hybrids as dual-stage antimalarial agents with enhanced metabolic stability. Eur. J. Med. Chem. 2018, 149, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Miranda, D.; Capela, R.; Albuquerque, I.S.; Meireles, P.; Paiva, I.; Nogueira, F.; Amewu, R.; Gut, J.; Rosenthal, P.J.; Oliveira, R.; et al. Novel endoperoxide-based transmission-blocking antimalarials with liver- and blood-schizontocidal activities. ACS Med. Chem. Lett. 2014, 5, 108–112. [Google Scholar] [CrossRef]
- Capela, R.; Cabal, G.G.; Rosenthal, P.J.; Gut, J.; Mota, M.M.; Moreira, R.; Lopes, F.; Prudêncio, M. Design and evaluation of primaquine-artemisinin hybrids as a multistage antimalarial strategy. Antimicrob. Agents Chemother. 2011, 55, 4698–4706. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Technical Strategy for Malaria 2016–2030; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Rosenthal, P.J. The interplay between drug resistance and fitness in malaria parasites. Mol. Microbiol. 2013, 89, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Antony, H.A.; Parija, S.C. Antimalarial drug resistance: An overview. Trop. Parasitol. 2016, 6, 30–41. [Google Scholar] [CrossRef]
- Mathews, E.S.; Odom John, A.R. Tackling resistance: Emerging antimalarials and new parasite targets in the era of elimination. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.D.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Coronado, L.M.; Nadovich, C.T.; Spadafora, C. Malarial hemozoin: From target to tool. Biochim. Biophys. Acta-Gen. Sub. 2014, 1840, 2032–2041. [Google Scholar] [CrossRef]
- Ross, L.S.; Dhingra, S.K.; Mok, S.; Yeo, T.; Wicht, K.J.; Kumpornsin, K.; Takala-Harrison, S.; Witkowski, B.; Fairhurst, R.M.; Ariey, F.; et al. Emerging Southeast Asian PfCRT mutations confer Plasmodium falciparum resistance to the first-line antimalarial piperaquine. Nat. Commun. 2018, 9, 13. [Google Scholar] [CrossRef]
- Cubides, J.R.; Camargo-Ayala, P.A.; Nino, C.H.; Garzon-Ospina, D.; Ortega-Ortegon, A.; Ospina-Cantillo, E.; Orduz-Duran, M.F.; Patarroyo, M.E.; Patarroyo, M.A. Simultaneous detection of Plasmodium vivax dhfr, dhps, mdr1 and crt-o resistance-associated mutations in the Colombian Amazonian region. Malar. J. 2018, 17, 9. [Google Scholar] [CrossRef]
- Cui, L.W.; Mharakurwa, S.; Ndiaye, D.; Rathod, P.K.; Rosenthal, P.J. Antimalarial Drug Resistance: Literature Review and Activities and Findings of the ICEMR Network. Am. J. Trop. Med. Hyg. 2015, 93, 57–68. [Google Scholar] [CrossRef]
- Antony, H.A.; Topno, N.S.; Gummadi, S.N.; Sankar, D.S.; Krishna, R.; Parija, S.C. In silico modeling of Plasmodium falciparum chloroquine resistance transporter protein and biochemical studies suggest its key contribution to chloroquine resistance. Acta Trop. 2019, 189, 84–93. [Google Scholar] [CrossRef]
- Voumbo-Matoumona, D.F.; Kouna, L.C.; Madamet, M.; Maghendji-Nzondo, S.; Pradines, B.; Lekana-Douki, J.B. Prevalence of Plasmodium falciparum antimalarial drug resistance genes in Southeastern Gabon from 2011 to 2014. Infect. Drug Res. 2018, 11, 1329–1338. [Google Scholar] [CrossRef]
- Veiga, M.I.; Dhingra, S.K.; Henrich, P.P.; Straimer, J.; Gnadig, N.; Uhlemann, A.C.; Martin, R.E.; Lehane, A.M.; Fidock, D.A. Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat. Commun. 2016, 7, 12. [Google Scholar] [CrossRef]
- Xu, C.; Wei, Q.K.; Yin, K.; Sun, H.; Li, J.; Xiao, T.; Kong, X.L.; Wang, Y.B.; Zhao, G.H.; Zhu, S.; et al. Surveillance of Antimalarial Resistance Pfcrt, Pfmdr1, and Pfkelch13 Polymorphisms in African Plasmodium falciparum imported to Shandong Province, China. Sci. Rep. 2018, 8, 9. [Google Scholar] [CrossRef]
- Kavishe, R.A.; van den Heuvel, J.M.W.; van de Vegte-Bolmer, M.; Luty, A.J.F.; Russel, F.G.M.; Koenderink, J.B. Localization of the ATP-binding cassette (ABC) transport proteins PfMRP1, PfMRP2, and PfMDR5 at the Plasmodium falciparum plasma membrane. Malar. J. 2009, 8, 9. [Google Scholar] [CrossRef]
- Mok, S.; Liong, K.Y.; Lim, E.H.; Huang, X.M.; Zhu, L.; Preiser, P.R.; Bozdech, Z. Structural polymorphism in the promoter of pfmrp2 confers Plasmodium falciparum tolerance to quinoline drugs. Mol. Microbiol. 2014, 91, 918–934. [Google Scholar] [CrossRef]
- Veiga, M.I.; Osorio, N.S.; Ferreira, P.E.; Franzen, O.; Dahlstrom, S.; Lum, J.K.; Nosten, F.; Gil, J.P. Complex Polymorphisms in the Plasmodium falciparum Multidrug Resistance Protein 2 Gene and Its Contribution to Antimalarial Response. Antimicrob. Agents Chemother. 2014, 58, 7390–7397. [Google Scholar] [CrossRef]
- Petersen, I.; Eastman, R.; Lanzer, M. Drug-resistant malaria: Molecular mechanisms and implications for public health. Febs Lett. 2011, 585, 1551–1562. [Google Scholar] [CrossRef] [Green Version]
- Andriantsoanirina, V.; Menard, D.; Rabearimanana, S.; Hubert, V.; Bouchier, C.; Tichit, M.; Le Bras, J.; Durand, R. Association of Microsatellite Variations of Plasmodium falciparum Na+/H+ Exchanger (Pfnhe-1) Gene with Reduced In Vitro Susceptibility to Quinine: Lack of Confirmation in Clinical Isolates from Africa. Am. J. Trop. Med. Hyg. 2010, 82, 782–787. [Google Scholar] [CrossRef] [Green Version]
- Medicines for Malaria Venture. Available online: https://www.mmv.org/research-development/project-portfolio/cipargamin (accessed on 10 February 2019).
- Nzila, A. Antifolates: Pyrimethamine, Proguanil, Sulphadoxine and Dapsone. In Treatment and Prevention of Malaria, 1st ed.; Krishna, S., Staines, H.M., Eds.; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Verhoef, H.; Veenemans, J.; Mwangi, M.N.; Prentice, A.M. Safety and benefits of interventions to increase folate status in malaria-endemic areas. Br. J. Haematol. 2017, 177, 905–918. [Google Scholar] [CrossRef] [Green Version]
- Heinberg, A.; Kirkman, L.; New York Acad, S. The molecular basis of antifolate resistance in Plasmodium falciparum: Looking beyond point mutations. In Malaria: Advances in Pathophysiology, Biology, and Drug Development; Blackwell Science Publ.: Oxford, UK, 2015; Volume 1342, pp. 10–18. [Google Scholar]
- Patel, P.; Bharti, P.K.; Bansal, D.; Ali, N.A.; Raman, R.K.; Mohapatra, P.K.; Sehgal, R.; Mahanta, J.; Sultan, A.A.; Singh, N. Prevalence of mutations linked to antimalarial resistance in Plasmodium falciparum from Chhattisgarh, Central India: A malaria elimination point of view. Sci. Rep. 2017, 7, 8. [Google Scholar] [CrossRef]
- Abugri, J.; Ansah, F.; Asante, K.P.; Opoku, C.N.; Amenga-Etego, L.A.; Awandare, G.A. Prevalence of chloroquine and antifolate drug resistance alleles in clinical isolates from three Plasmodium falciparum areas in Ghana. AAS Open Res. 2018, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tarnchompoo, B.; Chitnumsub, P.; Jaruwat, A.; Shaw, P.J.; Vanichtanankul, J.; Poen, S.; Rattanajak, R.; Wongsombat, C.; Tonsomboon, A.; Decharuangsilp, S.; et al. Hybrid Inhibitors of Malarial Dihydrofolate Reductase with Dual Binding Modes That Can Forestall Resistance. ACS Med. Chem. Lett. 2018, 9, 1235–1240. [Google Scholar] [CrossRef] [Green Version]
- Gogtay, N.; Brand, F.; Olliaro, P.; Sinclair, D. Artemisinin-based combination therapy for the treatment of uncomplicated Plasmodium vivax malaria and the prevention of relapses (Protocol). In Cochrane Database of Systematic Reviews; The Cochrane Library: New York, NY, USA, 2010; pp. 1–9. [Google Scholar]
- Li, Y. Qinghaosu (artemisinin): Chemistry and pharmacology. Acta Pharmacol. Sin. 2012, 33, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Konstat-Korzenny, E.; Ascencio-Aragon, J.A.; Niezen-Lugo, S.; Vazquez-Lopez, R. Artemisinin and Its Synthetic Derivatives as a Possible Therapy for Cancer. Med. Sci. (Basel, Switzerland) 2018, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Ismail, H.M.; Barton, V.E.; Panchana, M.; Charoensutthivarakul, S.; Biagini, G.A.; Ward, S.A.; O’Neill, P.M. A Click Chemistry-Based Proteomic Approach Reveals that 1,2,4-Trioxolane and Artemisinin Antimalarials Share a Common Protein Alkylation Profile. Angew. Chemie-Int. Ed. 2016, 55, 6401–6405. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.Z.; Moily, N.S.; Bridgford, J.L.; Wood, R.J.; Radwan, M.; Smith, T.A.; Song, Z.G.; Tang, B.Z.; Tilley, L.; Xu, X.H.; et al. A thiol probe for measuring unfolded protein load and proteostasis in cells. Nat. Commun. 2017, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Pelfrene, E.; Pinheiro, M.H.; Cavaleri, M. Artemisinin-based combination therapy in the treatment of uncomplicated malaria: Review of recent regulatory experience at the European Medicines Agency. Int. Health 2015, 7, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Nsanzabana, C. Resistance to Artemisinin Combination Therapies (ACTs): Do Not Forget the Partner Drug! Trop. Med. Infect. Dis. 2019, 4, 26. [Google Scholar] [CrossRef] [Green Version]
- Thriemer, K.; Hong, N.V.; Rosanas-Urgell, A.; Phuc, B.Q.; Ha, D.M.; Pockele, E.; Guetens, P.; Van, N.V.; Duong, T.T.; Amambua-Ngwa, A.; et al. Delayed Parasite Clearance after Treatment with Dihydroartemisinin-Piperaquine in Plasmodium falciparum Malaria Patients in Central Vietnam. Antimicrob. Agents Chemother. 2014, 58, 7049–7055. [Google Scholar] [CrossRef] [Green Version]
- Chookajorn, T. How to combat emerging artemisinin resistance: Lessons from “The Three Little Pigs”. PLoS Path. 2018, 14, 8. [Google Scholar] [CrossRef]
- Mukherjee, A.; Bopp, S.; Magistrado, P.; Wong, W.; Daniels, R.; Demas, A.; Schaffner, S.; Amaratunga, C.; Lim, P.; Dhorda, M.; et al. Artemisinin resistance without pfkelch13 mutations in Plasmodium falciparum isolates from Cambodia. Malar. J. 2017, 16, 11. [Google Scholar] [CrossRef]
- Breglio, K.F.; Amato, R.; Eastman, R.; Lim, P.; Sa, J.M.; Guha, R.; Ganesan, S.; Dorward, D.W.; Klumpp-Thomas, C.; McKnight, C.; et al. A single nucleotide polymorphism in the Plasmodium falciparum atg18 gene associates with artemisinin resistance and confers enhanced parasite survival under nutrient deprivation. Malar. J. 2018, 17, 16. [Google Scholar] [CrossRef] [Green Version]
- Demas, A.R.; Sharma, A.I.; Wong, W.; Early, A.M.; Redmond, S.; Bopp, S.; Neafsey, D.E.; Volkman, S.K.; Hartl, D.L.; Wirth, D.F. Mutations in Plasmodium falciparum actin-binding protein coronin confer reduced artemisinin susceptibility. Proc. Natl. Acad. Sci. USA 2018, 115, 12799–12804. [Google Scholar] [CrossRef] [Green Version]
- Isozumi, R.; Uemura, H.; Kimata, I.; Ichinose, Y.; Logedi, J.; Omar, A.H.; Kaneko, A. Novel Mutations in K13 Propeller Gene of Artemisinin-Resistant Plasmodium falciparum. Emerg. Infect. Dis. 2015, 21, 490–492. [Google Scholar] [CrossRef] [Green Version]
- Nixon, G.L.; Moss, D.M.; Shone, A.E.; Lalloo, D.G.; Fisher, N.; O’Neill, P.M.; Ward, S.A.; Biagini, G.A. Antimalarial pharmacology and therapeutics of atovaquone. J. Antimicrob. Chemother. 2013, 68, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Staines, H.M.; Burrow, R.; Teo, B.H.Y.; Ster, I.C.; Kremsner, P.G.; Krishna, S. Clinical implications of Plasmodium resistance to atovaquone/proguanil: A systematic review and meta-analysis. J. Antimicrob. Chemother. 2018, 73, 581–595. [Google Scholar] [CrossRef]
- Bakshi, R.P.; Tatham, L.M.; Savage, A.C.; Tripathi, A.K.; Mlambo, G.; Ippolito, M.M.; Nenortas, E.; Rannard, S.P.; Owen, A.; Shapiro, T.A. Long-acting injectable atovaquone nanomedicines for malaria prophylaxis. Nat. Commun. 2018, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Cottrell, G.; Musset, L.; Hubert, V.; Le Bras, J.; Clain, J.; Atovaquone-Proguanil, T. Emergence of Resistance to Atovaquone-Proguanil in Malaria Parasites: Insights from Computational Modeling and Clinical Case Reports. Antimicrob. Agents Chemother. 2014, 58, 4504–4514. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.; Lopes, F.; Moreira, R. Inhibitors of the Mitochondrial Electron Transport Chain and de novo Pyrimidine Biosynthesis as Antimalarials: The Present Status. Curr. Med. Chem. 2010, 17, 929–956. [Google Scholar] [CrossRef]
- World Health Organization. Global Leishmaniasis Update, 2006–2015: A Turning Point in Leishmaniasis Surveillance; World Health Organization: Geneva, Switzerland, 2017; pp. 557–572. [Google Scholar]
- Perez-Cabezas, B.; Cecilio, P.; Gaspar, T.B.; Gartner, F.; Vasconcellos, R.; Cordeiro-da-Silva, A. Understanding Resistance vs. Susceptibility in Visceral Leishmaniasis Using Mouse Models of Leishmania infantum Infection. Front. Cell. Infect. Microbiol. 2019, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Alemayehu, B.; Alemayehu, M. Leishmaniasis: A Review on Parasite, Vector and Reservoir Host. Health Sci. J. 2017, 11, 6. [Google Scholar] [CrossRef]
- Ghorbani, M.; Farhoudi, R. Leishmaniasis in humans: Drug or vaccine therapy? Drug Des. Dev. Ther. 2018, 12, 25–40. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Control of the Leishmaniases; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Diro, E.; Lynen, L.; Ritmeijer, K.; Boelaert, M.; Hailu, A.; van Griensven, J. Visceral Leishmaniasis and HIV Coinfection in East Africa. PLoS Negl. Trop. Dis. 2014, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Berg, M.; Mannaert, A.; Vanaerschot, M.; Van der Auwera, G.; Dujardin, J.C. (Post-) Genomic approaches to tackle drug resistance in Leishmania. Parasitology 2013, 140, 1492–1505. [Google Scholar] [CrossRef]
- Turcano, L.; Torrente, E.; Missineo, A.; Andreini, M.; Gramiccia, M.; Di Muccio, T.; Genovese, I.; Fiorillo, A.; Harper, S.; Bresciani, A.; et al. Identification and binding mode of a novel Leishmania Trypanothione reductase inhibitor from high throughput screening. PLoS Negl. Trop. Dis. 2018, 12, 21. [Google Scholar] [CrossRef] [Green Version]
- Maltezou, H.C. Drug Resistance in Visceral Leishmaniasis. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Pund, S.; Joshi, A. Nanoarchitectures for Neglected Tropical Protozoal Diseases: Challenges and State of the Art. In Nano-and Microscale Drug Delivery Systems, 1st ed.; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.C.; Barrett, M.P.; Lopez-Velez, R.; Garcia-Hernandez, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, 24. [Google Scholar] [CrossRef]
- Garcia-Salcedo, J.A.; Unciti-Broceta, J.D.; Valverde-Pozo, J.; Soriano, M. New Approaches to Overcome Transport Related Drug Resistance in Trypanosomatid Parasites. Front. Pharmacol. 2016, 7, 14. [Google Scholar] [CrossRef]
- Kumar, G.A.; Roy, S.; Jafurulla, M.; Mandal, C.; Chattopadhyay, A. Statin-induced chronic cholesterol depletion inhibits Leishmania donovani infection: Relevance of optimum host membrane cholesterol. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 2088–2096. [Google Scholar] [CrossRef]
- Ouellette, M.; Drummelsmith, J.; Papadopoulou, B. Leishmaniasis: Drugs in the clinic, resistance and new developments. Drug Res. Updat. 2004, 7, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Hombach-Barrigah, A.; Bartsch, K.; Smirlis, D.; Rosenqyist, H.; MacDonald, A.; Dingli, F.; Loew, D.; Spath, G.F.; Rachidi, N.; Wiese, M.; et al. Leishmania donovani 90 kD Heat Shock Protein—Impact of Phosphosites on Parasite Fitness, Infectivity and Casein Kinase Affinity. Sci. Rep. 2019, 9, 16. [Google Scholar] [CrossRef]
- Kaur, P.; Garg, M.; Hombach-Barrigah, A.; Clos, J.; Goyal, N. MAPK1 of Leishmania donovani interacts and phosphorylates HSP70 and HSP90 subunits of foldosome complex. Sci. Rep. 2017, 7, 11. [Google Scholar] [CrossRef]
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.N.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent Developments in Drug Discovery for Leishmaniasis and Human African Trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Shaha, C. Apoptotic death in Leishmania donovani promastigotes in response to respiratory chain inhibition—Complex II inhibition results in increased pentamidine cytotoxicity. J. Biol. Chem. 2004, 279, 11798–11813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Padmanabhan, P.K.; Sahani, M.H.; Barrett, M.P.; Madhubala, R. Roles for mitochondria in pentamidine susceptibility and resistance in Leishmania donovani. Mol. Biochem. Parasitol. 2006, 145, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, J.; Sundar, S. Drug Resistance in Leishmaniasis. J. Global Infect. Dis. 2010, 2, 10. [Google Scholar] [CrossRef]
- Yang, G.; Choi, G.; No, J.H. Antileishmanial Mechanism of Diamidines Involves Targeting Kinetoplasts. Antimicrob. Agents Chemother. 2016, 60, 6828–6836. [Google Scholar] [CrossRef] [Green Version]
- Cortazar, T.M.; Coombs, G.H.; Walker, J. Leishmania panamensis: Comparative inhibition of nuclear DNA topoisomerase II enzymes from promastigotes and human macrophages reveals anti-parasite selectivity of fluoroquinolones, flavonoids and pentamidine. Exp. Parasitol. 2007, 116, 475–482. [Google Scholar] [CrossRef]
- Bray, P.G.; Barrett, M.P.; Ward, S.A.; de Koning, H.P. Pentamidine uptake and resistance in pathogenic protozoa: Past, present and future. Trends Parasitol. 2003, 19, 232–239. [Google Scholar] [CrossRef]
- Basselin, M.; Denise, H.; Coombs, G.H.; Barrett, M.P. Resistance to pentamidine in Leishmania mexicana involves exclusion of the drug from the mitochondrion. Antimicrob. Agents Chemother. 2002, 46, 3731–3738. [Google Scholar] [CrossRef] [Green Version]
- Pramanik, P.K.; Alam, M.N.; Roy Chowdhury, D.; Chakraborti, T. Drug Resistance in Protozoan Parasites: An Incessant Wrestle for Survival. J. Glob. Antimicrob. Res. 2019, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.C.; Messier, N.; Ouellette, M.; Cotrim, P.C. Role of the ABC transporter PRP1 (ABCC7) in pentamidine resistance in Leishmania amastigotes. Antimicrob. Agents Chemother. 2007, 51, 3030–3032. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.; Glover, L.; Munday, J.C.; Andres, D.A.; Barrett, M.P.; de Koning, H.P.; Horn, D. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc. Natl. Acad. Sci. USA 2012, 109, 10996–11001. [Google Scholar] [CrossRef] [Green Version]
- Munday, J.C.; Eze, A.A.; Baker, N.; Glover, L.; Clucas, C.; Andres, D.A.; Natto, M.J.; Teka, I.A.; McDonald, J.; Lee, R.S.; et al. Trypanosoma brucei aquaglyceroporin 2 is a high-affinity transporter for pentamidine and melaminophenyl arsenic drugs and the main genetic determinant of resistance to these drugs. J. Antimicrob. Chemother. 2014, 69, 651–663. [Google Scholar] [CrossRef]
- Jeacock, L.; Baker, N.; Wiedemar, N.; Maser, P.; Horn, D. Aquaglyceroporin-null trypanosomes display glycerol transport defects and respiratory-inhibitor sensitivity. PLoS Path. 2017, 13, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, I.L.K.; Chan, K.F.; Burkett, B.A.; Zhao, Y.Z.; Chai, Y.; Sun, H.; Chan, T.H.; Chow, L.A.C. Flavonoid dimers as bivalent modulators for pentamidine and sodium stiboglucanate resistance in Leishmania. Antimicrob. Agents Chemother. 2007, 51, 930–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, I.L.K.; Chan, K.F.; Zhao, Y.Z.; Chan, T.H.; Chow, L.M.C. Quinacrine and a novel apigenin dimer can synergistically increase the pentamidine susceptibility of the protozoan parasite Leishmania. J. Antimicrob. Chemother. 2009, 63, 1179–1190. [Google Scholar] [CrossRef] [Green Version]
- Légaré, D.; Ouellette, M. Drug Resistance in Leishmania. In Handbook of Antimicrobial Resistance; Gotte, M.B., Matlashewski, A., Wainberg, G., Mark, A., Donald, S., Eds.; Springer: New York, NY, USA, 2017; pp. 313–341. [Google Scholar]
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Research 2017, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Purkait, B.; Kumar, A.; Nandi, N.; Sardar, A.H.; Das, S.; Kumar, S.; Pandey, K.; Ravidas, V.; Kumar, M.; De, T.; et al. Mechanism of Amphotericin B Resistance in Clinical Isolates of Leishmania donovani. Antimicrob. Agents Chemother. 2012, 56, 1031–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohapatra, S. Drug resistance in leishmaniasis: Newer developments. Trop. Parasitol. 2014, 4, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Mbongo, N.; Loiseau, P.M.; Billion, M.A.; Robert-Gero, M. Mechanism of amphotericin B resistance in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 1998, 42, 352–357. [Google Scholar]
- Khan, I.; Khan, M.; Umar, M.N.; Oh, D.H. Attenuation and Production of the Amphotericin B-Resistant Leishmania tropica Strain. Jundishapur J. Microbiol. 2016, 9, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pountain, A.W.; Weidt, S.K.; Regnault, C.; Bates, P.A.; Donachie, A.M.; Dickens, N.J.; Barrett, M.P. Genomic instability at the locus of sterol C24-methyltransferase promotes amphotericin B resistance in Leishmania parasites. PLoS Negl. Trop. Dis. 2019, 13, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirve, S.; Boelaert, M.; Matlashewski, G.; Mondal, D.; Arana, B.; Kroeger, A.; Olliaro, P. Transmission Dynamics of Visceral Leishmaniasis in the Indian Subcontinent—A Systematic Literature Review. PLoS Negl. Trop. Dis. 2016, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Dorlo, T.P.C.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef]
- Pachioni, J.D.; Magalhaes, J.G.; Lima, E.J.C.; Bueno, L.D.; Barbosa, J.F.; de Sa, M.M.; Rangel-Yagui, C.D. Alkylphospholipids—A Promising Class of Chemotherapeutic Agents with a Broad Pharmacological Spectrum. J. Pharm. Pharm. Sci. 2013, 16, 742–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingartner, A.; Drobot, B.; Herrmann, A.; Sanchez-Canete, M.P.; Gamarro, F.; Castanys, S.; Pomorski, T.G. Disruption of the Lipid-Transporting LdMT-LdRos3 Complex in Leishmania donovani Affects Membrane Lipid Asymmetry but Not Host Cell Invasion. PLoS ONE 2010, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Mishra, J.; Gupta, A.K.; Singh, A.; Shankar, P.; Singh, S. Laboratory confirmed miltefosine resistant cases of visceral leishmaniasis from India. Parasit. Vectors 2017, 10, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto-Martinez, A.K.; Rodriguez-Duran, J.; Serrano-Martin, X.; Hernandez-Rodriguez, V.; Benaim, G. Mechanism of Action of Miltefosine on Leishmania donovani Involves the Impairment of Acidocalcisome Function and the Activation of the Sphingosine-Dependent Plasma Membrane Ca2+ Channel. Antimicrob. Agents Chemother. 2018, 62, 10. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.G.; Vacchina, P.; Robles-Murguia, M.; Wadsworth, M.; McDowell, M.A.; Morales, M.A. Fitness and Phenotypic Characterization of Miltefosine-Resistant Leishmania major. PLoS Negl. Trop. Dis. 2015, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Shaw, C.D.; Lonchamp, J.; Downing, T.; Imamura, H.; Freeman, T.M.; Cotton, J.A.; Sanders, M.; Blackburn, G.; Dujardin, J.C.; Rijal, S.; et al. In vitro selection of miltefosine resistance in promastigotes of Leishmania donovani from Nepal: Genomic and metabolomic characterization. Mol. Microbiol. 2016, 99, 1134–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prajapati, V.K.; Sharma, S.; Rai, M.; Ostyn, B.; Salotra, P.; Vanaerschot, M.; Dujardin, J.C.; Sundar, S. In vitro Susceptibility of Leishmania donovani to Miltefosine in Indian Visceral Leishmaniasis. Am. J. Trop. Med. Hyg. 2013, 89, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, E.; Bulte, D.; Van Bockstal, L.; Van den Kerkhof, M.; Cos, P.; Delputte, P.; Hendrickx, S.; Maes, L.; Caljon, G. Miltefosine enhances the fitness of a non-virulent drug-resistant Leishmania infantum strain. J. Antimicrob. Chemother. 2019, 74, 395–406. [Google Scholar] [CrossRef]
- Perez-Victoria, J.M.; Bavchvarov, B.I.; Torrecillas, I.R.; Martinez-Garcia, M.; Lopez-Martin, C.; Campillo, M.; Castanys, S.; Gamarro, F. Sitamaquine Overcomes ABC-Mediated Resistance to Miltefosine and Antimony in Leishmania. Antimicrob. Agents Chemother. 2011, 55, 3838–3844. [Google Scholar] [CrossRef] [Green Version]
- da Costa, K.M.; Valente, R.C.; Salustiano, E.J.; Gentile, L.B.; Freire-de-Lima, L.; Mendonca-Previato, L.; Previato, J.O. Functional Characterization of ABCC Proteins from Trypanosoma cruzi and Their Involvement with Thiol Transport. Front. Microbiol. 2018, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Armitage, E.G.; Alqaisi, A.Q.I.; Godzien, J.; Pena, I.; Mbekeani, A.J.; Alonso-Herranz, V.; Lopez-Gonzalvez, A.; Martin, J.; Gabarro, R.; Denny, P.W.; et al. Complex Interplay between Sphingolipid and Sterol Metabolism Revealed by Perturbations to the Leishmania Metabolome Caused by Miltefosine. Antimicrob. Agents Chemother. 2018, 62, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiwanitkit, V. Interest in paromomycin for the treatment of visceral leishmaniasis (kala-azar). Ther. Clin. Risk Manag. 2012, 8, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, B.; Jhingran, A.; Panigrahi, A.; Stuart, K.D.; Madhubala, R. Paromomycin Affects Translation and Vesicle-Mediated Trafficking as Revealed by Proteomics of Paromomycin-Susceptible-Resistant Leishmania donovani. PLoS ONE 2011, 6, 12. [Google Scholar] [CrossRef]
- Jhingran, A.; Chawla, B.; Saxena, S.; Barrett, M.P.; Madhubala, R. Paromomycin: Uptake and resistance in Leishmania donovani. Mol. Biochem. Parasitol. 2009, 164, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, M.M.; Malchiodi, E.L.; Algranati, I.D. Differential Effects of Paromomycin on Ribosomes of Leishmania mexicana and Mammalian Cells. Antimicrob. Agents Chemother. 2011, 55, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidari-Kharaji, M.; Taheri, T.; Doroud, D.; Habibzadeh, S.; Badirzadeh, A.; Rafati, S. Enhanced paromomycin efficacy by solid lipid nanoparticle formulation against Leishmania in mice model. Parasite Immunol. 2016, 38, 599–608. [Google Scholar] [CrossRef]
- Khan, W.; Sharma, S.S.; Kumar, N. Bioanalytical method development, pharmacokinetics, and toxicity studies of paromomycin and paromomycin loaded in albumin microspheres. Drug Test. Anal. 2013, 5, 453–460. [Google Scholar] [CrossRef]
- Banerjee, A.; De, M.; Ali, N. Combination Therapy with Paromomycin-Associated Stearylamine-Bearing Liposomes Cures Experimental Visceral Leishmaniasis through Th1-Biased Immunomodulation. Antimicrob. Agents Chemother. 2011, 55, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, S.; Van den Kerkhof, M.; Mabille, D.; Cos, P.; Delputte, P.; Maes, L.; Caljon, G. Combined treatment of miltefosine and paromomycin delays the onset of experimental drug resistance in Leishmania infantum. PLoS Negl. Trop. Dis. 2017, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, V.; Sundar, S.; Dujardin, J.C.; Salotra, P. Elucidation of Cellular Mechanisms Involved in Experimental Paromomycin Resistance in Leishmania donovani. Antimicrob. Agents Chemother. 2014, 58, 2580–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, P.G.E.; Rodgers, J. Clinical and Neuropathogenetic Aspects of Human African Trypanosomiasis. Front. Immunol. 2019, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Barrett, M.P. The elimination of human African trypanosomiasis is in sight: Report from the third WHO stakeholders meeting on elimination of gambiense human African trypanosomiasis. PLoS Negl. Trop. Dis. 2018, 12, 4. [Google Scholar] [CrossRef]
- Franco, J.R.; Cecchi, G.; Priotto, G.; Paone, M.; Diarra, A.; Grout, L.; Simarro, P.P.; Zhao, W.N.; Argaw, D. Monitoring the elimination of human African trypanosomiasis: Update to 2016. PLoS Negl. Trop. Dis. 2018, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Buscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- Franco, J.; Scarone, L.; Comini, M.A. Drugs and Drug Resistance in African and American Trypanosomiasis. In Neglected Diseases: Extensive Space for Modern Drug Discovery; Botta, M., Ed.; Elsevier: Amsterdam, The Netherlands; Academic Press Inc: San Diego, CA, USA, 2018; Volume 51, pp. 97–133. [Google Scholar]
- Alsford, S.; Kelly, J.M.; Baker, N.; Horn, D. Genetic dissection of drug resistance in trypanosomes. Parasitology 2013, 140, 1478–1491. [Google Scholar] [CrossRef]
- Thomas, J.; Baker, N.; Hutchinson, S.; Dominicus, C.; Trenaman, A.; Glover, L.; Alsford, S.; Horn, D. Insights into antitrypanosomal drug mode-of-action from cytology-based profiling. PLoS Negl. Trop. Dis. 2018, 12, 19. [Google Scholar] [CrossRef]
- Deeks, E.D. Fexinidazole: First Global Approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Munday, J.C.; Settimo, L.; de Koning, H.P. Transport proteins determine drug sensitivity and resistance in a protozoan parasite, Trypanosoma brucei. Front. Pharmacol. 2015, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.H.; Welburn, S.C. The Long Wait for a New Drug for Human African Trypanosomiasis. Trends Parasitol. 2018, 34, 818–827. [Google Scholar] [CrossRef]
- Quintana, J.F.; Del Pino, R.C.; Yamada, K.; Zhang, N.; Field, M.C. Adaptation and Therapeutic Exploitation of the Plasma Membrane of African Trypanosomes. Genes 2018, 9, 368. [Google Scholar] [CrossRef] [Green Version]
- Babokhov, P.; Sanyaolu, A.O.; Oyibo, W.A.; Fagbenro-Beyioku, A.F.; Iriemenam, N.C. A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis. Path. Glob. Health 2013, 107, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedemar, N.; Graf, F.E.; Zwyer, M.; Ndomba, E.; Renggli, C.K.; Cal, M.; Schmidt, R.S.; Wenzler, T.; Maser, P. Beyond immune escape: A variant surface glycoprotein causes suramin resistance in Trypanosoma brucei. Mol. Microbiol. 2018, 107, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairlamb, A.H.; Horn, D. Melarsoprol Resistance in African Trypanosomiasis. Trends Parasitol. 2018, 34, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Graf, F.E.; Baker, N.; Munday, J.C.; de Koning, H.P.; Horn, D.; Maser, P. Chimerization at the AQP2-AQP3 locus is the genetic basis of melarsoprol-pentamidine cross-resistance in clinical Trypanosoma brucei gambiense isolates. Int. J. Parasitol. Drugs Drug Res. 2015, 5, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Steverding, D. The development of drugs for treatment of sleeping sickness: A historical review. Parasites Vectors 2010, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Burri, C.; Brun, R. Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res. 2003, 90, S49–S52. [Google Scholar] [CrossRef]
- Vincent, I.M.; Creek, D.; Watson, D.G.; Kamleh, M.A.; Woods, D.J.; Wong, P.E.; Burchmore, R.J.S.; Barrett, M.P. A Molecular Mechanism for Eflornithine Resistance in African Trypanosomes. PLoS Path. 2010, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.; Alsford, S.; Horn, D. Genome-wide RNAi screens in African trypanosomes identify the nifurtimox activator NTR and the eflornithine transporter AAT6. Mol. Biochem. Parasitol. 2011, 176, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.T. Antiparasitic Agents. In Principles and Practice of Pediatric Infectious Diseases, 5th ed.; Long, S.S., Prober, C.G., Fischer, M., Eds.; Elsevier Health Sciences: Philadelphia, PA, USA, 2018; pp. 1567–1587. [Google Scholar]
- Hall, B.S.; Bot, C.; Wilkinson, S.R. Nifurtimox Activation by Trypanosomal Type I Nitroreductases Generates Cytotoxic Nitrile Metabolites. J. Biol. Chem. 2011, 286, 13088–13095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, S.R.; Taylor, M.C.; Horn, D.; Kelly, J.M.; Cheeseman, I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 5022–5027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyllie, S.; Foth, B.J.; Kelner, A.; Sokolova, A.Y.; Berriman, M.; Fairlamb, A.H. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J. Antimicrob. Chemother. 2016, 71, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollastri, M.P. Fexinidazole: A New Drug for African Sleeping Sickness on the Horizon. Trends Parasitol. 2018, 34, 178–179. [Google Scholar] [CrossRef] [PubMed]
- EMA. Fexinidazole Winthrop; European Medicines Agency: Amsterdam, The Netherlands, 2018; p. 177. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Use in [12] | Mode of Action | Mechanism of Described Resistance |
|---|---|---|---|
| Chloroquine | Uncomplicated non-falciparum malaria | Inhibition of heme detoxification | Pfcrt, Pfmdr1, Pfmrp1, Pfnhe, PfATP4 mutations |
| Amodiaquine | Uncomplicated P. falciparum or P.vivax infections in ACT Chemoprophylaxis with SP | ||
| Quinine | Severe and uncomplicated malaria Alternative when effective ACT is not available | ||
| Mefloquine | Uncomplicated malaria in combination with artesunate Chemoprophylaxis of malaria caused by all species | ||
| Primaquine | Radical cure of P. vivax or P. ovale Anti-relapse therapy for P. vivax and P. ovale Gametocytocidal agent | Possibly a unique mode of action involving CYP2D6 and CPR [13] | |
| Lumefantrine | Treatment of uncomplicated malaria (all species) in combination with artemether | Inhibition of heme detoxification | Pfcrt, Pfmdr1, Pfmrp1 mutations |
| Sulfadoxine | SP for the treatment of malaria in pregnant women and children SP in combination with amodiaquine for seasonal chemoprevention in children acute uncomplicated malaria in combination with artesunate | Competitive inhibition of PfDHPS | Pfdhps, Pfdhfr-ts mutations |
| Pyrimethamine | Inhibition on folate biosynthesis (PfDHFR) | ||
| Proguanil | |||
| Atovaquone | Prophylaxis of malaria and treatment of uncomplicated malaria in travellers outside endemic areas in combination with proguanil | Inhibits the respiratory function of parasite | Pfcytb mutation |
| Artemisinin Artesunate Artemether | Multidrug-resistant Pf infection Combination with other drugs to prevent drug resistance (ACT) Children and adults with uncomplicated P. falciparum malaria and severe malaria | Generation of free radicals and reactive species and alkylation of parasite target biomolecules | |
| PfK13 |
| Drug | Use in | Mode of Action | Resistance Described | Mechanism of Resistance |
|---|---|---|---|---|
| SodiumStibogluconate | All clinical forms of leishmaniasis Combination therapy (with PMM) | Trypanothione reductase Inhibition | Yes | Elevated intracellular thiols levels Overexpression of: TXNPx, MRP1, and ABC transporters |
| Pentamidine | Systemic CL Secondary prophylaxis of VL treatment in HIV co-infection | Not clear. Hypothesis: Interaction with kDNAs; interference with polyamine synthesis; inhibition of RNA polymerase; inhibition of TOPII; apoptotic death | Yes | Overexpression of PRP1AQP2 mutation |
| Amphotericin B and Liposomal Amphotericin B | VL Combination therapy (with MT and PMM) | Not clear. Hypothesis: Apoptotic death, depolarization of the membrane | No effective resistance | Several hypotheses based on laboratory-derived resistant strains |
| Miltefosine | VL, CL, combination therapy (with LAMB) | Not clear. Hypothesis: Alteration in alkyl-lipid metabolism and phospholipid biosynthesis, apoptotic death | No effective resistance | Several hypotheses based on laboratory-derived resistant strains |
| Paromomycin | CL, PKDL, combination therapy (with SSG, LAMB and MT) | Not clear. Hypothesis: Inhibition of protein synthesis, decreasing of mitochondrial membrane potencial, alteration in membrane fluidity and lipid metabolism, respiratory dysfunction | No effective resistance | Several hypotheses based on laboratory-derived resistant strains |
| Drug | Use in | Mode of action | Resistance Described | Mechanism of Resistance |
|---|---|---|---|---|
| Pentamidine | g-HAT and r-HAT 1st stage | Interferes with the nuclear mechanisms, inhibiting synthesis of DNA, RNA | Yes | Loss of function of P2 aminopurine transporter |
| Suramin | g-HAT and r-HAT 1st stage | Inhibition of glycolytic enzymes | No effective resistance | Several hypotheses based on laboratory-derived resistant strains |
| Melarsoprol | g-HAT and r-HAT 2nd stage | Not completely clear | Yes | Mutations in P2 and AQP2 transporters |
| Eflornithine | g-HAT 2nd stage Used in combination with nifurtimox (NECT) | Inhibition of ornithine decarboxylase, an enzyme involved in polyamine synthesis in trypanosomes | No effective resistance | Several hypotheses based on laboratory-derived resistant strains |
| Nifurtimox | g-HAT 2nd stage Used in combination with eflornithine (NECT) | Inhibition of trypaniothione reductase, generation of free radicals toxic for the trypanosome, and mitochondrial disruption | No effective resistance | Several hypotheses based on laboratory-derived resistant strains |
| Fexinidazole | g-HAT 1st stage and 2nd stage | DNA synthesis inhibitor | No effective resistance | Several hypotheses based on laboratory-derived resistant strains |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capela, R.; Moreira, R.; Lopes, F. An Overview of Drug Resistance in Protozoal Diseases. Int. J. Mol. Sci. 2019, 20, 5748. https://doi.org/10.3390/ijms20225748
Capela R, Moreira R, Lopes F. An Overview of Drug Resistance in Protozoal Diseases. International Journal of Molecular Sciences. 2019; 20(22):5748. https://doi.org/10.3390/ijms20225748
Chicago/Turabian StyleCapela, Rita, Rui Moreira, and Francisca Lopes. 2019. "An Overview of Drug Resistance in Protozoal Diseases" International Journal of Molecular Sciences 20, no. 22: 5748. https://doi.org/10.3390/ijms20225748