


Abstract
The Dock-and-Lock (DNL) method, which makes bioactive molecules with multivalency and multifunctionality, is a new approach to develop targeting molecules for improved cancer imaging and therapy. It involves the use of a pair of distinct protein domains involved in the natural association between cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) and A-kinase anchoring proteins (AKAPs). The dimerization and docking domain found in the regulatory subunit of PKA and the anchoring domain (AD) of an interactive AKAP are each attached to a biologic entity, and the resulting derivatives, when combined, readily form a stably tethered complex of a defined composition that fully retains the functions of the individual constituents. The DNL method has generated several trivalent, bispecific, binding proteins, each consisting of 2 identical Fab fragments linked site-specifically to a different Fab fragment. For example, 2 identical Fabs reacting with carcinoembryonic antigen (CEA) are bound to a Fab reacting with a hapten peptide that bears a diagnostic or therapeutic radionuclide. Using a 2-step, pretargeting method that separates the bivalent anti-CEA antibody targeting of tumor from the delivery of the radioactive peptide that is captured by the second Fab of the tri-Fab construct, an improved method of cancer imaging and therapy has been developed and shows very sensitive and specific targeting of CEA-expressing tumors for either diagnostic imaging, such as with immunoSPECT and immunoPET, or radioimmunotherapy. Improved therapeutic efficacy is shown with pretargeting in a pancreatic cancer xenograft model given a tri-Fab to a pancreatic cancer MUC1 and the hapten peptide labeled with 90Y.
Monoclonal antibodies (mAbs) are in use and in clinical development for cancer imaging when conjugated with radionuclides and as naked mAbs, isotope-conjugates, or drug-conjugates for therapy (1,2). When used as naked antibodies, antitumor effects are achieved by direct cytotoxicity or indirect effector functions related to complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC) (2). As a naked mAb that can inhibit tumor proliferation by one or more of these mechanisms, increasing the residence time on the tumor by reducing off-rates, such as via increasing sites on the tumor bound by the antibody, may enhance the therapeutic efficacy. When conjugated to an isotope or drug, the slow localization to tumor while also distributing to various normal compartments supplied by circulating blood reduces the selective and high tumor accretion desired for the effector molecule. Both approaches (i.e., naked mAbs and conjugated mAbs) can potentially benefit by the design of multispecific, multifunctional mAbs. In the case of naked mAbs, using multispecific fusion proteins could improve tumor targeting and binding by reacting with multiple antigens on the tumor cell surface, whereas in the application of conjugated antibodies, the use of bispecific, multimeric mAbs can provide improved tumor targeting by the method of pretargeting. Here, antibody localization to the target is separated from delivery of the effector or therapeutic molecules, so that higher and more selective delivery of the effector molecules is achieved (3–5). In our own pretargeting technology, we focus on bispecific mAbs (bs-mAbs) consisting of one or more binding arms to the tumor and a different one to a carrier hapten. Once this localizes at the tumor and clears from blood and other normal tissues, the hapten bearing an imaging or therapeutic radionuclide is given and shows high accretion and high tumor-to-background ratios because it only binds to the mAb's extra arm at the tumor or clears rapidly from the body (6–14). Although this bs-mAb pretargeting technology is successful in both preclinical and clinical experiments (4), the use of chemical or recombinant constructs has presented certain production challenges that stimulated the search for versatile new methods, such as the Dock-and-Lock (DNL) platform technology, which is now described.
DOCK-AND-LOCK PLATFORM TECHNOLOGY
Protein Kinase A (PKA) and A-Kinase Anchoring Protein (AKAP)
The basis of the DNL method is the exploitation of the specific protein–protein interactions occurring in nature between the regulatory (R) subunits of protein kinase (PKA) and the anchoring domain (AD) of A-kinase anchoring proteins (AKAPs) (15,16). PKA, which plays a central role in one of the best-studied signal transduction pathways triggered by the binding of the second messenger cyclic adenosine monophosphate (cAMP) to the R subunits, was first isolated from rabbit skeletal muscle in 1968 (17). The structure of the holoenzyme consists of 2 catalytic subunits held in an inactive form by the R subunits (18). Isozymes of PKA are found with 2 types of R subunits (RI and RII), and each type has α- and β-isoforms (19). The R subunits have been isolated only as stable dimers, and the dimerization domain has been shown to consist of the first 44 amino-terminal residues (20). Binding of cAMP to the R subunits leads to the release of active catalytic subunits for a broad spectrum of serine/threonine kinase activities, which are oriented toward selected substrates through the compartmentalization of PKA via its docking with AKAPs (21).
Since the first AKAP, microtubule-associated protein-2, was characterized in 1984 (22), >50 AKAPs that localize to various subcellular sites—including plasma membrane, actin cytoskeleton, nucleus, mitochondria, and endoplasmic reticulum—have been identified with diverse structures in species ranging from yeast to humans (16). The AD of AKAPs for PKA is an amphipathic helix of 14–18 residues (23). The amino-acid sequences of the AD are quite varied among individual AKAPs, with the binding affinities reported for RII dimers ranging from 2 to 90 nM (24). Interestingly, AKAPs will bind only to dimeric R subunits. For human RIIα, the AD binds to a hydrophobic surface formed by the 23 amino-terminal residues (25). Thus, the dimerization domain and AKAP binding domain of human RIIα are both located within the same amino-terminal 44-amino-acid sequence, which is termed the dimerization and docking domain (DDD) herein (20,26).
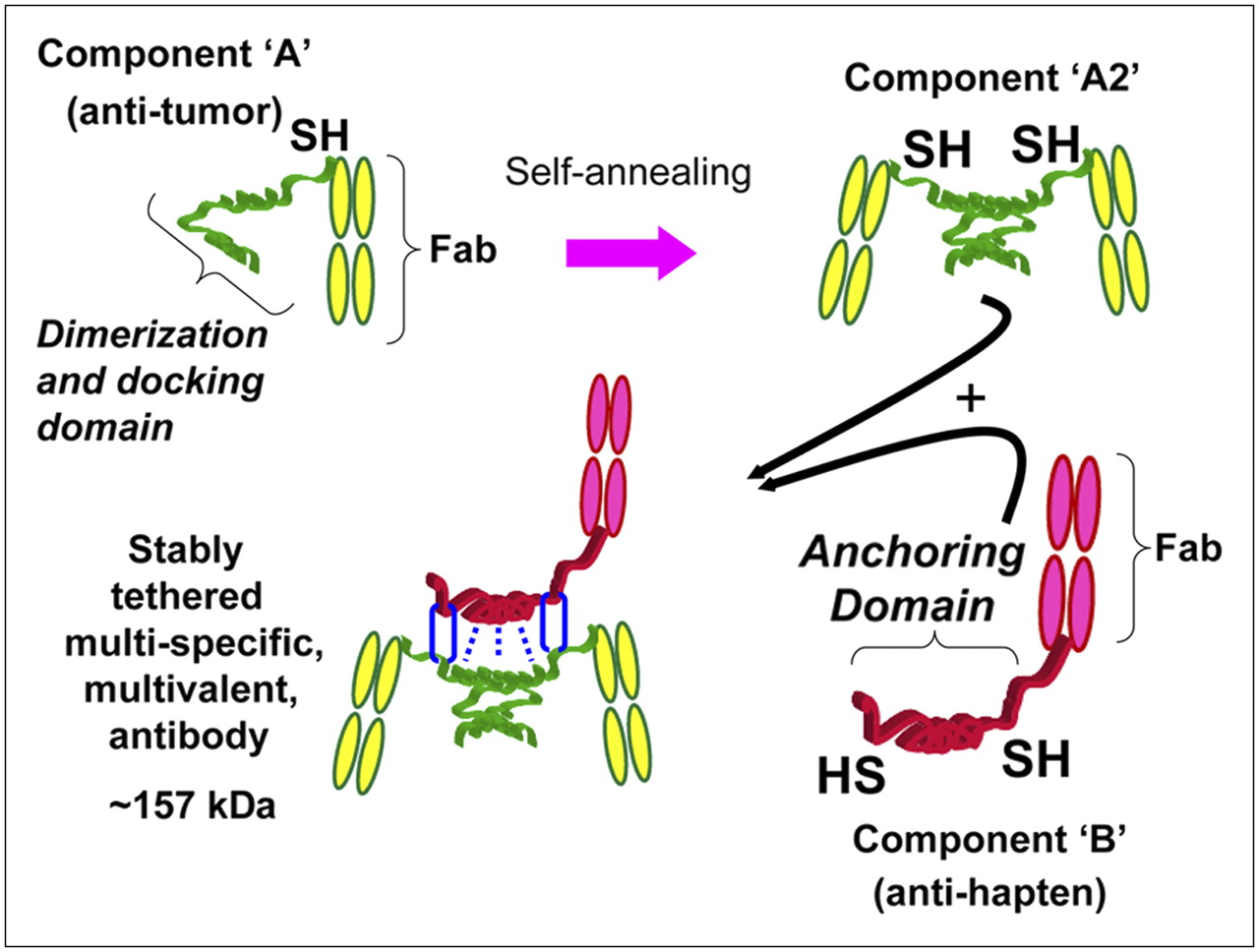
This platform technology exploits the DDD of human RIIα and the AD of a certain amino acid sequence as an excellent pair of linker modules for docking any 2 entities into a noncovalent complex, which could be further locked into a stably tethered structure through the introduction of cysteine residues into both the DDD and the AD at strategic positions to facilitate the formation of disulfide bonds, as illustrated in Figure 1. The general methodology of our DNL approach is as follows. A recombinant protein is constructed by linking a DDD sequence to the compound of interest, which, in the example shown in Figure 1, is a Fab fragment of an antitumor antibody. Because the DDD sequence effects the spontaneous formation of a dimer, the resulting recombinant protein is a divalent Fab fragment. To make the end product bispecific, a second recombinant protein is prepared by fusing an AD sequence, in this case, to an antihapten Fab fragment.

Schematic representation of how a tri-Fab (TF) bispecific antibody is formed by the DNL method. Component A links a cysteine-modified DDD to a Fab of an antitumor antibody, which results in spontaneous formation of component A2. Component B links an anchoring domain (AD) to a Fab of an antihapten antibody. The AD is modified with cysteine on each end. AD will naturally “dock” with the DDD when components A and B are mixed, which brings the 2 molecules together in a well-defined orientation and also results in disulfide bonds across the 2 proteins.
The dimeric motif of DDD in the first recombinant protein creates a docking site for binding to the AD sequence, thus facilitating a ready association of the dimeric antitumor Fab construct with the monomeric antihapten to form a binary, trimeric complex. This binding event is made irreversible with a subsequent reaction to secure the 2 entities covalently via disulfide bridges between the inserted cysteine residues. This reaction occurs very efficiently, because the initial binding interactions bring the reactive thiol groups on both the DDD and AD into proximity to ligate site-specifically (27). By attaching the DDD and AD away from the functional groups of the 2 precursors, such site-specific ligations preserve the original activities of the 2 precursors. This approach is modular in nature and potentially can be applied to link, site-specifically and covalently, a wide range of substances, including peptides, proteins, and nucleic acids, and in fact could provide a new method of specific localization to tumors and other disease targets of cytokines, vaccines, interference RNAs, and genes (28).
Bispecific Trivalent Structures Composed of 3 Stably Linked Fab Fragments
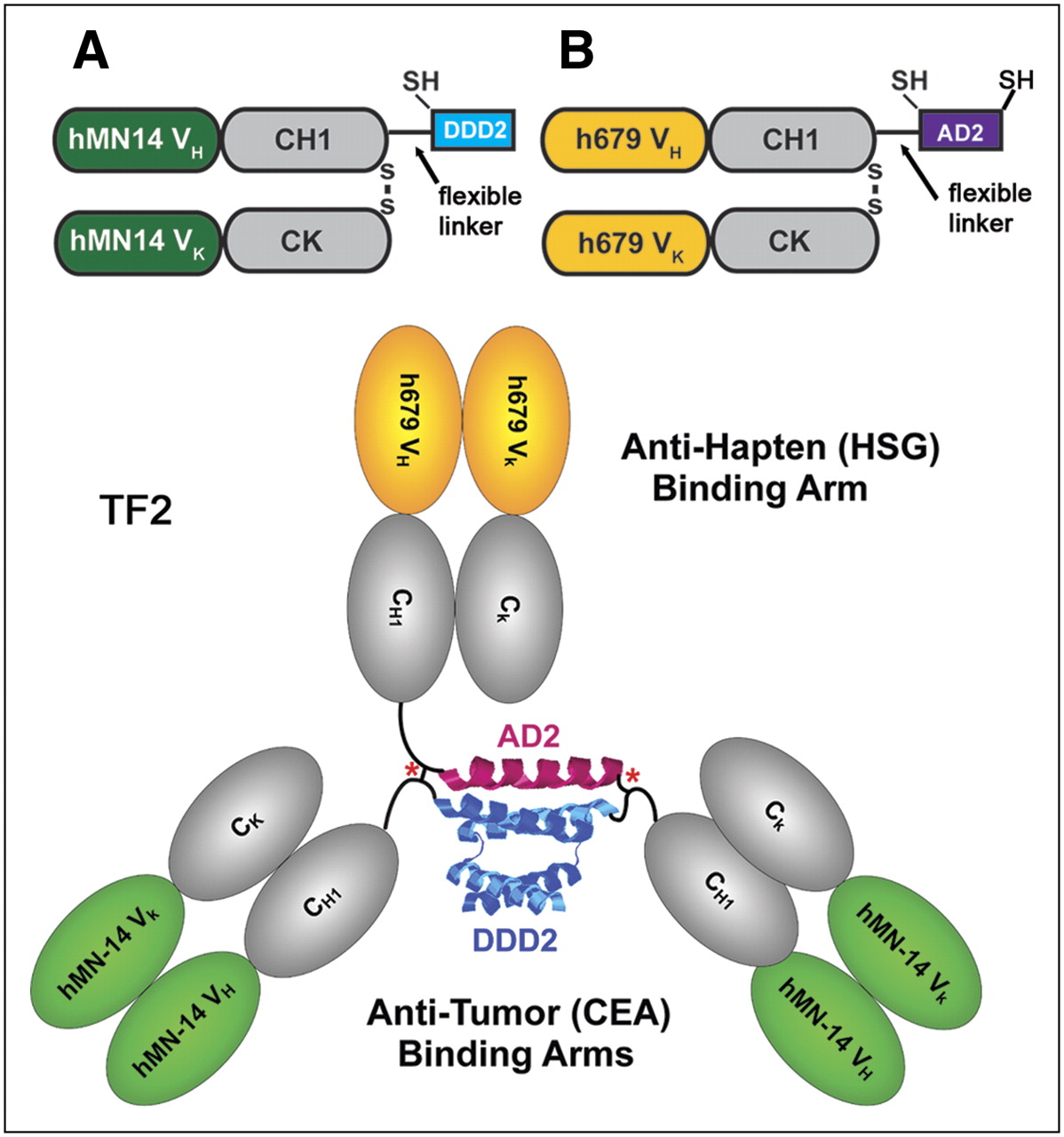
For proof-of-concept, we used the DNL method to assemble bispecific complexes comprising 3 Fab fragments. A DDD-containing entity was made recombinantly using as a precursor the Fab fragment of the humanized mAb hMN-14, which has binding specificity for human carcinoembryonic antigen ([CEA] CEACAM5) (29). The fusion protein, designated C-DDD2-Fab-hMN-14, was generated by linking the DDD2 peptide sequence, which is composed of an amino-terminal cysteine residue followed by amino acids 1–44 of human RIIα, to the carboxyl-terminal end of the Fd chain (Fd is the heavy chain portion of a Fab, consisting of the VH and CH1 domains) via a 14-residue flexible peptide linker (Fig. 2A). The AD-containing entity was generated recombinantly using as a precursor the Fab fragment of the humanized mAb h679, which has binding specificity for histamine-succinyl-glycine (HSG) (12). C-AD2-Fab-h679 was generated by linking the 17-residue amino-acid sequence derived from AKAP-IS, a synthetic peptide optimized for RII-selective binding with a reported dissociation constant (Kd) of 0.4 nM (24), to the carboxyl-terminal end of the Fd chain via a 15-residue flexible peptide linker (Fig. 2B).
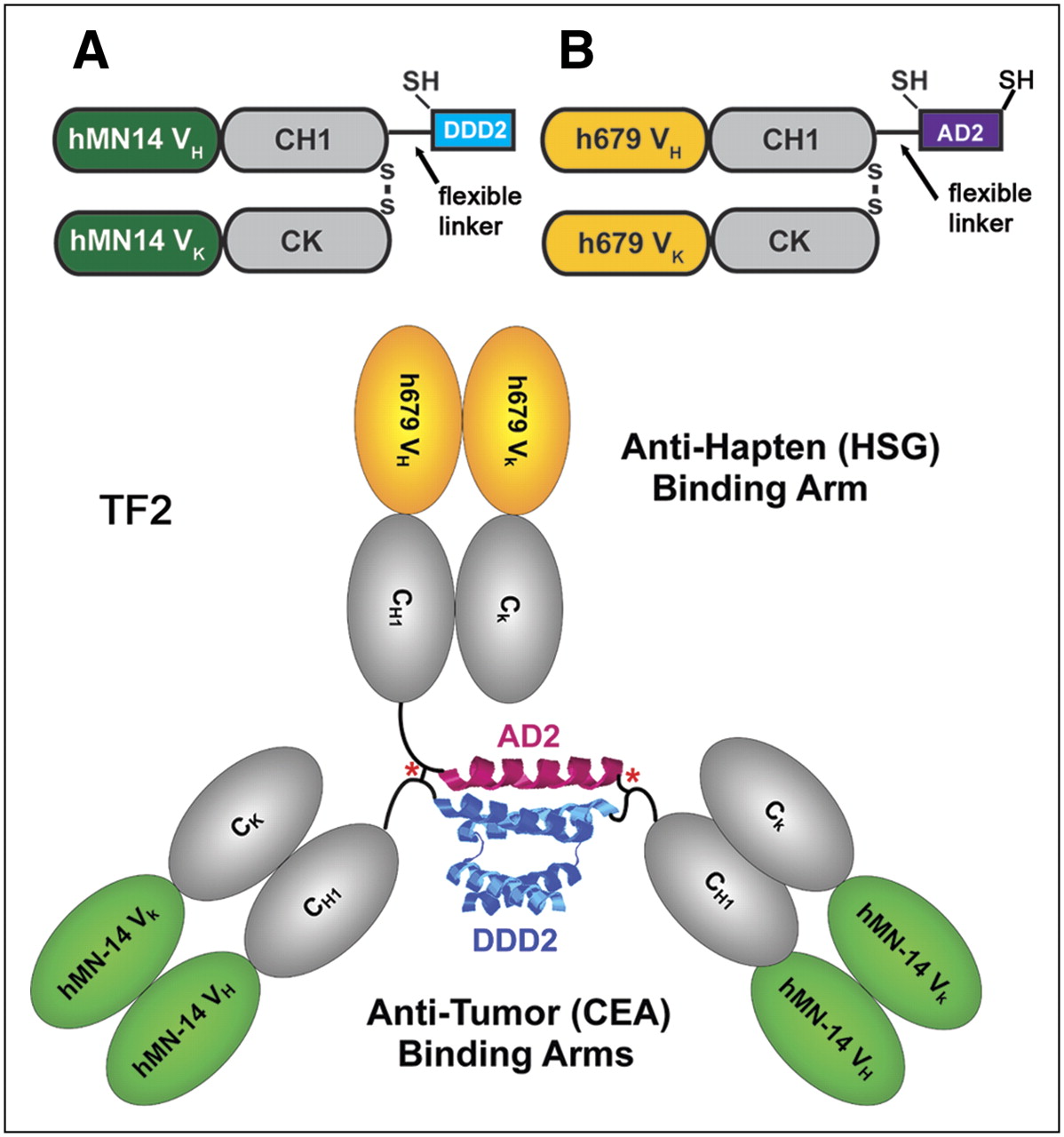
Components of TF2 anti-CEA × anti-HSG bs-mAb. (A) C-DDD2-Fab-hMN-14 was formed by linking a cysteine-modified peptide sequence (DDD2), which is composed of amino acids 1–44 of human RIIα, to the carboxyl-terminal end of the Fd chain via a 14-residue flexible peptide linker. (B) Antihapten binding arm was made by linking the 17-residue amino-acid sequence derived from AKAP-IS, a synthetic peptide optimized for RII-selective binding, to the carboxyl end of the Fd via a 15-residue flexible peptide linker with the addition of cysteine residues to both the amino- and carboxyl-terminal ends of AD2. TF2 is the anti-CEA × anti-HSG recombinant humanized protein formed by combining A and B.
To prevent the dissociation of a noncovalent complex at lower concentrations—thus allowing in vivo applications—as mentioned earlier, cysteine residues were added into the DDD and AD sequences. We anticipated that on mixing of the cysteine-modified entities, a dimer–monomer complex would promptly form, which could be further stabilized by the formation of disulfide bridges. A stably tethered trivalent bispecific structure, referred to as TF2, was obtained in nearly quantitative yields after tris(2-carboxyethyl)phosphine hydrochloride (TCEP) reduction, dimethyl sulfoxide oxidation, and affinity purification. Similar results were achieved by substituting TCEP and dimethyl sulfoxide with reduced and oxidized glutathione, respectively. TF2 was shown by size-exclusion high-performance liquid chromatography to be a single peak of the expected molecular size (∼150 kDa), by BIAcore to be bispecific for both histamine-succinyl-glycine and a rat antiidiotype mAb to hMN-14 (30), and by competition enzyme-linked immunosorbent assay to be equivalent to hMN-14 IgG and h679 Fab, reflecting the full retention of valency and binding affinity. Furthermore, TF2 was found to be stable for at least 7 d when incubated at 37°C in human or mouse serum, and the superiority of TF2 as a pretargeting agent for diagnostic imaging has been demonstrated in nude mice bearing CEA-expressing, human colonic cancer xenografts.
PRETARGETING AS A NEW PARADIGM
The major problem with the delivery of antibody-targeting radionuclides is that the efficiency of selective targeting is relatively poor. Maximum radiation-absorbed doses measured in solid tumors most often are much < 2,000 cGy at the maximum tolerated dose (31). The difference between the continuous low-dose-rate radiation delivered by a radioconjugate and conventional external-beam radiation makes it difficult to be certain what threshold might be required to achieve objective responses, but there is little doubt that directly radiolabeled antibodies have not reached this level, even with hematopoietic support. Thus, alternative strategies that could substantially increase the radiation dose to tumors are necessary.
Solving the problem associated with the deficiencies in targeting directly radiolabeled antibodies requires a separation of the targeting moiety from the effector bearing the radionuclide, so as to permit administration of the radionuclide when optimal targeting has been accomplished. To accomplish this, the targeting was first achieved with a bs-mAb that binds to a target antigen as well as to the effector molecule with the radiometal–chelate complex (32,33). By separating the targeting steps, the bs-mAb does not need to be treated with reagents used for conjugation to a suitable radionuclide, which ensures its immunoreactivity is retained. Other pretargeting methods have been developed and applied, particularly involving biotin-avidin (or streptavidin) binding, with encouraging results shown in certain situations (4). Unlike approaches based on avidin/streptavidin that rely on a clearing step, bs-mAb pretargeting simply delays administering the radionuclide until the bs-mAb has cleared sufficiently from the blood. Chemically conjugated bs-mAbs composed of 2 linked Fab′ (e.g., ∼100 kDa) were smaller in size than the larger IgG-based streptavidin conjugates (e.g., 220 kDa), which allowed them to clear more quickly from the blood. Other recombinant bs-mAbs used for pretargeting have been even smaller in size (34,35). The current DNL tri-Fab constructs are ∼157 kDa but, because they lack the Fc portion of the IgG, they are expected to have a more rapid clearance than IgG-based constructs. In addition, streptavidin's ultra-high affinity would require the concentration of the avidin-based conjugate to be reduced to far lower levels in the blood than that required for a bs-mAb; therefore, the use of a clearing agent is very important for those methods. In avidin-biotin approaches, radiolabeled biotin is strongly bound to streptavidin localized at the tumor site. bs-mAb pretargeting relies on a radiolabeled effector that bears 2 haptens capable of binding to the antihapten binding arm of the bs-mAb. Divalent haptens have been shown to have a binding advantage over monovalent hapten structures, originally described as the affinity-enhancement system (36–38).
What is the potential role of pretargeting for the molecular imaging of cancer, and how does the DNL platform technology contribute to this technology? Although pretargeting methods could be considered to be more complicated than a single-agent targeting method, once the optimal conditions are established, the injection sequence is straightforward. Irrespective of the pretargeting method used, patients would be able to receive the injection of the bs-mAb or antibody conjugate by their oncologist and then return 1 or more days later to receive the radiolabeled product in a nuclear medicine facility. Imaging could be performed within a few hours of this injection. Thus, for a pretargeting procedure, it will take a few days before an image is acquired, whereas it may be possible to image in a shorter period of time with some of the smaller, directly radiolabeled antibody constructs. In our experience, preclinical data suggest that image quality with pretargeting is superior to that of a directly radiolabeled antibody fragment (8,13,14).
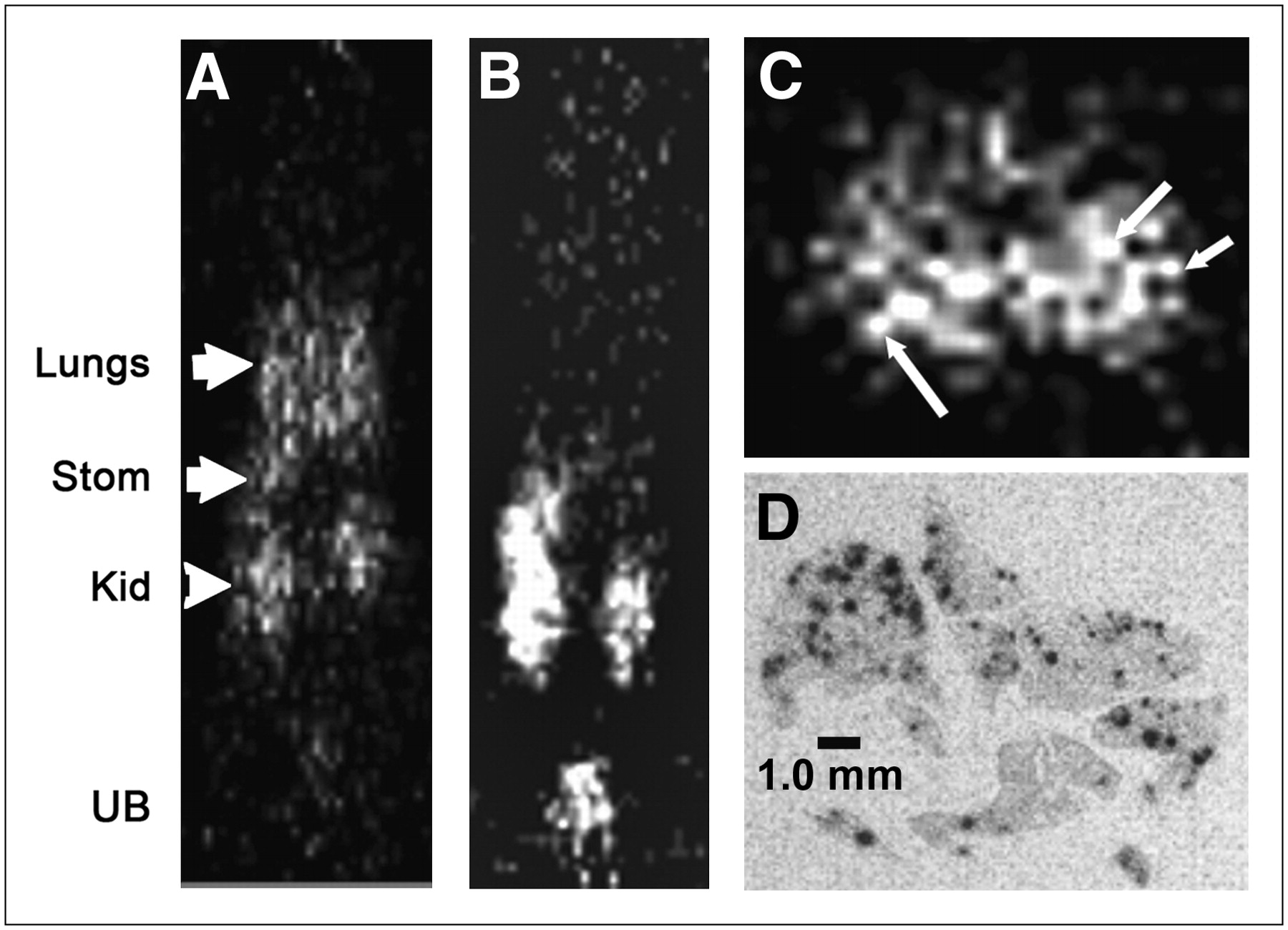
Using the DNL method to construct tri-Fab forms having 2 Fab arms binding to the tumor-associated antigen CEA, and the third Fab binding to the HSG hapten, we have been able to gain preclinical evidence of superior tumor targeting and imaging, by both SPECT and PET (14,39). These results confirm our prior experience with trivalent, bispecific antibodies against the same targets, but using other types of recombinantly made reagents (8,13). In recent studies with DNL-constructed pretargeting molecules for imaging with an 124I-labeled HSG hapten-peptide, microdisseminated human colon tumor nodules—no larger than 0.3 mm in diameter—in the lungs of nude mice were detected using pretargeted small-animal PET (Fig. 3), whereas 18F-FDG was unable to disclose evidence of tumor involvement (Fig. 4) (14). At necropsy, the microscopic involvement could be seen, but the tumor-bearing lungs taken from the pretargeted animals had 9-fold more radioactivity than lungs taken from pretargeted animals not implanted with tumor. In contrast, uptake in the lungs from animals given 18F-FDG or the 124I-hapten-peptide alone (no pretargeting) was only 1.5-fold higher in the tumor-bearing animals than that in animals without tumor. Separate autoradiography studies showed that the radioactivity in the lungs of the pretargeted animals was concentrated around the small colonies of tumor cells (Fig. 3D).

Localization of a micrometastatic human colon cancer cell line disseminated in lungs of nude mice using small-animal PET. Necropsy data showed all animals with ∼80 to 120 tumor nodules in lungs, ranging in size from a few cells to colonies that were <0.3 mm in diameter. (A) Activity is seen in lungs of coronal section 1.5 h after anti-CEA pretargeted 124I-hapten-peptide injection. Additionally, uptake was seen in kidneys (Kid) due to peptide clearance through this organ and stomach (Stom) as a result of catabolized iodide. (B) A tumor-bearing animal given only 124I-hapten peptide without bs-mAb showed no selective uptake in chest. In this slice, activity is also seen in kidneys, stomach, and urinary bladder (UB). (C) Transverse section through the chest shows uptake, with appearance of individual nodules (arrows). (D) Autoradiography study was performed in separate animals pretargeted with an 111In-labeled peptide at a later time after tumor implantation, but, even in these animals, individual tumor nodules did not exceed 0.5 mm. Autoradiography revealed selective uptake in and around tumor nodules within lungs.
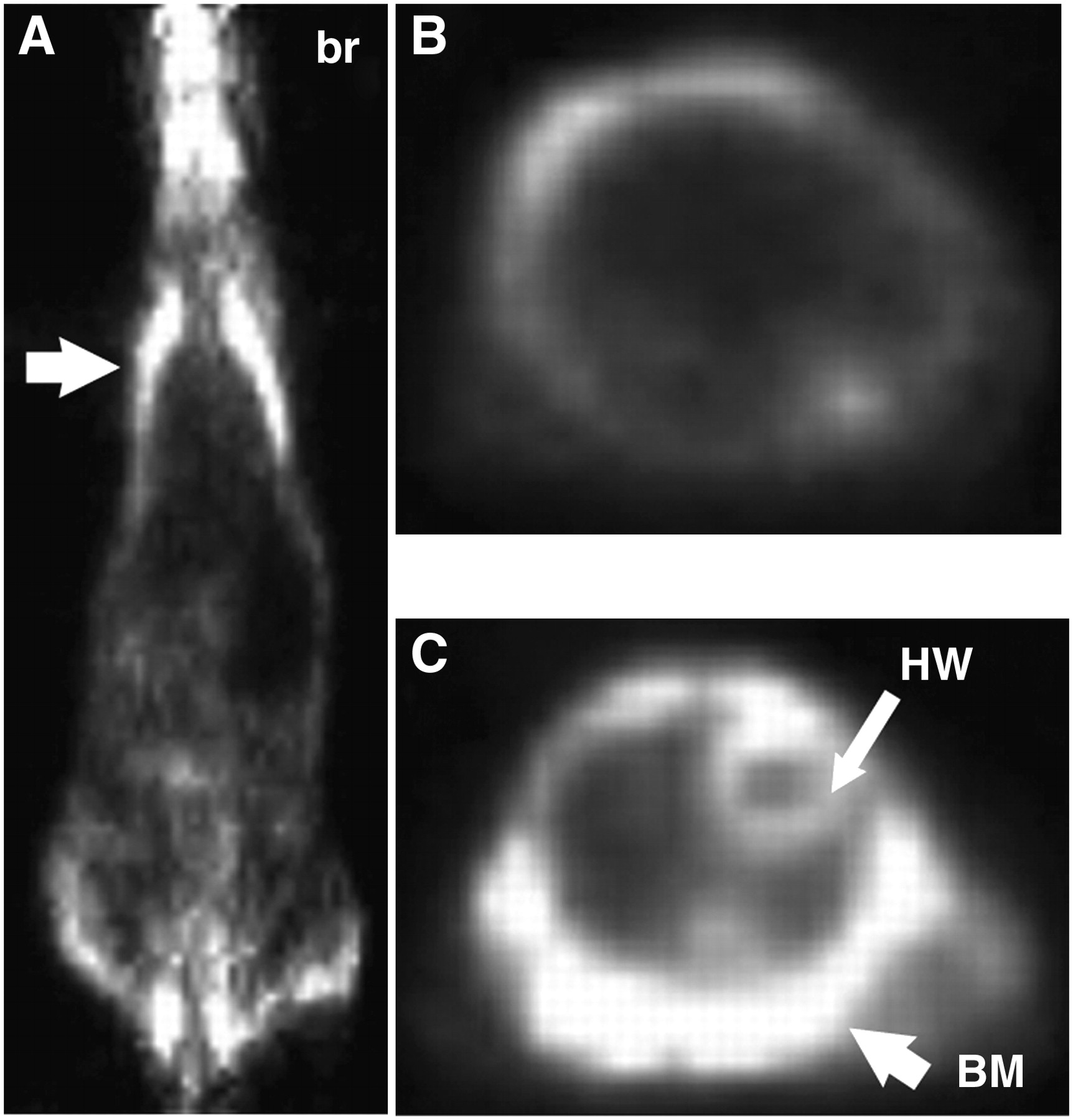
From the same study described in Figure 3, (A) is coronal section of an animal 1.5 h after 18F-FDG injection. Arrow indicates the lung plane, where no evidence of tumor involvement was seen. br = brain. (B and C) Transverse sections through different region of chest also failed to show tumor. Strong uptake was seen in bone marrow (BM) of ribs and shoulder blades surrounding the chest and, as expected, in heart wall (HW) and brain.
For therapy, improved antitumor responses, longer survival, and higher cure rates have been observed with bs-mAb pretargeting in animals bearing solid tumors (colorectal and pancreatic cancers) and lymphomas using DNL agents. Figure 5 illustrates initial studies examining the pretargeting ability of a DNL construct that binds to a MUC1 epitope that is highly selective for pancreatic cancer (40) and therapeutic efficacy of a 90Y-labeled hapten-peptide administered to nude mice bearing subcutaneous pancreatic tumor xenografts previously pretargeted with this anti-MUC1 bs-mAb. Biodistribution studies showed that at 3 h after the radiolabeled hapten-peptide injection, there was a 125-fold higher uptake of the pretargeted peptide in the tumor compared with that in the peptide alone (Table 1). Tumor-to-nontumor ratios were more than 100:1 for the normal tissues, except the kidneys, where the ratio at 3 h after injection was already 7.3 ± 1.2. Tumor-to-blood ratios averaged about 1,000:1, ranging from ∼45:1 to 2,500:1 in the 5 animals examined. Therapeutically, a single injection of 18.5 MBq (0.5 mCi) of the 90Y-hapten-peptide produced complete regression of 8 of 10 tumors. Prior experience indicated that the maximum tolerated dose for a similar pretargeting procedure was 25.9 MBq (0.7 mCi) (7); thus, further escalation is possible.
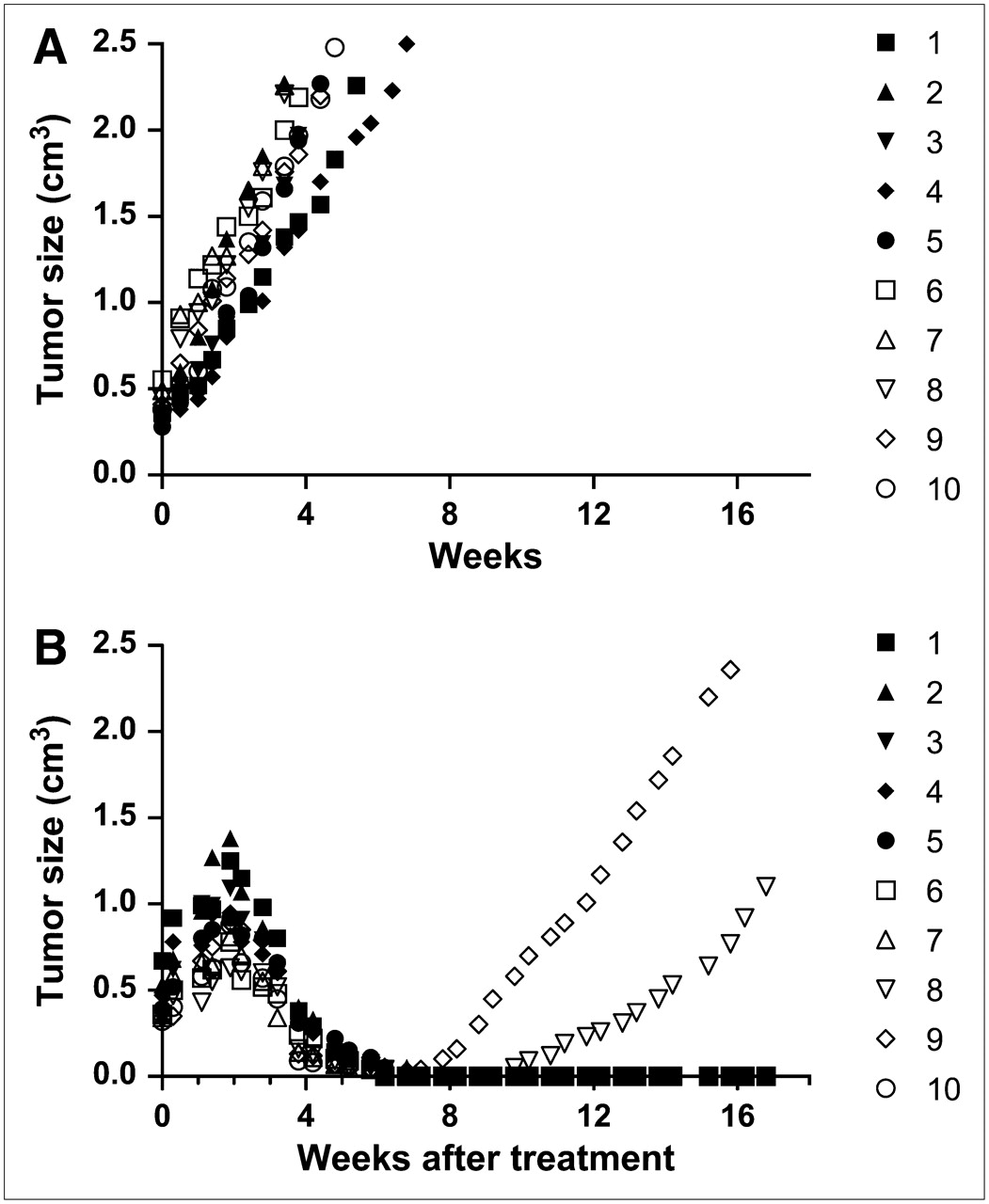
Therapeutic efficacy of 90Y-labeled peptide pretargeted with an anti-MUC1 DNL bs-mAb. Nude mice bearing ∼0.5 cm3 CaPan1 human pancreatic cancer subcutaneous xenografts were untreated (A; n = 10 mice) or were treated with 18.5 MBq (0.5 mCi) of a pretargeted 90Y-labeled hapten-peptide (B; n = 10 mice).
Tumor and Organ Uptake of 111In-Peptide 3 Hours After Intravenous Injection
CONCLUSION
The limitations experienced by directly labeling antibodies with radionuclides for radioimmunodetection and radioimmunotherapy appear to be overcome considerably by pretargeting methodologies, as reviewed earlier (4). However, to have a facile and convenient process for bispecific antibody production involving modules that can be adapted to different targeting systems and antigens, as well as a virtually universal peptide for radionuclide conjugation, the DNL platform technology has been invented and reduced to practice, as shown here for immunoPET and radioimmunotherapy in 2 preclinical cancer models. On the basis of the clinical success of first-generation bispecific antibody–pretargeting agents (4,41–43), we are now ready to evaluate DNL-produced agents for imaging and therapy of cancer in patients.
Acknowledgments
This research was supported in part by U.S. Public Health Service grants P01 CA10395, R01 CA107088, and R01 CA115755 from the NIH and by grant 06-1853-FS-N0 from the New Jersey Department of Health and Senior Services.
Footnotes
-
Dr. Goldenberg is the recipient of the 2005 Paul C. Aebersold award of the SNM. He declares a financial interest in Immunomedics, Inc., and IBC Pharmaceuticals, Inc., as a stockholding officer of both corporations. Drs. Rossi, McBride, and Chang are employees and stockholders of Immunomedics and/or IBC Pharmaceuticals. Dr. Sharkey declares no financial interests.
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication August 8, 2007.
- Accepted for publication September 21, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Pretargeting: A Path Forward for Radioimmunotherapy
- A Self-Assembling and Disassembling (SADA) Bispecific Antibody (BsAb) Platform for Curative Two-step Pretargeted Radioimmunotherapy
- {alpha}- Versus {beta}-Emitting Radionuclides for Pretargeted Radioimmunotherapy of Carcinoembryonic Antigen-Expressing Human Colon Cancer Xenografts
- Development of a Radiolabeled Irreversible Peptide Ligand for PET Imaging of Vascular Endothelial Growth Factor
- Advances in the treatment of hematologic malignancies using immunoconjugates
- Quantitative Immuno-SPECT Monitoring of Pretargeted Radioimmunotherapy with a Bispecific Antibody in an Intraperitoneal Nude Mouse Model of Human Colon Cancer
- Phase II Trial of Anticarcinoembryonic Antigen Pretargeted Radioimmunotherapy in Progressive Metastatic Medullary Thyroid Carcinoma: Biomarker Response and Survival Improvement
- Pretargeted 177Lu Radioimmunotherapy of Carcinoembryonic Antigen-Expressing Human Colonic Tumors in Mice
- Pretargeted Immuno-Positron Emission Tomography Imaging of Carcinoembryonic Antigen-Expressing Tumors with a Bispecific Antibody and a 68Ga- and 18F-Labeled Hapten Peptide in Mice with Human Tumor Xenografts
- CD20-targeted tetrameric interferon-{alpha}, a novel and potent immunocytokine for the therapy of B-cell lymphomas
- Pretargeted Radioimmunotherapy Using Anti-CD45 Monoclonal Antibodies to Deliver Radiation to Murine Hematolymphoid Tissues and Human Myeloid Leukemia
- A Novel Bispecific, Trivalent Antibody Construct for Targeting Pancreatic Carcinoma