

Abstract
Cholecystokinin (CCK), a neuropeptide originally discovered in the gastrointestinal tract, is abundantly distributed in the mammalian brains including the hippocampus. Whereas CCK has been shown to increase glutamate concentration in the perfusate of hippocampal slices and in purified rat hippocampal synaptosomes, the cellular and molecular mechanisms whereby CCK modulates glutamatergic function remain unexplored. Here, we examined the effects of CCK on glutamatergic transmission in the hippocampus using whole-cell recordings from hippocampal slices. Application of CCK increased AMPA receptor-mediated EPSCs at perforant path-dentate gyrus granule cell, CA3-CA3 and Schaffer collateral–CA1 synapses without effects at mossy fiber-CA3 synapses. CCK-induced increases in AMPA EPSCs were mediated by CCK-2 receptors and were not modulated developmentally and transcriptionally. CCK reduced the coefficient of variation and paired-pulse ratio of AMPA EPSCs suggesting that CCK facilitates presynaptic glutamate release. CCK increased the release probability and the number of readily releasable vesicles with no effects on the rate of recovery from vesicle depletion. CCK-mediated increases in glutamate release required the functions of phospholipase C, intracellular Ca2+ release and protein kinase Cγ. CCK released endogenously from hippocampal interneurons facilitated glutamatergic transmission. Our results provide a cellular and molecular mechanism to explain the roles of CCK in the brain.
Introduction
Cholecystokinin (CCK) interacts with two G-protein-coupled receptors: CCK-1 and CCK-2. Whereas CCK-1 receptors are present in peripheral tissues and in a few discrete brain regions including postrema, interpeduncular nucleus and nucleus tractus solitarius (Moran et al., 1986; Hill et al., 1987, 1990), CCK-2 receptors are the predominant form found in the brain (Van Dijk et al., 1984). Activation of CCK-1 and CCK-2 receptors increases the activity of phospholipase C (PLC) which in turn enhances the hydrolysis of phosphatidylinositol 4, 5-bisphosphate (PIP2) into inositol trisphosphate (IP3) to increase intracellular Ca2+ release, and diacylglycerol to activate protein kinase C (PKC) (Wank, 1995). Activation of CCK-1, but not CCK-2 receptors, also increases adenylyl cyclase activity which enhances the generation of cAMP and subsequent activation of protein kinase A (Wank, 1995).
CCK is extensively expressed in the hippocampus. CCK-like immunoreactive perikarya are located in the dentate hilus and all layers of Ammon's horn (Greenwood et al., 1981). The CCK-immunoreactive neurons represent a subpopulation of GABAergic interneurons (Somogyi et al., 1984; Nunzi et al., 1985). CCK-immunoreactive fibers are located around the cell bodies in the stratum pyramidale of Ammon's horn and among the granule cells and especially the inner molecular layer of the dentate gyrus (Greenwood et al., 1981; Hefft and Jonas, 2005) suggesting that significant amounts of endogenous CCK are released in the hippocampus. The hippocampus also expresses a high density of CCK receptors. The dentate gyrus exhibits the highest numbers of binding sites for CCK and moderate labeling has been detected in the stratum pyramidale of CA3 and CA1 regions (Köhler and Chan-Palay, 1988; Kritzer et al., 1988) suggesting that CCK plays an important role in the hippocampus. Indeed, CCK has been shown to increase the excitabilities of dentate gyrus granule cells (Brooks and Kelly, 1985; Sinton, 1988), CA3 (Gronier and Debonnel, 1995) and CA1 (Dodd and Kelly, 1981; Jaffe et al., 1987; Boden and Hill, 1988; Böhme et al., 1988) pyramidal neurons and modulates GABAergic transmission in the hippocampus (Miller et al., 1997; Deng and Lei, 2006; Földy et al., 2007; Karson et al., 2008). In addition, CCK has been shown to increase glutamate release in the perfusate of hippocampal slices (Migaud et al., 1994) and from purified rat hippocampal synaptosomes (Breukel et al., 1997). However, the cellular and molecular mechanisms whereby CCK modulates glutamatergic synaptic transmission remain unexplored. In this study, we examined the effects of CCK on glutamatergic transmission in the hippocampus and our results demonstrate that CCK facilitates glutamate release in the hippocampus by increasing the number of readily releasable vesicles and releasing probability via PLC, intracellular Ca2+ and PKC signals. We also showed that endogenously released CCK from hippocampal interneurons facilitates glutamate release. CCK-mediated enhancement of glutamate release may be involved in CCK-induced facilitation of anxiety.
Materials and Methods
Slice preparation.
Horizontal brain slices (400 μm) including the hippocampus, subiculum and entorhinal cortex were cut using a vibrating blade microtome (VT1000S; Leica) from 15- to 22-d-old Sprague Dawley rats as described previously (Deng and Lei, 2006, 2007; Deng et al., 2009; Xiao et al., 2009). After being deeply anesthetized with isoflurane, rats were decapitated and their brains were dissected out in ice-cold saline solution that contained (in mm) 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 5.0 MgCl2 and 10 glucose, saturated with 95% O2 and 5% CO2, pH 7.4. Slices were initially incubated in the above solution at 35°C for 40 min for recovery and then kept at room temperature (∼24°C) until use. All animal procedures conformed to the guidelines approved by the University of North Dakota Animal Care and Use Committee.
Recordings of synaptic currents.
Whole-cell patch-clamp recordings using an Axopatch 200B or two Multiclamp 700B amplifiers in current- or voltage-clamp mode from in vitro hippocampal slices were used for experiments. Cells in the slices were visually identified with infrared video microscopy and differential interference contrast optics (Deng et al., 2006; Lei et al., 2007). Recording electrodes were filled with the solution containing (in mm) 100 Cs-gluconate, 0.6 EGTA, 5 MgCl2, 8 NaCl, 2 ATP2Na, 0.3 GTPNa, 40 HEPES and 1 QX-314, pH 7.3. The extracellular solution comprised (in mm) 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 2.5 CaCl2, 1.5 MgCl2 and 10 glucose, saturated with 95% O2 and 5% CO2, pH 7.4. Bicuculline (10 μm) was included in the extracellular solution to block GABAA receptors. The holding potential was at −60 mV. AMPA receptor-mediated EPSCs were evoked by placing a stimulation electrode in the middle to the inner one third of molecular layer of dentate gyrus to stimulate the medial perforant path or in the stratum radiatum of the CA3 region to stimulate the recurrent fibers or in the stratum radiatum of the CA1 region to stimulate the Schaffer collateral fibers in different experiments. Under these conditions, the recorded currents were completely blocked by application of DNQX (10 μm) or GYKI 52466 (100 μm) at the end of experiments confirming that they were mediated by AMPA receptors. Series resistance was rigorously monitored by the delivery of 5 mV voltage steps after each evoked current. Experiments were discontinued if the series resistance changed by >10%. Miniature AMPA EPSCs (mEPSCs) were recorded from dentate gyrus granule cells in the presence of TTX (0.5 μm). Data were filtered at 2 kHz, digitized at 10 kHz, acquired on-line and analyzed after-line using pCLAMP 9 software (Molecular Devices). The recorded mEPSCs were analyzed afterward using Mini Analysis 6.0.1 (Synaptosoft Inc.). To avoid potential desensitization induced by repeated bath applications of CCK, one slice was limited to only one application of CCK and only one cell was recorded from each slice.
Dual-electrode recordings and immunocytochemistry.
The experiments examining the effects of endogenously released CCK on glutamate release (see Fig. 7) were conducted using a Multiclamp 700B amplifier. The stimulation electrode containing the extracellular solution was first placed into the internal molecular layer for stimulation of the perforant path. The patch electrode sealed to the interneuron contained (in mm) 100 K-gluconate, 0.6 EGTA, 5 MgCl2, 8 NaCl, 2 ATP2Na, 0.3 GTPNa, 40 HEPES, pH 7.2–7.3. The patch electrode sealed to the granule cell contained the same Cs-gluconate solution described above. Both electrodes contained 0.2% biocytin. Data were collected only from the recordings of which the holding currents of the granule cells at −60 mV were less than −100 pA and the resting membrane potentials of the interneurons were negative to −60 mV. The extracellular solution contained bicuculline (10 μm) to block GABAA receptors, CGP55845 (5 μm) to block GABAB receptors, AM 251 (10 μm) to block cannabinoid receptors and amastatin (10 μm) to inhibit amino-peptidase (Su et al., 2002). After recordings, slices were fixed in 4% paraformaldehyde for 24 h. After extensive washing in 0.1 m PBS, slices were incubated with PBS containing 5% normal donkey serum and 1% Triton X-100 for 2 h. Slices were then incubated with goat anti-CCK antibody (sc-21615, Santa Cruz Biotechnology, CA) at a dilution of 1:100 for 48 h at 4°C. Slices were incubated with donkey anti-goat IgG-FITC (sc-2024, Santa Cruz Biotechnology, 1:200, for detecting CCK) and Texas red-conjugated streptavidin (SA-5006, Vector Laboratories, 1:200, for detecting biocytin-filled neurons) for 2 h at room temperature. After washing, slices were mounted on slides and coverslipped. Finally, slides were visualized with an Olympus Fluoview 300 confocal microscope and photographed. The specificity of the CCK immunoreactivity was confirmed by pretreatment of the CCK antibody with the corresponding CCK blocking peptide (1:5 by weight, sc-21615P, Santa Cruz Biotechnology Inc.) in PBS for ∼2 h at room temperature. The mixture was then diluted at 1:100 (according to the CCK primary antibody) and used for immunostaining with the same procedures described above.
Perforated patch-recordings of action potential (AP) firing from dentate granule cells.
Perforated-patch recordings were used to record AP firing from dentate granule cells as described previously (Deng and Lei, 2007). Recording pipettes were tip-filled with the above K+-gluconate intracellular solution and then back-filled with freshly prepared K+-gluconate intracellular solution containing amphotericin B (200 μg/ml, Calbiochem). Patch pipettes had resistance of 6–8 MΩ when filled with the preceding solution. A 5 mV hyperpolarizing test pulse was applied every 5 s to monitor the changes of the series resistance and the process of perforation. Stable series resistances (50–70 MΩ) were usually obtained ∼30 min after the formation of gigaohm seals. For those cells showing abrupt reduction in series resistance during membrane perforation suggesting the simultaneous formation of whole-cell configuration, experiments were terminated immediately. Perforated-patch configurations were verified by examining the series resistance again at the end of the experiments. Data were included for analysis only from those cells showing <15% alteration of series resistance.
Recordings of calcium and potassium currents from identified stellate neurons in the entorhinal cortex.
Depolarizing voltage sag in response to hyperpolarizing current injections and K+ channel currents were recorded from stellate neurons in layer II of the entorhinal cortex using K+-containing intracellular solution comprising (in mm) 100 K-gluconate, 0.6 EGTA, 5 MgCl2, 8 NaCl, 40 HEPES, 2 MgATP, 0.3 NaGTP, 0.1 leupeptin, 20 phosphocreatine and 50 U/ml phosphocreatine kinase, pH 7.3. K+ currents were isolated by including, in the above extracellular solution, TTX (0.5 μm) to block Na+ channels and CdCl2 (100 μm) to block Ca2+ channels. Ca2+ currents were recorded with intracellular solution containing (in mm) Cs-gluconate 100, NaCl 5, MgCl2 2, CaCl2 0.5, TEA-Cl 20, EGTA 2, HEPES 10, phosphocreatine 20, phosphocreatine kinase 50 U/ml, MgATP 2, NaGTP 0.1 and leupeptin 0.1, pH 7.3. The extracellular solution was supplemented with 10 mm TEA-Cl and 0.5 μm TTX. Ca2+ currents were induced by depolarization from −70 to 0 mV for 200 ms.
Tissue culture.
Primary hippocampal neurons were cultured from embryonic C57BL/6 mice at 16 d of gestation. Briefly, the pregnant mice were killed by asphyxiation with CO2 and the fetuses were removed and decapitated. The hippocampi were isolated and incubated with 0.25% trypsin-EDTA for 10 min, dissociated by trituration with fire-polished glass pipettes, and plated on poly-d-lysine-coated glass-bottom 35 mm tissue culture dishes. Neurons were grown in Neurobasal medium with l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 0.25 μg/ml amphotericin B and B27 supplement (Invitrogen) at 37°C in 5% CO2 for 11–14 d before experiments.
Calcium image.
Cultured hippocampal neurons were loaded with Fura-2 AM (1 μm) and then washed twice with prewarmed HBSS buffer (5 mm KCl, 125 mm NaCl, 1 mm MgCl2, 25 mm HEPES, 5 mm glucose, 1 mg/ml BSA, pH 7.4). The neurons were bathed in the extracellular solution containing dl-APV (50 μm), DNQX (10 μm), Cd2+ (100 μm) and TTX (0.5 μm) to block the effects of CCK on synaptic transmission. Cells were excited alternatively at 340 and 380 nm and images were captured at 510 nm with an Axiocam MRm camera (Carl Zeiss GmbH). Background fluorescence was subtracted from total fluorescence measured. The intracellular calcium concentration was determined using the formula: [Ca2+]i = K d × ((R − R min)/(R max − R)) × β, after establishing the values for R min = 0.17, R max = 3.53, β = 4.86, and K d = 140 nm by using in vitro standard calibration curves (Invitrogen) and validating the fitting by in situ calibration with the Ca2+ ionophore 4-bromo-A23187 (10 μm, Invitrogen) in HBSS with known concentrations of Ca2+.
Breeding and genotyping of mutant mice.
Heterozygous mating pairs (F1 hybrid crosses from 129 PLC-β1+/− × C57BL/6J PLC-β1+/−) were used to derive wild-type, heterozygous and homozygous pups for experimental analysis. PCR genotyping from purified genomic DNA was performed as described previously (Kim et al., 1997; Deng et al., 2006). Detailed methods for the generation and genotyping of homozygous knock-out mice for CCK-2 receptors (Nagata et al., 1996), PKCα (Leitges et al., 2002) and PKCβ (Leitges et al., 1996) were described previously. Homozygous PKCγ knock-out mating pairs (002466B6; 129P2-Prkcc<tm1Stl>/J) were purchased from The Jackson Laboratory. Pups derived from these homozygous mating pairs were bred in the animal facility of the University of North Dakota and used for experiments.
Data analysis.
Data are presented as the means ± SEM. Concentration–response curve of CCK was fit by Hill equation: I = I max ×{1/[1 + (EC50/[ligand]) n ]}, where I max is the maximum response, EC50 is the concentration of ligand producing a half-maximal response, and n is the Hill coefficient. Student's paired or unpaired t test or ANOVA was used for statistical analysis as appropriate; P values are reported throughout the text and significance was set as p < 0.05. N numbers in the text represent the number of cells examined unless stated otherwise.
Results
CCK increases excitatory synaptic transmission at hippocampal synapses
Whereas application of CCK or a CCK-2 receptor agonist has been shown to increase glutamate release in the perfusate of hippocampal slices (Migaud et al., 1994) and in purified rat hippocampal synaptosomes (Breukel et al., 1997), these experiments did not address the synapse type(s) at which CCK facilitates glutamate release. We therefore initially asked at which synapse(s) CCK increases glutamate release in the hippocampus. Afferents from the entorhinal cortex (perforant path) make glutamatergic synapses onto dentate gyrus granule cells (PP-GC synapse) and the highest density of CCK receptors are detected in the region adjacent to the granule cell layer (Kritzer et al., 1988). We recorded, from dentate gyrus granule cells, AMPA receptor-mediated EPSCs evoked by placing a stimulation electrode in the middle to the inner one third of the molecular layer of the dentate gyrus to stimulate the medial perforant path. Bath application of the sulfated CCK octapeptide (CCK-8S, 0.3 μm) increased AMPA EPSCs to 166 ± 10% of control (n = 30, p < 0.001, paired t test, Fig. 1 A) whereas application of the vehicle (0.004% NH4OH in the extracellular solution) used to dissolve CCK did not significantly change AMPA EPSCs (98 ± 5% of control, n = 8, p = 0.78, Fig. 1 A).

CCK increases AMPA EPSCs at hippocampal synapses via CCK-2 receptors. A , Bath application of CCK increased AMPA EPSCs at PP-GC synapses whereas application of vehicle (0.004% NH4OH) had no effects. The vertical axis was truncated to show the magnitude of the effect of CCK. Left panel shows the current traces averaged from 10 AMPA EPSCs before ( a ) and after ( b ) application of vehicle (top) or CCK (bottom). B , Amplitude distribution of AMPA EPSCs recorded at PP-GC synapses. Arrow indicates the criterion used to separate the responsive and the unresponsive synapses. C , Concentration–response curve of CCK by plotting the percentage of increases in the amplitudes of AMPA EPSCs recorded at the PP-GC synapses versus the concentrations of CCK. Numbers in the parenthesis are numbers of cells examined. D , Application of CCK-2 receptor antagonist, LY225910 (1 μm), not CCK-1 receptor antagonist, lorglumide (1 μm), blocked CCK-induced increases in AMPA EPSCs. Left, Top, Current traces averaged from 10 AMPA EPSCs in the presence of lorglumide alone ( a ) and after coapplication of CCK ( b ). Left, Bottom, Current traces averaged from 10 AMPA EPSCs in the presence of LY225910 alone ( a ) and after coapplication of CCK ( b ). E , Application of CCK increased AMPA EPSCs recorded from 11 cells in slices cut from 3 wild-type [CCK-2(+/+)] mice whereas CCK failed to change AMPA EPSCs recorded from 13 cells in slices cut from 4 knock-out [CCK-2(−/−)] mice. Left panel shows the current traces averaged from 10 AMPA EPSCs before ( a ) and after ( b ) application of CCK from wild-type (top) and knock-out (bottom) mice. F , There were no age-dependent changes in CCK-mediated increases in AMPA EPSCs and application of the transcriptional inhibitor, anisomycin (25 μm), failed to block CCK-induced increases in AMPA EPSCs. Numbers on the top of the bars indicate the number of cells recorded.
Figure 1 B shows the distribution of the effect of CCK on the peak of AMPA EPSCs. Because the normal variation of the averaged AMPA EPSC was ∼10% under our recording conditions, we defined cells exhibiting a response >115% of control as responsive cells (Fig. 1 B). By this criterion, 87% of the cells at the PP-GC synapses were responsive. AMPA EPSC amplitudes began to increase in <5 min after the beginning of CCK application, but it took ∼15–20 min to observe the maximal effect of CCK. The maximal effect of CCK was independent of the duration of CCK application because application of CCK for 3 min (time required for complete solution exchange in the recording chamber) still significantly augmented AMPA EPSCs to 159 ± 11% of control (n = 8, p < 0.001, data not shown) after 30 min washing in CCK-free external solution. The holding current before and during the application of CCK was not significantly altered (111 ± 9% of control, n = 26, p = 0.23, data not shown) suggesting that CCK had no effects on the resting membrane potential of the recorded cells under our recording conditions. CCK dose-dependently increased AMPA EPSCs recorded at the PP-GC synapses with an EC50 value of 27 nm (Fig. 1 C), a value close to that obtained from binding experiments (Van Dijk et al., 1984).
We also examined the effects of CCK on other three hippocampal synapses: mossy fiber-CA3, CA3-CA3 and CA3-CA1 pyramidal neuron synapses. Application of CCK in the concentration range of 0.03 to 3 μm failed to alter significantly AMPA EPSCs at the mossy fiber-CA3 synapses (supplemental Fig. S1A 1,A 2, available at www.jneurosci.org as supplemental material). However, application of CCK significantly increased AMPA EPSCs in 74% of the cells at the CA3-CA3 (supplemental Fig. S1B 1,B 2, available at www.jneurosci.org as supplemental material) and 65% of the cells at the CA3-CA1 (supplemental Fig. S1C 1,C 2, available at www.jneurosci.org as supplemental material) pyramidal neuron synapses. These results demonstrate that CCK increases AMPA receptor-mediated synaptic transmission at PP-GC, CA3-CA3 and CA3-CA1 synapses with no effects at mossy fiber-CA3 pyramidal neuron synapses. The highest responsive ratio was found at PP-GC synapses, consistent with previous immunostaining results showing that the highest density of CCK receptors is in the dentate gyrus, but moderate expressions of CCK receptors are in CA3 and CA1 area (Kritzer et al., 1988). Because the highest responsive ratio to CCK was observed at the medial PP-GC synapses, we recorded AMPA EPSCs at this synapse for the rest of the experiments to further determine the involved cellular and molecular mechanisms.
CCK increases AMPA EPSCs via activation of CCK-2 receptors
Whereas CCK-8S is the major form of CCK in the brain (Rehfeld et al., 1985; You et al., 1994), we also examined the effects of the unsulfated CCK octapeptide (CCK-8U). Application of CCK-8U (0.3 μm) increased AMPA EPSCs to 164 ± 23% of control (n = 11, p = 0.02, data not shown). Because CCK-8U is a weak agonist for CCK-1 receptors, but it is almost as potent as CCK-8S for CCK-2 receptors (Wank, 1995), these results suggest the involvement of CCK-2 receptors. We further tested the role of CCK-2 receptors. Application of a CCK-2 receptor antagonist, LY225910 (1 μm) (Yu et al., 1991) alone did not significantly alter AMPA EPSCs (101 ± 5% of control, n = 10, p = 0.79, data not shown), but blocked CCK-mediated increases in AMPA EPSCs (104 ± 5% of control, n = 10, p = 0.43, Fig. 1 D). Similarly, application of another CCK-2 receptor inhibitor, YM022 (1 μm) (Nishida et al., 1994) alone did not significantly change AMPA EPSCs (95 ± 7% of control, n = 10, p = 0.51), but blocked CCK-induced increases in AMPA EPSCs (98 ± 7% of control, n = 10, p = 0.78, data not shown). However, application of a selective CCK-1 receptor antagonist, lorglumide (1 μm) (de Tullio et al., 1999), did not block CCK-induced increases in AMPA EPSCs (157 ± 10% of control, n = 6, p = 0.002, Fig. 1 D). We further used the CCK-2 receptor knock-out (KO) mice (Nagata et al., 1996) and tested the role of CCK-2 receptors. Application of CCK failed to change AMPA EPSCs recorded from 13 cells in slices cut from 4 CCK-2 receptor KO mice (p = 0.6, Fig. 1 E) whereas it significantly increased AMPA EPSCs recorded from 11 cells in slices cut from 3 wild-type mice (p = 0.001, Fig. 1 E). All these results unanimously demonstrate that the effects of CCK on AMPA EPSCs are mediated by CCK-2 receptors.
CCK-mediated increases in AMPA EPSCs are not modulated developmentally or transcriptionally
The above experiments were performed in slices cut from 15- to 22-d-old rats simply because rats of this age produce better slices. Because we previously observed that CCK-mediated modulation of GABA release was limited to juvenile rats (<31 d) (Deng and Lei, 2006), we also examined whether CCK-mediated increases in AMPA EPSCs were developmentally regulated. We extended the age of the rats to 8 d after birth and adult (>2 months). CCK still increased AMPA EPSCs to the same extent at these ages (Fig. 1 F, p > 0.05 for each group) suggesting that CCK-mediated increases in AMPA EPSCs are not developmentally modulated. Because it took ∼15 min to observe the maximal effect of CCK, we examined whether protein synthesis was required for the effects of CCK. We pretreated slices with a transcription inhibitor, anisomycin (25 μm), and the same concentration of anisomycin was bath-applied. Application of CCK in the presence of anisomycin still increased AMPA EPSCs to 172 ± 18% of control (n = 7, p = 0.007, Fig. 1 F) suggesting that protein synthesis is not required for CCK-mediated increases in AMPA EPSCs.
CCK facilitates presynaptic glutamate release
The effects of CCK could be attributable to an increase in presynaptic glutamate release or to an upregulation of postsynaptic AMPA receptor function. We next differentiated between the presynaptic and postsynaptic effects of CCK. First, we calculated the coefficient of variation (CV) of AMPA EPSCs recorded before and during the application of CCK because changes in presynaptic transmitter release are usually concomitant with an alteration in CV. CCK significantly reduced the value of CV (Fig. 2 A). Second, we compared the paired-pulse ratio (PPR) before and during the application of CCK. CCK significantly reduced PPR (Fig. 2 B). These results indicate that CCK increases AMPA EPSCs by facilitating presynaptic glutamate release.

CCK facilitates presynaptic glutamate release by increasing quantal content. A , CCK reduced the coefficient of variation (CV = SD/mean) of AMPA EPSCs. SD and mean were obtained by averaging 15 consecutive EPSCs. Top shows 15 consecutive EPSCs recorded before (left) and during (right) the application of CCK. Bottom shows the calculated CVs from 20 cells (open circles) and their averages (solid circles). B , CCK reduced paired-pulse ratio (PPR = P2/P1, P1 and P2 are the EPSCs evoked by two stimuli at an interval of 20 ms). Top left, EPSCs averaged from 20 to 30 current traces before (bold) and during (thin) the application of CCK. Top right, EPSCs recorded before (bold) and during (thin) application of CCK were scaled to the first EPSC. Note that the second EPSC during the application of CCK is smaller than control. Bottom, PPRs recorded from 11 cells (open circles) and their averages (solid circles). C , mEPSCs recorded in the presence of TTX before and during the application of CCK. D , Summarized mEPSC frequency and amplitude (n = 20). E , Evoked AMPA EPSCs recorded from the same synapse in the presence of Sr2+ (6 mm) before and during application of CCK. Note that AMPA EPSCs recorded as asynchronous events in the extracellular solution containing Sr2+. Also note that application of CCK increased the frequency of asynchronous events recorded in the presence of Sr2+. F , Cumulative frequency distribution of asynchronous EPSCs in the presence of Sr2+ before and during the application of CCK. G , Cumulative amplitude distribution of asynchronous EPSCs in the presence of Sr2+ before and during the application of CCK. H , Summarized asynchronous EPSC frequency and amplitude (n = 7).
We next probed whether CCK facilitates glutamate release via activation of presynaptic or postsynaptic CCK-2 receptors at the PP-GC synapses because it is still possible that CCK interacts with postsynaptic CCK-2 receptors to generate some retrograde messengers to enhance glutamate release. If so, intracellular application of GDP-β-S, a G-protein inactivator, should block CCK-mediated increases in AMPA EPSCs. We therefore included GDP-β-S (4 mm) in the recording pipettes and waited for >25 min after the formation of whole-cell configuration. Under this condition, application of CCK still increased AMPA EPSC amplitudes significantly (149 ± 7% of control, n = 8, p < 0.001, supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). To exclude the possibility that the incapability of GDP-β-S to block CCK-induced increases in AMPA EPSCs was due to the loss of the biological activity of GDP-β-S, we performed a positive control experiment. As would be demonstrated below (see Fig. 5 E), bath application of CCK inhibited the delayed rectifier K+ channels (I K) recorded from the stellate neurons in layer II of the entorhinal cortex in slices. Intracellular application of GDP-β-S (4 mm) blocked CCK-induced depression of I K (n = 5, p = 0.89, supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). Together, these results suggest that CCK increases glutamate release via activation of presynaptic CCK-2 receptors. Consistent with our electrophysiological results, enriched CCK-binding sites have been detected in layer II of the entorhinal cortex where the cell bodies of the perforant pathway reside in (Köhler and Chan-Palay, 1988).
CCK increases quantal content with no effects on quantal size
We then tested whether CCK-mediated increases in glutamate release are AP-dependent or not by recording AMPA receptor-mediated miniature EPSCs (mEPSCs) in the presence of TTX. Bath application of CCK did not change the frequency (n = 20, p = 0.37) or the amplitude (n = 20, p = 0.84, Fig. 2 C,D) of mEPSCs suggesting that CCK-mediated increases in glutamate release are AP-dependent. However, we cannot exclude the possibility that the effect of CCK on mEPSCs is diluted by the fact that mEPSCs are originated from all the glutamatergic terminals impinging on the recorded cell.
We next measured the effects of CCK on the quantal properties of glutamate release at the PP-GC synapses using the method of replacing extracellular Ca2+ with strontium (Sr2+) to induce asynchronous transmitter release (Goda and Stevens, 1994; Oliet et al., 1996). After forming whole-cell recordings, AMPA EPSCs evoked by stimulation of perforant path in the presence of extracellular Ca2+ were initially recorded. To avoid the confounding effects of spontaneous EPSCs on the asynchronous events when extracellular Ca2+ was replaced with Sr2+, experiments were limited to those cells exhibiting spontaneous events <0.5 Hz. The extracellular Ca2+ was then replaced by Sr2+ (6 mm) to induce asynchronous release from the same synapse. After recording sufficient events to analyze the quantal properties in control, we applied CCK in the extracellular solution containing the same concentration of Sr2+ for 20 min to observe the maximal effect of CCK. We then recorded the asynchronous events in the presence of Sr2+ and CCK. The results are shown in Figure 2 E–H. When extracellular Ca2+ was replaced by Sr2+, asynchronous release occurred and CCK significantly increased the frequency of asynchronous EPSCs (control: 4.5 ± 0.3 Hz, CCK: 6.2 ± 0.6 Hz, n = 7, p = 0.01, Fig. 2 E,F,H) without altering the amplitude of asynchronous EPSCs (control: 5.5 ± 0.6 pA, CCK: 5.9 ± 0.4 pA, n = 7, p = 0.21, Fig. 2 E,G,H) suggesting that CCK increases quantal content without altering quantal size.
CCK increases the number of releasable vesicles and release probability without changing the rate of recovery from vesicle depletion
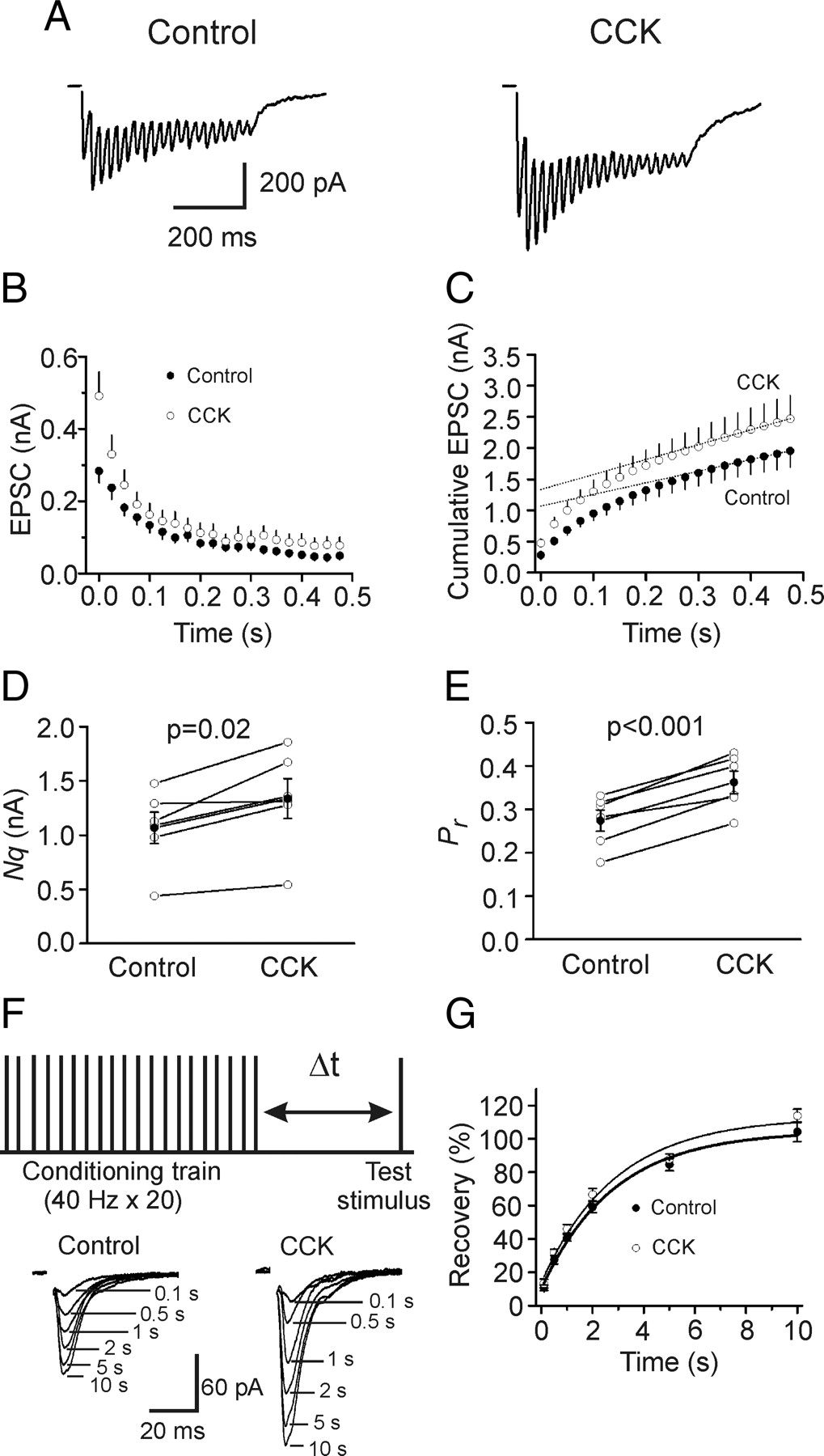
Increases in presynaptic transmitter release can result from an increase in the number of readily releasable quanta (synaptic vesicles) (N) or an increase in release probability (Pr ). We next used the method of high-frequency stimulation (Schneggenburger et al., 1999; Taschenberger et al., 2002) to evaluate CCK-induced changes in N and Pr . This method is based on the assumption that high-frequency stimulation induced depression is primarily caused by the depletion of readily releasable quanta which could be estimated by calculating the cumulative EPSC amplitude for time intervals that are short with respect to the time required for recovery from depression. The zero time intercept of a line fitted to a cumulative amplitude plot of EPSCs equals to the product of N and the quantal size (q). Pr can be estimated from the first EPSC amplitude divided by Nq. To exclude potential contaminations from the activation of other receptors, we included, in the extracellular solution, a “cocktail” of inhibitors to block NMDA (dl-APV, 100 μm), metabotropic glutamate (MCPG, 1 mm), kainate (UBP296, 10 μm), GABAA (bicuculline, 10 μm), GABAB (CGP55845, 5 μm), cannabinoid (AM 251, 10 μm), muscarinic (atropine, 10 μm), nicotinic (mecamylamine, 100 μm) and adenosine (A1, DPCPX, 1 μm; A2A, SCH442416, 1 μm; A2B, MRS1706, 1 μm; A3, MRS1220, 10 μm) receptors. Figure 3 A shows the EPSC trains evoked by 20 stimuli at 40 Hz before and during the application of CCK. The average data from 6 cells for the 20 stimuli are shown in Figure 3 B. Figure 3 C shows the cumulative amplitude histogram. CCK increased Nq by 26 ± 6% (n = 6, p = 0.02, Fig. 3 D) and Pr by 34 ± 6% (n = 6, p < 0.001, Fig. 3 E). Because CCK did not change quantal size (q) (Fig. 2), these results suggest that CCK increases both the number of readily releasable quanta (N) and release probability (Pr ).

CCK increases the number of releasable vesicles and release probability without changing the rate of recovery from vesicle depletion. A , EPSC trains averaged from 15 traces evoked by 20 stimuli at 40 Hz before (left) and during (right) the application of CCK. Stimulation artifacts were blanked for clarity. B , EPSC amplitudes averaged from 6 cells in response to 20 stimuli at 40 Hz before and during the application of CCK. The amplitude of EPSC evoked by each stimulus was measured by resetting the base line each time at a point within 0.5 ms before the beginning of each stimulation artifact. C , Cumulative amplitude histogram of EPSCs. For each cell, the last 6 EPSC amplitudes were fit with a linear regression line and extrapolated to time 0 to estimate the readily releasable pool size (Nq). D , CCK increases Nq (n = 6). E , CCK increases release probability (Pr , n = 6). For each cell, Pr was calculated as the ratio of the first EPSC amplitude divided by its Nq obtained by linear fitting of the cumulative EPSC histogram. F , Top, Experimental protocol. A conditioning train (20 stimuli at 40 Hz) was followed by a test stimulus at various intervals (Δt = 0.1–10 s). This protocol was repeated every 30 s. Bottom, EPSCs evoked by the test pulse from the same synapse at different intervals were aligned and superimposed before (left) and during (right) application of CCK. Stimulation artifacts were blanked for clarity. G , time course of recovery from depletion before and during the application of CCK expressed as percentage recovery = (I test − I ss)/(I first − I ss) × 100, where I test is the EPSC evoked by the test pulse, I ss is the steady-state current left after the conditioning train (the average of the last 5 EPSC evoked by the conditioning train), I first is the EPSC evoked by the first stimulus of the conditioning train. Data before (thick line) and during (thin line) the application of CCK from 6 cells were fit by a single exponential function.
Increases in the number of readily releasable quanta can occur with or without a concomitant increase in the rate of vesicle replenishment. We next tested whether CCK increases the rate of recovery from vesicle depletion. We used a protocol comprising a train of stimulation (40 Hz, 20 stimuli) to deplete the readily releasable pool followed by a test pulse at various intervals (0.1 s, 0.5 s, 1 s, 2 s, 5 s, 10 s) to evaluate the replenishment of synaptic vesicles from depletion (Fig. 3 F). The time course of recovery after the 40 Hz train could be fitted by a single exponential function with a time constant of 3.6 ± 1.0 s before and 3.7 ± 1.1 s during the application of CCK (n = 6, p = 0.96, Fig. 3 G) indicating that CCK does not increase the rate of recovery from vesicle depletion.
CCK does not directly modulate voltage-dependent Ca2+ channels
We next examined the ionic mechanisms underlying CCK-induced increases in glutamate release. We first examined whether CCK facilitates glutamate release by increasing Ca2+ channel function, although CCK slightly inhibits Ca2+ channels in CA1 pyramidal neurons (Shinohara and Kawasaki, 1997). P/Q-type Ca2+ channels are the predominant Ca2+ channels involved in transmitter release whereas N-type Ca2+ channels contribute a small amount of Ca2+ for glutamate release at PP-GC synapses (Qian and Noebels, 2001). If CCK increases glutamate release by directly enhancing Ca2+ channel function, application of specific P/Q- or N-type Ca2+ channel inhibitors would block the effects of CCK. We therefore used toxins specific for those two types of Ca2+ channels and examined their roles in CCK-mediated increases in glutamate release. Application of P/Q-type Ca2+ channel inhibitor, ω-agatoxin TK (250 nm) inhibited AMPA EPSCs to 28 ± 4% of control (n = 6, p < 0.001) and subsequent application of CCK still increased AMPA EPSCs to 161 ± 13% of control (n = 6, p = 0.006, Fig. 4 A). Application of N-type Ca2+ channel inhibitor, ω-conotoxin GVIA (250 nm) inhibited AMPA EPSCs to 77 ± 4% of control (n = 6, p = 0.004) and subsequent application of CCK increased AMPA EPSCs to 148 ± 13% of control (n = 6, p = 0.02, Fig. 4 B). CCK-induced increases in AMPA EPSCs in the presence of ω-agatoxin TK or ω-conotoxin GVIA were not significantly different from the effect of CCK in the absence of those toxins (p > 0.05, Students' unpaired t test). Application of both toxins completely blocked transmitter release (inhibition by 98 ± 3%, n = 3) suggesting that it is unlikely that other types of Ca2+ channels are involved. These results suggest that CCK is not acting selectively on P/Q- or N-type Ca2+ channels.

CCK does not increase glutamate release by direct interaction with Ca2+ channels. A , Inhibition of P/Q-type Ca2+ channels failed to block CCK-mediated increases in AMPA EPSCs (n = 6). Top shows the averaged AMPA EPSCs recorded at the time points indicated in the figure. B , Inhibition of N-type Ca2+ channels failed to block CCK-mediated increases in AMPA EPSCs (n = 6). Insets are the averaged AMPA EPSCs recorded at the time points indicated in the figure. C , Hyperpolarizing current pulse injection caused the membrane potential to attain an early peak and then “sag” to a steady-state level in a stellate neuron. D , CCK did not modulate Ca2+ currents (I Ca) recorded from stellate neurons (n = 6). Insets show the current traces recorded before and during application of CCK for 20 min. E , F , CCK failed to alter I Ca recorded from stellate neurons after P/Q-type (n = 6) or N-type (n = 7) Ca2+ channels were blocked.
If CCK acts on both types of Ca2+ channels, blocking one type would still permit CCK to interact with the other to increase glutamate release. To test this possibility, we directly recorded Ca2+ channel currents (I Ca) from identified stellate neurons in layer II of the entorhinal cortex, the cell bodies of the perforant path. The horizontal slices contain both the hippocampus and the entorhinal cortex. Stellate neurons were identified by their morphology and electrophysiological properties described previously (Deng and Lei, 2007; Deng et al., 2007, 2009). Stellate neurons have larger and polygonal soma with variable number of main dendrites radiating out from the cell bodies, but are devoid of an evidently dominant dendrite. These neurons show an electrophysiological property, i.e., hyperpolarizing current pulse injections always caused the membrane potential to attain an early peak and then “sag” to a steady-state level (Fig. 4 C). Application of CCK failed to modulate I Ca (106 ± 5% of control, n = 6, p = 0.26, Fig. 4 D). We also examined the effects of CCK on isolated N- and P/Q-type I Ca recorded from stellate neuron soma. Application of ω-agatoxin TK (250 nm) reduced I Ca to 41 ± 3% of control (n = 6, p < 0.001, Fig. 4 E). Subsequent application of CCK failed to further change I Ca (95 ± 2% of control, n = 6, p = 0.12, Fig. 4 E). Similarly, application of ω-conotoxin GVIA (250 nm) reduced I Ca to 75 ± 3% of control (n = 7, p < 0.001, Fig. 4 F) and CCK did not significantly change I Ca after application of ω-conotoxin GVIA (96 ± 2% of control, n = 7, p = 0.06, Fig. 4 F). All those data strongly indicate that CCK does not directly modulate Ca2+ channels.
CCK inhibits K+ channels to increase glutamate release
We then examined the roles of K+ channels. If CCK inhibits K+ channels resulting in an increase in Ca2+ influx, transmitter release can also be increased. Because the types of K+ channels expressed on the axons of perforant path and the soma of the stellate neurons in layer II of the entorhinal cortex are Kv1 subtypes (Monaghan et al., 2001) that are sensitive to 4-aminopyridine (4-AP) (Coetzee et al., 1999), we used 4-AP to examine the roles of K+ channels in CCK-mediated increases in glutamate release. Application of 4-AP (40 μm) increased AMPA EPSCs to 239 ± 27% of control (n = 9, p < 0.001, Fig. 5 A) when the external Ca2+ concentration was 2.5 mm. Under these conditions, application of CCK failed to further increase AMPA EPSCs (Fig. 5 A). To exclude the possibility that Ca2+ influx produced by the inhibition of K+ channels might have saturated the release machinery and overwhelmed the effects of CCK, we reduced the external Ca2+ concentration from 2.5 mm to 0.5 mm to limit Ca2+ influx in the presence of 4-AP (Fig. 5 B). AMPA EPSCs recorded in the presence of 4-AP with 0.5 mm Ca2+ were reduced almost to the initial level recorded in the absence of 4-AP with 2.5 mm Ca2+ (Fig. 5 B) suggesting that the release machinery is unlikely to be saturated in this condition. However, application of CCK failed to increase AMPA EPSCs in the presence of 4-AP with 0.5 mm Ca2+ (106 ± 14% of control, n = 11, p = 0.7, Fig. 5 B) whereas CCK still increased AMPA EPSCs to 173 ± 13% of control (n = 5, p = 0.005) in the extracellular solution containing 0.5 mm Ca2+ without 4-AP (Fig. 5 C) suggesting that the binding of CCK to CCK-2 receptors is not affected by lower extracellular Ca2+, consistent with previous results (Bondy et al., 1989). Together, these data indicate that the function of K+ channels is required for the effects of CCK.

CCK increases glutamate release via inhibition of K+ channels. A , Application of 4-AP (40 μm) significantly increased AMPA EPSCs and blocked CCK-induced increases in AMPA EPSCs in the presence of 2.5 mm Ca2+ (n = 9). Top shows the current traces averaged from 10 AMPA EPSCs as indicated in the figure. B , CCK failed to significantly increase AMPA EPSCs when the Ca2+ concentration of extracellular solution was switched from 2.5 mm to 0.5 mm in the presence of 4-AP (40 μm, n = 8). C , CCK still increased AMPA EPSCs in the extracellular solution containing 0.5 mm Ca2+ (n = 5). D , CCK significantly inhibited I K recorded from stellate neurons (n = 5). Insets are I K recorded from a stellate neuron before and during the application of CCK (0.3 μm). Steady currents within 10 ms before the end of the depolarization pulse were measured and used to evaluate the effects of CCK on I K. E , Time course of the effect of CCK or vehicle (0.004% NH4OH) on I K recorded by depolarization from −60 mV to +40 mV every 30 s. Inset shows the currents recorded before ( a ) and during ( b ) the application of CCK or vehicle at the time points indicated in the figure. F , Summarized data showing that application of 4-AP (40 μm) inhibited I K and blocked CCK-mediated inhibition of I K (n = 10).
To further examine the involvement of K+ channels, we directly recorded from the identified stellate neurons. CCK did not change the resting membrane potential (control: −60.1 ± 0.8 mV, CCK: −59.8 ± 1.6 mV, n = 7, p = 0.76). We then recorded the voltage-dependent K+ channel currents by depolarization from the holding potential of −60 mV (resting membrane potential) to +60 mV at an interval of 10 mV. The extracellular solution contained TTX (0.5 μm) to block Na+ channels and CdCl2 (100 μm) to block Ca2+ channels. Under these conditions, only I K was recorded (Fig. 5 D). Application of CCK (0.3 μm) for 15 min significantly inhibited I K (53 ± 9% of control, n = 5, p = 0.003, Fig. 5 D,E) whereas application of the vehicle (0.004% NH4OH) in the extracellular solution did not significantly alter I K (n = 5, p = 0.94, Fig. 5 E). To test whether CCK and 4-AP interact with the same type of K+ channels, we examined the effects of CCK in the presence of 4-AP. Application of 4-AP (40 μm) significantly inhibited I K (56 ± 6% of control, n = 10, p = 0.001) and blocked the effect of CCK (99 ± 3% of control, n = 10, p = 0.4, Fig. 5 F) suggesting that 4-AP and CCK interact with the same type of K+ channels.
Signal transduction mechanisms underlying CCK-mediated increases in glutamate release
We then examined the role of PLC pathway in CCK-mediated increases in glutamate release. Initially, we used U73122, a PLC inhibitor. Slices were pretreated with U73122 (10 μm) and the extracellular solution contained the same concentration of U73122. Under these conditions, application of CCK failed to significantly increase AMPA EPSCs (Fig. 6 A) whereas application of U73343 (10 μm), the inactive analog of U733122, did not block CCK-mediated increases in AMPA EPSCs (Fig. 6 A) suggesting that PLC activity is required. To further confirm the involvement of PLC, we used PLC knock-out mice (Kim et al., 1997; Deng and Lei, 2007; Lei et al., 2007). Among the 4 isoforms of PLCβ, PLCβ1 is expressed in the hippocampus (Watanabe et al., 1998). However, CCK failed to significantly increase AMPA EPSCs recorded from the PLCβ1 KO mice (102 ± 4% of control, n = 13 cells in slices cut from 4 mice, p = 0.66) whereas CCK still increased AMPA EPSCs in wild-type mice (156 ± 7% of control, n = 9 cells in slices cut from 3 mice, p < 0.001, Fig. 6 B). Together, these results indicate that PLCβ1 is required for CCK-induced increases in glutamate release.

Signal transduction pathway underlying CCK-mediated increases in glutamate release. A , Pretreatment of slices with and bath application of U73122 (10 μm) blocked CCK-induced increases in AMPA EPSCs (n = 8) whereas the same treatment of slices with U73343 (10 μm) did not significantly change the effects of CCK (n = 10). Left panel shows the averaged AMPA EPSCs taken at the time points indicated in the figure in the presence of U73343 (upper) or U73122 (lower). B , Application of CCK significantly increased AMPA EPSCs in wild-type [PLCβ1(+/+)] mice (n = 9 cells in slices cut from 3 mice) but failed to change AMPA EPSCs in PLCβ1 knock-out [PLCβ1(−/−)] mice (n = 13 cells in slices cut from 4 mice). Left panel shows the averaged AMPA EPSCs recorded at the time points indicated in the figure from wild-type (top) and PLCβ1 knock-out (bottom) mice. C , CCK increased intracellular Ca2+ concentration in a cultured hippocampal neuron. D , Application of IP3 receptor inhibitor, 2-APB (50 μm), partially inhibited CCK-induced increases in AMPA EPSCs (n = 10) whereas application of PKC inhibitors, GF109203X (n = 7) and Ro318220 (n = 10), completely blocked CCK-induced increases in AMPA EPSCs (*p < 0.05, **p < 0.01 vs CCK alone). E , Application of CCK failed to increase AMPA EPSCs in PKCγ knock-out mice (n = 8 cells in slices cut from 3 mice) but still significantly increased AMPA EPSCs from wild-type (n = 7 cells in slices cut from 3 mice), PKCα (n = 8 cells in slices cut from 3 mice) and PKCβ (n = 7 cells in slices cut from 3 mice) knock-out mice.
Activation of PLC leads to an increase in intracellular Ca2+ release and activation of PKC. We also examined the roles of intracellular Ca2+ release and PKC in CCK-mediated increases in glutamate release. We first measured CCK-induced change in intracellular Ca2+ concentration in cultured hippocampal neurons. Application of CCK significantly increased intracellular Ca2+ concentration (control: 29.9 ± 7.4 nm, CCK: 461.6 ± 87.7 nm, n = 10, p < 0.001, Fig. 6 C) indicating that CCK induces intracellular Ca2+ release. We then tested the roles of intracellular Ca2+ release in CCK-mediated increases in glutamate release. Application of 2-aminoethoxydiphenyl borate (2-APB, 50 μm), an IP3 receptor antagonist, did not alter AMPA EPSCs (97 ± 5% of control, n = 10, p = 0.37), but significantly reduced CCK-mediated increases in AMPA EPSCs (121 ± 3% of control, n = 10, p = 0.02, Fig. 6 D). These results suggest that intracellular Ca2+ release via IP3 receptors is partially responsible for CCK-mediated increases in glutamate release. We then examined the roles of PKC by using two PKC inhibitors, GF109203X (0.5 μm) and Ro318220 (0.5 μm). Bath application of either of these inhibitors completely blocked CCK-induced increases in AMPA EPSCs demonstrating that the activity of PKC is fully required (Fig. 6 D). Together, these data suggest that the conventional, Ca2+-dependent PKCs are involved in CCK-mediated increases in glutamate release. Consistent with our results, activation of PKC increases glutamate release at PP-GC synapses (Chen and Roper, 2003).
The conventional PKCs include PKCα, PKCβ and PKCγ. We next examined the roles of these PKC subtypes in CCK-mediated facilitation of glutamate release using PKC knock-out mice. Application of CCK failed to significantly increase AMPA EPSCs (3.3 ± 3.2%) recorded from 8 cells in slices cut from 3 PKCγ knock-out mice (The Jackson Laboratory, p < 0.01 vs wild-type mice, Fig. 6 E), but still increased AMPA EPSCs in slices cut from wild-type (74.6 ± 8.1%, n = 7 cells in slices cut from 3 mice, p < 0.001), PKCα (60.5 ± 8.6%, n = 8 cells in slices cut from 3 mice, p = 0.26 vs wild-type mice) or PKCβ (80.2 ± 17.6%, n = 7 cells in slices cut from 3 mice, p = 0.78 vs wild-type mice) knock-out mice indicating that CCK increases glutamate release via activation of PKCγ.
Endogenously released CCK increases AMPA EPSCs at PP-GC synapses
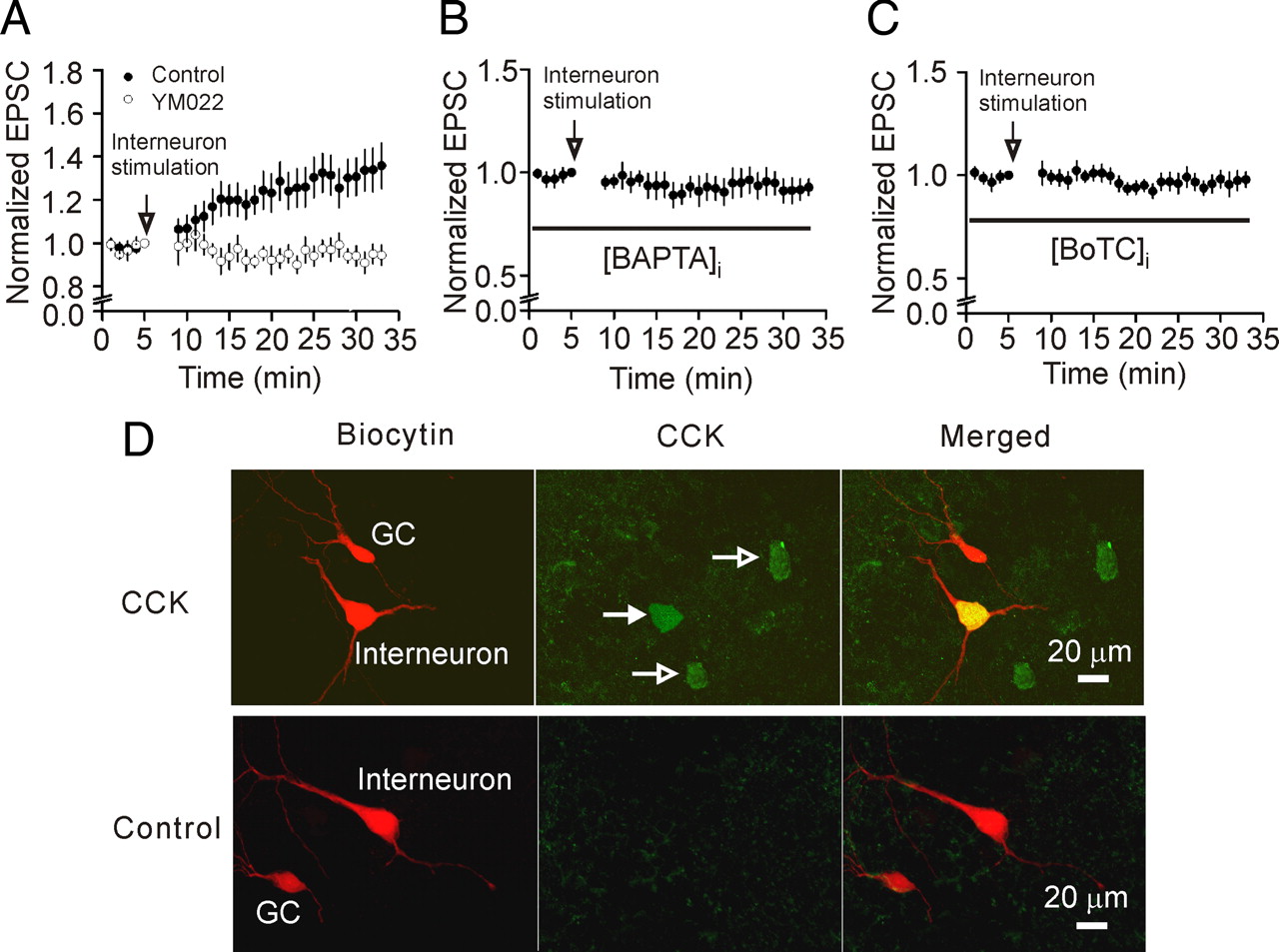
The above experiments were performed by exogenous application of CCK. We next tested the role of endogenously released CCK in modulating glutamate release at the PP-GC synapses by taking the advantage that the axons of CCK-containing interneurons form a band in the inner molecular layer of the dentate gyrus (Hefft and Jonas, 2005) where PP-GC synapse forms. We tested the hypothesis that CCK released locally increases glutamate release at PP-GC synapses. As described in the method, three electrodes (one field stimulation electrode for stimulation of perforant path, two patch-clamp recording electrodes for a granule cell and an interneuron) were used. The patch-clamp electrodes contained 0.2% biocytin for post hoc immunocytochemical detection of CCK expression in interneurons (Fig. 7 D). We took the advantage that the pyramidal-shaped interneurons located at the stratum granulosum/hilus border contain CCK (Acsády et al., 2000; Morozov et al., 2006) and selectively recorded from this population of interneurons. The identity of CCK-containing interneurons was verified by post hoc immunostaining for CCK (Fig. 7 D, top). Analyses were limited to those interneurons showing detectable immunoreactivity for CCK. We initially recorded the basal AMPA EPSCs at the PP-GC synapses. We depolarized the interneuron via the patch pipette from −60 to 0 mV for 100 ms at a frequency of 2 Hz for 250 times to induce CCK release. This kind of protocol has been used successfully to induce opioid peptide release in the hypothalamus (Iremonger and Bains, 2009). After stimulation of the interneuron, recording of AMPA EPSCs at the PP-GC synapses was reassumed. AMPA EPSCs were increased to 125 ± 9% of control (n = 10, p = 0.02, Fig. 7 A) after interneuron depolarization. Of the 10 interneurons showing positive staining for CCK, increases in AMPA EPSCs (>115% of control as the criterion used above) were observed in 7 cells resulting in a responsive ratio of 70%. However, in the presence of YM022 (1 μm), a CCK-2 receptor antagonist, depolarization of interneuron failed to significantly change AMPA EPSCs (99 ± 6% of control, n = 7, p = 0.89, Fig. 7 A). Of the 7 interneurons showing positive staining for CCK, none of the recordings showed an increase in AMPA EPSCs resulting in a responsive ratio of 0% (p < 0.001 vs control in the absence of YM022). To confirm the specificity of the CCK antibody, we pretreated the CCK antibody with the corresponding blocking peptide and stained 5 slices in which interneuron stimulation evoked increases in AMPA EPSCs. However, pretreatment of the CCK antibody with the corresponding blocking peptide prevented the detection of CCK immunoreactivity in interneurons in all the 5 slices (Fig. 7 D, bottom) confirming the specificity of the CCK antibody.

Endogenously released CCK increases glutamate release at PP-GC synapses. A , An electrode filled with the extracellular solution was first placed in the molecular layer of the dentate gyrus for stimulation of perforant path. Two patch-clamp electrodes filled with intracellular solution containing 0.2% biocytin were sealed to a dentate granule cell and a pyramidal-like interneuron in the border of granular layer and the hilus, respectively. Depolarizing stimulation of interneurons generated increases in AMPA EPSCs recorded from granule cells evoked by stimulation of perforant path (n = 10). The same experiment was performed in the presence CCK-2 receptor inhibitor, YM022 (1 μm, n = 7). Note that interneuron stimulation in the presence of YM022 did not lead to an increase in AMPA EPSCs. B , C , The same experiments were performed except that the intracellular solution of the electrode sealed to the interneurons contained 20 mm BAPTA ( B ) or botulinum toxin C (BoTC, 5 μg/ml) ( C ). D , The recorded dentate gyrus granule cells (GC) and pyramidal-like interneurons located at the border of granular layer and hilus filled with biocytin were stained with Texas red-conjugated streptavidin (in red). Top, The expression of CCK in the interneurons (arrows) was detected by goat anti-CCK primary antibody and anti-goat IgG-FITC secondary antibody (in green). The merged image shows the recorded interneuron expressing CCK (in yellow). Bottom, No CCK immunoreactivity was detected when the primary CCK antibody was preabsorbed with a CCK blocking peptide in slices from which interneuron stimulation evoked an increase in AMPA EPSCs recorded at the PP-GC synapses confirming the specificity of CCK antibody.
We performed the following experiments to test whether endogenous CCK release was Ca2+-dependent and exocytosis was involved in CCK release. We first included BAPTA (20 mm) in the intracellular solution of the electrode sealed to the interneurons and repeated the above experiment. Under this condition, stimulation of interneurons failed to increase AMPA EPSCs significantly (90.4 ± 6.5% of control, n = 8, p = 0.18, Fig. 7 B). Of the 8 interneurons showing immunoreactivity to CCK, none of the recordings showed an increase in AMPA EPSCs resulting in a responsive ratio of 0% (p < 0.001 vs control). This result suggests that an increase in intracellular Ca2+ is required for endogenous CCK release. Furthermore, inclusion of botulinum toxin C (BoTC, 5 μg/ml) in the intracellular solution of the electrode sealed to the interneurons to cleave syntaxin prevented the increases of AMPA EPSCs induced by stimulation of interneurons (96.7 ± 4.1% of control, n = 8, p = 0.46, Fig. 7 C). Of the 8 interneurons showing CCK-positive immunostaining, one recording demonstrated an increase (119% of control) of AMPA EPSC amplitude resulting in a responsive ratio of 12.5% which was still significantly lower than that of the control (p = 0.01). This result suggests that SNARE-dependent exocytosis is required for CCK release from hippocampal interneurons.
Neuronal excitability is controlled by CCK-mediated modulations of both glutamate and GABA release
In addition to increasing glutamate release, CCK also transiently facilitates, followed by a reduction in GABA release in the dentate gyrus (Deng and Lei, 2006). Because the outcome of CCK-mediated modulation of transmitter release is to modify neuronal excitability, we tested the effect of CCK on neuronal excitability by recording AP firing from the dentate granule cells. Suprathreshold positive current was continuously injected to induce AP firing at a frequency of 0.5–1 Hz. However, granule cells could not sustain stable AP firing in the conventional whole-cell configuration. The frequency of APs was gradually reduced and eventually disappeared in ∼10 min. We therefore used perforated-patch recording. In this recording configuration, the firing frequency of APs was stable and lasted for at least 50 min (109 ± 9% of control, n = 5, p = 0.49, data not shown). We then assayed the effect of CCK on AP firing in four distinct conditions. First, experiments were performed in the normal extracellular solution to render both glutamatergic and GABAergic transmission functional. Because the maximal effect of CCK-induced facilitation of GABAergic transmission occurs in ∼3–5 min after the beginning of application of CCK (Deng and Lei, 2006), we paid specific attention to the action of CCK in this period (early phase). Bath application of CCK (0.3 μm) failed to alter the AP firing frequency significantly at the early phase (91 ± 10% of control, n = 12, p = 0.39, Fig. 8 A 1,A 2) but significantly increased that recorded at the 30th min after the beginning of its application (late phase, 192 ± 19% of control, n = 12, p < 0.001, Fig. 8 A 1,A 2). This result suggests that CCK-induced initial transient facilitation of GABA release is overwhelmed by its effect on glutamatergic transmission. Second, we tested the contribution of CCK-induced increases in glutamate release by recording APs in the extracellular solution supplemented with bicuculline (20 μm) to block GABAA and CGP55845 (5 μm) to block GABAB receptors. Under these circumstances, application of CCK significantly increased the firing frequency recorded at both early (129 ± 9% of control, n = 10, p = 0.018, Fig. 8 B 1,B 2) and late (209 ± 40% of control, n = 10, p = 0.022, Fig. 8 B 1,B 2) phase. Third, we assessed the effect of CCK-mediated modulation of GABAergic transmission on neuronal excitability by recording AP firing in the extracellular solution containing DNQX (20 μm) to block AMPA/kainate, dl-APV (100 μm) to block NMDA and MCPG (1 mm) to block metabotropic glutamate receptors. In this condition, CCK had a transient inhibition in the early phase (63 ± 7% of control, n = 11, p < 0.001, Fig. 8 C 1,C 2) but a persistent enhancement of the late phase (152 ± 9% of control, n = 11, p < 0.001, Fig. 8 C 1,C 2). This result suggests that dentate gyrus granule cells are still under considerable tonic inhibition by GABA when glutamatergic functions are blocked. To test this possibility, we recorded AP firing in the presence of glutamate receptor inhibitors (DNQX + APV + MCPG). We then applied bicuculline and CGP55845 to block GABAergic functions. Bath application of bicuculline (20 μm) and CGP55845 (5 μm) in the presence of glutamate receptor inhibitors (DNQX + APV + MCPG) still increased AP firing frequency to 183 ± 12% of control (n = 4, p = 0.006, data not shown). Finally, we recorded AP firing in the presence of inhibitors for both glutamatergic and GABAergic transmission to exclude the possibilities that CCK could directly alter the excitability of granule cells or affect the functions of other neurotransmitters. CCK did not significantly change AP firing frequency in this situation (3 min: 101 ± 3% of control, n = 9, p = 0.67; 30 min: 93 ± 6% of control, n = 9, p = 0.28, Fig. 8 D 1,D 2). These data together demonstrate that CCK-mediated modifications of glutamatergic and GABAergic transmission contribute to neuronal network activity.
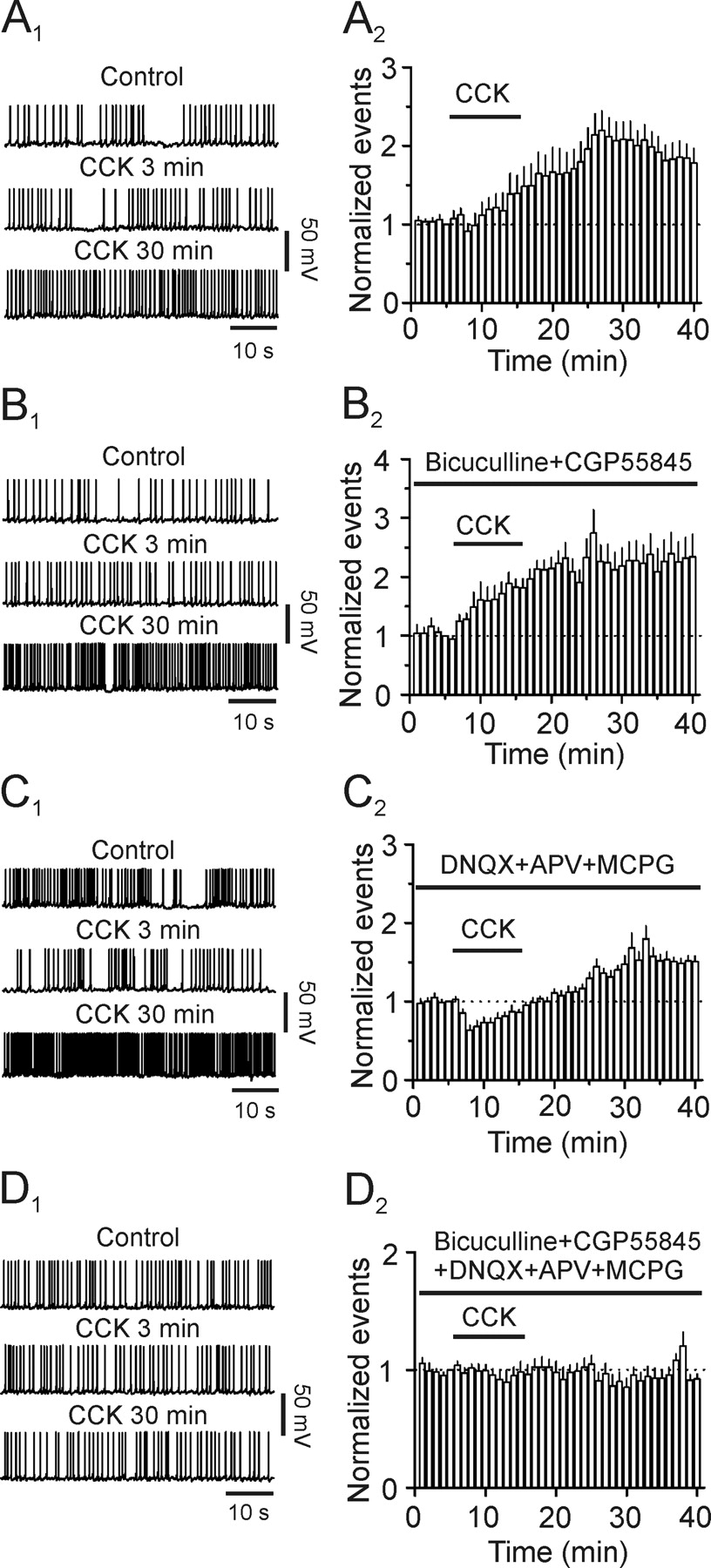
Contributions of CCK-induced modulation of GABAergic and glutamatergic transmission to the excitability of dentate granule cells recorded with perforated patches. A 1 , A 2 , CCK increased the firing frequency of APs recorded in normal extracellular solution. A 1 , APs recorded before (upper) and at the third (middle) and 30th (lower) minute after the beginning of CCK application. A 2 , Summarized time course of AP firing frequency (n = 12). B 1 , B 2 , CCK facilitated the firing frequency of APs recorded in the extracellular solution supplemented with inhibitors for GABAA (bicuculline) and GABAB (CGP55845) receptors. B 1 , APs recorded before (upper) and at the third (middle) and 30th (lower) minute after the beginning of CCK application. B 2 , Pooled time course of AP firing frequency (n = 10). C 1 , C 2 , CCK transiently inhibited the early phase but augmented the late phase of AP firing frequency recorded in the extracellular solution containing inhibitors for glutamate receptors (DNQX, APV and MCPG). C 1 , APs recorded before (upper) and at the third (middle) and 30th (lower) minute after the beginning of CCK application. C 2 , Pooled time course of AP firing frequency (n = 11). D 1 , D 2 , CCK failed to alter AP firing frequency in the presence of inhibitors for glutamatergic and GABAergic transmission (n = 9). Figures were arranged in the same fashion.
Discussion
Our results indicate that CCK augments glutamate release at PP-GC, CA3-CA3 and CA3-CA1 synapses with no effects at MF-CA3 synapses. CCK increases the number of readily releasable vesicles and release probability without effects on the rate of recovery from vesicle depletion. The effects of CCK on glutamate release are mediated by inhibition of a 4-AP-sensitive K+ channel and require the functions of CCK-2 receptors, PLC, intracellular Ca2+ release and PKC. We further demonstrate that CCK endogenously released from interneurons increases glutamatergic transmission in the hippocampus.
Whereas CCK has been shown to activate a cationic conductance in rat neostriatal neurons (Wu and Wang, 1996) and supraoptic nucleus neurons (Chakfe and Bourque, 2001), our results demonstrate that CCK does not facilitate glutamate release in the hippocampus by activating cationic channels because CCK failed to modulate mEPSCs recorded from dentate granule cells and CCK had no effects on the resting membrane potentials recorded from stellate neurons in the entorhinal cortex. Our results also demonstrate that CCK does not increase glutamate release by direct interaction with and facilitation of presynaptic Ca2+ channels because application of the selective P/Q- and N-type Ca2+ channel blockers did not block CCK-induced increases in AMPA EPSCs and application of CCK failed to alter Ca2+ channel currents recorded from stellate neurons, the cell body of the perforant pathway. Actually, CCK slightly inhibits Ca2+ channels in CA1 pyramidal neurons (Shinohara and Kawasaki, 1997). Our results support the scenario that CCK inhibits I K resulting in increases in presynaptic Ca2+ influx via voltage-gated Ca2+ channels to facilitate glutamate release in the hippocampus because application of 4-AP at micromolar concentration blocked CCK-induced increases in AMPA EPSCs and application of CCK inhibited I K recorded from stellate neurons in the entorhinal cortex. In agreement with our results, CCK has been shown to inhibit other K+ channels in a variety of neurons (Branchereau et al., 1993; Cox et al., 1995; Miller et al., 1997; Deng and Lei, 2006; Yang et al., 2007; Chung et al., 2009). Furthermore, CCK-induced increases in glutamate release gauged by measuring glutamate concentration in the perfusate of hippocampal slices (Migaud et al., 1994) and in purified rat hippocampal synaptosomes (Breukel et al., 1997) are Ca2+-dependent. CCK-induced augmentation of presynaptic Ca2+ concentration likely contributes to its increased effects on the number of readily releasable vesicles and release probability because both the mobilization of vesicles from the reserve pool to the readily releasable pool and the release probability are Ca2+-dependent (Zucker and Regehr, 2002).
We have shown that CCK-mediated facilitation of glutamate release requires the functions of CCK-2 receptors. This conclusion is consistent with the results obtained by measuring glutamate concentration in the perfusate of hippocampal slices (Migaud et al., 1994) and in purified rat hippocampal synaptosomes (Breukel et al., 1997) after application of CCK. The following three lines of evidence indicate that presynaptic CCK-2 receptors should be responsible for CCK-induced glutamate release in the hippocampus. First, enriched CCK-binding sites have been detected in layer II of the entorhinal cortex where the cell bodies of the perforant path are located in (Köhler and Chan-Palay, 1988). Second, inclusion of GDP-β-S, a G-protein inhibitor, in the recording pipettes failed to change CCK-induced increases in AMPA EPSCs whereas it blocked CCK-induced depression of I K recorded from the stellate neurons in layer II of the entorhinal cortex. Third, Breukel et al. (1997) have shown that CCK increases glutamate release in purified hippocampal synaptosomes because in this preparation the continuity of presynaptic and postsynaptic structures should be disconnected. We further demonstrate that the down-stream targets of CCK-2 receptors, PLC, intracellular Ca2+ and PKCγ are involved in CCK-induced increases in glutamate release. Because our results demonstrate that CCK augments glutamate release by inhibiting presynaptic I K, it is reasonable to speculate that the activated PKCγ phosphorylates I K or I K-associated proteins resulting in an inhibition of I K. Consistent with our results, activation of PKC has been shown to inhibit I K in neurons cultured from rat hypothalamus and brainstem (Pan et al., 2001) and in cerebrocortical synaptosomes (Barrie et al., 1991) and I K expressed in Xenopus oocytes (Peretz et al., 1996).
CCK has been shown to transiently increase GABA release in hippocampal CA1 region (Miller et al., 1997; Deng and Lei, 2006; Földy et al., 2007; Karson et al., 2008). However, these studies did not examine the late effect of CCK on GABAergic transmission. We have previously shown that CCK exerts bidirectional modification, with initial transient enhancement followed by a persistent depression, of GABAergic transmission onto the granule cells of the dentate gyrus (Deng and Lei, 2006). Here, we further assessed the contribution of glutamatergic and GABAergic transmission to CCK-mediated modification of the excitability of granule cells. Our results demonstrate that CCK-mediated initial transient enhancement of GABA release does not lead to a perceptible inhibition of the excitability of granule cells when both GABAergic and glutamatergic transmissions are functional. The possible reason is that the inhibition derived from CCK-induced transient augmentation of GABA release is inundated by its action on glutamate release because the initial inhibitory effect of CCK on neuronal excitability was unfolded when glutamatergic transmission was inhibited. However, these results do not deny a potential function for CCK-induced initial increase in GABAergic transmission because it is possible that this kind of modification may exert a fine-tuning on neuronal network activity. Nonetheless, CCK-induced late phase inhibition on GABAergic transmission is in line with its effect on glutamatergic transmission. The concerted actions of CCK on GABAergic and glutamatergic transmissions likely lead to an increase in neuronal excitability. CCK has long been known to exert anxiogenic effects in both animal models and humans (Rehfeld, 2000) and the generation of anxiety can be due to a reduction in GABAergic (Rupprecht et al., 2006; Whiting, 2006) and/or an increase of glutamatergic (Bergink et al., 2004) function. Our results are agreeable with this scenario and may serve as the cellular and molecular mechanisms to explain the anxiogenic effects of CCK. In addition, CCK has also been implicated in modulating other important brain functions including satiety, analgesia, learning and memory processes (Sebret et al., 1999; Rehfeld, 2000; Beinfeld, 2001), which are closely related to both glutamate and GABA. Our results therefore provide a basis to explain the roles of CCK in these physiological functions as well.
Footnotes
-
This work was supported by National Institutes of Health Grant R01MH082881 (S.L.).
- Correspondence should be addressed to Saobo Lei, Department of Pharmacology, Physiology and Therapeutics, School of Medicine and Health Sciences, University of North Dakota, Grand Forks, ND 58203. slei{at}medicine.nodak.edu




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}