

Abstract
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease resulting from the expansion of a glutamine repeat in the huntingtin (Htt) protein. Current therapies are directed at managing symptoms such as chorea and psychiatric disturbances. In an effort to develop a therapy directed at disease prevention we investigated the utility of highly specific, anti-Htt intracellular antibodies (intrabodies). We previously showed that VL12.3, an intrabody recognizing the N terminus of Htt, and Happ1, an intrabody recognizing the proline-rich domain of Htt, both reduce mHtt-induced toxicity and aggregation in cell culture and brain slice models of HD. Due to the different mechanisms of action of these two intrabodies, we then tested both in the brains of five mouse models of HD using a chimeric adeno-associated virus 2/1 (AAV2/1) vector with a modified CMV enhancer/chicken β-actin promoter. VL12.3 treatment, while beneficial in a lentiviral model of HD, has no effect on the YAC128 HD model and actually increases severity of phenotype and mortality in the R6/2 HD model. In contrast, Happ1 treatment confers significant beneficial effects in a variety of assays of motor and cognitive deficits. Happ1 also strongly ameliorates the neuropathology found in the lentiviral, R6/2, N171-82Q, YAC128, and BACHD models of HD. Moreover, Happ1 significantly prolongs the life span of N171-82Q mice. These results indicate that increasing the turnover of mHtt using AAV-Happ1 gene therapy represents a highly specific and effective treatment in diverse mouse models of HD.
Introduction
Huntington's disease (HD) results from the expansion of a glutamine repeat in exon 1 (HDx-1) of the huntingtin protein (Htt) (The Huntington's Disease Collaborative Research Group, 1993). Although the disease-causing mutation is limited to the glutamine repeat, the flanking domains modulate toxicity of the mutant protein (Duennwald et al., 2006). Exon 1 of Htt consists of 17 N-terminal amino acids, the glutamine repeat, two polyproline (polyP) domains separated by a proline-rich (P-rich) domain [collectively known as the proline-rich region (PRR)], and 13 C-terminal amino acids.
The simple, autosomal dominant nature of HD should allow for neuroprotective strategies rather than more complicated restorative strategies. The therapies currently available to HD patients are, however, aimed at symptom management rather than the early stages of the disease. Although HD has a single genetic cause, it has a very complex pathology with detrimental effects on a wide variety of cellular processes. Therefore, it is advantageous in terms of specificity and efficacy to direct therapy toward the most upstream HD targets including the mutant Htt (mHtt) protein itself. One approach of this type is the use of therapeutic anti-Htt intrabodies (iAbs).
Intrabodies are intracellularly expressed, recombinant antibody fragments that provide a powerful therapeutic tool well suited to treatment of protein misfolding neurodegenerative diseases. Intrabodies recognizing different epitopes of the same protein can have vastly different effects (Khoshnan et al., 2002), and they are capable of distinguishing between highly homologous proteins and even different conformations of the same protein (Zhou et al., 2004; Emadi et al., 2007). Intrabodies may also be used to ectopically target proteins to particular cellular compartments using localization sequences (Lecerf et al., 2001; Paganetti et al., 2005).
A number of anti-Htt iAbs with therapeutic potential have been generated and characterized. C4, an iAb that recognizes the N terminus of Htt, reduces aggregation and toxicity in cell culture, brain slice, and Drosophila HD models (Murphy and Messer, 2004; Wolfgang et al., 2005; McLear et al., 2008). This iAb increases turnover of both wild-type (wt) and mutant HDx-1 (mHDx-1) in 293 cells (Miller et al., 2005). MW7 and Happ1, iAbs recognizing the polyP and P-rich domains of Htt, respectively, reduce mHDx-1 aggregation and toxicity in cell culture, brain slice, and Drosophila models of HD, and increase turnover of mutant but not wtHDx-1 (Khoshnan et al., 2002; Southwell et al., 2008). VL12.3, an iAb that recognizes the N terminus of Htt, potently reduces mHDx-1-induced toxicity in cell culture and brain slice HD models (Colby et al., 2004; Southwell et al., 2008). However, this iAb increases nuclear Htt (Southwell et al., 2008). EM48, an iAb recognizing an epitope C-terminal to the PRR of Htt, preferentially binds mHtt and increases turnover of mHDx-1 in a cell culture model of HD. Adenoviral delivery of EM48 to the brains of R6/2 and N171-82Q HD model mice improves some aspects of neuropathology as well as motor performance in N171-82Q mice (Wang et al., 2008). This adenoviral delivery strategy is limited to the study of short life span HD models because of the transient transgene expression of the adenoviral vector. While both vectors can infect neurons (depending on the serotype), the adeno-associated virus (AAV) vector is preferable due to its more stable expression.
Due to the clear mechanistic differences of VL12.3 and Happ1 as potential therapeutics and the lack of a single, ideal mouse HD model, we tested both iAbs for therapeutic efficacy in a lentiviral model and four transgenic HD mouse lines using intrastriatal AAV delivery.
Materials and Methods
Lentivirus production and purification.
Ten 15 cm plates of ∼80% confluent 293 GPG cells (Ory et al., 1996) were triple transfected by calcium phosphate with 20 μg of pFugW lentiviral genome encoding either mHDx-1Q103-GFP or GFP (A. Khoshnan, Caltech, Pasadena, CA), 15 μg of Δ8.9, and 10 μg of VSV-G plasmids (Naldini et al., 1996). Sixteen hours post-transfection, medium was removed and replaced with medium supplemented with 2% FBS. Medium containing lentivirus was collected at 48 and 72 h post-transfection, filtered at 0.45 μm, and centrifuged for 90 min at 25,000 rpm at 4°C to pellet lentivirus. Pellets were then dissolved overnight at 4°C in sterile PBS. Viral solutions were buffer exchanged into sterile saline (0.9% NaCl) and concentrated using Amicon ultra 4 ml tubes [MW cutoff (MWCO) 100,000] (Millipore) until a final volume of 150–250 μl was reached, aliquotted, and stored at −80°C until use. Viral biological titer was determined by infection of HEK 293 cells with a dilution series and counting colonies of GFP-positive cells (Tiscornia et al., 2006). Biological titer was used rather than quantitation of p24 to ascertain the number of infectious particles. Before injection, all lentiviruses were adjusted to a titer of 5 × 108 transducing units/ml.
AAV production and purification.
Fifty 15 cm plates of ∼80% confluent HEK 293 cells were triple transfected by calcium phosphate with 12.5 μg of AAV serotype 2 genome (Stratagene) with a modified chicken β-actin promoter plus CMV promoter enhancer regions (CBA) (Northwestern University Viral Vector Core) encoding GFP, VL12.3, or Happ1, 25 μg of pHelper (Stratagene), and 37.5 μg of AAV serotype 1 rep/cap (University of Pennsylvania Viral Vector Core) plasmids per plate. Medium was replaced 16 h post-transfection. At 48 h post-transfection, medium was removed and cells were dislodged by pipetting, collected by centrifugation, and washed with and resuspended in 10 mm Tris buffer. Cells were lysed by two rounds of freeze/thaw with vortexing and treated with DNase I for 30 min. at 37°C. The viral fraction was then isolated by ultracentrifucation at 40,000 rpm for 2 h at 4°C in an optiprep gradient (15–60%) (Sigma). The fraction containing the virus (40–60% interface) was collected by syringe, diluted 1:5 in 20 mm Tris buffer, pH 8.0, and further purified by Mustang-Q membrane ion exchange (Acrodisc, Pall). Virus was eluted in 500 μl of 20 mm Tris buffer, pH 8.0, with 400 mm NaCl and stored at −80°C in 50 μl aliquots. Before use each aliquot was dialyzed in a slide-a-lyzer mini dialysis unit (MWCO 7000, Pierce) for 1 h at 4°C in 1 L of sterile saline with gentle stirring. Viral titer was determined by qPCR of a dilution series using primers that recognize the inverted terminal repeats and comparison to AAV of a known titer obtained from the University of Iowa Viral Vector Core. Before injection, all AAVs were adjusted with sterile saline to a titer of 1 × 1013 viral particles/ml.
Animals.
Mice were obtained from the Jackson Laboratory [C57BL/6 and BACHD (through the laboratory of W. Yang, University of California, Los Angeles, Los Angeles, CA)] or bred in the Caltech facility from breeding pairs obtained from the Jackson Laboratory (R6/2, N171-82Q, and YAC128). Due to the infertility of the R6/2 line, ovarian transplant hemizygote females and wt males were used. Wild-type females and hemizygote males were used for mating in the N171-82Q and YAC128 lines. Genotyping PCR was performed as specified on the Jackson Laboratory website. Due to the high demand for the relatively new BACHD model, an all-male cohort with wt littermates could not be obtained. However, this model is on the same genetic background as the YAC128 model, so wt mice from the BACHD line are expected to perform similarly to wt mice from the YAC128 line. Experimental data were analyzed independently for each gender and combined only if there was no significant difference between male and female mice. Mice were housed in ventilated cages and all surgical and experimental procedures were performed according to Institute Animal Care and Use Committee specifications.
Surgeries.
Adult mice were anesthetized with 500 mg/kg ketamine and 100 mg/kg xylazine and placed in a stereotaxic frame. After shaving and disinfection of the scalp, an incision was made along the midline. A dental drill was used to make a burr hole at 0.75 mm anterior and 2 mm lateral to bregma. Injections were done at a depth of 3.5 mm from the dura using a 5 μl Hamilton syringe and removable 30-gauge needle with a 20° bevel tip attached to an ultra micropump III and micro4 controller (World Precision Instruments). After injection, the needle was left in place for 5 min and withdrawn slowly. Incisions were closed using vet bond glue (3M) and mice were allowed to recover on a heating pad. Mice were checked for general health daily for 3 d following surgery.
For the lentiviral model, four-week-old C57BL/6 mice were injected unilaterally in the striatum on alternating sides with 4 μl of mHDx-1 or GFP lentivirus plus 1 μl of GFP, VL12.3, or Happ1 AAV at a rate of 0.5 μl/min (10 mice per group). Four-week-old male N171-82Q and wt littermates were injected bilaterally in the striatum with 1 μl of GFP or Happ1 AAV followed by 4 μl of sterile saline at a rate of 0.5 μl/min (10 mice per group). The needle was loaded first with saline and then with virus so that saline is injected after the virus and serves to push virus into the brain, increasing spread. Two-month-old male YAC128 and wt littermates were injected bilaterally in the striatum with 1 μl of GFP, VL12.3, or Happ1 AAV followed by 4 μl of sterile saline at a rate of 0.5 μl/min (10 mice per group). Two-month-old male and female BACHD mice were injected bilaterally in the striatum with 1 μl of either GFP or Happ1 AAV followed by 4 μl of sterile saline at a rate of 0.5 μl/min (five females and four males per group).
Postnatal day 3 male R6/2 and wt littermates were anesthetized by hypothermia by submersion in ice water inside a latex glove for 5 min. Pups were then placed on an ice pack with their heads stabilized in a putty mold for the duration of the surgery. After disinfecting the scalp, a 5 μl Hamilton syringe with a removable 33-gauge needle with a 20° bevel tip was pushed through the skin and skull at approximately the center of each forebrain hemisphere to a depth of 2.5 mm. Bilateral injections of 1 μl of GFP, VL12.3, or Happ1 AAV at a rate of 0.1 μl/min were performed using an ultra micropump III and micro4 controller. The needle was left in place for an additional 5 min and withdrawn slowly. Pups were allowed to recover on a 37°C water circulating heating pad, and a small amount of the dam's urine was rubbed on their rump to prevent subsequent cannibalism. Pups were checked for the presence of a milk spot 2 h after surgery and for general health daily for three subsequent days. After weaning, genotyping was performed to determine how many mice from each of the six groups had been injected until a minimum of 20 per group was reached. Ten mice per group were killed for histology at 10 weeks of age, while the remaining mice were studied until the point of mortality.
Behavioral assays.
All behavioral experiments were conducted during the light cycle by a researcher blind to genotype and treatment group. Repeated tests were conducted at approximately the same time of day. Single-time-point behaviors were compared using ANOVA. Repeated behavior tests were compared using repeated-measures ANOVA for lines in which all animals were killed at the same age (YAC128 and BACHD) and two-way ANOVA at individual time points for lines in which animals were tested until a disease-related endpoint was reached and therefore not killed at the same age (R6/2 and N171-82Q). Bonferroni post hoc tests were used to determine significance.
Rotarod.
The latency to fall from an accelerating rotarod (Ugo Basile) beginning at 5 rpm and accelerating to 40 rpm over 240 s was scored. Mice were allowed to stay on the rotarod for a maximum of 300 s. Mice were trained before the initial test for two consecutive days for R6/2, N171-82Q, and YAC128 lines and for three consecutive days for the BACHD line. Two trials were performed per training day with a 10 min intertrial interval (ITI). On testing days, two trials were performed separated by a 10 min ITI. Only the second trial was scored.
Beam crossing.
The time to cross the center 80 cm of a 1 m beam was scored. Three square beams of different widths were used (28, 12, and 6 mm). The beams were mounted atop poles (50 cm above the tabletop) with a bright light at the starting end and a dark box containing the animal's home cage nest material at the far end. A nylon hammock 7.5 cm above the tabletop was used to prevent injury to mice falling from the beam. Mice were placed at the end of the beam with the bright light and the time from when the entire body of the mouse entered to the time that the nose of the mouse exited the center 80 cm was measured using an electronic infrared interrupt sensor. The 80 cm section is used because the mice sometimes hesitate before starting and before entering the dark box. Mice were trained before the initial test for three consecutive days on all beams with a 10 min ITI between beams. During training, mice were encouraged to keep moving across the beam by nudging and tail pinching. Training trials were repeated until each animal crossed the beam three times without stopping or turning around. On testing days, three trials were performed per mouse for each beam with a 10 min ITI between beams. Trials in which the animal took longer than 60 s to cross or fell off the beam were scored as 60 s. Trials in which the animal stopped or turned around were repeated. The average of the trials was scored.
Climbing.
Mice were placed at the bottom end of a closed-top wire mesh cylinder (10 × 15 cm) on the tabletop and recorded with a video camera for 5 min. The time from when a mouse's fourth foot left the tabletop to the time when the first foot was replaced on the tabletop was scored as time spent climbing. The sum of climbing time for the 5 min trial was compared.
Clasping.
Animals were suspended by the tail ∼30 cm above the tabletop for 1 min and recorded with a video camera. No clasping was scored as 0, forelimb clasping was scored as 1, and hindlimb clasping was scored as 2.
Amphetamine-induced rotation.
Mice were injected intraperitoneally with 10 mg/kg amphetamine in sterile saline and placed in a 50 × 50 cm open white Plexiglas box with 16 cm sides. Activity was recorded for 30 min beginning 5 min after amphetamine injection by a ceiling-mounted video camera. The number of ipsilateral rotations was counted. Only rotations with a diameter similar to the animal's body length or smaller (as opposed to large circles around the edge of the box) were scored.
Open field, novel object, novel object location, and novel object preference.
Mice were placed in the lower left corner of a 50 × 50 cm open white Plexiglas box with 16 cm sides in a room brightly lit by fluorescent ceiling lights. Open field activity was recorded for 10 min by a ceiling-mounted video camera. Center entries and time spent in the center were scored. Mice were then removed from the box for a 5 min ITI and two different novel objects were placed in the upper two corners of the box, far enough from the sides so as to not impede movement around the outer edge (∼7 cm). Mice were reintroduced to the box in the lower left corner and recorded for 5 min, during which the number of investigations of the novel objects was scored. Episodes involving the mouse in close proximity to the objects but not facing or sniffing them were not considered investigations. Circling or rearing on the objects with continued sniffing was considered a single investigation, while episodes in which the mouse sniffed the object, turned away, or reared against the wall and subsequently turned back to sniff the object again were considered separate investigations. Mice were then removed from the box for a 5 min ITI, and the object at the top right corner of the box was moved to the lower right corner of the box. Mice were reintroduced to the box and recorded for 5 min, and the number of investigations of the objects was scored. For novel object location testing, the percentage of the investigations, or nose pokes, to the target object (the one in the new location) was computed. For novel object preference testing, the experiment was repeated on the subsequent day with the exception that rather than repositioning the object in the top right corner, it was replaced by a different, unfamiliar object in the same location. The percentage of the investigations of the target object (the unfamiliar one) was computed.
Histology.
Brain tissue was fixed by transcardiac perfusion of 4% PFA in PBS. Brains were postfixed in ice-cold 4% PFA in PBS for 45 min and then placed in 30% sucrose at 4°C overnight until they sunk. Brains were then embedded in optimal cutting temperature embedding medium and cut in 16 μm slide-mounted coronal sections. Sections were stored at −20°C until immunohistochemistry (IHC). For IHC, sections were blocked for 30 min at room temperature in 3% BSA, 10% normal goat serum, 0.1% Triton X-100, and 0.02% sodium azide in PBS. Antibodies were diluted in the blocking solution. Primary antibodies were incubated on sections overnight or for 48 h (anti-HA) at 4°C. Secondary antibodies were incubated on sections for 2 h at room temperature. Primary antibodies used include rabbit anti-DARPP-32 (1:500, Millipore Bioscience Research Reagents), mouse anti-Htt MW8 (Ko et al., 2001), mouse anti-Htt EM48 (1:1000, Millipore Bioscience Research Reagents), rabbit anti-GFP (1:400, Invitrogen), mouse anti-NeuN (1:500, DAKO), rabbit anti-GFAP (1:500 Millipore Bioscience Research Reagents), and mouse anti-HA (1:200, Covance). Secondary antibodies used include Alexa-fluor 350 (blue)-, 488 (green)-, and 568 (red)-labeled goat anti-mouse or anti-rabbit (1:250, Invitrogen). Topro-3 iodide nuclear stain (Invitrogen) was incubated at 1:5000 in PBS for 5 min at RT after secondary antibody. Animals with <50% of the striatal area transduced at 0.75 mm anterior to bregma were eliminated from all behavioral and histological analyses. Quantification of IHC was compared using two-way ANOVA and Bonferroni post hoc tests for significance.
Lentiviral HD model.
Three sections per mouse selected from the anterior region of the forebrain (1.7–2 mm anterior to bregma), the injection site (0.75 mm anterior to bregma), and the posterior region of the forebrain (0.8–1 mm posterior to bregma) were stained for GFP (green) and HA (iAb, red) to determine the extent of viral transgene spread. In the case of the iAb-treated groups, these sections were also used to determine the similarity of spread and cotransduction efficiency of the mHDx-1 lentivirus and iAb AAV. Three sections per mouse selected near the injection site were stained for Htt inclusions (MW8, green), DARPP-32 (red), and all nuclei (blue). MW8 is an Ab that recognizes a unique epitope in the C terminus of HDx-1 and preferentially binds aggregated Htt (Ko et al., 2001). This Ab was used to quantify inclusions. Striatal Htt inclusions were quantified by counting green foci larger than 2 pixels at 40× magnification for each section. Striatal DARPP-32 staining was assessed both for the area of the lesion and for the intensity of staining within the lesion. For lesion area, the ratio of the area of total DARPP-32 loss to the area of striatal lentiviral transgene spread was calculated. For staining intensity, the ratio of DARPP-32 (red fluorescence) staining in the transduced area of the injected striatum to DARPP-32 staining in the same area of the contralateral, uninjected striatum was calculated. ImageJ was used for all three quantifications. To further characterize lesions, two sections per mouse were stained for DARPP-32 (red), GFAP (green), and NeuN (blue) or all nuclei (topro-3 iodide, blue).
R6/2 HD model.
Three sections per mouse selected from the anterior region of the forebrain, the injection site (∼0.75 mm anterior to bregma), and the posterior region of the forebrain from both the group killed at 10 weeks of age and the group used to assess survival were stained for GFP (green) and HA (red) to determine the extent of viral transgene spread. Three sections per mouse at approximately bregma 0.0 mm from the group killed at 10 weeks of age were stained for Htt inclusions (MW8, green), DARPP-32 (red), and all nuclei (topro-3-iodide, blue). Small Htt aggregates were quantified by counting green foci 2–8 pixels in size within a 250 μm2 area of the transduced striatum. Intranuclear inclusions were quantified by counting green foci 9–15 pixels in size that colocalized with topro-3-iodide staining. DARPP-32 was quantified by mean red fluorescence intensity of the entire striatal area of both hemispheres. Ventricle area was also measured for both hemispheres of these sections by tracing its outline (area computed in arbitrary units). ImageJ was used for all three of these measures.
N171-82Q HD model.
Three sections per mouse selected from the anterior region of the forebrain, the injection site (0.75 mm anterior to bregma), and the posterior region of the forebrain were stained for GFP (green) and HA (red) to determine the extent of viral transgene spread. Neuropathological markers were not assessed for these animals as they were used to assess survival and were therefore not killed at the same age but at the same disease stage.
YAC128 and BACHD HD models.
Three sections per mouse selected from the anterior region of the forebrain, the injection site (0.75 mm anterior to bregma), and the posterior region of the forebrain were stained for GFP (green) and HA (red) to determine the extent of viral transgene spread. Three sections per mouse at approximately bregma 0.0 mm were stained for Htt inclusions (EM48, green), DARPP-32 (red), and all nuclei (blue). DARPP-32 was quantified by mean red fluorescence intensity of the entire striatal area of both hemispheres. Ventricle size was also measured for both hemispheres of these sections. ImageJ was used for these measures.
Survival.
When animals became obviously ill, they were assessed twice daily for the loss of righting reflex. If animals failed to right themselves immediately after being laid on their side in the home cage, they were killed.
Results
Widespread striatal transduction is achieved with the lentiviral vector in adult mice and with the AAV vector in adult and neonatal mice
In the lentiviral model, adult wt mice were unilaterally injected with a mixture of mHDx-1 or GFP lentivirus, plus GFP-, VL12.3-, or Happ1-AAV. Mice were killed 6 weeks later and coronal sections throughout the striatum were stained for mHDx-1 and iAb. Similar striatal spread is seen with both lentivirus and AAV (Fig. S1A, available at www.jneurosci.org as supplemental material). Colocalization of mHDx-1 and iAb in confocal images shows that these viruses cotransduce cells (Fig. S1B, available at www.jneurosci.org as supplemental material). There are a larger number of iAb-positive cells than mHDx-1-positive cells, suggesting greater transduction efficiency for AAV2/1. Adult injections of AAV2/1 were also used bilaterally in N171-82Q, YAC128, and BACHD HD transgenic mice. Striatal iAb spread in these mice is similar to that seen in the lentiviral model (data not shown). The R6/2 model has an accelerated time frame that includes Htt inclusions at birth and onset of behavioral symptoms at <3 weeks of age (Mangiarini et al., 1996; Stack et al., 2005). Because of this and the 3 week delay before AAV delivered transgene expression reaches its peak (Tenenbaum et al., 2004), postnatal day 3, rather than adult, injections were used in this line. Striatal spread equivalent to or better than that seen with adult injections is apparent (Fig. S1C, available at www.jneurosci.org as supplemental material).
Happ1 treatment improves rotarod performance in four transgenic HD models
The ability to stay on a rotating rod is often used as a test of sensory-motor ability. Mice were assessed for accelerating rotarod performance as follows: weekly from 4 weeks of age until death in R6/2 and wt littermates (Fig. 1A), every 2 weeks from 6 weeks of age until death in N171-82Q and wt littermates (Fig. 1B), monthly from 3 until 7 months of age in YAC128 and wt littermates (Fig. 1C,D), and monthly from 3 until 6 months of age in BACHD mice (Fig. 1E). Compared with wt littermates, R6/2 mice display a deficit in rotarod performance from 5 weeks of age. Compared with GFP treatment, Happ1 treatment ameliorates this deficit between 9 and 12 weeks of age (Fig. 1A). In contrast, compared with GFP treatment, VL12.3 treatment reduces latency to fall in R6/2 mice between 10 and 12 weeks of age. Although rotarod performance is also impaired below GFP controls at 13 weeks of age, statistics cannot be performed, as only one VL12.3-treated mutant remained alive at this time point. Compared with wt littermates, N171-82Q mice display a motor deficit on the rotarod from 20 weeks of age. From 22 weeks of age performance is significantly improved by Happ1 treatment (Fig. 1B). Happ1-treated mutants do not differ from wt animals in their performance until 40 weeks of age.

Happ1 treatment improves rotarod performance in four HD mouse models. Mice were tested on an accelerating rotarod for a maximum of 300 s. A, Male R6/2 and wt littermates were tested weekly: Happ1 treatment significantly improves performance during weeks 9–12, while VL12.3 treatment degrades performance during weeks 10–12. B, Male N171-82Q and wt littermates were tested every other week. Happ1 treatment improves performance to a level not different from wt animals from 22 weeks until 38 weeks. C, Male YAC128 and wt littermates were tested monthly and Happ1 (but not VL12.3) treatment significantly improves performance at months 3, 4, and 7. D, YAC128 rotarod performance only for GFP- and Happ1-treated mutant groups is shown for simplicity. E, Male and female BACHD mice were tested monthly: Happ1 treatment significantly improves performance at months 5 and 6. Asterisks indicate difference between GFP- and iAb-treated mutants. *p < 0.05, **p < 0.01, ***p < 0.001.
Happ1 treatment also improves rotarod performance in 3-, 4-, and 7-month-old YAC128 mice (Fig. 1C,D). The lack of significant difference at 5 and 6 months of age appears to be related to an improvement in the performance of GFP-treated animals rather than a decline in the performance of Happ1-treated mice. YAC128 mice display a learning deficit that includes impaired learning of the rotarod task from 2 months of age (Van Raamsdonk et al., 2005). As rotarod training was performed at 3 months of age with the same number of training trials for all mice, the difference in performance between young GFP- and Happ1-treated mice could be the result of enhanced learning of the task by Happ1 treatment. By 7 months of age, the performance of GFP-treated YAC128 mice begins to decline, presumably as a result of declining motor ability. Happ1 treatment also ameliorates the motor deficit in the BACHD line. Compared with GFP treatment, Happ1 treatment increases the latency to fall from the rotarod in 5- and 6-month-old BACHD mice (Fig. 1E). In summary, Happ1 attenuates the loss of motor function in the rotarod test induced by mHtt in four HD mouse models.
Happ1 treatment improves beam-crossing performance in four transgenic HD models
Similar to the rotarod, beam crossing is an objective assay for balance and motor function. Mice were assessed for beam-crossing performance as follows: weekly from 4 weeks of age until death in R6/2 and wt littermates (Fig. 2A; Fig. S2A–C, available at www.jneurosci.org as supplemental material), every 2 weeks from 6 weeks of age until death in N171-82Q and wt littermates (Fig. 2B; Fig. S2D–F, available at www.jneurosci.org as supplemental material), monthly from 3 until 7 months of age in YAC128 and wt littermates (Fig. 2C; Fig. S2G–I, available at www.jneurosci.org as supplemental material), and monthly from 3 until 6 months of age in BACHD mice (Fig. 2D; Fig. S2J–L, available at www.jneurosci.org as supplemental material). Three widths of square beam were used: 28, 12, and 6 mm. The data for the former two widths are shown in Fig. S2, available at www.jneurosci.org as supplemental material. Compared with wt littermates, R6/2 mice display increased time to cross the beams from 10, 7, and 5 weeks, respectively. Being the easiest to traverse, there are no significant effects of iAb treatment on time to cross the 28 mm beam. However, compared with GFP treatment, Happ1 treatment does reduce the time to cross the 12 mm beam in 10- and 11-week-old R6/2 mice, and the 6 mm beam between 9 and 11 weeks of age. Conversely, compared with what was seen for GFP-treated littermates, VL12.3 treatment increases the time to cross the 12 mm beam at 11 and 12 weeks of age and the 6 mm beam at 9 and 10 weeks of age (Fig. S2A–C, available at www.jneurosci.org as supplemental material).

Happ1 treatment improves beam-crossing performance in four HD mouse models. Time to cross the center 80 cm of a square, 1-m-long, 6-mm-wide beam was measured. A, Male R6/2 and wt littermates were tested weekly: Happ1 treatment improves performance during weeks 9–11, while VL12.3 treatment degrades performance at 9 and 10 weeks. B, Male N171-82Q and wt littermates were tested every other week: Happ1 treatment improves performance at 18 weeks of age and older. C, Male YAC128 and wt littermates were tested monthly: Happ1 treatment improves performance at 3 and 4 months. D, Male and female BACHD mice were tested monthly: Happ1 treatment improves performance at 6 months. Asterisks indicate difference between GFP- and iAb-treated mutants. *p < 0.05, **p < 0.01, ***p < 0.001.
N171-82Q mice display increased time to cross the three beams at 22, 16, and 12 weeks, respectively (Fig. S2D–F, available at www.jneurosci.org as supplemental material). Happ1 treatment significantly reduces time to cross the 28 mm beam from 22 weeks of age on, the 12 mm beam in mutants 16, 18, and 24 weeks old and older, and the 6 mm beam in mutants 18, 20, and 26 weeks old and older. Happ1-treated mutants only show a deficit compared with wt mice in time to cross the 12 mm beam at 30 and 40 weeks of age and the 6 mm beam at 28, 30, 38, and 40 weeks of age.
Compared with wt littermates, YAC128 mice display impaired beam-crossing performance at 7 months of age for the 28 mm beam and 3 and 4 months of age for the 12 and 6 mm beams (Fig. S2G–I, available at www.jneurosci.org as supplemental material). Compared with GFP treatment, Happ1 treatment reduces the time to cross the 28 mm beam in 7-month-old YAC128 mice and the 12 and 6 mm beams at 3 and 4 months (Fig. 2C; Fig. S2G–I, available at www.jneurosci.org as supplemental material). As in the rotarod test, the performance of the GFP-treated YAC128 mice appears to improve over the earliest time points, consistent with the motor task learning deficit reported for these mice (Van Raamsdonk et al., 2005). The Happ1-induced improvement at the early time points suggests an improved learning of the task. In contrast, VL12.3 treatment appears to exacerbate this early learning deficit in 3-month-old mice. Happ1 treatment of BACHD mice reduces the time to cross the beams at 5 and 6 months for the 28 mm beam, at 4 and 6 months for the 12 mm beam, and at 6 months for the 6 mm beam (Fig. 2D; Fig. S2J–L, available at www.jneurosci.org as supplemental material). In summary, Happ1 attenuates the loss of motor function in the beam-crossing test induced by mHtt in four HD mouse models.
Happ1 treatment improves climbing performance in YAC128 and BACHD mice
Climbing in a single episode is a useful measure of motor ability that is not complicated by potential learning deficits, and is thus well suited to the YAC128 model. Climbing time was assessed in 7-month-old YAC128 and wt littermates (Fig. 3A) and in 6-month-old BACHD mice (Fig. 3B). Mice were placed at the bottom of a closed-top wire mesh cylinder and observed for 5 min. Time spent climbing with all four feet off of the tabletop was scored. Compared with wt littermates, GFP- and VL12.3-treated YAC128 mice investigate the wire mesh and rear on two or three legs frequently, but climbing time is reduced (Movie S3). Happ1 treatment increases climbing time in these mice to a level not significantly different from wt mice (Fig. 3A). Compared with GFP treatment, Happ1 treatment also increases climbing time in BACHD mice (Fig. 3B). Thus, Happ1 attenuates the loss of motor function in the climbing test induced by mHtt in two HD mouse models.

Happ1 treatment improves climbing and prevents clasping in HD transgenic mice, and VL12.3 or Happ1 treatment prevents the amphetamine-induced rotation phenotype caused by mHDx-1 lentivirus. A, B, Mice were placed at the bottom of a vertical wire mesh tube and observed for 5 min. The time when all four feet were off the ground was scored as climbing time. A, Seven-month-old GFP- and VL12.3-treated YAC128 mice have impaired climbing compared with wt mice. This is rescued by Happ1 treatment. B, Happ1 treatment improves climbing time in 6-month-old BACHD mice. C, Twenty-week-old male N171-82Q and wt littermates were suspended by the tail, observed for 1 min, and given a clasping score as follows: no clasping = 0, forelimb clasping = 1, hindlimb clasping = 2. D, Wild-type mice were injected unilaterally at 4 weeks of age with mHDx-1 or GFP lentivirus plus GFP-, VL12.3-, or Happ1-AAV. At 10 weeks of age mice were assayed for amphetamine-induced rotation. Uninjected (naive) animals were tested as a negative control. Animals injected with mHDx-1 lentivirus and GFP-AAV exhibit many ipsilateral rotations in response to amphetamine. VL12.3 or Happ1 prevents this phenotype. *p < 0.05, **p < 0.01, ***p < 0.001.
Happ1 treatment ameliorates clasping in N171-82Q mice
Since clasping is seen in several neurodegenerative disease models, it is often used as a test for potential therapeutics. The pathology underlying clasping is not understood, however. Twenty-week-old N171-82Q and wt littermates were assayed for clasping. Clasping was not assessed in YAC128 or BACHD mice, as this behavior has not been reported in these lines. Mice were suspended by the tail and observed for 1 min. Mice with normal limb extension were given a score of 0. Forelimb clasping was scored as 1, and hindlimb clasping was scored as 2 (Fig. 3C). Wild-type mice do not exhibit clasping. All GFP-treated N171-82Q mice exhibit clasping, with approximately half displaying forelimb-only clasping and half displaying hindlimb clasping. The majority of Happ1-treated mutants display forelimb-only clasping, while a few display hindlimb clasping or no clasping (Fig. S4A, available at www.jneurosci.org as supplemental material). Thus, Happ1 attenuates the abnormal motor behavior induced by mHtt in this HD mouse model.
Treatment with VL12.3 or Happ1 prevents abnormal amphetamine-induced rotation behavior in the lentiviral HD model
Four-week-old C57BL/6 mice were injected unilaterally with mHDx-1 or GFP lentivirus plus GFP-, VL12.3-, or Happ1-AAV. Six weeks after surgery, animals were injected with 10 mg/kg amphetamine, intraperitoneal. After 5 min, animals were placed in an open field for 30 min and ipsilateral rotations counted (Fig. 3D). Age-matched, uninjected mice were also tested as a control for surgery effects. Naive mice were randomly assigned to right turn or left turn groups for scoring ipsilateral turns. Compared with what was seen for naive mice, injection of GFP lentivirus along with any of the AAVs does not significantly increase the number of ipsilateral rotations. In contrast, animals injected with mHDx-1 lentivirus plus GFP-AAV display a major increase in ipsilateral rotations. Coinjection of VL12.3- or Happ1-AAV strongly reduces the number of ipsilateral rotations in mHDx-1 lentivirus-injected animals to levels not significantly different from GFP lentivirus-injected or naive animals. Thus, the iAbs attenuate the dopamine-driven motor abnormality induced by mHtt in the lentiviral HD mouse model.
Happ1 treatment normalizes open field behavior in full-length Htt transgenic models of HD
Although increased anxiety has not previously been reported for YAC128 and BACHD mice, we observed that some of the mice exhibit progressive, researcher-induced anxiety. As a result, 7-month-old YAC128 and wt littermates and 6-month-old BACHD mice were assayed for anxiety during open field exploration (Fig. 4). Compared with wt littermates, GFP-treated YAC128 mice display reduced entries into, and time spent in, the center of the field, which suggests enhanced anxiety. In contrast, Happ1 treatment increases center entries and time in the center to levels not significantly different from wt mice (Fig. 4A,B). In the BACHD mice, there is a trend toward an increased number of center entries as a result of Happ1 treatment, but it does not reach significance (Fig. 4C). However, compared with what was seen for GFP-treated BACHD mice, Happ1 treatment significantly increases the time spent in the center of the open field (Fig. 4D). Thus, Happ1 attenuates anxiety in two full-length HD models.

Happ1 treatment normalizes open field behavior in full-length transgenic models of HD. Mice were observed for 10 min during exploration of an open field. Anxiety was inferred by scoring entries into, and time spent in, the center of the open field. A, C, There was a trend toward increased center entries for both models in response to Happ1 treatment, but it was not significant. B, D, Happ1 treatment increases time spent in the center of the open field in 7-month-old YAC128 mice (B) and 6-month-old BACHD mice (D). *p < 0.05, **p < 0.01.
Happ1 treatment improves learning in YAC128 mice
Due to the learning deficit reported for YAC128 mice (Van Raamsdonk et al., 2005), and the indication from motor testing that this deficit may be ameliorated by Happ1 treatment, learning was assayed in the full-length transgenic models. Although iAb was delivered to the striatum, extensive viral spread also results in cortical and hippocampal transgene expression (Fig. S1A,D, available at www.jneurosci.org as supplemental material), which could affect learning. The HD-like phenotype of these models includes altered motor performance and motivation. Therefore, learning was assayed using the novel object location and novel object preference tests (Mumby et al., 2002, Mumby et al., 2007), which do not require training or depend on the speed of swimming or running, allowing the testing of learning with less influence of motor ability and motivation.
To assess hippocampal-dependent learning, 7-month-old YAC128 and wt littermates and 6-month-old BACHD mice were tested for preference for the novel location of a known object (Fig. 5A) (Mumby et al., 2002). On day 1, mice were habituated to an open field. After a 5 min ITI they were reintroduced to the same field now containing two novel objects in the upper corners of the box located far enough from the sides to allow free movement around the outer edge. Investigations of each object were scored for 5 min (trial 1, T1). In trial 2 (T2) after another 5 min ITI, they were reintroduced to the same field with the object previously located in the upper right corner moved to the lower right corner. Investigation of each object was scored for 5 min, and the percentage of the investigations, or nose pokes, to the target object (the one that was moved) was computed. A score of 50% represents no preference. As expected, wt mice of the YAC128 line display no preference in T1 and a preference for the target object in T2 (Fig. 5C). In contrast, GFP- and VL12.3-treated YAC128 mice display no preference in either trial, indicating impaired spatial learning. However, Happ1 treatment restores significant spatial learning (Fig. 5C). BACHD mice in both treatment groups show no preference for the target object, indicating impaired spatial learning and no effect of iAb treatment (Fig. 5E).

Happ1 treatment ameliorates the learning deficit of YAC128 mice. A, To assay for preference of a known object in a novel location, mice were habituated to an open field for 10 min. After a 5 min ITI, they were exposed for 5 min to novel objects in the upper corners of the open field (T1). Investigation of the novel objects was scored. After another 5 min ITI, the mice were reintroduced to the same field with the object previously in the upper right corner moved to the lower right corner (T2) for 5 min. The percentage of the investigations of the target object (the one in the new location) was scored. A score of 50% would indicate no preference. B, On the next day mice were tested for preference for a novel object. Mice were rehabituated to the open field for 10 min. After a 5 min ITI, they were exposed for 5 min to two objects in the upper corners of the open field (T1). Investigation of the objects was scored. After another 5 min ITI the mice were reintroduced to the same field with the object in the upper right corner replaced with a completely novel object in the same location. The percentage of the investigations of the target object (the completely novel one) was scored. A score of 50% would indicate no preference. C, D, Seven-month-old YAC128 and wt littermates were tested. Wild-type mice display a preference for the novel object location (C) and a trend toward a preference for the novel object (D). This does not reach significance, but when data from the three wt treatment groups are pooled, the preference is significant (p < 0.01). GFP- and VL12.3-treated YAC128 mice show no preference for either object in either paradigm, indicating a learning deficit. Happ1 treatment improves this deficit. E, F, GFP- and Happ1-treated BACHD mice show no preference for either object in either paradigm. There is a trend toward a preference for the novel object in Happ1-treated BACHD mice, but it does not reach significance. *p < 0.05, ***p < 0.001.
To assess cortical-dependent learning, mice were tested for preference for a novel object (Fig. 5B) (Mumby et al., 2007). On day 2, mice were rehabituated to the same field and T1 from day 1 was repeated. In T2 they were reintroduced to the field with the object in the upper right corner replaced with a completely novel object. Investigation of each object was scored for 5 min, and the percentage of the investigations of the target object (the completely novel object) was computed. Wild-type mice of the YAC128 line display no preference in T1 and trended toward a preference for the target object in T2, although the difference is not significant. There are no differences between the treatment groups in wt mice, and when all wt groups are combined, the preference for the target object reaches significance (p < 0.01) (data not shown) indicating that the lack of significant preference is related to small sample size. GFP- and VL12.3-treated YAC128 mice display no preference for the target object, indicating impaired cortical learning. In contrast, Happ1-treated YAC128 mice display a preference for the target object, indicating preserved cortical learning (Fig. 5D). In the BACHD line, GFP-treated mice display no preference for the target object in T2 while Happ1-treated mice trended toward a preference for the target object, although this does not reach significance (Fig. 5F). In summary, Happ1 attenuates a cognitive deficit in a full-length HD model.
In the lentiviral HD model, treatment with VL12.3 or Happ1 ameliorates neuron-specific mHDx-1 toxicity
DARPP-32 is a useful, specific marker of the medium spiny neurons (MSNs) that are lost in HD. Four-week-old C57BL/6 mice were injected unilaterally with mHDx-1 or GFP lentivirus plus GFP-, VL12.3-, or Happ1-AAV. Six weeks after surgery, animals were perfused for histology. Mice injected with mHDx-1 lentivirus plus GFP-AAV display large areas of strongly diminished DARPP-32 immunostaining. Areas of DARPP-32 loss also display loss of NeuN-positive cells, indicating death of striatal neurons (Fig. 6A). Topro-3 iodide nuclear staining does, however, show the presence of cells within lesioned areas, indicating hypersensitivity of neurons to the toxicity of mHDx-1 lentivirus (Fig. 6B). Lesioned areas also display reactive gliosis, as evidenced by an increase in GFAP staining (Fig. 6C). Coinjection of VL12.3- or Happ1-AAV with mHDx-1 lentivirus reduces both the size and intensity of DARPP-32 loss (Fig. 6D). To quantify lesion size, the ratio of the area of total DARPP-32 loss to the area of lentivirus transduction was computed (Fig. 6E). Animals injected with GFP lentivirus and any of the AAVs display very small if any lesion areas. Injection of mHDx-1 lentivirus with GFP-AAV causes a major lesion, which is reduced by coinjection of VL12.3- or Happ1-AAV to a level not significantly different from animals injected with GFP lentivirus plus any of the AAVs.
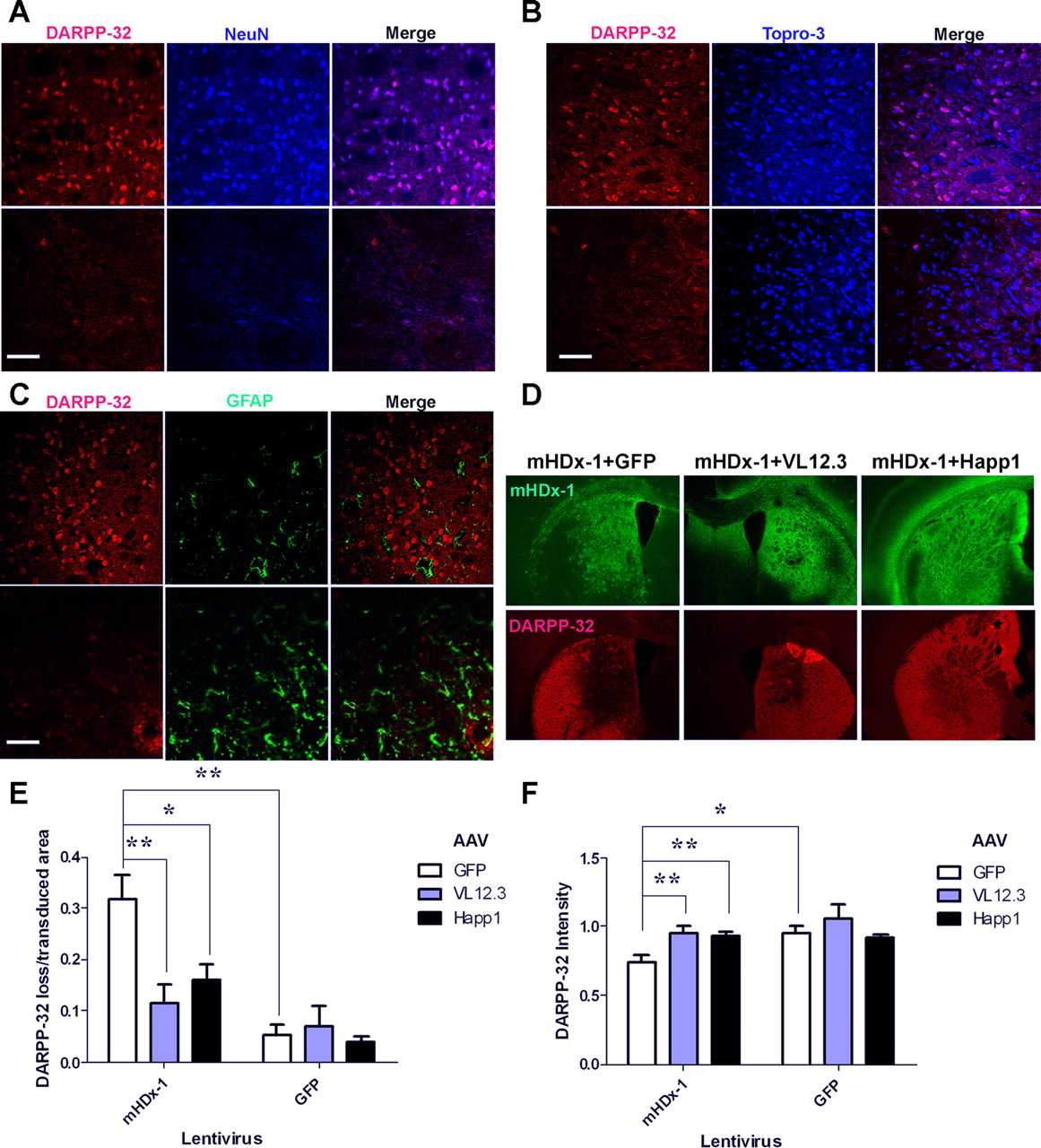
mHDx-1 lentivirus causes neuron-specific toxicity in the striatum, which is reduced by VL12.3 or Happ1 treatment. Mice were injected unilaterally with mHDx-1 or GFP lentivirus plus GFP-, VL12.3-, or Happ1-AAV. Areas with loss of DARPP-32 staining were analyzed. A, Areas of DARPP-32 loss (bottom) also show loss of NeuN-positive cells, indicating death of neurons in these areas. B, Topro-3 iodide nuclear stain shows the presence of cells in lesioned areas, indicating that toxicity is neuron specific. C, Areas of DARPP-32 loss show increased GFAP staining, indicating increased inflammation in lesioned areas. In A–C, the top shows GFP lentivirus and the bottom shows mHDx-1 lentivirus. D–F, Coinjection of either Happ1 or VL12.3 with mHDx-1 reduces the area and intensity of DARPP-32 loss. D, Adjacent coronal sections stained either for mHDx-1 (green) or DARPP-32 (red). DARPP-32 loss is reduced in the presence of either intrabody. E, The ratio of the area of total DARPP-32 loss to the transduced area was compared to assess lesion size. Lesions are significantly smaller in the presence of either intrabody. Three sections per mouse were analyzed. F, The ratio of DARPP-32 staining fluorescence intensity in the transduced area of the striatum to DARPP-32 staining fluorescence intensity in the same size area of the uninjected striatum was compared to assess the severity of the lesion. Lesions are less severe in the presence of iAb. Three sections per mouse were analyzed. *p < 0.05, **p < 0.01, Scale bars, 50 μm.
To assess the severity of DARPP-32 staining loss within the lesion, the ratio of DARPP-32 intensity within the transduced striatum to DARPP-32 intensity in the noninjected side was computed (Fig. 6F). Injection of mHDx-1 lentivirus plus GFP-AAV causes a decrease in DARPP-32 intensity of the transduced striatum, which is rescued by VL12.3- or Happ1-AAV to the level of animals injected with GFP lentivirus. Thus, both iAbs very effectively prevent the mHtt-induced loss of MSNs in the lentiviral model.
Treatment with VL12.3 or Happ1 reduces mHtt aggregation in the lentiviral and R6/2 HD models
Coronal sections from mHDx-1 lentivirus-injected animals 6 weeks after surgery (Fig. S5A, available at www.jneurosci.org as supplemental material) and 10-week-old R6/2 mice (Fig. S5B, available at www.jneurosci.org as supplemental material) were stained for Htt. Most of the Htt staining in both models was in the form of aggregates, although some diffuse staining in neurons is seen (Fig. S5A, arrows in left panel, available at www.jneurosci.org as supplemental material). VL12.3 or Happ1 treatment reduces aggregates and increases the level of diffuse Htt, both in neuronal somas (arrows) and in the neuropil (Fig. S5A, available at www.jneurosci.org as supplemental material). The larger areas of bright staining in both the Happ1- and VL12.3-treated R6/2 brains appear to represent neuronal cytoplasm (Fig. S5B, available at www.jneurosci.org as supplemental material). Happ1-treated brains appear to display lower overall levels of Htt staining in both HD models, although this was not quantified. To quantify aggregation in the lentiviral model, three sections per animal were stained with MW8, an anti-Htt antibody that recognizes the C terminus of HDx-1 and preferentially labels aggregated Htt in R6/2 mouse brains (Ko et al., 2001). Striatal MW8-positive foci were counted and normalized to the area of HDx-1 transduction (Fig. 7A). Treatment with VL12.3 or Happ1 dramatically reduces mHDx-1 aggregates. To quantify small neuropil aggregates in the R6/2 model, three sections per animal were stained with MW8 and topro-3 iodide nuclear marker. MW8-positive foci of 2–8 pixels in size that do not colocalize with topro-3 iodide in a 250 μm2 area of the transduced striatum were counted (Fig. 7B). Treatment with VL12.3 or Happ1 dramatically reduces the number of these neuropil aggregates as well. To quantify intranuclear inclusions, MW8-positive foci of 9–15 pixels in size that colocalize with topro-3 iodide staining were counted (Fig. 7C). There is a trend toward a reduction of intranuclear inclusions in VL12.3-treated animals, and a significant reduction in Happ1-treated animals. Htt inclusions are not observed in YAC128 or BACHD brains. This is not surprising as inclusion formation has been reported to begin at 12 months of age in YAC128 mice (Slow et al., 2005) and between 12 and 18 months of age in BACHD mice (Gray et al., 2008). In summary, Happ1 strongly prevents the accumulation of mHtt aggregates in the R6/2 and lentiviral models.
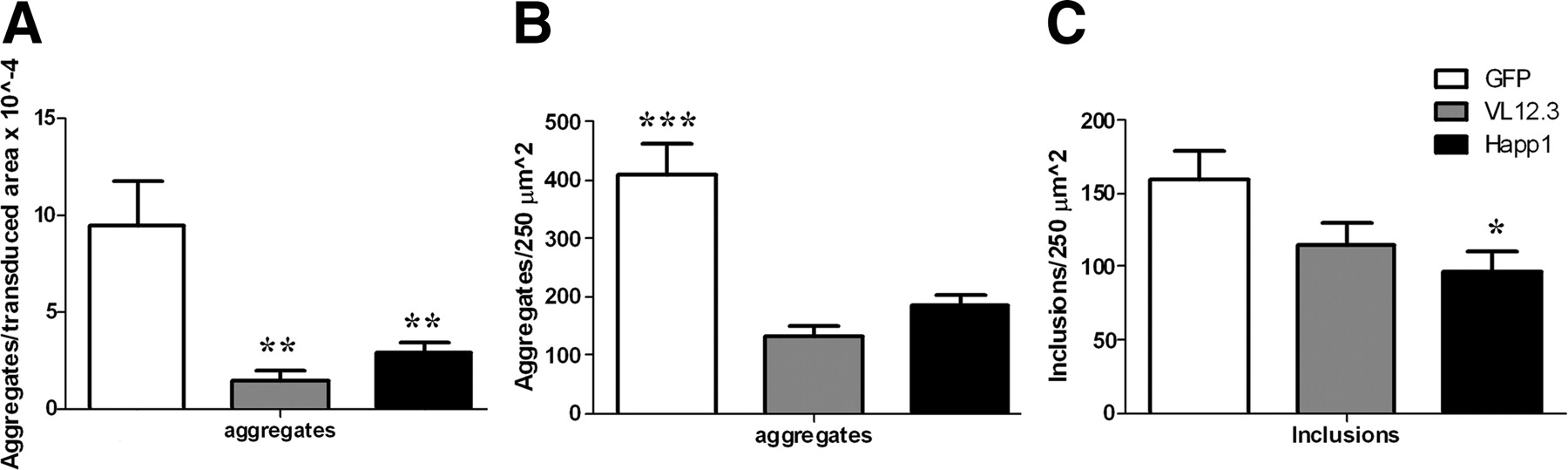
VL12.3 or Happ1 decreases Htt aggregation in the lentiviral and R6/2 HD models. A–C, Three sections each of 6-weeks-postsurgery mHDx-1 lentivirus-injected (A) and 10-week-old (B, C) R6/2 brains were stained for Htt. A, The ratio of striatal aggregates to transduced area is reduced by both intrabodies. B, The number of small Htt aggregates per 250 μm2 is reduced by both intrabodies. C, Happ1 treatment reduces the number of intranuclear inclusions per 250 μm2. *p < 0.05, **p < 0.01, ***p < 0.001.
Happ1 treatment reduces ventricular enlargement in three transgenic HD models
In R6/2 mice, ventricular enlargement is reported to begin at 3 weeks of age (Stack et al., 2005) and at 9 and 12 months in YAC128 mice (Slow et al., 2003). This pathology has not yet been reported in BACHD mice. We assessed ventricle size in 10-week-old R6/2 and wt littermates (Fig. 8A,B), in 7-month-old Yac128 and wt littermates (Fig. 8C), and in 6-month-old BACHD mice (Fig. 8D). Compared with wt littermates, GFP- and VL12.3-treated R6/2 mice display very significant ventricular enlargement. Happ1 treatment reduces ventricle size to a level not significantly different from wt animals (Fig. 8A,B). Compared with wt littermates, GFP- and VL12.3-treated YAC128 mice display ventricular enlargement while Happ1 treatment reduces ventricle size (Fig. 8C). In addition, Happ1-treated BACHD mice also display smaller ventricles than GFP-treated BACHD mice (Fig. 8D). As with DARPP-32 neuropathology, Happ1 is highly effective in preventing ventricular enlargement in three HD transgenic models.

Happ1 treatment reduces ventricular enlargement in three HD mouse models. A–D, Ventricle area was measured at approximately bregma 0.0 mm in three sections each from 10-week-old male R6/2 and wt littermates (A, B), 7-month-old male YAC128 and wt littermates (C), and 6-month-old male and female BACHD mice (D). Both R6/2 and YAC128 mice display increased ventricle size compared with wt littermates. B–D, Happ1 treatment reduces ventricular enlargement in R6/2 mice (B), YAC128 mice (C), and BACHD mice (D). *p < 0.05, **p < 0.01, ***p < 0.001.
Happ1 treatment improves body weight of N171-82Q mice
Body weight was assessed as follows: weekly from 4 weeks of age until death in R6/2 and wt littermates (Fig. 9A), every 2 weeks from 6 weeks of age until death in N171-82Q and wt littermates (Fig. 9B), monthly from 3 until 7 months of age in YAC128 and wt littermates (Fig. 9C), and monthly from 3 until 6 months of age in BACHD mice (Fig. 9D). N171-82Q mice weigh significantly less than wt littermates from 16 weeks of age onward. Happ1-treated mutants, although still weighing less than wt littermates, display increased weight compared with GFP-treated mutants from 22 weeks of age (Fig. 9; Fig. S4B, available at www.jneurosci.org as supplemental material). Compared with wt littermates from 10 weeks of age, R6/2 mice display decreased body weight. In contrast, YAC128 mice trend toward greater body weight than wt littermates. BACHD males display greater body weight than females. There is no effect of iAb treatment on this parameter in any of these three models.

Happ1 treatment improves body weight and survival of N171-82Q mice. A, R6/2 mice weigh significantly less than wt mice from 10 weeks of age until death, and there is no effect of iAb treatment. Asterisks indicate a difference between GFP-treated wt and R6/2 mice. B, Happ1-treated N171-82Q mice weigh less than wt littermates, but they also weigh more than GFP-treated mutants. Asterisks indicate a difference between GFP- and Happ1-treated N171-82Q mice. C, While YAC128 mice trend toward weighing more than wt mice, there is no effect of iAb treatment. D, While male BACHD mice weigh more than female mice, there is no effect of iAb treatment. Asterisks indicate a difference between GFP-treated male and female mice. E, Happ1 treatment has no effect, while VL12.3 decreases survival of R6/2 mice. F, Happ1 treatment increases maximum survival of N171-82Q mice by 33% from 30 weeks to 40 weeks of age. *p < 0.05, **p < 0.01, ***p < 0.001.
Happ1 treatment increases survival in N171-82Q mice while VL12.3 treatment decreases survival in the R6/2 HD model
Life span was assessed in R6/2 mice (Fig. 9E) and N171-82Q mice (Fig. 9F). Once mice became visibly ill, they were assayed twice daily for loss of the righting reflex. Mice who did not immediately right themselves after being laid on their side were killed. Happ1 treatment has no effect on life span while VL12.3 treatment decreases survival time of R6/2 mice. However, Happ1 treatment increases maximum life span of N171-82Q mice 33%, from 30 weeks of age in GFP-treated mutants to 40 weeks of age in Happ1-treated mutants. Thus, Happ1 is effective in prolonging life span in the less severe transgenic HD model.
Discussion
Viral vectors for gene therapy
AAV is a promising vector for gene therapy in humans. Wild-type AAV is widespread in human populations, which reduces the probability of host immune activation complications (Chirmule et al., 1999; Peel and Klein, 2000). It is nonpathogenic in humans and unable to replicate in the absence of a helper virus. A number of clinical trials using AAV vectors to treat neurodegenerative diseases have found no pathology related to the vector (McPhee et al., 2006; Kaplitt et al., 2007; Marks et al., 2008). AAV can infect both dividing and nondividing cells and is capable of generating long-term transgene expression, which has been monitored for up to six years in nonhuman primates (Klein et al., 2002; Bankiewicz et al., 2006). Although wt AAV commonly integrates into genomic DNA at specific sites, integration events for recombinant AAV (rAAV), which lacks 96% of the viral genome, are rare, reducing the probability of oncogenic complications (Schnepp et al., 2003). The existence of >120 different capsid proteins also confers a wide range of tropisms allowing customization of rAAV gene therapy vectors (Mueller and Flotte, 2008).
AAV2 is the most common serotype found in humans. As a result, most early AAV gene therapy studies employ this serotype. Although AAV2 is capable of long-term gene expression, this serotype has low transduction efficiency, low viral spread in the CNS, as well as an inability to transduce non-neuronal cells (McCown et al., 1996; Bartlett et al., 1998; Klein et al., 1998; Tenenbaum et al., 2000). Chimeric AAV2/1, consisting of the AAV2 genome and the AAV1 capsid protein, exhibits increased spread and transduction efficiency, and the ability to transduce glial and ependymal cells as well as neurons, while retaining the long-term expression capabilities of AAV2 (Wang et al., 2003). Chimeric AAV2/1 also has a decreased lag time between infection and transgene expression (Auricchio et al., 2001).
Traditional AAV gene therapy vectors use the high-expressing CMV promoter. However, gene expression from this promoter in AAV infected cells in the CNS declines over time (McCown et al., 1996; Klein et al., 1998; Tenenbaum et al., 2000). Use of CBA stabilizes gene expression in AAV infected cells in the CNS (Klein et al., 2002). Therefore, we used a chimeric AAV2/1 vector, consisting of the AAV2 genome under the control of the CBA promoter and the AAV1 capsid. We observe extensive striatal iAb expression for at least 8 months (the longest postsurgical experimental time point).
HD models
Despite the large number of available HD mouse models, no one model completely recapitulates the human disease (Ehrnhoefer et al., 2009). Models of HD induced by viral vectors coding for mHtt, such as the lentiviral model, exhibit the striatal MSN loss characteristic of human HD, which makes these models very attractive for studying this key aspect of the HD phenotype. However, these animals show only certain motor deficits and no change in body weight or survival (de Almeida et al., 2002; Régulier et al., 2003).
Transgenic mice expressing N-terminal mHtt fragments (which are much more toxic than full-length mHtt), such as R6/2 and N171-82Q, exhibit rapid onset of progressive motor and cognitive deficits, weight loss, Htt inclusion formation, and striatal atrophy accompanied by ventricular enlargement, but no loss of MSNs (Rubinsztein, 2002). These models are also limited to therapeutic strategies directed at the N terminus of mHtt, so they cannot be used to study modifiers of mHtt toxicity with sites of action outside of this area such as caspase-6 cleavage (Graham et al., 2006).
Full-length Htt transgenic models, such as the YAC128 and BACHD lines, exhibit more human-like, slower, progressive cognitive and motor deficits along with striatal atrophy, ventricular enlargement, and some loss of MSNs at later stages. However, unlike in the human disease, the YAC128 and BACHD mice gain, rather than lose, weight during disease progression (Slow et al., 2003; Van Raamsdonk et al., 2005; Gray et al., 2008). The background FVB strain of these models also undergoes retinal degeneration leading to blindness, which confounds behavior testing in older mice (Taketo et al., 1991).
Efficacy of intrabody treatment
Despite the likely utility of iAb gene therapy for neurodegenerative diseases, and the many potential therapeutic iAbs that have been developed for treating Alzheimer's disease, Parkinson's disease, and HD, only one study using iAb gene therapy for neurodegenerative disease in mice has been published. In that study, the anti-Htt iAb EM48 was delivered via adenovirus to the striatum of R6/2 and N171-82Q mice (Wang et al., 2008). EM48 treatment reduced neuropil aggregates in the brains of both models, but had no effect on intranuclear inclusions. This is most likely due to the fact that, unlike Happ1, which distributes freely in cells (Southwell et al., 2008), EM48 is excluded from the nucleus. EM48 treatment improved both gait and rotarod performance in 18-week-old N171-82Q mice but had no effect on body weight or survival. Behavioral assays were not reported for the R6/2 line. Although promising, these results suggest that more efficacious iAbs, possibly directed against other Htt epitopes, coupled with more effective vectors, could improve therapeutic outcome even further.
Regarding mechanism of iAb action, previous work showed that both Happ1 and VL12.3 reduce mHtt-induced toxicity and aggregation in cell culture and brain slice models of HD, although by different means (Southwell et al., 2008). VL12.3 appears to prevent aggregation by blocking an epitope required for aggregate seeding (Thakur et al., 2009). On the other hand, the mechanism by which VL12.3 prevents mHtt toxicity remains unclear, as this iAb also causes an increase in nuclear Htt levels, which can increase toxicity under some circumstances (Saudou et al., 1998; Hackam et al., 1999; Peters et al., 1999; Schilling et al., 2004). Conversely, Happ1 has no effect on Htt localization. This iAb appears to reduce toxicity by increasing clearance of mutant but not wtHtt. The clearance of soluble mHtt could then lead to reduced aggregation by a shift in the equilibrium between the soluble and aggregated state. Reducing soluble mHtt would be predicted to also decrease the level of oligomeric species of the mutant protein, which are thought to be responsible for mHtt toxicity (Bucciantini et al., 2002; Sánchez et al., 2003; Behrends et al., 2006; Kitamura et al., 2006; Takahashi et al., 2008). In fact, Abs are proving effective for the identification of the particular conformations of oligomers that are responsible for the toxicity of amyloid-forming proteins in a variety of diseases (Kayed et al., 2003; Legleiter et al., 2009).
We find that VL12.3 treatment results in improved behavior and neuropathology in the lentiviral mouse model of HD. In transgenic HD models, however, VL12.3 either has no effect or adverse effects on symptoms. Perhaps the presence of VL12.3 before the introduction of mHtt is required for beneficial effects of this iAb, and some of the transgenic models (particularly R6/2) develop pathology before the iAb is introduced. Conversely, the negative effects of VL12.3, i.e., stabilizing soluble mHtt and increasing its nuclear localization (Southwell et al., 2008), may have detrimental effects in long-term studies.
Happ1 treatment, while equally as beneficial as VL12.3 in the lentiviral model, also improves motor and cognitive performance as well neuropathology in the transgenic models. In the lentiviral model, Happ1 prevents amphetamine-induced rotation and reduces striatal lesion size and intensity, as well as Htt aggregation. Happ1 also increases body weight and survival of N171-82Q but not R6/2 mice. The lack of effect in R6/2 may be related to the extremely early onset and severity of symptoms in this model. Happ1 treatment improves rotarod and beam-crossing performance in all four transgenic models. Unlike the other three models, the improvement in YAC128 motor performance is most significant at the earliest time points, suggesting a beneficial effect of Happ1 on the motor task learning deficit of this model (Van Raamsdonk et al., 2005). A climbing test was performed on the full-length transgenic models as a way to test motor performance independent of learning ability, as this behavior is spontaneous and does not require training. Happ1 treatment increased climbing time of both YAC128 and BACHD mice. Both full-length transgenic models exhibit anxiety in the open field and deficits in spatial and cortical learning tasks. Happ1 treatment normalizes open field exploration in both models and strongly improves learning in the YAC128 model.
Like VL12.3, Happ1 treatment reduces mHDx-1 aggregates in the R6/2 model. The similar effect of these iAbs on aggregation is particularly interesting as Happ1 ameliorates while VL12.3 exacerbates the HD behavioral phenotype in these mice. This indicates that, in this case at least, preventing aggregation is not predictive of therapeutic benefit. There is, however, evidence that Htt aggregation can be correlated with enhanced neuronal survival (Arrasate et al., 2004). Unlike VL12.3, Happ1 treatment also reduces intranuclear inclusions in these mice. This suggests that intranuclear inclusions may play a greater role in mHtt toxicity than neuropil aggregates (Hackam et al., 1999; Peters et al., 1999; Iwata et al., 2009).
Striatal atrophy resulting from neuronal shrinkage and/or death and concomitant ventricular enlargement is common in human HD patients as well as the four transgenic models used in this study. Happ1 treatment decreases ventricular enlargement in R6/2, YAC128, and BACHD mice. Aggregation and ventricle size were not assessed in N171-82Q mice as they were used to study survival and thus killed at the same disease stage rather than at the same age.
In summary, Happ1 treatment restores normal motor performance in N171-82Q, YAC128, and BACHD mice in rotarod and beam-crossing tests, YAC128 and BACHD mice in climbing tests, and the lentiviral model in the amphetamine-induced rotation test. Significant improvement is also seen in R6/2 mice in the rotarod and beam-crossing tests, as well as N171-82Q mice in the clasping test (summarized in Fig. S6, available at www.jneurosci.org as supplemental material). Happ1 treatment also restores normal cognitive performance in Yac128 and BACHD mice in open field tests and in Yac128 mice in novel object location and novel object preference tests. Although learning deficits were detected in BACHD mice by these tests, there is no effect of Happ1 treatment (summarized in Fig. S7, available at www.jneurosci.org as supplemental material). Happ1 treatment dramatically reduces striatal neuron loss in the lentiviral model, Htt aggregation in the lentiviral and R6/2 models, and normalizes ventricle size in R6/2, YAC128, and BACHD mice (summarized in Fig. S8, available at www.jneurosci.org as supplemental material). While Happ1 has no effect on R6/2, YAC128, or BACHD body weight or R6/2 survival, it does significantly increase both body weight and life span of N171-82Q mice.
The highly beneficial effects of Happ1 treatment on numerous aspects of the HD phenotype in all five mouse models of HD supports the idea that therapies directed at the specific degradation of mHtt represent a selective and effective strategy for the treatment of HD, with a very low probability of off-target effects. This approach can, however, still be improved. Although currently the most efficacious anti-Htt iAb in vivo, Happ1 does not completely prevent HD symptoms in mice. The therapeutic benefits of Happ1 most likely derive from increased clearance of mHtt as a result of Happ1 binding. Re-engineering to increase stability and binding efficiency, as was done with VL12.3 (Colby et al., 2004), could increase clearance of mHtt and subsequently increase the degree of disease prevention.
Another therapeutic approach directed at reducing levels of mHtt is gene silencing. AAV-mediated striatal silencing of both wt and mHtt RNAs in N171-82Q mice results in significantly enhanced motor performance as well as improved survival, but no effect on body weight loss (Boudreau et al., 2009). Although no detrimental effects have been observed thus far as a result of decreased wtHtt expression in these mice, side effects caused by loss of wtHtt function remain a possibility in human patients due to the extended time frame that would be required for suppressing mHtt expression. Importantly, allele-specific silencing of mHtt has been achieved in cellular models of HD either by the use of SNPs, which would be effective only for the portion of the affected population with the appropriate haplotype (Pfister et al., 2009; Warby et al., 2009), or by single stranded oligomers targeting expanded CAG, which would only be effective for HD patients with a significant repeat difference between disease and wt alleles (Hu et al., 2009). Nonetheless, like iAb-induced mHtt reduction, gene silencing is a therapeutic strategy with a great deal of potential.
Footnotes
-
This work was funded by the Hereditary Disease Foundation, the CHDI Foundation, and the National Institute of Neurological Disorders and Stroke (RO1 NS055298). We thank Ali Khoshnan for the mHtt lentiviral genome and for much advice and guidance, David Colby and K. Dane Wittrup for VL12.3, David Anderson for 293-GPG cells, David Baltimore for lentiviral production plasmids, Elio Vanin and Martha Bohn at Northwestern University for the AAV2 genome plasmid with modified CBA promoter, the University of Pennsylvania Viral Vector Core for the AAV1 rep/cap plasmid, Beverly Davidson and the University of Iowa Viral Vector Core for AAV1-GFP of known titer and for protocols and technical support for AAV production and purification, Jeffrey Cantle and William Yang for BACHD mice, Michael Hayden for YAC128 mice, and Wally Bugg for discussion and support.
- Correspondence should be addressed to Paul H. Patterson, Division of Biology, California Institute of Technology, 216-76, Pasadena, CA 91125. php{at}caltech.edu




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}