

Abstract
Endocannabinoids function as retrograde messengers and modulate synaptic transmission through presynaptic cannabinoid CB1 receptors. The magnitude and time course of endocannabinoid signaling are thought to depend on the balance between the production and degradation of endocannabinoids. The major endocannabinoid 2-arachidonoylglycerol (2-AG) is hydrolyzed by monoacylglycerol lipase (MGL), which is shown to be localized at axon terminals. In the present study, we investigated how MGL regulates endocannabinoid signaling and influences synaptic transmission in the hippocampus. We found that MGL inhibitors, methyl arachidonoyl fluorophosphonate and arachidonoyl trifluoromethylketone, caused a gradual suppression of cannabinoid-sensitive IPSCs in cultured hippocampal neurons. This suppression was reversed by blocking CB1 receptors and was attenuated by inhibiting 2-AG synthesis, indicating that MGL scavenges constitutively released 2-AG. We also found that the MGL inhibitors significantly prolonged the suppression of both IPSCs and EPSCs induced by exogenous 2-AG and depolarization-induced suppression of inhibition/excitation, a phenomenon known to be mediated by retrograde endocannabinoid signaling. In contrast, inhibitors of other endocannabinoid hydrolyzing enzymes, fatty acid amide hydrolase and cyclooxygenase-2, had no effect on the 2-AG-induced IPSC suppression. These results strongly suggest that presynaptic MGL not only hydrolyzes 2-AG released from activated postsynaptic neurons but also contributes to degradation of constitutively produced 2-AG and prevention of its accumulation around presynaptic terminals. Thus, the MGL activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus.
- 2-arachidonoylglycerol
- monoacylglycerol lipase
- diacylglycerol lipase
- CB1 cannabinoid receptor
- depolarization-induced suppression of inhibition
- hippocampus
Introduction
Endocannabinoids are released from neurons in activity-dependent manners, act retrogradely on presynaptic CB1 cannabinoid receptors, and modulate transmitter release (Alger, 2002; Freund et al., 2003; Diana and Marty, 2004; Ohno-Shosaku et al., 2005). This retrograde signaling is involved in short-term and long-term forms of synaptic plasticity (Chevaleyre et al., 2006) and contributes to certain forms of learning and memory (Marsicano et al., 2002; Kishimoto and Kano, 2006). Endocannabinoid release can be triggered by either strong depolarization of neurons (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001) or activation of Gq-coupled receptors such as group I metabotropic glutamate receptors and M1/M3 muscarinic acetylcholine receptors (Maejima et al., 2001; Fukudome et al., 2004) through activating phospholipase Cβ (PLCβ) (Hashimotodani et al., 2005; Maejima et al., 2005).
The magnitude and time course of intercellular signaling by diffusible molecules are generally determined by the balance between the generation and clearance of the molecules. Therefore, to understand endocannabinoid signaling, mechanisms of endocannabinoid degradation have to be elucidated. Biochemical studies have identified the enzymes that metabolize 2-arachidonoylglycerol (2-AG) and anandamide, the two major endocannabinoids. Fatty acid amide hydrolase (FAAH) and cyclooxygenase-2 (COX-2) metabolize both 2-AG and anandamide (Cravatt et al., 1996; Yu et al., 1997; Goparaju et al., 1998; Kozak et al., 2000), whereas monoacylglycerol lipase (MGL) hydrolyzes 2-AG but not anandamide (Dinh et al., 2002). Previous studies have suggested that 2-AG, rather than anandamide, is responsible for several forms of endocannabinoid-mediated retrograde signaling (Chevaleyre and Castillo, 2003; Kim and Alger, 2004; Melis et al., 2004; Hashimotodani et al., 2005; Maejima et al., 2005; Safo and Regehr, 2005). Biochemical, pharmacological, and genetic studies have suggested that 2-AG is degraded in the brain primarily by MGL (Freund et al., 2003; Sugiura et al., 2006). Anatomical studies have clearly demonstrated that MGL is densely distributed on CB1-expressing presynaptic terminals (Dinh et al., 2002; Gulyas et al., 2004). All of these findings strongly suggest that presynaptic MGL activity is crucial for terminating the endocannabinoid signal. This possibility was supported by the study showing that the newly reported MGL inhibitors URB754 (6-methyl-2-[(4-methylphenyl)amino]-4H-3,1-benzoxazin-4-one) and URB602 ([1,1′-biphenyl]-3-yl-carbamic acid, cyclohexyl ester) prolonged the hippocampal depolarization-induced suppression of inhibition (DSI) (Makara et al., 2005). However, a recent study has shown that both URB754 and URB602 fail to inhibit 2-AG hydrolysis in rat brain preparations (Saario et al., 2006). Therefore, roles of MGL in regulating endocannabinoid signaling remain unclear. In the present study, we addressed this issue by using URB754 and two other classical MGL inhibitors, methyl arachidonoyl fluorophosphonate (MAFP) and arachidonoyl trifluoromethylketone (ATFMK). With cultured hippocampal neurons, we found that MAFP and ATFMK, but not URB754, markedly influenced the CB1-dependent presynaptic modulation of synaptic transmission. The data to be presented clearly show that MGL activity plays an important role not only in termination of endocannabinoid signaling but also in degradation of constitutively released endocannabinoids.
Materials and Methods
Preparation of neurons.
All experiments were performed according to the guidelines laid down by the animal welfare committees of Kanazawa University and Osaka University. Cultured hippocampal neurons were prepared from newborn Sprague Dawley rats for most experiments and C57BL/6 mice for the experiments shown in Figure 6D, as described previously (Ohno-Shosaku et al., 2001). Briefly, cells were mechanically dissociated from the hippocampi and plated onto culture dishes (35 mm) pretreated with poly-l-ornithine (0.01%). The cultures were kept at 36°C in 5% CO2 for 12–15 d before use.
Electrophysiology.
Each neuron of a pair was whole-cell voltage clamped at −80 mV using a patch pipette (3–5 MΩ) filled with the internal solution containing the following (in mm): 130 K-gluconate, 15 KCl, 10 HEPES, 0.2 EGTA, 6 MgCl2, 5 Na2ATP, and 0.2 Na2GTP, pH 7.3 adjusted with KOH. The presynaptic neuron was stimulated by applying positive voltage pulses (to 0 mV, 2 ms) at 0.5 Hz, and IPSCs or EPSCs were measured from the postsynaptic neuron with a patch-clamp amplifier (EPC9/3 or EPC10/2; HEKA Elektronik, Lambrecht/Pfalz, Germany). The external solution contained the following (in mm): 140 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, and 1 kynurenic acid, pH 7.3 adjusted with NaOH. The recording chamber was perfused with the external solution with or without drugs at a flow rate of 1–3 ml/min. For recording of EPSCs, the concentration of kynurenic acid was decreased to 0.5 mm. The cannabinoid sensitivity of IPSCs was checked by 2-AG application. The solution containing 2-AG was locally applied through a capillary tube (250 μm inner diameter) located near the neurons using a perfusion valve controller (VC-6M; Warner Instruments, Hamden, CT). The applied 2-AG was rapidly washed out by continuous bath perfusion. DSI was induced by postsynaptic depolarization from −80 to 0 mV. For each neuron pair, the duration of depolarization was adjusted to induce DSI >50% within a range of 1–5 s. Magnitudes of DSI were measured as the percentage of the mean amplitude of IPSCs acquired between 4 and 14 s after the end of depolarization relative to that acquired for 30 s before the depolarization. For experiments to monitor depolarization-induced suppression of excitation (DSE), neurons prepared from mouse hippocampi were used. Because DSE is less prominent than DSI (Ohno-Shosaku et al., 2002), longer duration of depolarization (5 or 10 s) was required for inducing DSE, and neuron pairs exhibiting >20% of DSE magnitude were selected (four of seven neuron pairs). In Figure 3A, the postsynaptic neuron was dialyzed with an internal solution containing the following (in mm): 120 K-gluconate, 15 KCl, 10 HEPES, 5 EGTA, 6 MgCl2, 5 Na2ATP, and 0.2 Na2GTP, pH 7.3 adjusted with KOH. Magnitudes of suppression caused by the muscarinic agonist oxotremorine-M (oxo-M) were calculated as the percentage of the mean amplitudes of 10 consecutive IPSCs during oxo-M application relative to that before application. All data are expressed as mean ± SEM. All experiments were performed at room temperature.
Chemicals.
MAFP, ATFMK, tetrahydrolipstatin (THL), and meloxicam were purchased from Sigma (St. Louis, MO). AM281 [1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide] and oxo-M were from Tocris Cookson (Bristol, UK). 2-AG was from Cayman Chemical (Ann Arbor, MI). URB597 [3′-(aminocarbonyl)[1,1′-biphenyl]-3-yl)-cyclohexylcarbamate] was purchased from Calbiochem (Darmstadt, Germany) and Cayman Chemical. URB754 was purchased from Biomol (Plymouth Meeting, PA) and Cayman Chemical. We obtained the same results by using the chemicals from different companies, and therefore we pooled the data. All of these chemicals except for oxo-M were dissolved in DMSO as stock solutions and added to the external solution just before use. The concentration of DMSO did not exceed 0.1%.
Results
Neither DSI nor 2-AG-induced suppression is affected by URB754
We began by examining effects of the newly reported MGL inhibitor URB754 (Makara et al., 2005) on hippocampal DSI. We recorded cannabinoid-sensitive IPSCs and induced DSI by postsynaptic depolarization. We also monitored transient suppression of IPSCs induced by exogenous 2-AG applied locally around the postsynaptic neuron. When 2-AG (0.1 μm) was applied for 10 s, the IPSC amplitude rapidly declined and recovered to the original level (Fig. 1A). The time course of 2-AG-induced suppression, especially the recovering phase, was dependent on the concentration of applied 2-AG but also exhibited a small variation among neuron pairs. However, in the same neuron pair, the time course of 2-AG-induced suppression was stable over repeated trials and thus can be used as an indicator of 2-AG degradation. If presynaptic 2-AG degradation is suppressed, the recovery phase is expected to be prolonged. As exemplified in Figure 1B, we induced DSI (open arrows) and 2-AG-dependent suppression (filled arrows) in individual neuron pairs. The amplitude of IPSCs slightly increased to 118.8 ± 5.3% (n = 4) of control after bath application of URB754 (5 μm) (Fig. 1B). This result was unexpected, because Makara et al. (2005) reported previously a decrease in IPSC amplitude by 3 μm URB754 in hippocampal slices. Furthermore, contrary to the previous study, we could not observe any changes in time course of DSI after URB754 treatment (Fig. 1C). We also tested three other concentrations of URB754 (0.5, 10, and 50 μm) but observed no prolongation of DSI (data not shown). Furthermore, we observed no prolongation of the 2-AG-induced suppression of IPSCs by URB754 (Fig. 1D). If URB754 does inhibit 2-AG hydrolysis by MGL, as shown by Makara et al. (2005), the present results indicate that MGL has little contribution to endocannabinoid signaling in our preparation. Alternatively, if URB754 does not inhibit MGL, as demonstrated recently by Saario et al. (2006), the results in Figure 1 are inconclusive and a possibility remains that MGL contributes to DSI termination. To test this possibility, we examined effects of the classical MGL inhibitors MAFP (Goparaju et al., 1999; Dinh et al., 2002, 2004; Savinainen et al., 2003; Saario et al., 2004; Walter et al., 2004) and ATFMK (Dinh et al., 2002; Walter et al., 2004) on DSI and 2AG-induced suppression of IPSCs.

URB754 has no effect on DSI and 2-AG-induced suppression of IPSCs. A, Schematic drawing showing local application of 2-AG to a pair of voltage-clamped neurons with cannabinoid-sensitive synapses (left). Right shows a representative experiment, in which locally applied 2-AG (0.1 μm for 10 s) reversibly decreased the amplitudes of cannabinoid-sensitive IPSCs. B, Examples of IPSC traces (left) and the time course of IPSC amplitude (right) from a representative experiment on the effects of URB754. DSI and 2-AG-induced suppression were induced repeatedly by postsynaptic depolarization (0 mV, 5 s; open arrows) and by local 2-AG application (0.1 μm, 10 s; filled arrows), respectively, before and during application of URB754 (5 μm). Sample IPSC traces (left) were acquired at the time points indicated in the graph (a–l, right). C, D, Averaged time courses of DSI (C) and 2-AG-induced suppression (D) obtained before and 5–10 min after the initiation of URB754 application (5 μm; n = 4).
Accumulation of endocannabinoids by MGL inhibitors
We found that bath application of MAFP (0.1 μm) caused a gradual decrease in the amplitude of cannabinoid-sensitive IPSCs (Fig. 2A,B,E). The time course of this suppression varied from cell to cell (Fig. 2A,B), but, in most cases, the IPSC amplitude reached a stable level within 5 min. Importantly, the MAFP-induced suppression was reversed by the CB1 antagonist AM281 (Fig. 2A,B,E). The same MAFP treatment induced no effects on cannabinoid-insensitive IPSCs (Fig. 2C,E) and only a slight suppression of EPSCs (Fig. 2D,E) that are less sensitive to cannabinoids than cannabinoid-sensitive IPSCs (Ohno-Shosaku et al., 2002). The small suppression of EPSC was also reversed by AM281 (Fig. 2E). Similar results were obtained in experiments with another MGL inhibitor, ATFMK. Application of ATFMK (10 μm) induced a gradual suppression of cannabinoid-sensitive IPSCs, which was reversed by AM281 (Fig. 2E). The most likely explanation for these results is that the inhibition of MGL caused accumulation of 2-AG around presynaptic terminals and activation of CB1 receptors. Alternatively, these drugs might activate CB1 receptors directly. This possibility, however, was excluded by the following experiments, in which effects of MGL inhibitors were examined under suppression of 2-AG synthesis. 2-AG is produced from membrane lipids mainly through two enzymatic steps by PLC and diacylglycerol (DAG) lipase (Stella et al., 1997; Freund et al., 2003; Sugiura et al., 2006). THL inhibits DAG lipase and suppresses 2-AG production under certain conditions (Bisogno et al., 2003; Melis et al., 2004; Maejima et al., 2005; Safo and Regehr, 2005). We confirmed that THL treatment blocked DSI almost completely (Fig. 3A,B, left). We also confirmed that THL blocked the endocannabinoid release induced by application of the muscarinic agonist oxo-M (Fukudome et al., 2004) (Fig. 3B, right). When neurons were pretreated with 5 μm THL for 5–7 min, the MAFP-induced suppression was significantly reduced (Fig. 3C). The effect of ATFMK was also reduced after the THL treatment (Fig. 3C). These data suggest that 2-AG is constitutively produced at basal conditions, at least partly through a THL-sensitive pathway.

MGL inhibitors specifically suppress cannabinoid-sensitive synaptic transmissions in a CB1-dependent manner. A, B, Two representative experiments showing the effects of bath-applied MAFP (0.1 μm) on cannabinoid-sensitive IPSCs. The IPSC traces acquired at the indicated time points (a–c) (top) and the IPSC amplitudes plotted as a function of time (bottom) are shown. The IPSC amplitude declined slowly (A) or rapidly (B) after MAFP application and recovered to the initial level after addition of the CB1 antagonist AM281 (0.3 μm). C, Effect of bath-applied MAFP (0.1 μm) on cannabinoid-insensitive IPSCs. Examples of IPSC traces before and 5 min after the initiation of MAFP application (top) and averaged data for the time course of IPSC amplitude (bottom; n = 4) are shown. D, Effects of bath-applied MAFP (0.1 μm) on EPSCs. Examples of EPSC traces before and 5 min after the initiation of MAFP application (top) and averaged data for the time course of EPSC amplitude (bottom; n = 9) are shown. E, From left to right, Averaged data showing percentage changes in the amplitudes of cannabinoid-sensitive IPSCs, cannabinoid-insensitive IPSCs, and EPSCs by application of indicated drugs. Numbers of tested cells are indicated in parentheses. *p < 0.05; **p < 0.01; ***p < 0.001 by paired t test.

The DAG lipase inhibitor THL eliminates endocannabinoid release and reduces IPSC suppression caused by MGL inhibitors. A, Blockade of DSI by THL. The graph shows averaged time course of DSI before and 5 min after bath application of 10 μm THL. B, Summary bar graphs showing the effects of THL on DSI (left) and endocannabinoid-mediated suppression of IPSCs by the muscarinic agonist oxo-M (3 μm). Oxo-M (3 μm) was bath applied for 1 min before (Control) and after (THL) treatment with THL (5 μm) for 5 min. C, Pretreatment with THL attenuates the suppression of IPSCs by MGL inhibitors. Effects of MAFP (0.1 μm) or ATFMK (10 μm) on cannabinoid-sensitive IPSCs were examined in the neuron pairs with (+THL) or without (−THL) pretreatment of THL (5 μm) for 5–7 min. Averaged data showing percentage changes in the amplitudes of cannabinoid-sensitive IPSCs 5 min after the initiation of MAFP or ATFMK application. Numbers of tested cells are indicated in parentheses. **p < 0.01; ***p < 0.001 by paired (A, B) or unpaired (C) t test.
MGL inhibitors prolong recovery from 2-AG-induced suppression
We next examined effects of MGL inhibitors on 2-AG-induced suppression of IPSCs. To minimize the endogenous 2-AG accumulation, the neurons were treated with THL before application of MGL inhibitors (Fig. 4A). We applied 2-AG (0.1 μm) three times, before and after THL pretreatment and during additional application of MAFP (Fig. 4A,B) or ATFMK (Fig. 4C). The time course of 2-AG-induced suppression was not influenced by THL but markedly changed by the two MGL inhibitors (Fig. 4B,C). Whereas the time course of initial falling phase and the peak level of suppression were unchanged, the recovery from suppression was markedly prolonged.
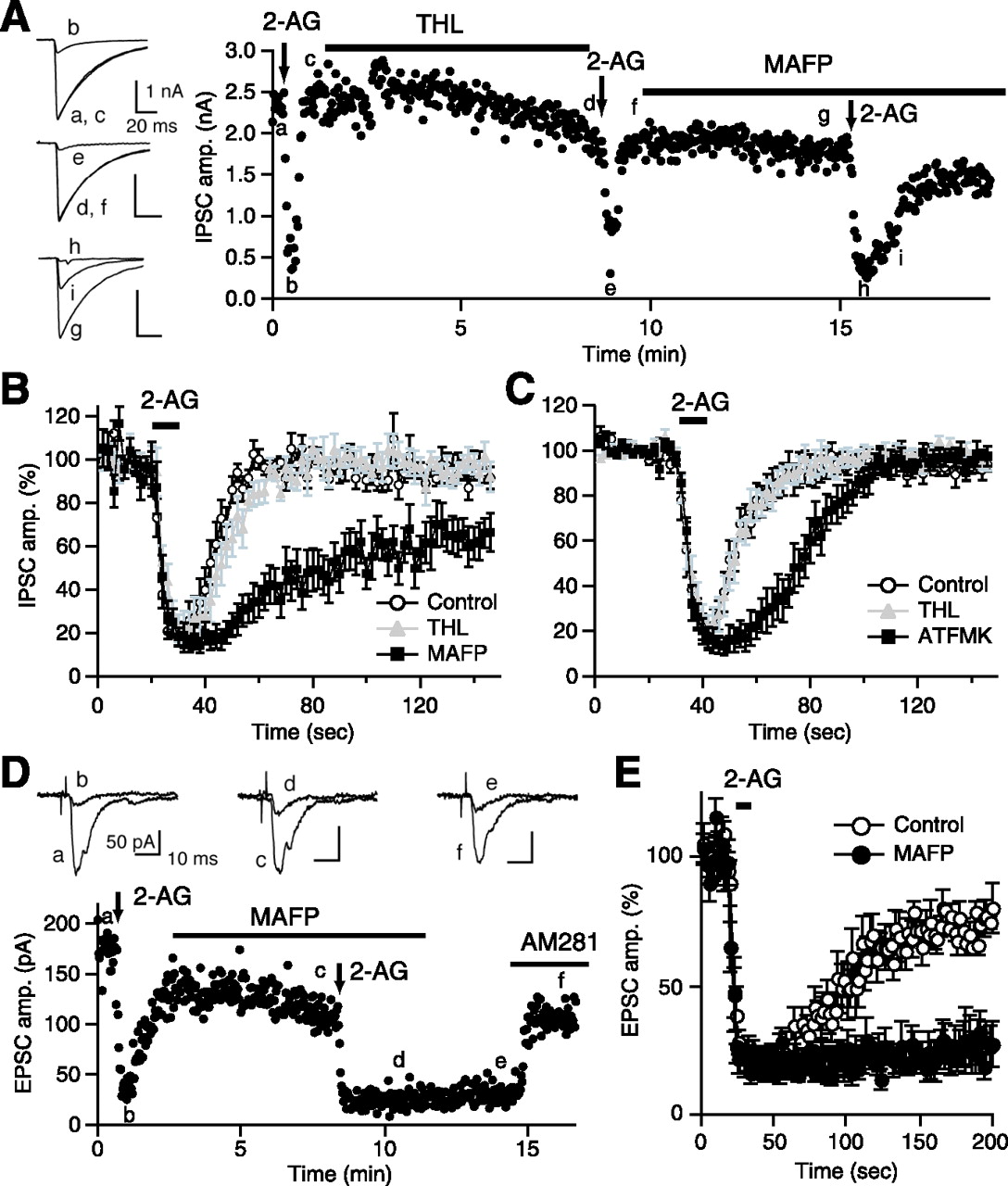
MGL inhibitors prolong the 2-AG-induced suppression. A, A representative experiment showing effects of a sequential treatment with THL (5 μm) and MAFP (0.1 μm) on 2-AG-induced IPSC suppression. Downward arrows indicate the periods of 2-AG application. IPSC traces acquired at the indicated time points (a–i) are shown on the left. B, Average data for the time course of 2-AG-induced IPSC suppression before (Control) and after THL treatment and during additional treatment with MAFP (0.1 μm; n = 10). C, Average data for the time course of 2-AG-induced IPSC suppression before and after THL treatment and during additional treatment with ATFMK (10 μm; n = 6). D, E, An example (D) and the averaged data (E) showing effects of MAFP treatment on 2-AG-induced suppression of EPSCs. EPSC traces acquired at the indicated time points (a–f) are shown on the top. 2-AG (25 μm) was applied for 10 s (vertical arrows in D or a horizontal bar in E) before and after application of 0.1 μm MAFP. The 2-AG-induced persistent suppression of EPSC after MAFP application was reversed by addition of AM281 (0.3 μm). The averaged time courses of 2-AG-induced EPSC suppression (E) were obtained before and 5 min after the initiation of MAFP application (n = 5).
A prolongation of 2-AG-induced suppression by MAFP was also observed at excitatory synapses. In this series of experiments, THL pretreatment was omitted and a high dose of 2-AG (25 μm) was used, because of the low cannabinoid sensitivity of EPSCs (Ohno-Shosaku et al., 2002). The EPSC amplitude was decreased after 2-AG application and slowly recovered after washout of 2-AG. The difference in recovery time course between IPSCs and EPSCs (Fig. 4B,E) can be attributable to the difference in the concentrations of applied 2-AG. The treatment with MAFP did not affect the falling phase and the peak value of suppression but almost completely suppressed the recovery of EPSCs from suppression (Fig. 4D,E). In these conditions, the EPSC suppression lasted for at least >5 min (four of five cells) and was reversed by AM281 (Fig. 4D). Together, these results indicate that the MGL activity is required for decreasing the 2-AG level around presynaptic CB1 receptors when 2-AG is applied exogenously.
FAAH and COX-2 do not contribute to 2-AG degradation at presynaptic sites
We next examined whether other endocannabinoid hydrolyzing enzymes, FAAH and COX-2, are involved in degradation of exogenously applied 2-AG. In contrast to MGL inhibitors, the selective FAAH inhibitor URB597 and the selective COX-2 inhibitor meloxicam failed to affect the 2-AG-induced suppression of IPSCs. Treatment with 1 μm URB597 for 5 min caused no significant changes in basal IPSC amplitude (92.5 ± 4.1% of control; n = 8) and peak value and time course of 2-AG-induced IPSC suppression (Fig. 5A). Treatment with 30 μm meloxicam for 5 min reduced the basal IPSC amplitude to 75.2 ± 5.0% of control (n = 12) but did not influence the time course of 2-AG-induced suppression (Fig. 5B). In contrast, meloxicam (30 μm) significantly prolonged DSI (Fig. 5C), as has been reported in hippocampal slices (Kim and Alger, 2004). These results are consistent with the hypothesis that COX-2 is distributed postsynaptically and inactivates 2-AG before it is released (Kim and Alger, 2004). Together, our data strongly suggest that FAAH and COX-2 are not involved in degradation of 2-AG at presynaptic terminals.

FAAH and COX-2 inhibitors have no effect on the time course of 2-AG-induced IPSC suppression. A, B, Average data for the time course of 2-AG-induced IPSC suppression obtained before and 5 min after the initiation of application of the FAAH inhibitor URB597 (1 μm) (A; n = 8) or the COX-2 inhibitor meloxicam (30 μm) (B; n = 4). C, Meloxicam slightly but significantly prolongs DSI. Average data for the time course of DSI before and after treatment with meloxicam for 5 min (30 μm; n = 8). The normalized IPSC amplitudes from 10 to 28 s after the depolarization were significantly smaller after the meloxicam treatment when compared with those before the treatment (p < 0.05 by paired t test).
Prolongation of DSI by MGL inhibitors
Finally, we determined whether MGL activity is required for the termination of phasic endocannabinoid signal generated by postsynaptic activity. We induced DSI by postsynaptic depolarization and examined effects of MGL inhibitors on DSI. Because THL blocks DSI (Fig. 3A), we could not use THL to prevent accumulation of 2-AG, which we adopted in the experiments shown in Figure 4. Therefore, we had to use MGL inhibitor at much lower concentrations to minimize 2-AG accumulation. We found that treatment with 7 nm MAFP caused relatively mild suppression of IPSC amplitude (64.9 ± 10.0% of control; n = 6) but markedly prolonged the recovery phase of DSI (Fig. 6A,B). In the same neuron pairs, we confirmed that the recovery from 2-AG-induced suppression was similarly prolonged (Fig. 6A,C). We also examined whether inhibition of MGL could prolong DSE. For this purpose, we used mouse hippocampal neurons, because DSE is larger in magnitude and can be induced more constantly in mice and than in rats (Ohno-Shosaku et al., 2002). As shown in Figure 6D, DSE was significantly prolonged by treatment with 7 nm MAFP. These results indicate that MGL activity is crucial for termination of DSI/DSE as well as degradation of constitutively produced 2-AG.

The MGL inhibitor MAFP prolongs DSI, DSE, and 2-AG-induced IPSC suppression. A, Examples of IPSC traces (top) and the time course (bottom) of IPSC amplitude from a representative experiment. DSI and 2-AG-induced suppression were induced repeatedly by postsynaptic depolarization (0 mV, 5 s; open arrows) and by local 2-AG application (0.1 μm, 10 s; filled arrows), respectively, before and during application of MAFP (7 nm). IPSC traces were acquired at the indicated time points (a–l). B, C, Average data for the time course of DSI (B) and 2-AG-induced IPSC suppression (C) obtained before and 5–8 min after the initiation of 7 nm MAFP application (n = 6). D, Examples of EPSC traces (left) and the averaged time course of DSE (right; n = 4). Treatment with MAFP (7 nm) significantly prolonged DSE induced by 5 or 10 s postsynaptic depolarization of mouse hippocampal neurons. Thin, bold, and gray traces were acquired before, 2 s after, and 40 s after depolarization, respectively.
Discussion
In the present study, we examined roles of MGL, which has been shown to be localized in presynaptic terminals, in termination of endocannabinoid signaling in cultured hippocampal neurons. Our data with URB754 are at variance from those in the previous study that URB754 prolonged DSI and caused gradual suppression of spontaneous IPSCs. We found that URB754 failed to prolong DSI and rather increased the IPSC amplitude. The reasons for this discrepancy are unknown. One possibility is that URB754 used in this study was accidentally inactive or that the commercially available URB754 is structurally different from the substance that Makara et al. (2005) reported. This possibility is, however, unlikely because we obtained the same results by using URB754 purchased from two different companies (Cayman Chemical and Biomol). Importantly, Saario et al. (2006) confirmed that URB754 from Cayman Chemical is identical to that reported by Makara et al. (2005) by 1H nuclear magnetic resonance spectroscopy. Another possibility is that the reported effects of URB754 (Makara et al., 2005) might be caused by a different molecular identity, as suggested in the recent report (Saario et al., 2006).
Our results with MAFP and ATFMK demonstrated a crucial role of MGL in the regulation of endocannabinoid signaling. These MGL inhibitors caused a gradual suppression of IPSC/EPSC in a CB1-dependent manner and prolonged the recovery phase of DSI/DSE. Furthermore, we found that the recovery of IPSC/EPSC from suppression after a brief application of 2-AG is highly sensitive to MGL inhibitors but not to FAAH and COX-2 inhibitors. Together, our data indicate that MGL activity plays an important role not only in terminating the phasic endocannabinoid signal generated by postsynaptic activity but also in scavenging the constitutively produced 2-AG and preventing its accumulation around CB1 receptors.
In this study, we used MAFP and ATFMK as MGL inhibitors (Goparaju et al., 1999; Dinh et al., 2002, 2004; Savinainen et al., 2003; Saario et al., 2004; Walter et al., 2004). However, the possibility remains that the effects of MAFP and ATFMK are caused by the actions on MAFP/ATFMK-sensitive cellular components other than MGL. One possible target is phospholipase A2. MAFP was first reported as an inhibitor of phospholipase A2. However, it has been revealed that MGL is more sensitive to MAFP than phospholipase A2 (Goparaju et al., 1999). In the present study, we showed that MAFP caused marked effects on DSI/DSE even at 7 nm, which is comparable with the IC50 (2–3 nm) to MGL (Goparaju et al., 1999; Saario et al., 2004). Therefore, it is most likely that the effects of MAFP were caused by inhibition of MGL but not phospholipase A2. The second possible target is FAAH, because it is reported that MAFP inhibits FAAH (De Petrocellis et al., 1997; Deutsch et al., 1997). However, this possibility can be excluded by the finding that the selective FAAH inhibitor URB597 failed to mimic the effects. The third possible target is the CB1 receptor. MAFP and ATFMK might depress IPSC/EPSC by acting directly on CB1 receptors. This possibility is, however, inconsistent with our finding that the effects of MAFP/ATFMK on basal IPSCs were primarily suppressed by THL that inhibits 2-AG synthesis and also with the biochemical study showing that MAFP has neither agonist nor antagonist activity at CB1 receptors (Savinainen et al., 2003). Thus, it is reasonable to conclude that the effects of MAFP and ATFMK presented here result mostly from the inhibition of MGL activity, although contribution of some unknown nonspecific effects of MAFP/ATFMK cannot be excluded completely.
Our experimental data are consistent with the anatomical studies showing the distributions of endocannabinoid degradation enzymes. FAAH and COX-2 are expressed at the postsynaptic sites (Kaufmann et al., 1996; Egertova et al., 2003), whereas MGL is distributed at presynaptic sites (Dinh et al., 2002; Gulyas et al., 2004). We found that the recovery from 2-AG-induced suppression was affected by MGL inhibitors but not by FAAH and COX-2 inhibitors, suggesting a dominant role of MGL in inactivating 2-AG at presynaptic terminals. In line with this notion, we found that the recovery phase of DSI/DSE was markedly prolonged by MAFP. In hippocampal slices, it has been reported that the DSI recovery is not affected by the FAAH inhibitor URB597 but prolonged by the COX-2 inhibitor meloxicam (Kim and Alger, 2004). We also confirmed that DSI was prolonged by meloxicam in cultured hippocampal neurons. These results suggest that COX-2 regulates 2-AG production at the postsynaptic site, whereas MGL degrades 2-AG at the presynaptic site.
Anandamide is degraded in the brain primarily by FAAH, which is densely distributed at postsynaptic sites but not presynaptic terminals. Therefore, it is expected that anandamide-mediated presynaptic effects are long lasting when compared with those mediated by 2-AG. In fact, it has been reported that exogenously applied anandamide induced a long-lasting depression of excitatory transmission in cultured hippocampal neurons (Straiker and Mackie, 2005). In our culture system, we also observed that anandamide application induced a long-lasting depression of IPSCs (data not shown), making a striking contrast to the transient suppression by 2-AG. It is therefore possible that 2-AG and anandamide might function as short-lived and long-lived retrograde messengers, respectively, in the brain.
There are several reports suggesting that endocannabinoids are tonically released in the nervous tissues and their constitutive synthesis is balanced by their degradation. In hippocampal slices, application of the endocannabinoid uptake inhibitor AM404 [N-(4-hydroxyphenyl)-arachidonamide] depressed IPSCs and occluded DSI (Wilson and Nicoll, 2001), and the COX-2 inhibitor meloxicam induced a gradual depression of field EPSPs (Slanina and Schweitzer, 2005). In the hypothalamic arcuate nucleus, application of AM251 increased the amplitude of IPSCs in proopiomelanocortin neurons (Hentges et al., 2005). These authors reasoned their results as the presence of constitutive endocannabinoid tone, in agreement with the present study.
Our data suggest that presynaptic MGL activity plays an important role in scavenging the constitutively produced and accumulated 2-AG around CB1 receptors, which is consistent with the biochemical studies showing that inhibition of MGL increased basal 2-AG levels in mouse brain (Quistad et al., 2006) or mouse cultured astrocytes (Walter et al., 2004). Then, where is 2-AG produced and released? As illustrated in Figure 7, we assume that the most likely candidate is the postsynaptic sites facing presynaptic terminals, because one of the major 2-AG synthetic enzymes, DAG lipase α, is densely distributed at postsynaptic sites (Katona et al., 2006; Yoshida et al., 2006). Additionally, astrocytes might also contribute to 2-AG production as shown previously (Walter et al., 2004). The third possibility remains that 2-AG is produced within presynaptic terminals. Nevertheless, 2-AG appears to be produced constitutively and released tonically at basal conditions in the nervous tissue. The released 2-AG then diffuses into presynaptic terminals and is inactivated by MGL. The concentration of 2-AG around presynaptic CB1 receptors should be determined by the balance between production and degradation of 2-AG. When presynaptic MGL is functional, the 2-AG level appears to be kept so low that CB1 receptors are not activated (Fig. 7A). When presynaptic MGL is blocked, 2-AG will be accumulated at presynaptic terminals, resulting in tonic activation of CB1 receptors (Fig. 7B). When a large amount of 2-AG is produced transiently depending on postsynaptic activity, the released 2-AG can activate CB1 receptors and cause transient suppression of transmitter release, which will then be terminated after 2-AG degradation by presynaptic MGL activity (Fig. 7C).

A model for the roles of presynaptic MGL in regulating basal endocannabinoid tone and terminating phasic endocannabinoid actions dependent on postsynaptic neuronal activity. A, At a resting state, a certain amount of 2-AG is constitutively produced and released from neurons or glial cells. The released 2-AG then diffuses into presynaptic terminal membranes and is rapidly inactivated by MGL so that CB1 receptors are not activated. B, When MGL is blocked at a resting state, 2-AG is accumulated around presynaptic terminals, and its local concentration is elevated high enough to tonically activate CB1 receptors. C, When a large amount of 2-AG is produced and released from activated postsynaptic neurons, the released 2-AG activates CB1 receptors, and the 2-AG signal is terminated by its degradation by presynaptic MGL.
In this model, any changes in presynaptic MGL activity are expected to influence both tonic and phasic endocannabinoid signaling. Therefore, it is important to elucidate how MGL activity is regulated under physiological conditions. In the hippocampal CA3 area, persistently active CB1 receptors is reported to mute subpopulation of interneurons (Losonczy et al., 2004). It is possible that low MGL activities at presynaptic terminals may underlie persistent activation of CB1 receptors. It is reported that induction of endocannabinoid-mediated long-term depression at hippocampal inhibitory synapses requires CB1 receptor activation lasting for 5–10 min (Chevaleyre and Castillo, 2003). How can short tetanic stimulation (for example, 100 Hz, 1 s, twice) cause such a long-lasting activation of CB1 receptors? If presynaptic MGL is downregulated by tetanic stimulation, activation of CB1 receptors can last for several minutes. Although regulatory factors of MGL activity in neurons are not yet identified, Ca2+ is reported to inhibit MGL activity in microglial cells (Witting et al., 2004). The same group, however, demonstrated that MGL activity in astrocytes is not affected by Ca2+ (Walter et al., 2004). In the neocortex, timing-dependent long-term depression is reported to require simultaneous activation of presynaptic NMDA and CB1 receptors (Sjostrom et al., 2003). If Ca2+ is a negative regulator of MGL, it is possible that presynaptic activation of NMDA receptors inactivates MGL and thereby enhances the endocannabinoid signaling. Despite such circumstantial evidence, our current knowledge about the regulation of MGL activity is rather limited. Additional studies are needed to elucidate mechanisms of regulation of MGL activity and its physiological relevance.
Footnotes
-
This work was supported by Grants-in-Aid for Scientific Research 18-08582 (Y.H.), 17650112, 17024021, and 18022016 (T.O.-S.), and 17023021 and 17100004 (M.K.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. Y.H. is a recipient of Research Fellowships of the Japan Society for the Promotion of Science for Young Scientists.
- Correspondence should be addressed to Masanobu Kano, Department of Cellular Neuroscience, Graduate School of Medicine, Osaka University, Suita 565-0871, Japan. mkano{at}cns.med.osaka-u.ac.jp




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}