

Abstract
Axonal mRNA transport is robust in cultured neurons but there has been limited evidence for this in vivo. We have used a genetic approach to test for in vivo axonal transport of reporter mRNAs. We show that β-actin's 3′-UTR can drive axonal localization of GFP mRNA in mature DRG neurons, but mice with γ-actin's 3′-UTR show no axonal GFP mRNA. Peripheral axotomy triggers transport of the β-actin 3′-UTR containing transgene mRNA into axons. This GFP-3′-β-actin mRNA accumulates in injured PNS axons before activation of the transgene promoter peaks in the DRG. Spinal cord injury also increases axonal GFP signals in mice carrying this transgene without any increase in transgene expression in the DRGs. These data show for the first time that the β-actin 3′-UTR is sufficient for axonal localization in both PNS and CNS neurons in vivo.
Introduction
In cultured neurons, axonally synthesized proteins contribute to growth cone turning, growth cone formation, and axon survival (Verma et al., 2005; Leung et al., 2006; Yao et al., 2006; Aschrafi et al., 2008; Cox et al., 2008; Andreassi et al., 2010). There has been limited evidence for axonal mRNA localization and translation in vivo. The 3′-UTR of EphA2 mRNA has been shown to localize a reporter mRNA into embryonic spinal cord axons (Brittis et al., 2002). Localization of ribosomes and β-actin mRNA in mature myelinated PNS axons has also been reported (Koenig et al., 2000; Sotelo-Silveira et al., 2008). The Fainzilber laboratory provided evidence for synthesis of proteins in mature PNS axons in vivo (Hanz et al., 2003; Perlson et al., 2005; Yudin et al., 2008). Although these studies support the hypothesis that mature PNS axons contain mRNAs and translational machinery, a report on ribosome transfer from Schwann cells to axons raises the possibility that these molecules may derive from sources other than transport from the neuronal cell body (Court et al., 2008). Ultrastructural studies of rodent hippocampus also suggested that ribosomes are excluded from mature CNS axons in vivo (for review, see Steward and Schuman, 2003). To date there has been no direct evidence that neuronal derived mRNAs are transported into and translated within mature PNS or CNS axons in vivo. Here, we have used a transgenic approach to directly test for axonal mRNA transport in vivo. Our data indicate that the 3′-UTR of β-actin is sufficient to target a reporter mRNA for transport into distal axons in vivo.
Materials and Methods
Generation of transgenic mice.
All animal procedures were approved by the respective Institutional Animal Care and Use Committees. cDNA for destabilized GFP (dzGFP) with a myristoylation motif and 5′- and 3′-UTRs of CaMKIIα (dzGFPmyrCaMKIIα) was used for transgenic constructs. The 3′-UTRs of rat β-actin and γ-actin mRNAs were amplified by reverse transcription (RT)-coupled PCR to add Not1 and Xho1 restriction sites. Primer sequences for transgene preparation and PCR analyses (see below) are available upon request. PCR products were cloned into pTOPO vector (Invitrogen), sequence verified, and then subcloned into dzGFPmyrCaMKIIα replacing the CaMKIIα 3′-UTR. Mouse Tα1 promoter was amplified adding 5′- and 3′-AflII sites, and cloned into the above constructs to generate transgenes designated Tα1-GFP-3′-β-actin and Tα1-GFP-3′-γ-actin, respectively. Transgenes were digested with Xho1, gel purified, and microinjected into zygotes of FVB/N inbred mice (Taconic Farms) (Nagy et al., 2003). Three Tα1-GFP-3′-β-actin and one Tα1-GFP-3′-γ-actin founders were generated.
Genotyping.
PCR and Southern blotting tested GFP transgene insertion. Genomic DNA from tail clippings was used for duplex PCR for GFP and MeCP2 to confirm transgene integration and determine relative copy number (i.e., through ratio of the GFP and X-linked MeCP2 products). Copy number was further verified by Southern blot and quantitative PCR (qPCR).
For Southern blotting, genomic DNA was digested with Xho1 and BamH1, electrophoresed on agarose gels, denatured, and transferred to Hybond-N (GE Healthcare Life Sciences). Hybridization was performed using 32P-labeled GFP probe generated by random priming (Stratagene). Blots were imaged using a Typhoon 9400 (GE Healthcare Life Sciences). For qPCR, a standard curve was generated with serial dilutions of the transgene template. DNA from 3 animals for each line was diluted to 30 ng/μl (∼1 × 104 genome copies) and three 1:5 serial dilutions were made. qPCR was performed for GFP on each dilution using the Prism 7900HT (Applied Biosystems) with 2× SybrGreen Master Mix (Qiagen).
Mice homozygous for transgenes were used for all studies, including the four copy Tα1-GFP-3′-β-actin mice that were generated by crossing homozygous Tα1-GFP-3′-β-actin2a and Tα1-GFP-3′-β-actin2b mice.
Neuronal cultures.
DRG cultures were prepared as described previously (Zheng et al., 2001). For immunostaining, neurons were cultured at low density on poly-lysine/laminin-coated glass coverslips. For isolation of axons, neurons were cultured on polyethylene tetrapthalate membranes, and axons were isolated and tested for purity by RT-PCR as described previously (Vuppalanchi et al., 2010).
Animal surgeries.
Mice were subjected to sciatic nerve crush at mid-thigh as previously described (Zheng et al., 2001). The nerve grafting procedure was performed for the common fibular nerve (CF) as described previously (English et al., 2005). CF segment from wild-type mice was grafted into transected CF of transgenic mice. For acellular grafts, wild-type CF segment was repeatedly frozen in liquid nitrogen before grafting (English et al., 2007).
The Infinite Horizon Spinal Cord Impactor (Precision Systems and Instrumentation) was used for T9 contusive spinal cord injury at 65 kdyn force. Sham mice underwent laminectomy alone. Mice received intraperitoneal injections of saline and Baytril for 7 d after injury and were given vitamin C-supplemented drinking water throughout the study. Bladder expression was performed twice daily for injured animals until voluntary control was resumed.
Analyses of RNAs.
RNA was isolated from tissues using a motorized homogenizer (Omni International) and an RNAqueous kit (Ambion). RNA was isolated from cultured cells using an RNAqueous-Micro kit (Ambion). DNase-treated RNA (100 ng) was used as template for the iScript RT kit (Bio-Rad). Reactions were diluted fivefold for PCR. Standard PCR (≤ 30 cycles) was analyzed by ethidium bromide stained agarose gels. Prism 7900HT with 2× SybrGreen MasterMix was used for qPCR with quadruplicate reactions for all samples.
Immunostaining.
Tissue samples were fixed in 4% paraformaldehyde (PF), cryoprotected overnight in 30% sucrose at 4°C and then cryosectioned at 10 μm. Immediately before use, sections were warmed to room temperature, washed in PBS and incubated in 20 mm glycine for 30 min followed by 0.25 m NaBH4 for 30 min. For cultured neurons, coverslips were fixed in 4% PF for 20 min and then rinsed in PBS. Samples were processed identically for subsequent steps as described previously (Vuppalanchi et al., 2010). The following primary antibodies were used: chicken anti-Neurofilament Heavy (NFH, 1:1000; Millipore), mouse anti-neurofilaments (NFH, NFM, and NFL, 1:1500 each; Sigma), Alexa Fluor 546 goat anti-chicken (1:1000; Invitrogen), and Alexa Fluor 555 goat anti-rabbit (1:1000; Invitrogen).
Fluorescence in situ hybridization.
Fluorescence in situ hybridization (FISH) on nerve tissues was performed as described previously (Muddashetty et al., 2007). Digoxigenin (DIG)-labeled oligonucleotide probes were used to detect GFP mRNA with Cy5-labeled mouse anti-DIG (Jackson ImmunoResearch) for detection. Rabbit anti-S100 antibody (1:200; Abcam) was used with secondary antibodies as above. Scrambled probes and primary antibody omission served as negative controls. These controls did not reveal any fluorescent signals (data not shown).
Results
In cultured neurons, β-actin mRNA is transported into axons and dendrites, while γ-actin mRNA remains in the cell body (Bassell et al., 1998; Zheng et al., 2001; Tiruchinapalli et al., 2003). This axonal localization is driven by the 3′-UTR of β-actin mRNA through a conserved “zip code” element (Kislauskis et al., 1994). We have taken advantage of this zip code element to test for axonal localization in vivo. For this, we generated transgenic mice expressing a destabilized, myristoylated GFP with the 3′-UTR of rat β-actin or γ-actin mRNAs (Fig. 1A). The 54 nt zip code region of rat β-actin shows 100% identity to the corresponding region of mouse β-actin mRNA 3′-UTR; rat γ-actin 3′-UTR shows 94% identity with the mouse γ-actin mRNA (data not shown). Myristoylation limits GFP diffusion in neuronal processes to provide a reporter for sites of translation (Aakalu et al., 2001; Willis et al., 2007; Yudin et al., 2008). The neuronal-specific Tα1 tubulin promoter, which is activated during periods of axon growth (Gloster et al., 1994), was used to drive transgene expression.

Transgenic model for axonal protein synthesis. A, Schematic of DNA constructs used to generate transgenic mice. B, Transgene copy number from qPCR studies is shown as average ± SD (n = 4). C, DRG cultures from transgenic mice show GFP or GFP, NF, and DAPI as indicated. Inset shows high magnification of GFP-3′-β-actin neurite. Scale bars, 50 μm. D, RT-PCR and immunoblotting from Schwann cell vs DRG cultures of transgenic mice show no signals for GFP in Schwann cells.
Genotyping by PCR and Southern blotting confirmed transgene integration (Fig. 1). Two β-actin lines and one γ-actin line were chosen for subsequent studies based on robust GFP expression in DRGs. Tα1-GFP-3′-γ-actin line has four transgene copies and Tα1-GFP-3′-β-actin2a and Tα1-GFP-3′-β-actin2b lines have two transgene copies (Fig. 1B). The two Tα1-GFP-3′-β-actin lines were crossed to generate a line carrying four transgene copies (Tα1-GFP-3′-β-actin2ax2b; Fig. 1B). DRG cultures from the Tα1-GFP-3′-β-actin lines showed GFP in cell bodies and neurites, but those from the Tα1-GFP-3′-γ-actin line showed GFP only in the neuronal cell bodies (Fig. 1C). RT-PCR of axonal isolates from DRGs cultured from the transgenic lines also confirmed axonal localization of GFP-3′-β-actin but not GFP-3′-γ-actin mRNA (data not shown). RT-PCR and immunoblotting showed no evidence for GFP expression in Schwann cells in these mice (Fig. 1D).
To determine whether the 3′-UTR of β-actin can support in vivo localization of GFP mRNA in the PNS, sciatic nerve was crushed to activate the Tα1 promoter and L4–5 DRGs and sciatic nerve were analyzed. Both transgenes showed increase in GFP fluorescence in the DRG after injury (Fig. 2A). GFP signals were highest in the DRGs over 5–14 d after crush injury, consistent with previous reports for Tα1 activation (Gloster et al., 1994). Only the nerve sections from the Tα1-GFP-3′-β-actin mice showed GFP signals (Fig. 2B). The Tα1-GFP-3′-γ-actin nerves did not show signals above the autofluorescence of wild-type nerve.

Tα1-GFP-3′-β-actin but not Tα1-GFP-3′-γ-actin mice show GFP in PNS axons. A, B, Immunolabeling of DRG (A) and sciatic nerve (B) sections from transgenic mice is shown at indicated times after nerve injury. GFP increases with injury in DRGs with both transgenes, but only sciatic nerve of Tα1-GFP-3′-β-actin mice shows any GFP signal. Images are matched for exposure, gain, and offset. Scale bar, 10 μm.
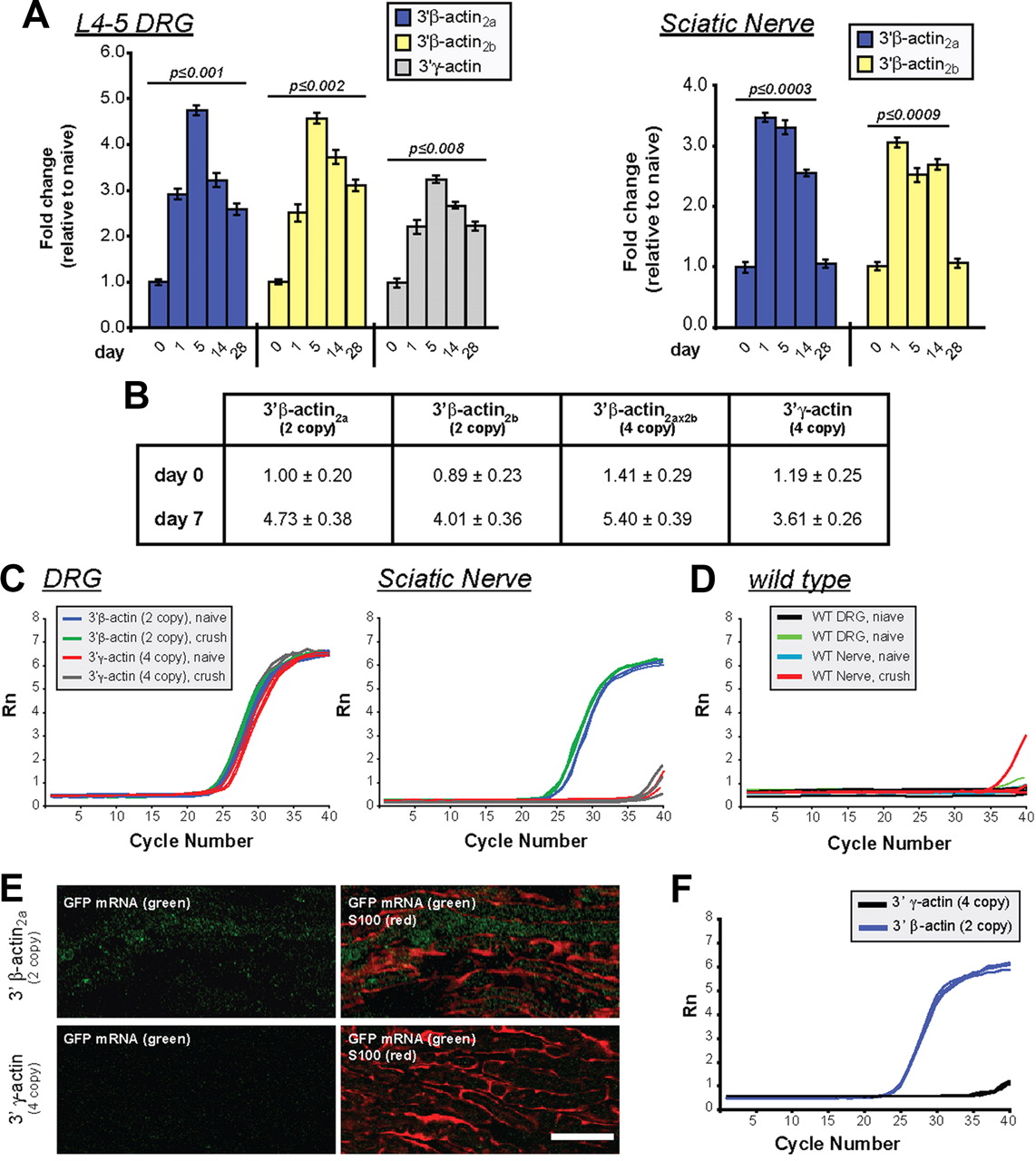
Since we saw no transgene expression in non-neuronal cells (Figs. 1D, 2A), we used RT-qPCR to analyze GFP mRNA levels. DRGs from both transgenic lines showed increase in GFP mRNA peaking 5 d after crush (Fig. 3A). Relative transgene induction in the DRGs was overall higher in Tα1-GFP-3′-β-actin than in Tα1-GFP-3′-γ-actin lines, but the absolute levels of the GFP mRNA were not significantly different before injury (Fig. 3B). Curiously, the sciatic nerve GFP-3′-β-actin mRNA levels showed peak accumulation at 1 d after injury, which precedes the 5 d peak in GFP-3′-β-actin mRNA levels in the DRG (Fig. 3A). Relative nerve GFP mRNA levels in Tα1-GFP-3′-γ-actin mice could not be calculated over 0–28 d samples since threshold values were not reached for this ΔΔCt analyses. Thus, we analyzed raw RT-qPCR amplification plots. DRG and sciatic nerve RT-qPCR plots for Tα1-GFP-3′-β-actin mice rose from baseline within a few cycles of one another (Fig. 3C,D). Plots for the DRG RNA from Tα1-GFP-3′-γ-actin mice were similar to the Tα1-GFP-3′-β-actin mice. However, the sciatic nerve RT-qPCR plots from Tα1-GFP-3′-γ-actin mice were essentially the same as wild type (Fig. 3C,D). Thus, GFP-3′-γ-actin mRNA does not appear to be transported into distal sciatic nerves.

Sciatic nerve injury increases axonal levels of β-actin reporter mRNA. A, L4–5 DRGs and sciatic nerves of transgenic mice were analyzed for GFP mRNA levels by RT-qPCR as indicated. The GFP signals were normalized to 18S rRNA. Maximal DRG GFP mRNA levels are reached at 5 d after injury with both transgenes. Nerve samples (taken 1 cm proximal to the crush site) show peak in GFP mRNA levels by 1 d after injury in Tα1-GFP-3′-β-actin lines. Mean ± SEM is shown (n ≥ 12; p values are indicated for significant difference vs t = 0 from one-way ANOVA and Tukey post hoc test). B, RT-qPCR for absolute GFP mRNA levels in L4–5 DRGs of transgenic lines at 0 and 5 d following sciatic nerve crush is shown as fold change relative to GFP-3′-β-actin2a line. Values indicate average ± SEM (n = 4). C, qPCR amplification plots for GFP mRNA for DRGs and sciatic nerve (naive and 5 d crush) are shown for transgenic mice in C and wild-type mice in D. Plots for the DRGs are comparable for the transgenic mice, but plots for naive and injured Tα1-GFP-3′-γ-actin nerves are essentially the same as wild type. E, FISH for GFP mRNA combined with S100 protein is shown for 14 d nerve graft. Tα1-GFP-3′-β-actin graft shows signal for GFP mRNA that is distinct from S100, while matched exposures for the Tα1-GFP-3′-γ-actin graft shows no GFP mRNA [Scale bar, 20 μm]. F, RT-qPCR plots for GFP mRNA in acellular nerve grafts show that GFP mRNA is only detected in the Tα1-GFP-3′-β-actin grafts.
The RT-qPCR amplification plots for the naive Tα1-GFP-3′-β-actin sciatic nerves show higher Ct values, indicative of less GFP mRNA, than the naive DRG and 5 d crushed nerve RNA samples, but clearly indicate presence of GFP-3′-β-actin mRNA in the mature uninjured nerve (Fig. 3C). Thus, the neuronally expressed transgene mRNA with the β-actin 3′-UTR also localizes into uninjured PNS axons.
As a more rigorous test for axonal localization of the GFP-3′-β-actin mRNA, we transected the CF of transgenic mice and provided a wild-type nerve graft as a regeneration substrate (English et al., 2005). FISH for GFP mRNA showed signals in 14 d grafts from Tα1-GFP-3′-β-actin mice that were clearly distinct from Schwann cells (Fig. 3E). The Tα1-GFP-3′-γ-actin lines only showed background GFP mRNA signals in grafts (Fig. 3E). Axons from transected nerves similarly regenerated into acellular allografts (data not shown); RT-qPCR for acellular grafts also showed Ct values for Tα1-GFP-3′-β-actin nerves indicative of GFP mRNA localization while grafts of Tα1-GFP-3′-γ-actin mice showed no evidence for GFP mRNA (Fig. 3F).
Axonal mRNA localization has been detected in the developing spinal cord (Brittis et al., 2002). Thus, we asked whether Tα1-GFP-3′-β-actin mice showed axonal GFP fluorescence in their spinal cord axons. Since PNS nerve injury increased axonal GFP signals in the Tα1-GFP-3′-β-actin mice, we reasoned that spinal cord injury might also lead to localization of the GFP-3′-β-actin mRNA into ascending DRG axons. Scattered GFP-positive axons of the sham Tα1-GFP-3′-β-actin mice were observed (Fig. 4A–D). Strong GFP fluorescence was seen in axons of the Tα1-GFP-3′-β-actin mice at 10 d after contusion injury (Fig. 4E–H). By confocal imaging, these GFP signals overlapped with axonal markers but were distinct from glial markers (Fig. 4F,G). No GFP fluorescence was seen in the Tα1-GFP-3′-γ-actin spinal cord (Fig. 4I–L). Lower thoracic DRGs in these animals showed no increase in GFP signals with spinal cord injury (Fig. 4B,F,G inset), suggesting that this injury did not activate the Tα1 promoter.

Axonal localization of GFP-3′-β-actin product in central axons. Confocal images from spinal cord of Tα1-GFP-3′-β-actin (A–H) and Tα1-GFP-3′-γ-actin (I–L) mice at 10 d after thoracic spinal cord injury (SCI) (E–L) or sham procedure (A–D) are shown. Images were taken caudal to the injury site to capture central processes of DRGs. Images are matched for laser energy, gain, offset, and postprocessing. Top two rows show average projections of Z stacks. Lower two rows are single optical planes through the center of the axoplasm of the axon indicated by the rectangles in A, E, and I (channel designations for each row are indicated on the left). No GFP signal was detectable in samples from Tα1-GFP-3′-γ-actin (I–L). GFP signals are easily seen in the Tα1-GFP-3′-β-actin after injury (E–H, arrows) and low GFP signals are seen in the sham samples from Tα1-GFP-3′-β-actin mice (A–D, arrows). These GFP signals overlap with NF and are distinct from GFAP. Insets in B, F, and J show matched images for GFP signals in caudal DRGs. Scale bars: (A, B, E, F, I, J) 10 μm, (C, D, G, H, K, L) 20 μm.
Discussion
The data presented here indicate that β-actin's 3′-UTR but not γ-actin's 3′-UTR can drive axonal mRNA localization in vivo, both in the peripheral and central processes of sensory neurons. The identical coding sequences of these transgenes make it highly unlikely that protein diffusion could account for the appearance of GFP selectively in the axons of the Tα1-GFP-3′-β-actin mice. The lack of any transgene expression in Schwann cells points to axonal transport of the GFP-3′-β-actin mRNA as the only plausible explanation for its presence in these axons. This is particularly true for the nerve grafts, where axons from the transgenic mice are regenerating into a wild-type substrate that contains no cells with the transgene. These data provide unequivocal proof for transport of mRNAs in adult mammalian sensory axons in vivo.
The vast majority of studies of axonal mRNA transport and local translation have focused on developing and regenerating axons (for review, see Willis and Twiss, 2010). The presence of GFP-3′-β-actin mRNA in the naive nerve combined with absence of any expression in Schwann cells indicates that this transgene mRNA is transported into adult axons in vivo, regardless of their growth status. Thus, the transgenic approach used here specifically overcomes the unavoidable limitations of in vitro models where neurons cannot be cultured without axotomy. This is consistent with works showing that both injury and neuropathic pain-invoking stimuli can activate translation in PNS axons, presumably by using mRNAs that reside in the mature axon before injury or stimulation (Hanz et al., 2003; Perlson et al., 2005; Jímenez-Díaz et al., 2008; Yudin et al., 2008; Melemedjian et al., 2010).
A multitude of mRNAs localize into axons of cultured CNS neurons (Taylor et al., 2009) and our data show that the 3′-UTR of β-actin mRNA can drive transport of the GFP mRNA into the CNS processes of DRG neurons. Although transgene expression was limited to the DRG neurons in these mouse lines, our data justify future genetic approaches to test for functionality of axonal RNA localization motifs in the mature CNS. A parallel genetic approach for visualizing RNA localization in mice was also recently taken by the Singer laboratory (Lionnet et al., 2011). In this study, MS2 recognition stem loops were placed into the endogenous β-actin gene, effectively tagging the mRNA with a nontranslated epitope. Our data complement this model, but also extend the findings to show in vivo axonal mRNA localization in adult mammals.
Immunolabeling studies have suggested that CNS axons have a much lower content of translational machinery than PNS axons, and Verma et al. (2005) hypothesized that this relates to the differing regenerative capacities of CNS vs PNS neurons. Future studies will be needed to determine the extent to which RNA transport differs between PNS and CNS projections of the DRG neurons studied here. mRNAs have been detected in hypothalamic and olfactory axons in vivo but there was no ultrastructural evidence for ribosomes in these processes (Mohr et al., 1991, 2001; Mohr and Richter, 1992; Wensley et al., 1995; Denis-Donini et al., 1998). Nonetheless, translationally active mRNAs were recently detected in olfactory axons suggesting that ultrastructural absence of ribosomes cannot be used to exclude the possibility for translation in vivo (Dubacq et al., 2009). This emphasizes the need for more sensitive tools and reagents to assess mRNA localization and translation in vivo as we report here.
The Tα1 promoter used for transgenic mice here generates an artificial situation since transgene expression is activated during periods of axonal growth (Gloster et al., 1994). Despite limitations introduced by this, our data suggest that injury can activate transport of mRNAs from the cell body rather than the axonal mRNA levels simply paralleling an increase in transcription. Consistent with this, tropic stimulation can mobilize transport of existing cellular mRNAs from the perikaryon into axons (Willis et al., 2007). CGRP mRNA also appears to be targeted for increased axonal delivery in injury-conditioned neurons despite a clear decline in expression of CGRP mRNA in the cell body after injury (Toth et al., 2009). It is intriguing to speculate that the injury-induced transport of GFP-3′-β-actin mRNA could be independent of transcriptional activation of the transgene. The peak in sciatic nerve GFP-3′-β-actin mRNA levels before the peak in transcriptional activation of the Tα1 promoter (Fig. 3A) and increased axonal GFP signals in ascending sensory axons without corresponding increase in DRG GFP expression after spinal cord injury (Fig. 4) are consistent with this notion.
Vogelaar et al. (2009) have recently shown that axons of cultured neurons have decreased regenerative capacity when β-actin mRNA is depleted (Vogelaar et al., 2009). However, it is not clear whether this is the case in vivo. As more is learned of the mRNAs transported into axons, the mechanisms regulating their transport and translation, and the roles of localized protein synthesis in axons in culture settings, it will be of critical importance to move from culture to in vivo models so that axonal protein synthesis can be assessed in the neuron's natural environment.
Footnotes
-
This work was supported by funds from NIH (Grants R21-NS045880 and R01-NS041596 to J.L.T.; Grant K99-NR010797 to D.E.W.; Grant R21-NS060098 to G.J.B.; and Grant P20-RR15588 to C.B.K.-S.) and the Christopher and Dana Reeve Foundation (Grant TB2-0602 to J.L.T. and S.O.Y.). The Transgenic Core Facility of the University of Delaware was used to generate mice. The Nemours Core facilities provided technical assistance (Nemours Foundation and P20-RR020173 COBRE funding). Erin Schuman provided the destabilized GFPmyr reporter used here. Freda Miller provided the Tα1 tubulin promoter.
-
The authors declare no competing financial interests.
- Correspondence should be addressed to Jeffery L. Twiss, Department of Biology, Drexel University, 3141 Chestnut Street, Stratton 118, Philadelphia, PA 19104. jeff.twiss{at}drexel.edu




{kind=link}
{kind=link}
{kind=link}
{kind=link}