

Abstract
Unlike the proposed role of reactive oxygen species in neurodegeneration, acute effects of reactive oxygen on synaptic plasticity are poorly understood. Using rat hippocampal slices, we found that exposure to a high concentration (0.5–5 mm) of H2O2 reduces EPSPs in both potentiated and nonpotentiated synapses. Exposure of the slices to 20 μmH2O2 did not affect expression of preestablished long-term potentiation (LTP) but prevented induction of new LTP and enhanced long-term depression (LTD). Surprisingly, 1 μm H2O2 caused a twofold increase in LTP compared with controls, and it further enhanced NMDA-independent LTP. A low concentration of H2O2 also suppressed LTD. Nifedipine, an L-type calcium channel blocker, did not affect control LTP but blocked effects of both 1 and 20 μm H2O2. Calcineurin inhibitors [FK506 (FR900506) and cyclosporin A but not rapamycin] acted similarly and also restored LTP in the presence of 20 μm H2O2. These results suggest that H2O2 alters NMDA-independent, voltage-gated calcium channel-mediated LTP by activating calcineurin.
Introduction
Reactive oxygen species (ROS) are the byproducts of cellular metabolism that have been implicated in neurodegeneration and age-associated cognitive and memory impairments. Thus far, ROS have been treated as harmful agents that cause damage to macromolecules through nucleophillic attack (Halliwell, 1992). However, there are recent indications that ROS may have an important role as signaling molecules that are used by cells as second messengers for altering the redox state of specific molecules that affect their functions (Klann,1998; Barrett et al., 1999). These regulatory roles can only occur under physiologically significant concentrations of ROS, which are strictly regulated in vivo by a myriad of antioxidant molecules. Recently, a link between ROS and modulation of synaptic plasticity has been proposed; high concentrations of ROS attenuate synaptic transmission and long-term potentiation (LTP) (Colton et al., 1986, 1989; Pellmar et al., 1991; Avshalumov et al., 2000). They also induce stress-related responses (O'Donnell et al., 2000). On the other hand, superoxide radicals are proposed to be involved in LTP induction (Klann, 1998; Thiels et al., 2000; Knapp and Klann, 2002).
We studied effects of hydrogen peroxide (H2O2), a membrane-permeable form of ROS that is normally produced in living cells, on the synapse between the Schaffer collateral projections in stratum radiatum and pyramidal cells in area CA1 of the rat hippocampus. LTP and long-term depression (LTD) of the EPSPs are valuable tools in the investigation of cellular processes affecting synaptic plasticity. These processes are induced by calcium influx through NMDA receptors but also through voltage-dependent calcium channels (VDCCs) (Morgan and Teyler, 1999). The transient changes in calcium are transduced by signal transduction cascades, including the calcium-dependent activation of calcium calmodulin, which activates the phosphatase calcineurin (CaN) (Foster et al., 2001). These processes cause a change in the AMPA receptor functions that underlie long-term changes in EPSPs. Blockade of CaN activity with the specific inhibitor FR900506 (FK506) in the postsynaptic cell induces LTP (Wang and Kelly, 1997). On the other hand, FK506 blocks VDCC-dependent LTP (Onuma et al., 1998). These seemingly contradictory results may be explained by the observation that CaN mediates LTD of the GABAergic IPSP in the postsynaptic cells (Lu et al., 2000). CaN activity is also increased in aged individuals (Foster et al., 2001), and Ermak et al. (2001) found an overexpression of a CaN inhibitory gene in brains of Alzheimer's disease patients.
We examined the effect of physiologically relevant concentrations of H2O2 on different measures of synaptic plasticity in the CA1 region of rat hippocampal slices. Subsequently, we began to elucidate the molecular sequence of events leading to these changes. These data shed new light on the understanding of redox contribution to mechanisms underlying synaptic plasticity in the hippocampus.
Materials and Methods
dl-2-Amino-5-phosphonopentanoic acid (APV), nifedipine, cyclosporin A (Cys A), and hydrogen peroxide were purchased from Sigma (St. Louis, MO). FK506 was a generous gift from Fujisawa Pharmaceuticals (Tokyo, Japan). Rapamycin was purchased from Alomone Labs (Jerusalem, Israel). Pharmaceuticals were added into the perfusion medium with special care to prevent changes in temperature, pH, flow rate, or degree of oxygenation of the artificial CSF (ACSF).
Electrophysiology. Hippocampal slices from 6- to 8-week-old male Wistar rats were prepared as follows. After decapitation, the hippocampus was removed, and 350 μm slices were made with a McIlwain tissue slicer and incubated for 1.5 hr in ACSF containing (in mm): 124 NaCl, 2 KCl, 26 NaHCO3, 1.24 KH2PO4, 2.5 CaCl2, 2 MgSO4, and 10 glucose, pH 7.4, at room temperature. The ACSF was saturated with 95% O2–5% CO2 gas mixture. After incubation, the slices were placed in a perfusion-type chamber for recording. Recordings were made with a glass pipette containing 0.75 m NaCl (4 MΩ) placed in stratum radiatum. Stimulation was delivered through two sets of bipolar nichrome electrodes placed on either side of the recording electrode such that two independent stimulation channels were used for each slice. Unless stated otherwise, LTP was induced by high-frequency stimulation (HFS) consisting of 100 pulses at twice the test intensity delivered at a frequency of 100 Hz, and stimulation and data acquisition and analysis was performed using the LTP Program (Anderson and Collingridge, 2001).
Enzyme assay. Five hippocampal slices were placed in the perfusion chamber and subjected to either ACSF or ACSF containing 1 or 20 μmH2O2. The slices were collected, washed with cold saline, and homogenized in lysis buffer supplied with the Calcineurin Assay kit (Biomol, Plymouth Meeting, PA), containing 50 mm Tris, pH 7.5, 0.1 mm EDTA, 0.1 mm EGTA, 1 mm DTT, and 0.2% NP-40. The samples were centrifuged (100,000 × g, 45 min), and the supernatant was filtered through a desalting column for the removal of free phosphates. The filtered samples were incubated at 30°C for 5 min in reaction buffer from the kit with or without 0.5 μm okadaic acid. The enzyme activity was expressed in nanomoles of orthophosphate (PO4) released per milligram of protein from the substrate. The calcineurin substrate sequence is Asp-Leu-Asp-Val-Pro-Ile-Pro-Gly-Arg-Phe-Asp-Arg-Arg-Val-pSer-Val-Ala-Ala-Glu. The sequence is from protein kinase A regulatory subunit type II. The protein concentration was calculated using the Bradford method (Bio-Rad, Hercules, CA).
Statistical analysis was done by using one-way or two-way ANOVA, Student's t test, or Mann–Whitney nonparametric test whenever relevant.
Results
H2O2 at 20 μm affects synaptic plasticity without disrupting baseline synaptic responses
Previous studies show that 0.5–10 mmH2O2 can dramatically suppress synaptic transmission and plasticity (Colton et al., 1986,1989; Pellmar et al., 1991; Avshalumov et al., 2000). EPSP slopes shown were normalized and are expressed as a fraction of mean baseline EPSP slope. When 5 mmH2O2 was added to the perfusion medium, the magnitude of EPSP slopes declined to 0.07 ± 0.07 in the previously potentiated channel and to 0.34 ± 0.2 in the nonpotentiated channel (n = 4). This decline in EPSP magnitude began 2–3 min after addition of H2O2 to the perfusing medium (Fig. 1A). The rapid decline in EPSP slope preceded a slower decline in the magnitude of the presynaptic volley, which was associated with an irreversible decline in slice viability.
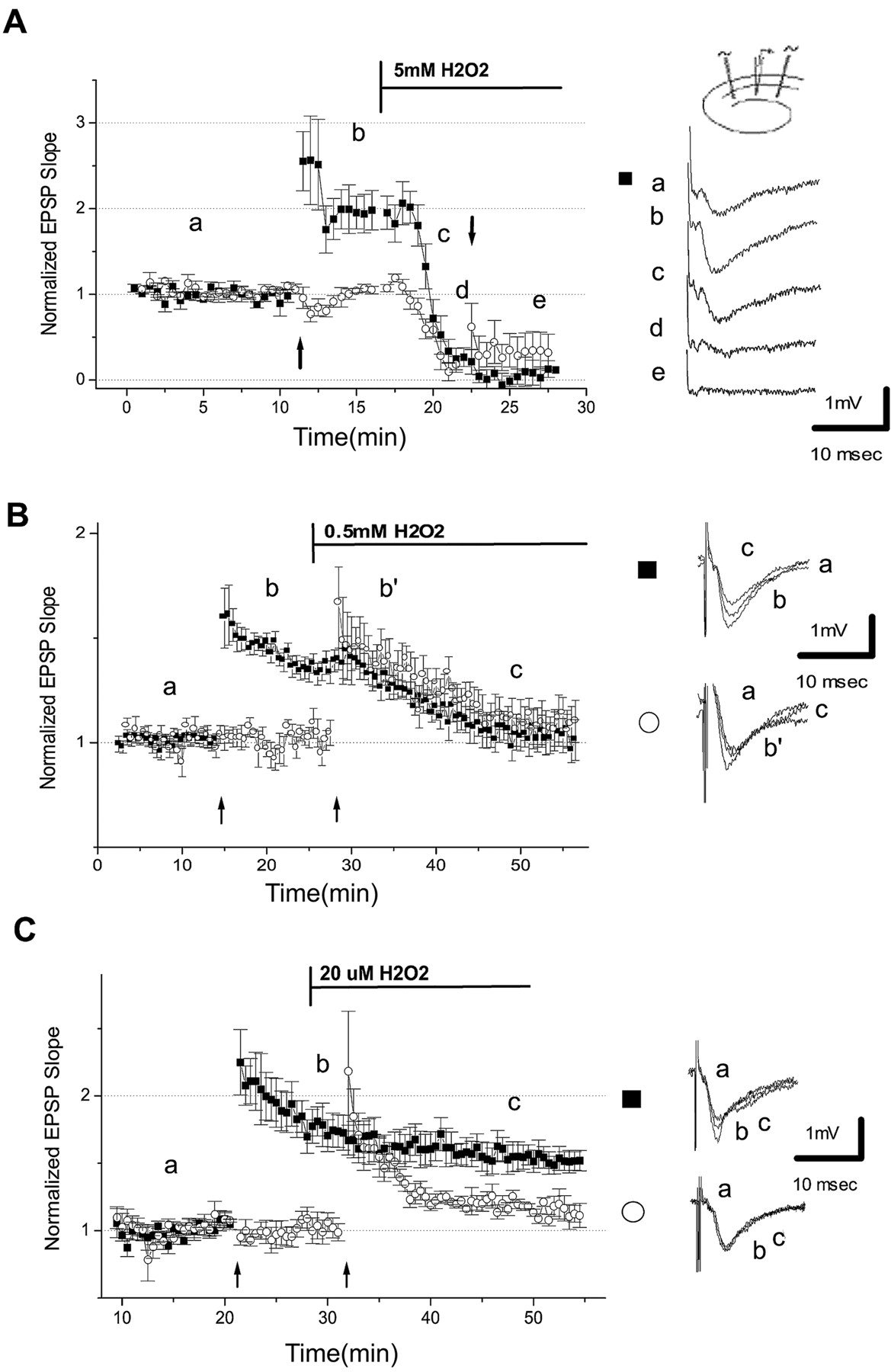
Long-term potentiation is inhibited by hydrogen peroxide. A, Application of 5 mmH2O2 (bar) depresses EPSP magnitude in both potentiated (▪) and unpotentiated (○) pathways. Schematic presentation of the two channels is in theinset on the right. Average normalized EPSP slope is plotted versus time. In the right,a–e are representative tracings at indicated times. Thearrows indicate the time of application of HFS (100 Hz for 1 sec at twice the test stimulus intensity). B, Application of 0.5 mm H2O2 causes a decrease in EPSP amplitude of the potentiated channel (▪), which declines 15 min after H2O2 application. This concentration of H2O2 also prevents the induction of new LTP in the previously unpotentiated channel (○).C, Application of 20 μmH2O2 does not affect the potentiated pathway; it does, however, inhibit the induction of new LTP in the previously unpotentiated pathway. (a–c on the rightcorrespond to the position of the EPSP peak.)
Exposure of the slices to 0.5 mmH2O2 (Fig.1B) did not change basal EPSP slope; however, preestablished LTP declined from 1.48 ± 0.04 10 min after HFS to 1.02 ± 0.07 15 min after addition of H2O2 (t = 7.24; p < 0.0001; n = 7). HFS that was applied in the presence of 0.5 mmH2O2 resulted in EPSP slope that was 1.11 ± 0.08 of control 15 min after HFS (t = 5.72; p < 0.0001;n = 7).
In the presence of 20 μmH2O2 applied for 10 min (Fig. 1C), there was no change in EPSP slope (0.98 ± 0.06). LTP that was induced before H2O2 perfusion remained at 1.6 ± 0.12, indicating that this concentration of H2O2 did not affect the ability to express larger and already potentiated EPSPs. HFS delivered in the presence of 20 μmH2O2 resulted in significantly reduced (t = 4.18; p < 0.001) potentiation to 1.18 ± 0.04 20 min after HFS (n = 6).
H2O2 at 1 μmenhances LTP
To determine the threshold of H2O2 effects on LTP, the tissue was exposed to lower concentrations of H2O2. Exposure to <20 μm of H2O2did not produce a reliable blockade of LTP. In the presence of 1 μm H2O2perfused for 10 min, EPSP slope was 1.02 ± 0.06. EPSP slope of the pathway that was potentiated by HFS before the addition of H2O2 was 1.48 ± 0.12 10 min after the addition of H2O2. Surprisingly, HFS applied 10 min after the addition of 1 μmH2O2 resulted in EPSP slope that was 1.99 ± 0.16 20 min after HFS (n = 10), which is a statistically significant (ANOVA; p < 0.05) twofold increase in LTP compared with the pathway that was potentiated before the addition of 1 μmH2O2 (Fig.2A).
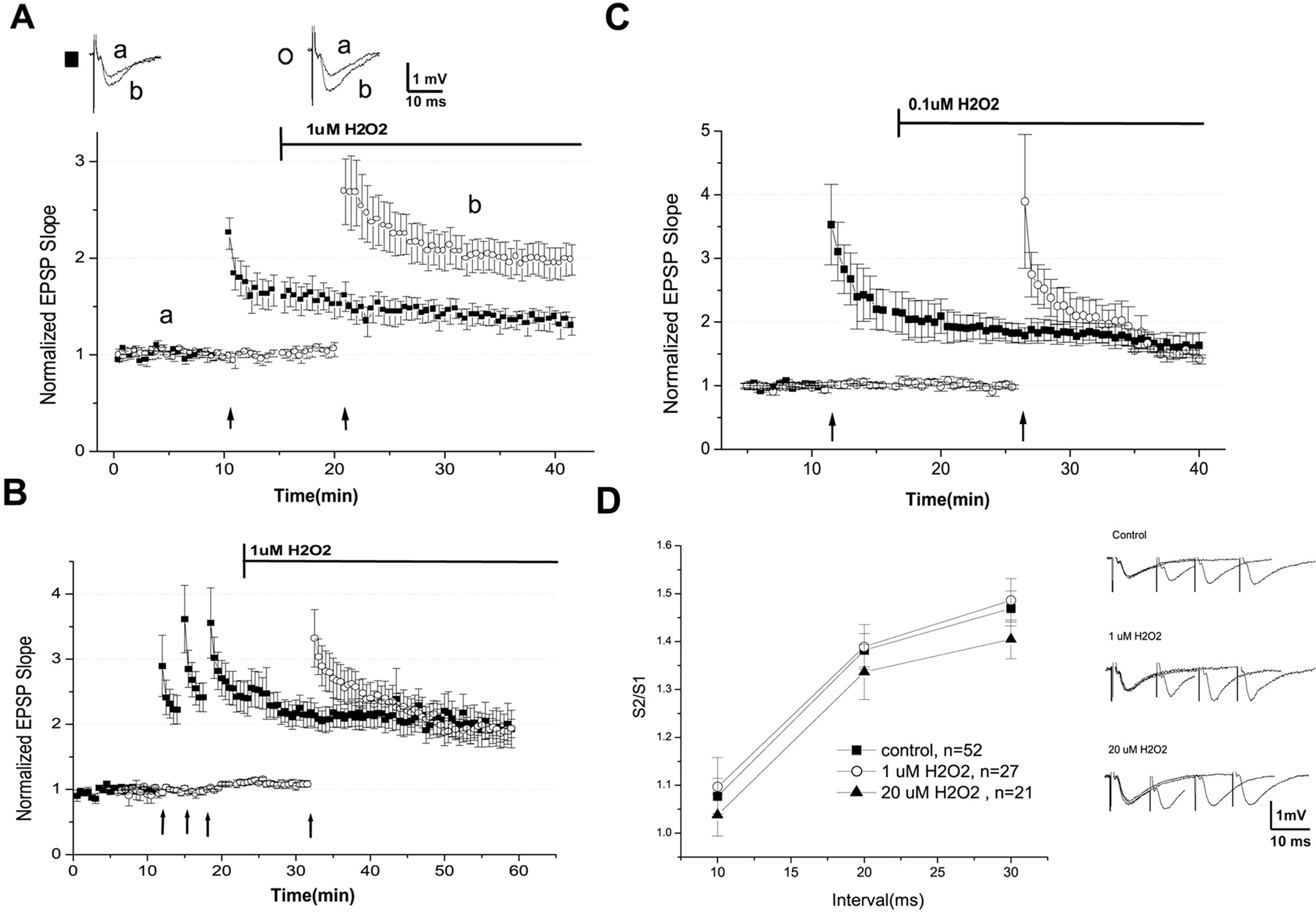
Lower concentrations of H2O2 have opposing effects on LTP.A, Application of 1 μmH2O2 (bar) increases LTP levels by twofold compared with control. HFS applied in the same slices before H2O2 (▪) resulted in a normal LTP, whereas HFS applied 10 min after addition of H2O2 (○) resulted in a significantly larger LTP. In the top,a and b are representative tracings at indicated times. B, Application of multiple trains of HFS resulted in LTP that is similar in size to LTP induced by one HFS train in the presence of 1 μmH2O2. HFS was applied three times with an intertrain interval of 2.5 min on one channel (▪). A single train of HFS was applied to the second channel (○) 10 min after onset of infusion of 1 μm H2O2(bar). C, Application of 0.1 μm H2O2 does not affect LTP. LTP induced before (▪) and 10 min after (○) the application of 0.1 μm H2O2 (bar) resulted in similar potentiation. D, Paired-pulse potentiation is not significantly altered by micromolar concentrations of hydrogen peroxide. The ratio of the second EPSP slope to the first is plotted as a function of the interval between them. In theright are representative traces of the data summarized on the left.
To establish that this increase was within normal physiological range and not a result of H2O2 causing hyperexcitability of the slice, more rigorous stimulation protocols were used before addition of H2O2. Three trains of HFS 2.5 min apart resulted in EPSP slope that was 2.16 ± 0.2 20 min after the last train. This potentiation was not significantly different from that induced in the same slices in the presence of 1 μm H2O2 (Fig.2B), which resulted in EPSP slope that was 1.95 ± 0.32, 20 min after the single HFS train (n = 20). This indicates that the enhanced LTP by H2O2 saturates at the level reached by the multiple trains of HFS.
Finally, in the presence of 0.1 μmH2O2, the EPSP slope was 1.66 ± 0.13 15 at minutes after HFS. This potentiation was not significantly different from that induced before the addition of 0.1 μmH2O2, which resulted in EPSP slopes that were 1.83 ± 0.15 at 15 min after the HFS (Fig.2C) (n = 5). Thus, 0.1 μmH2O2 did not seem to affect the plastic properties of the slice.
Changes in LTP can result from either presynaptic change in probability of release or postsynaptic changes in the response to the tetanic stimulation. To examine a possible presynaptic locus of action, paired-pulse stimulation protocol was used (Fig. 2D). Under control conditions, two pulses, at 10, 20, and 30 msec interpulse intervals, resulted in a second EPSP that was 1.08 ± 0.03, 1.38 ± 0.03, and 1.47 ± 0.04 of the first pulse, respectively. In the presence of 1 μmH2O2, the values were 1.09 ± 0.06, 1.39 ± 0.04, and 1.48 ± 0.04, respectively. In the presence of 20 μmH2O2, the comparative values were 1.03 ± 0.04, 1.33 ± 0.05, and 1.40 ± 0.04. There was no significant difference among the treatments. In addition, the size of the presynaptic volley did not change with either 1 or 20 μmH2O2 or after HFS (Figs.1C, 2A). These experiments indicate that the effects of H2O2 are not likely to take place presynaptically at the Schaffer collateral fibers and terminals.
Because the EPSP evoked in response to afferent stimulation is mediated primarily by activation of the AMPA receptor and there was no effect of H2O2 on EPSP, it is still possible that these effects are mediated by an interaction with the NMDA receptor, normally inactive at rest attributable to an Mg2+ block. To examine the effect of H2O2 on NMDAR EPSPs, slices were perfused with magnesium-free ACSF and in the presence of 10 μm glycine and 10 μm DNQX, an AMPA receptor antagonist. The addition of 1 or 20 μmH2O2 did not change NMDA EPSPs (Fig. 3).

Micromolar concentrations of H2O2 do not change NMDA receptor function.A, Hippocampal slices were placed in a perfusion chamber, and EPSPs were recorded in stratum radiatum in response to stimulation every 30 sec. After stable baseline conditions were established, the perfusion medium was changed to magnesium-free ACSF containing glycine and 10 μm DNQX. After stable NMDA receptor EPSPs were established, 1 μmH2O2 was added to the medium and EPSPs were recorded. On the top right, a–c are sample traces taken at the times indicated on the left. At the bottom right are input–output curves for NMDA receptor EPSP slopes before and after the addition of H2O2. B, Similar toA but with 20 μmH2O2.
H2O2 affects both LTP and LTD
To test the possibility that H2O2 shifts the optimal response to different stimulation frequencies, they were tested in conjunction with exposure to a medium containing 1 or 20 μm H2O2 (Fig.4). Under control conditions, 0.5 Hz stimulation depressed EPSP slopes to 0.79 ± 0.08 at 15 min after stimulation (n = 6), 5 Hz resulted in EPSP slopes that were not different from controls (0.97 ± 0.03; n= 6), whereas 100 Hz stimulation potentiated EPSP slopes to 1.54 ± 0.14 (n = 6). In the presence of 20 μmH2O2 0.5 Hz stimulation depressed EPSP slopes to 0.8 ± 0.06 (n = 6), 5 Hz also depressed EPSP slopes to 0.77 ± 0.06 (n = 6), thus expanding LTD to frequencies that do not produce it in the control case, whereas 100 Hz stimulation potentiated EPSP slopes only to 1.2 ± 0.2 (n = 6).

Micromolar concentrations of hydrogen peroxide affect both LTP and LTD. A, Under control conditions, a train of pulses at the frequency of 0.5 Hz results in LTD, stimulation with a 5 Hz train does not have an affect, and stimulation at 100 Hz results in LTP. B, When 20 μmH2O2 was added, a train of pulses at the frequency of 0.5 Hz resulted in LTD, stimulation with a 5 Hz train also caused LTD, and stimulation at 100 Hz caused reduced LTP.C, When 1 μm H2O2was added, a train of pulses at the frequency of 0.5 Hz did not induce LTD, stimulation with a 5 Hz train, similarly, did not have an effect, and stimulation at 100 Hz induced large LTP. D, The graph summarizes the data shown in A–C. The normalized EPSP slope at 15 min after the application of a train of pulses is plotted as a function of stimulation frequency (log scale) for the two concentrations of H2O2.
In the presence of 1 μmH2O2, 0.5 Hz stimulation failed to cause depression of EPSP slopes, and they were 1.03 ± 0.08 at 15 min after stimulation (n = 6); 5 Hz stimulation resulted in EPSP slopes that were 0.99 ± 0.05 (n = 6), whereas 100 Hz potentiated EPSP slopes to 2.12 ± 0.16 of controls (n = 6). Two-way ANOVA shows a significant variation attributable to stimulation frequency (p < 0.001) and also H2O2 concentration (p < 0.005).
Together, these findings demonstrate that H2O2 affects hippocampal plasticity in a concentration- and frequency-dependent manner.
The enhancement of LTP by 1 μmH2O2 is mediated by activation of VDCCs
In addition to NMDA receptors, calcium ions can permeate the membrane through VDCCs. These channels increase permeability to calcium when the neuronal membrane is depolarized during the EPSP. VDCC are assumed to be activated by higher-stimulation frequencies than those activating NMDA receptors (Morgan and Teyler, 1999). Because the effects of 1 μmH2O2 could be reproduced by using multiple trains of stimulation (Fig. 2), the possible mediation of the effects of 1 μmH2O2 by activation of VDCC was tested using nifedipine (50 μm), a specific L-type VDCC blocker. Nifedipine by itself did not affect EPSPs or LTP under control conditions (Fig. 5A). EPSPs that had been already potentiated before exposure to nifedipine were 1.5 ± 0.11 of controls at 20 min after HFS compared with those potentiated after the addition of nifedipine, which were 1.49 ± 0.07 (n = 6). However, in the presence of nifedipine, 1 μmH2O2 could no longer potentiate EPSP slope in response to HFS beyond the control level. EPSP slopes potentiated in the presence of 1 μmH2O2 before addition of nifedipine were 2.1 ± 0.09 at 20 min after HFS compared with those potentiated after the addition of nifedipine, which were 1.49 ± 0.06 (Fig. 5B) (n = 9). Interestingly, nifedipine could not reverse the suppressive action of 20 μmH2O2 on LTP (data not shown). However, nifedipine abolished LTD at 20 min after 5 Hz stimulation down to 0.98 ± 0.07 (n = 6) (see Fig.7B).

Potentiation of LTP modulated by 1 μm H2O2 is mediated through voltage-dependent calcium channels. A, Nifedipine alone did not change LTP. After LTP was induced by HFS (arrow) in one channel (▪), nifedipine (bar; 50 μm) was added and HFS was applied to the second channel (○), yielding similar LTP. B, Nifedipine dampened LTP modulation by H2O2. One micromolar H2O2 was applied for 10 min (long bar) before application of HFS on the first channel (▪). Nifedipine (50 μm) was added 5 min before application of HFS on the second channel (○), resulting in a much-reduced LTP.C, The NMDA component of LTP is not required for modulation by 1 μm H2O2. Non-NMDA LTP was induced in the first channel (▪) by three trains of 100 pulses at the same intensity as the test pulse at 200 Hz (arrows) in the presence of 50 μm APV (bars). The addition of 1 μmH2O2 (long bar) 10 min before application of the three-high frequency trains to the second channel resulted in a much larger non-NMDA LTP in that channel (○).
To further establish a connection between the effect of 1 μm H2O2 on LTP and VDCCs, we induced non-NMDA LTP in the presence of APV. LTP can still be induced in the presence of APV by three 200 Hz trains (Fig.5C). EPSP slopes in these conditions were 1.5 ± 0.12 of control at 25 min after stimulation. In the presence of 1 μmH2O2, EPSP slopes were 2.8 ± 0.3 of control at 25 min after stimulation (n = 7). These experiments indicate that NMDA receptors are not required for the potentiation induced by 1 μmH2O2 and that blocking VDCCs eliminated this effect.
Calcineurin mediates the changes in synaptic plasticity induced by 20 μm H2O2
Synaptic plasticity is assumed to be regulated through a balance between phosphorylation–dephosphorylation of ion channels. This balance is maintained by kinases and phosphatases, respectively. The phosphatase CaN has been found to be high in older animals that exhibit less LTP and more LTD (Foster et al., 2001). CaN has a redox-sensitive domain (Wang et al., 1996), which can be affected by H2O2 and can change the activity of the enzyme.
We tried to reverse the changes in plasticity seen with H2O2 by inhibiting CaN activity. To this end, the specific CaN inhibitors FK506 (n = 6 slices) and Cys A (n = 7 slices) were used. LTP was not altered by the presence of 20 μm FK506 (Fig.6A). EPSP slopes in the presence of FK506 were 1.64 ± 0.07 of controls when measured 20 min after HFS, which was not significantly different from EPSP slopes of a second channel in the same slices in which HFS was induced before FK506 perfusion (1.7 ± 0.15). Perfusing FK506 or Cys A before the addition of 20 μmH2O2 resulted in a return of LTP back to control levels, with EPSP slopes being 1.87 ± 0.18 and 1.52 ± 0.11 (n = 6), respectively (Fig.6B,D), compared with 20 μmH2O2 alone, which resulted in EPSP slopes of 1.2 ± 0.05 (n = 5) of controls.

Inhibitors of calcineurin reverse the effects of micromolar concentrations of H2O2 on LTP.A, FK506 alone does not change LTP. After the induction of LTP by HFS (arrow) in one channel (▪), FK506 (bar; 20 μm) was added and HFS was applied to the second channel, yielding similar LTP. B, FK506 restores LTP in slices treated with 20 μmH2O2. EPSP slopes for different sets of slices treated with either 20 μm H2O2alone (▪) or 20 μm H2O2 and 20 μm FK506 (○). C, FK506 suppresses LTP in slices treated with 1 μm H2O2. EPSP slopes for different sets of slices treated with either 1 μm H2O2 alone (▪) or 1 μm H2O2 and 20 μmFK506 (○), indicating that FK506 dampens the enhancing effect of 1 μm H2O2 on LTP. D, Summary results of the effects of both FK506 (FK) and cyclosporin A (CA) to reverse the action of micromolar concentrations of H2O2 on LTP. The normalized EPSP slope 15 min after HFS is shown for the various treatments.
Furthermore, perfusing FK506 or Cys A before the addition of 1 μm H2O2resulted in a decrease in LTP; EPSP slopes were 1.65 ± 0.17 and 1.7 ± 0.2 (n = 7), respectively (Fig.6C,D). These, again, were not different from untreated controls compared with 1 μmH2O2 alone, which resulted in EPSP slopes 1.99 ± 0.16 (n = 11).
The response to 5 Hz stimulation was not altered by the presence of FK506 (Fig. 7A). Under control conditions, EPSP slopes were 0.94 ± 0.06 at 20 min after 5 Hz stimulation (n = 10) and 0.93 ± 0.01 in the presence of FK506 (n = 10). Stimulation at 5 Hz in the presence of 20 μmH2O2 resulted in EPSP slopes that were depressed to 0.77 ± 0.04 of control at 20 min after 5 Hz stimulation. FK506 and Cys A were also effective in reducing LTD in the presence of 20 μmH2O2. Perfusing FK506 or Cys A before addition of 20 μmH2O2 resulted in a decrease in the response to 5 Hz stimulation; EPSP slopes were 0.98 ± 0.05 and 0.96 ± 0.01 at 20 min after 5 Hz stimulation, respectively (Fig. 7C,D) (n = 7). These experiments suggest that redox-regulated CaN activity underlies the plasticity changes seen with H2O2.

Inhibitors of calcineurin and VDCC reverse the effects of 20 μm H2O2 on LTD.A, FK506 alone does not facilitate LTD after 5 Hz stimulation. EPSP slopes for control slices (▪) or for slices treated with 20 μm FK506 (○) virtually overlap.B, Nifedipine prevents LTD induced by 5 Hz low-frequency stimulation in the presence of 20 μmH2O2. H2O2 at 20 μm was applied for 10 min (long bar) before application of low-frequency stimulation, and nifedipine (50 μm) was added 5 min before application of LFS.C, FK506 blocks the LTD produced in response to a 5 Hz stimulation in presence of 20 μmH2O2. Averaged EPSP slopes for separate sets of slices treated with 20 μm H2O2alone (▪) or with 20 μm H2O2and 20 μm FK506 (○). D, FK506 (FK), cyclosporin A (CA), and nifedipine (nif) reverse the effects of 20 μm H2O2 on LTD. The normalized EPSP slope at 15 min after low-frequency stimulation is shown for the various treatments.
FK506 inhibits calcineurin by binding to FK506 binding protein 12 (FKBP-12) and forming a complex that inhibits CaN. To better define the underlying process, we added rapamycin to the perfusion medium. Rapamycin binds FKBP-12, but the complex formed does not interact with CaN. Rapamycin alone did not affect LTP, and HFS in the presence of rapamycin resulted in potentiation to 1.48 ± 0.07 of control at 20 min after HFS (n = 12). In the presence of 20 μmH2O2 and rapamycin, potentiation was down to 1.18 ± 0.08 of control, showing no interaction between rapamycin and 20 μmH2O2 (n = 9). Interestingly, perfusion of rapamycin with 1 μmH2O2 resulted in no LTP (EPSP slope, 1.04 ± 0.04; n = 12) (Fig.8). These findings suggest that different mechanisms underlie the effects mediated by 1 and 20 μmH2O2.

Rapamycin blocks the effect of 1 μmH2O2 but not that of 20 μmH2O2 on LTP. Slices were exposed to different concentrations of H2O2 after preincubation with rapamycin. Rapamycin has no effect on LTP under control conditions (control records not shown). In the presence of 20 μmH2O2, LTP was reduced as before. Surprisingly, in the presence of 1 μmH2O2, rapamycin completely suppressed formation of LTP. On the right are representative traces at the time points indicated.
To find more direct evidence connecting H2O2 with CaN, we conducted an enzymatic assay. Hippocampal slices were perfused with ACSF containing H2O2, collected, and homogenized. The centrifuged supernatants were passed through a desalting column, and the phosphate-free samples were measured for phosphatase activity (Fig. 9). Phosphatase activity was also measured in the presence of 0.5 μm okadaic acid, which inhibits protein phosphatase 1 (PP1) and PP2A, resulting in net CaN activity. To achieve maximal validity, we conducted two sets of experiments in triplicate. Because incubation time with the substrate and temperature can affect the amount of free phosphate that is detected, we normalized the results as a fraction of the results obtained for the controls. The overall serine–threonine phosphatase activity can be the result of CaN activity, as well as that of PP1 and PP2A, which are inhibited by okadaic acid. We found a slight increase in total phosphatase activity in slices incubated with 20 μmH2O2 and a nonsignificant increase in specific CaN activity in slices incubated with 1 μm H2O2. A significant (42%; Mann–Whitney U test; p< 0.04) increase was seen in the okadaic acid-treated homogenate of slices treated with 20 μmH2O2, indicating a selective increase by hydrogen peroxide of CaN activity (Fig. 9).

Micromolar concentrations of H2O2 enhance the activity of CaN. Hippocampal slices (5 in each group) were incubated with different concentrations of H2O2, homogenized, and centrifuged. The supernatant was passed through a desalting column, the extract was incubated with Ser–Thr phosphatase substrate, and the released PO4 was detected with the Biomol Green reagent. The total phosphatase activity is presented in the left columns, and the specific activity of CaN in the presence of okadaic acid (OA), which inhibits other phosphatases (PP1 and PP2A), is presented in the right columns. Four to five measurements were pooled, and the results are expressed as a fraction of the phosphatase activity under control conditions. Statistical significance is indicated with anasterisk.
Discussion
In previous studies (Colton et al., 1986, 1989; Pellmar et al., 1991; Avshalumov et al., 2000), LTP was found to be blocked by exposure to high concentrations of H2O2. Similarly high concentrations, in the millimolar range, are being used to explore the neurotoxic action of H2O2in relation to neurodegenerative diseases in cell cultures andXenopus oocytes (Li et al., 1998; Kanno et al., 1999;Yermolaieva et al., 2000; Burlacu et al., 2001; Jang and Surh, 2001;Datta et al., 2002). Although there is no direct evidence showing the amount of ROS that is actually generated in aged brains, Hyslop et al. (1995) measured 200 μmH2O2 in rat forebrain after ischemia using microdialysis. Although studies using H2O2 may provide useful information, with few exceptions (Auerbach and Segal, 1997), they are applying far higher concentrations than those seen by normal neural tissue. We found that 20 μmH2O2 does not affect control level of synaptic transmission and does not affect an already potentiated pathway but blocks the induction of novel LTP by HFS. This indicates that H2O2interferes with one or more of the processes activated directly by the tetanic stimulation. This concentration also increases the range of frequencies that can induce LTD. These alterations in LTP and LTD could be reversed by pharmacological intervention, which is directed at CaN, a protein phosphatase that was shown to take part in signal transduction events underlying synaptic plasticity that we found to be more active in slices treated with 20 μmH2O2.
Synaptic plasticity is regulated by calcium influx into the postsynaptic cell. A “sliding rule” has been proposed to account for the fact that both LTP and LTD can be induced in the same pathway by different patterns of stimulation (Bear, 1995): low-frequency stimulation induces LTD, whereas high-frequency stimulation induces LTP. Young adult animals display both LTD and LTP with a characteristic crossing frequency of ∼5 Hz. Slices taken from old rats require more severe stimulation for inducing LTP but display a larger LTD to low frequencies and a wider range of frequencies that can induce LTD. It has been suggested that aged rats are impaired in the regulation of ROS (McGahon et al., 1999; O'Donnell et al., 2000), and their brains are exposed to higher concentrations of ROS than young adults. Norris et al. (1998a) suggested a mechanism by which an increase in VDCC permeability in aged individuals results in a larger calcium flux, which in turn activates calcium-dependent potassium channels, causing a longer afterhyperpolarization (AHP). Moreover, it has been suggested (Mermelstein et al., 2000) that L-type VDCCs can act as a kinetic filter differentiating between action potentials and EPSPs causing differential phosphorylation of cAMP response element-binding protein. The longer AHP in aged rats has been associated with higher VDCC permeability (Campbell et al., 1996; Thibault et al., 2001). Calcium-gated potassium channels remain open longer in aged neurons (Moyer et al., 1992). Furthermore, nifedipine, an L-type VDCC antagonist, can reduce LTD in aged neurons (Norris et al., 1998b) in a manner similar to that mediated by the addition of apamin, a potassium channel antagonist. Calcium channels have been found on hippocampal dendrites and have been shown to be regulated by phosphorylation (Hell et al., 1995). Wang and Kelly (1997) have shown that FK506 injected postsynaptically can induce LTP, which is blocked by the addition of the calcium chelator BAPTA, indicating a postsynaptic role for CaN. Recently, Norris et al. (2002) found that CaN can enhance VDCC activity in hippocampal neurons with an increased effect in aged cultures.Shirotani et al. (2001) found that hydroxyl radicals can suppress calcium influx through VDCCs, suggesting a direct effect of ROS on VDCC.
Inactivation of CaN by H2O2has been shown previously (Bogumil et al., 2000); however, this inactivation was demonstrated using higher concentrations than we used (>250 μmH2O2). Several studies demonstrated that even low concentrations of H2O2 can induce the release of calcium from internal stores (Gen et al., 2001; Nam et al., 2002), which can then affect the activation of CaN. Moreover, Huang et al. (2001) demonstrated that vanadium can induce the activation of NFAT (nuclear factor of activated T cells), a CaN substrate, in a manner that is reversible by the addition of H2O2 scavengers and suggested a direct role for H2O2 in the activation of CaN.
Together, these studies are consistent with our observations on the effects of 20 μmH2O2 and suggest that a similar phenotype is found in slices taken from older rats and that the way to restore their normal function is similar, i.e., by blocking CaN.
The effect of 1 μmH2O2 is more complex and less consistent with the assumed toxic action of ROS predicted in other studies. This very low concentration of H2O2 actually potentiates the ability to express long-lasting LTP, producing an effect similar to that seen after a strong (three times) tetanic stimulation protocol. The effect of 1 μmH2O2 is also blocked by CaN and calcium channel antagonists; however, it is completely reduced by rapamycin, indicating a different molecular mechanism from that affected by the higher concentration of H2O2, which may explain how the effect of the lower concentration of H2O2 is opposite to that of the higher concentration. Both FK506 and rapamycin act by forming a complex with FK506 binding protein 12 (FKBP-12), but, whereas the FK506–FKBP-12 complex inhibits CaN, the rapamycin–FKBP-12 does not. The binding of FKBP-12 by these agents detaches it from ryanodine receptors and facilitates the release of calcium from internal stores (Terashima et al., 2000). Moreover, the rapamycin–FKBP-12 complex, but not the FK506–FKBP-12 complex, inhibits the Ser–Thr kinase mTOR (mammalian target of rapamycin) (Huang and Houghton, 2001). Together, it is hypothesized that 1 μmH2O2 acts via FKBP-12, affecting internal calcium stores or kinase activity.
It may then be hypothesized that 1 μmH2O2 “primes” the postsynaptic neuron by activation of kinases, resulting in a larger reaction to the calcium flux induced by HFS. In that sense, our results are congruent with those of Klann (1998), who proposed that formation of ROS is a necessary phase in the formation of LTP. Subsequently, clamping down levels of endogenous H2O2 may reduce the ability to express LTP rather than enhance it.
Together, these findings suggest a complex modulatory role for ROS in synaptic plasticity through the activation of specific signal transduction cascades in a reversible manner.
Footnotes
This work was supported by Alzheimer's Association Grant IIRG-00-2152. We thank Shay Covo for advice regarding CaN assay.
Correspondence should be addressed to Menahem Segal, Department of Neurobiology, The Weizmann Institute, Rehovot 76100, Israel. E-mail:menahem.segal{at}weizmann.ac.il.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}