

Abstract
Dendritic arborization and spine formation are critical for the functioning of neurons. Although many proteins have been identified recently as regulators of dendritic morphogenesis, the intracellular signaling pathways that control these processes are not well understood. Here we report that the Ras–phosphatidylinositol 3-kinase (PI3K)–Akt–mammalian target of rapamycin (mTOR) signaling pathway plays pivotal roles in the regulation of many aspects of dendrite formation. Whereas the PI3K–Akt–mTOR pathway alone controlled soma and dendrite size, a coordinated activation together with the Ras-mitogen-activated protein kinase signaling pathway was required for increasing dendritic complexity. Chronic inhibition of PI3K or mTOR reduced soma and dendrite size and dendritic complexity, as well as density of dendritic filopodia and spines, whereas a short-term inhibition promoted the formation of mushroom-shaped spines on cells expressing constitutively active mutants of Ras, PI3K, or Akt, or treated with the upstream activator BDNF. Together, our data underscore the central role of a spatiotemporally regulated key cell survival and growth pathway on trophic regulation of the coordinated development of dendrite size and shape.
Introduction
Dendrites are the primary sites in which neurons receive, process, and integrate inputs from their multiple presynaptic partners. The functions of dendrites are critically dependent on the branching pattern of the dendritic tree and the dendritic specializations called spines. Dendritic spines, highly specialized actin-based protrusions, are the primary site of excitatory synapses and are thought to function as the basic unit of synaptic integration. Dendritic morphogenesis requires an intrinsic differentiation program that is further guided by extracellular cues and electrical activity. In due course, these different forces converge to control gene expression and cytoskeletal dynamics that specify dendritic growth and branching, as well as dendritic spine formation (Whitford et al., 2002; Jan and Jan, 2003; Miller and Kaplan, 2003; Scott et al., 2003; Van Aelst and Cline, 2004).
Previous studies have underscored the importance of a balance of neurotrophins and neural activity to dendrite formation (for review, see McAllister et al., 1999); however, the precise molecular mechanisms are not fully understood. It is conceivable that these distinct cues might converge onto some key signaling pathways to regulate the size and shape of dendritic trees. Indeed, members of the Rho family of proteins, Rho A, Rac 1, and cell division cycle 42 (CDC42), key regulators of actin cytoskeleton, have been shown to function as key regulators for dendrite and spine formation and maintenance (Luo, 2002). The Ras–Raf–MAP kinase kinase (MEK)–mitogen-activated protein kinase (MAPK) pathway, which transduces extracellular signals at the cell membrane into nuclear events in regulating many important cellular processes, has been shown recently to be involved in activity-dependent formation of dendritic filopodia and dendrites (Wu et al., 2001; Koh et al., 2002; Redmond et al., 2002; Vaillant et al., 2002; Goldin and Segal, 2003; Alonso et al., 2004). Among the other likely candidates, the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway has emerged as a key molecular regulator for size control of a variety of organs in mammals as well as yeast and Drosophila (Schmelzle and Hall, 2000; Hanada et al., 2004). Notably, PI3K–Akt, acting through or in coordination with the mammalian target of rapamycin (mTOR), a serine-threonine kinase, has found a special place in coupling of growth stimuli with cell hypertrophy (Kwon et al., 2003) and, more recently, in the regulation of synaptic plasticity and memory formation (Tang et al., 2002; Cammalleri et al., 2003; Si et al., 2003). In a simplified scheme, it can be generalized that PI3K–Akt via mTOR plays a central role in growth control by controlling ribosomal biogenesis and activity of the translation machinery, whereas other downstream effectors play dominant roles in cell survival through phosphorylation of the cytoskeleton and transcription factors to regulate proapoptotic/antiapoptotic proteins (Brunet et al., 2001; Hanada et al., 2004).
There is evidence that PI3K can induce neurite outgrowth as well as neurite retraction in neuronal cell lines (for review, see Rodgers and Theibert, 2002; Leemhuis et al., 2004). More recently, the vital roles of PI3K and Akt signaling in the determination of neuronal polarity and axonal growth (Markus et al., 2002; Shi et al., 2003; Jiang et al., 2005) have been revealed. Here, we have used well tested molecular and pharmacological tools to systematically elucidate the role of the Ras–PI3K–Akt–mTOR signaling pathway in dendrite formation and regulation of spine growth in cultured hippocampal neurons. Our results support a notion that neurons use the same conserved signaling cascades to regulate their cell physiology according to their developmental stages and specialized functions.
Materials and Methods
Cell culture, transfection, immunocytochemistry, and Western blot. Calcium phosphate transfections, immunocytochemistry, and culture of dissociated hippocampal CA3/CA1 pyramidal neurons were performed as described previously (Wu et al., 2001). Enhanced green fluorescent protein (EGFP) was cotransfected with hemagglutinin (HA)- or myc-tagged constructs to visualize the detailed cell morphology. Hippocampal slice culture was done from 7–8 d postnatal rats and transfected biolistically at 2 d in vitro (DIV) as described previously (Lo et al., 1994). A modified procedure was used for Western blot experiments using cells growing on 12 mm coverslips. In brief, coverslips were placed in 1.5 ml microcentrifuge tubes containing 40 μl of 9 m urea and were crushed, and, after vigorous vortexing, samples were centrifuged (18,000 × g for 2 min). The protein concentration was determined by BCA assay (Pierce, Rockford, IL), and volumes were adjusted with 9 m urea so that all samples were 0.3–1.0 μg/μl. Five times SDS/PAGE sample buffer was added to each sample and boiled, and 5–10 μg/lane was loaded onto 10% polyacrylamide gels.
Pharmacological agents in DMSO or water were added in culture medium. Drugs were obtained from Calbiochem (San Diego, CA) (K252a, LY294002, LY303511, rapamycin, U0126, and wortmannin), and Alomone Labs (Jerusalem, Israel) (BDNF). The EGFP and Discosoma red (DsRed) expression plasmids were obtained from Clontech (Mountain View, CA); expression plasmids for wild-type and constitutively active (CA) Ras (Ras L61) were purchased from Upstate Biotechnology (Charlottesville, VA). The RasV12 and Ras effector domain mutations (RasL61S35, RasL61C40, RasV12S35, and RasV12C40) were generated, and sequences were confirmed in the laboratory. Dominant-negative (DN) PI3K (Δp85, which lacks the binding site for the catalytic subunit p110) or Akt [HA-Akt(R25C), which contains a point mutation of Arg to Cys in the Akt PH domain]; constitutively active PI3K (p110*, in which p110α is fused to a c-Src myristoylation sequence) or Akt (myr Akt, in which the catalytic domain of Akt is fused to a c-Src myristoylation sequence) (Franke et al., 1997; Rodriguez-Viciana et al., 1997) were generous gifts from Drs. Alex Toker (Beth Israel Deaconess Medical Center, Boston, MA) and Lewis Williams (Five Prime Therapeutics, San Francisco, CA). Antibody to dually phosphorylated extracellular signal-regulated kinase ½ (ERK1/2), pAkt473, phosphorylated ribosomal S6 protein (pS6), total ERK1/2, and Akts were obtained from Cell Signaling Technology (Beverly, MA) and used at 1:100 to 1:500. Pan-Ras antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). In this study, we did not further quantify pAkt immunostaining because the antibody also significantly recognized nonactive Akt in our hands (data not shown), preventing an accurate assessment of the extent of Akt activation in neurons expressing CA Akt. Because of the same reason, immunostaining with this antibody only produced a small LY294002-sensitive signal change in response to stimulation by 20–50 ng/ml BDNF or 3 min 90 mm K+ (data not shown). Nevertheless, the immunocytochemical approach allowed us, at the single-cell level, to verify whether the different Ras mutants and CA PI3K constructs activate downstream pathways as expected.
Imaging. To image and quantify cell morphology, confocal microscopy was performed as described previously (Wu and Cline, 1998). For fixed samples, high-resolution confocal images were obtained using a Zeiss (Oberkochen, Germany) LSM 510 Meta system with sequential acquisition setting to minimize “bleeding-through” between different channels. A z series projection of ∼7–15 images with 0.5–1 μm depth interval, each averaged two times, was taken at 1024 × 1024 pixel resolutions to cover the entire z dimension of the labeled neurons. A 20× lens [0.75 numerical aperture (NA)] was used to image whole-cell and dendritic morphology. A 40× oil lens (1.0 NA) with 2× electronic zoom was used to image fine structures such as dendritic spines. In analysis of filopodia formation, standard confocal microscopy techniques using stacked z series data were used to rule out any artifacts from shifting focus or changes in dendrite positioning.
Image analysis. To quantify dendritic spine morphology, a ∼50 μm portion of dendritic segment was used, and each individual spine present on the dendrites was manually traced onto an acetate sheet. The drawings were then scanned into a computer, and the maximum length (measured from tip of head to shaft), head width (widest portion perpendicular to length axis), and shape factor of each spine was calculated automatically with the MetaMorph software (Molecular Devices, Downingtown, PA) and logged into Microsoft (Seattle, WA) Excel. The experimental group was kept blind until all analysis was done, and then the data from the same group of experiments were pooled together. The limit set for considering dendritic outgrowth as filopodia was length >4 μm (Fiala et al., 1998) without a bulbous head. Spines were defined as a headless dendritic protrusion <4 μm long or a headed protrusion of any length up to 10 μm. To quantify the total dendritic branch length (TDBL), number of primary-order dendrite, and dendritic tips, 20× images were used and whole-cell morphology was traced onto acetate sheet and scanned as above. TDBL was calculated using Scion (Frederick, MD) software, whereas primary-order dendrite number and tips were counted manually. For each condition, all neurons that were distinguishable from neighboring neurons were drawn until 30–40 neurons were collected. Dendritic caliber (measured 20 μm from the soma in randomly selected proximal dendritic segments) and soma area were measured from 2× zoomed 20× differential interference contrast (DIC) images using NIH Image J software. We combined the use of DIC and GFP fluorescence images to outline and distinguish the cell body from the adjacent proximal dendritic region. This method reduced the subjective variability in selection of the soma area. Statistical significance was calculated using one-way ANOVA with Dunnett's or Tukey's correction factor for multiple groups.
Results
Activation of PI3K–Akt signaling increases dendrite size and complexity as well as soma size
To study the signaling mechanisms underlying dendrite formation, we used dissociated postnatal hippocampal CA1/CA3 neuronal cultures as our model system. The cell culture paradigm offers several advantages, such as great accessibility, ready amenability to molecular and pharmacological manipulations, and relative ease of monitoring individual synaptic structural components and activity with high resolution. We cotransfected EGFP with HA- or myc-tagged version of constructs to identify the transfected neurons and to visualize the morphological details by confocal microscopy (Wu et al., 2001). In some experiments, we also used a double-transfection protocol that allows sequential transfection with EGFP and a red version of fluorescent protein (DsRed). This protocol has enabled us to directly compare the morphology of neurons transfected with a gene of interest with neurons transfected with a control empty vector in the same coverslip, thereby reducing the variability among different coverslips in the sister cultures. Furthermore, it greatly facilitates the assessment of whether an effect is through a cell-autonomous mechanism or through secretory processes (Fig. 1A–C).
In the first series of experiments, we overexpressed constitutively active mutants of PI3K (CA PI3K) or Akt (CA Akt) at 5–7 DIV hippocampal neurons, at a stage in which the neurons had acquired their initial cell type-specific dendritic patterns and started to form dendritic filopodia (Ziv and Smith, 1996) (data not shown). We then examined their effects on dendrite morphology at 15 DIV, a stage in which the highly dynamic dendritic filopodia were gradually replaced by protrusions with mushroom-shaped mature spines. As shown in Figure 1, neurons overexpressing CA PI3K or CA Akt showed consistent increases in the soma size and dendritic caliber. Both the total dendritic branching tips and the number of primary dendrites are significantly increased, resulting in a net increase of TDBL.
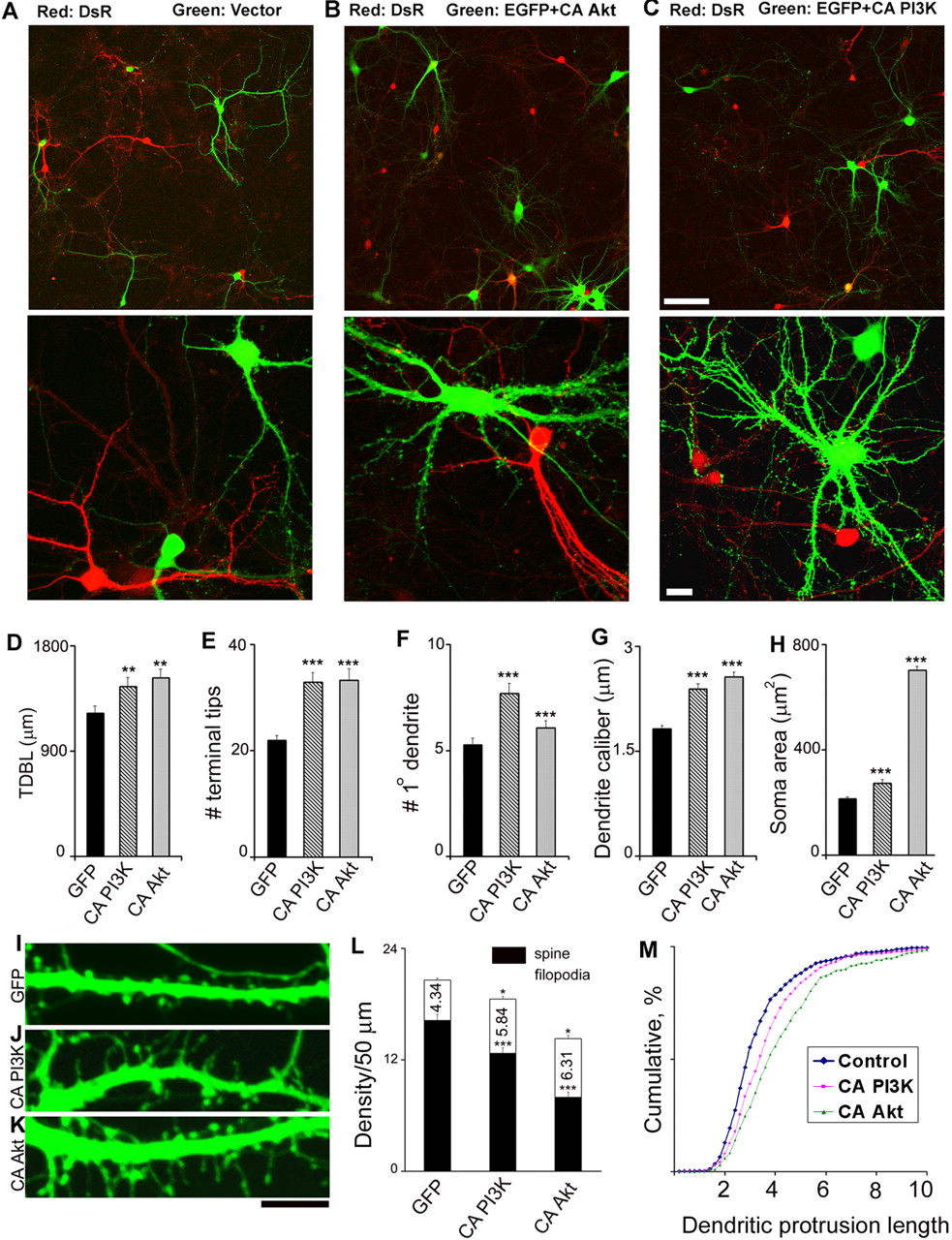
Constitutively active PI3K and Akt expression increases dendritic complexity and alters spine morphology. A–C depict overall and close-up view of hippocampal neurons double-transfected with EGFP, DsRed, and active mutants of Akt(CA Akt) and PI3K (CA PI3K). The empty vector (A), CA Akt (B), and CA PI3K (C) constructs were cotransfected with EGFP on 5–7 DIV, followed by DsRed transfection 12–24 h later. The graphs show mean ± SEM for TDBL (D), terminal tip number (E), primary-order dendrite number (F), dendritic caliber (G), and soma area (H). The bottom panels illustrate dendritic protrusions at 15 DIV in GFP (I), CA PI3K (J), and CA Akt (K) expressing cells. L illustrates spine and filopodia density of the representative groups (mean ± SEM). M shows cumulative distribution of dendritic protrusion length. Data are fromat least three independent experiments (n = 49 GFP, 58 CA PI3K, and 94 CA Akt cells; n = 600 GFP, 455 CA PI3K, and 540 CA Akt dendritic protrusions). There is no significant difference in width among the three groups (data not shown). *p < 0.05, **p < 0.005, and ***p < 0.0001 demonstrates statistical significance by one-way ANOVA (Tukey' stest). Scale bars: C, 100 μm; I, 20μm. Note that the fold overexpression for CA Akt was quantitated using immunostaining for Akt1 antibody and was found to be increased by fourfold to fivefold (n = 50 neurons from 3 independent experiments).
To verify that these transfected neurons indeed had increased PI3K–Akt signaling, we used immunocytochemical staining for an antibody specific for the active Akt that is phosphorylated at Ser473 (pAkt). As expected, the nontransfected cells or cells transfected with empty vector showed a low immunoreactivity, mostly confined to the nucleus, which is in agreement with a previous report (Brami-Cherrier et al., 2002). Cells expressing CA PI3K showed clearly increased pAkt staining throughout the dendritic trees as well as slightly increased staining in the nucleus, whereas cells expressing CA Akt showed very strong staining throughout the whole cell with a prominent membrane distribution (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Thus, expression of CA PI3K and CA Akt leads to PI3K–Akt activation as expected (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Activation of PI3K–Akt signaling is sufficient to alter spine morphology
In addition to the significant changes in overall dendritic morphology, neurons expressing CA PI3K and CA Akt also displayed prominent alterations in dendritic spine morphology. Overall, these neurons showed a significant increase in the number of filopodia-like protrusions with a concomitant significant reduction in the density of mushroom-shaped spines. Because the total number of spines/filopodia for each transfected neuron was difficult to assess, we limited our quantification of spine/filopodia density in our current study to the middle segments ∼50 μm from the soma (Fig. 1L). These quantitative data further supported our overall conclusion that overexpression of CA PI3K or CA Akt significantly reduced the density of spine and increased the filopodia-like protrusions. We could not distinguish whether these filopodia-like protrusions are genuine filopodia as seen during early developmental stages or some of those are “transformed” long dendritic spines that have made functional connections.
We further quantitated these changes by measuring the length and width of all individual protrusions, including both spines and filopodia-like protrusions (Fig. 1M). These results reiterated the fact that chronic activation of PI3K–Akt signaling promoted an elongation and growth in dendritic protrusion length and thus caused an increase in protrusions that resembled filopodia. Noticeably, some of the spines in the CA PI3K, especially in CA Akt-expressing neurons, showed very unusual shapes with a very long neck or a prominent head in the middle of a filopodium-like protrusion (Fig. 1J,K). This was never observed in control neurons (Fig. 1I).
Critical role of PI3K–Akt signaling in the regulation of dendrite size, complexity, and soma size: a loss-of-function study
Having established that constitutively active PI3K and Akt overexpression increases dendrite size and dendritic complexity, we next sought to determine whether these effects are indeed attributable to alteration of PI3K–Akt signaling and, more importantly, whether endogenous PI3K–Akt activity plays a role in dendrite formation. To this end, we used selective pharmacological inhibitors of PI3K and DN PI3K and DN Akt mutants (Franke et al., 1995; Rodriguez-Viciana et al., 1997).

Inhibition of PI3K–Akt signaling reduces dendritic complexity, soma size, and dendritic protrusions. A, Overall view of 15 DIV neurons transfected with GFP, CA PI3K, and CA Akt cells on 6 DIV and treated with 50μm LY294002 (LY) starting on 6 DIV. Also shown are DNPI3K- and DNAkt-expressing cells. The bar graphs show mean ± SEM for TDBL (B), number of terminal tips (C), and soma area (D). The data were quantified from three independent experiments (n = 49GFP, 58CAPI3K, 94CAAkt, 35GFPplus LY294002, 40 CA PI3K plus LY294002, 58 CA Akt plus LY294002, 25 DN PI3K, and 35 DN Akt cells). E, The micrograph panel represents the dendritic protrusions in 15 DIV neurons transfected with GFP, CA PI3K, and CA Akt on 6 DIV and treated with LY294002 (50μm) starting on 7 DIV and cells expressing DN PI3K and DN Akt. The bar graph (F) shows mean ± SEM for spine and filopodia densities. Quantifications were performed from at least three independent experiments (n = 600 GFP, 455 CA PI3K, 540 CA Akt, 350 GFP plus LY294002, 350 CA PI3K plus LY294002, 300 CA Akt plus LY294002, 400 DN PI3K, and 390 DN Akt dendritic protrusions). Statistical difference within group: *p < 0.05, **p < 0.01, ***p < 0.001. Difference with control group: ##p < 0.001, one-way ANOVA (Dunnett's test). Scale bars: A, 50μm; E, 10μm. Note that the control set used for this set of experiments was treated with DMSO (vehicle) but was not found to be statistically different compared with control data shown in Figure 1. Also, the dataset for CA PI3K and CA Akt from Figure 1 was used here with proper statistical post hoc test.
Because the PI3K–Akt pathway has been shown to play an essential role in neuronal survival, one obvious concern is that any loss of function of this pathway may also affect the survival of the neurons. Indeed, inhibition of PI3K during an early stage of dendritic formation (3–6 DIV) caused profound cell death, precluding assessment of the potential role of this pathway in this fast phase of dendritic initiation and growth (data not shown). Remarkably, from 6–7 DIV on, we did not detect any significant effects on cell survival attributable to inhibition of the PI3K activity. These were supported by the comparable cell density, the healthy cell morphology (smooth and bright DIC and phase imaging) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), and no significant changes in terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling and propidium iodide staining (assays for DNA and plasma membrane damage, respectively) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). These data indicate that, at this late stage, the functional role of this pathway has switched from an antiapoptotic to a growth-promoting role to stimulate outgrowth of dendrites. To further verify this hypothesis, we then examined the effects of the chronic inhibition of PI3K signaling on dendritic morphology in neurons expressing CA PI3K and CA Akt. The neurons were transfected at 6–7 DIV, and the PI3K-specific inhibitor LY294002 was applied into culture medium starting from 12–24 h after transfection and throughout the next 6–7 d. The efficacy and specificity of the chronic drug applications was verified using Western blots and immunostaining for pAkt and dually phosphorylated MAPK (pMAPK) (Wu et al., 2001). As expected, LY294002 specifically blocked the pAkt and one of its downstream targets, pS6 (supplemental Fig. 4E,F, available at www.jneurosci.org as supplemental material).
Figure 2A–D shows that chronic inhibition of PI3K significantly reduced the total dendritic branch length, terminal tip number, and soma size in control cells, underscoring an important role of endogenous PI3K activity in controlling the overall development of dendritic morphology. Furthermore, LY294002 completely eliminated the effects of the CA PI3K on dendrite morphology. To our surprise, LY294002 also blocked the effects of CA Akt on terminal tips and soma area. Because Akt lies downstream to the PI3K, it was first puzzling as to why a specific inhibitor to an upstream molecule would affect constitutive downstream activity. However, as will be demonstrated in the following sections, the concentration of LY294002 used in our experiments has been shown to be effective in inhibiting mTOR (Brunn et al., 1996), a key downstream target for mediating these effects (see below). Another likely mechanism is that there exists a positive feedback loop, and the effects of CA Akt may also involve the regulation of endogenous Akt through PI3K. An analogous case has been found for the effects of Ras V12 in cell transformation (Peyssonnaux et al., 2000).
As an independent approach, we transfected neurons with DN PI3K or DN Akt at 6–7 DIV and examined dendrite morphology at 15 DIV. Although we did not observe a significant reduction in the number of transfected neurons compared with cells transfected with empty vector, we could not conclusively exclude a possible selective loss of neurons with high expression of the DN constructs, because transfection efficiency was very low (∼1%). Nevertheless, both the DN PI3K and DN Akt significantly reduced the TDBL, whereas terminal tips were reduced only by DN PI3K and not by DN Akt. The soma size demonstrated a moderate but not a significant reduction (Fig. 2). The somewhat less significant effects of the DN constructs possibly were attributable to the less expression level compared with suppression by the pharmacological inhibitors. We, therefore, concluded that the combination of loss-of-function studies by the pharmacological and molecular approaches strongly suggest a critical role of PI3K–Akt signaling in dendrite formation.
Chronic inhibition of PI3K–Akt signaling reduces both dendritic spines and dendritic filopodia
We next examined dendritic spine and filopodia morphology by the chronic inhibition of PI3K–Akt signaling. As for the overall dendritic development, chronic inhibition of PI3K by LY294002 caused a moderate reduction of both dendritic filopodia and spines, indicating that PI3K activity is involved in maintaining spines and filopodia in basal state in control cells (Fig. 2 E). The effect of inhibition of PI3K signaling in cells expressing CA PI3K and CA Akt was next evaluated. Cells expressing CA PI3K showed a moderate reduction in spines (Fig. 2F), whereas chronic inhibition of effect of CA PI3K brought the filopodia number to a level close to control level but still significantly higher than that in control cells treated with LY294002. Unlike in the overall dendritic morphological development, no significant change in either spine density or filopodia-like protrusions was observed when CA Akt-expressing neurons were chronically treated with LY294002 (Fig. 2G).
Consistent with gross dendritic morphological changes, cells transfected with DN PI3K or DN Akt also did not show dramatic changes in fine dendritic morphology compared with control, except for a moderate but significant reduction of filopodia-like protrusions in cells expressing DN PI3K and spine with DN Akt (Fig. 2E,G).
Together, the data reported above indicate that activation of the PI3K–Akt signaling pathway is sufficient to increase the dendrite and soma size and dendritic complexity, as well as to produce filopodia-like protrusions. Conversely, chronic inhibition of this pathway results in smaller soma and dendritic trees and a reduction of dendritic complexity, as well as a loss of dendritic filopodia and spines.
Distinct and cooperative roles of the Ras–PI3K–Akt and Ras-MAPK signaling pathways in the regulation of dendritic complexity, soma growth, and spine morphology
What are the upstream and downstream signaling mechanisms mediating these changes? Ras-dependent activation of PI3K has been shown to be a major pathway by which neurotrophins convey cell survival-promoting signals (for review, see Brunet et al., 2001). Furthermore, transgenic mice overexpressing a constitutively active mutant RasV12 driven by a synapsin I promoter show similar increases in dendritic size and complexity, as well as soma size (Gartner et al., 2004), suggesting that Ras may be well positioned as an upstream signal leading to the PI3K–Akt-dependent morphological changes during dendrite formation in vivo. In addition, Ras is well known to activate the Raf–MEK–MAPK pathway to regulate long-term synaptic plasticity and memory formation (Weeber and Sweatt, 2002; Thomas and Huganir, 2004). The critical question as to whether the Ras–PI3K–Akt pathway plays a role in dendrite formation and whether and how this signaling pathway plays an independent or a cooperative role with the Ras–MAPK pathway during dendrite formation have not yet been addressed.
To address these questions, we combined several specific pharmacological inhibitors and the expression of Ras effector domain mutations (Rodriguez-Viciana et al., 1997) to dissect the signaling mechanisms for dendrite formation. As shown in Figure 3, overexpression of RasL61 or RasV12 (data not shown), which activates multiple parallel downstream pathways including the PI3K–Akt and MAPK pathways, indeed caused a significant increase in total dendritic branch length, terminal tip number, and soma size compared with control cells (Fig. 3E–G). Interestingly, inhibition of either PI3K with LY294002 or MEK, the immediate upstream kinase of MAPK, with U0126 essentially blocked the effects of Ras on dendrite size and complexity. Remarkably, the soma increase by RasL61 was only blocked by LY294002 but not by U0126. Therefore, Ras signals to both PI3K/Akt and MAPK pathways to cooperatively regulate the development of dendrites, whereas the soma size is essentially regulated by the Ras–PI3K–Akt pathway.
Similar to cells expressing CA Akt or CA PI3K, cells overexpressing RasL61 also displayed profound changes in the development of fine dendritic structures. Generally, an increase in filopodia-like protrusions and dramatic reduction of dendritic spines were seen in RasL61-expressing cells (Fig. 3I,L,M). Chronic inhibition of PI3K by LY294002 or MEK by U0126 both significantly reduced the filopodia-like protrusions in RasL61-expressing neurons, although not to the control level (Fig. 3M). In contrast, no noticeable difference with LY294002 treatment on spine density was observed (Fig. 3L), whereas U0126 caused a moderate but significant recovery in spine density compared with RasL61-expressing neurons. Interestingly, U0126 did not change any of the parameters for dendrites in control neurons. We also found that a combination of both LY294002 and U0126 caused significant cell death, precluding an additional assessment on dendrite development (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
A central role of mTOR in the regulation of soma and dendrite size and dendritic complexity, as well as spine formation
The above results suggest that Ras signals through the PI3K–Akt and MAPK pathways, both of which can independently and coordinately regulate dendrite morphology, whereas soma size is controlled by PI3K–Akt signaling alone. To verify these results by an independent approach, two well characterized Ras effector domain mutants on dendritic development were further tested. RasL61S35 interacts preferentially with Raf, thereby selectively activating the MAPK pathway, whereas RasL61C40 selectively activates PI3K pathway attributable to specific amino acid changes in the effector loop of the Ras protein (Rodriguez-Viciana et al., 1997).

Increased dendritic complexity, soma size, and alteration of spine morphology in RasL61-overexpressing neurons. Overall views of 15 DIV neurons expressing GFP (A), RasL61 (B), and RasL61 cells treated with 50μm LY294002 (LY) (C) or 10μm U0126 (U0) (D). The neurons were transfected on 6 DIV, and the inhibitors were added on 7 DIV. U0126 were replaced every 2 d, whereas LY294002 was found to remain stable for the whole period. E–G represent mean ± SEM for TDBL, number of terminal tips, and soma area, respectively. H–K depict dendritic protrusions in the respective groups. L and M show quantification for spine and filopodia densities. Quantifications are from at least three independent experiments (n = 50 GFP, 35 U0126, 40 RasL61, 60 RasL61 plus LY294002, 46 RasL61 plus U0126 neurons; and n = 600 GFP, 350 U0126, 350 RasL61, 300 RasL61 plus LY294002, and 350 RasL61 plus U0126 dendritic protrusions). Statistical difference with RasL61 group: ***p < 0.0001. Difference with control group: ###p < 0.0001, one-way ANOVA (Dunnett's test). Scale bars: D, 50 μm; K, 10 μm. Note that the fold overexpression for RasL61 was quantitated using pan-Ras antibody (n = 35 neurons from 2 independent experiments) and was found to be twofold increased. Similar expression level was also found for other RasL61 mutants. The same data for GFP controls with LY294002 as in Figure 2 are plotted here for easy comparison.
We first confirmed that RasL61S35 selectively activated the MAPK pathway, whereas RasL61C40 only activated the PI3K–Akt pathway, and RasL61 activated both pathways in the HeLa cell line by Western blots (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Immunostaining for pAkt or pMAPK was then further used to confirm their specificity in producing hyperactive pAkt and/or pMAPK in the transfected hippocampal neurons (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material).
Figure 4 shows that cells expressing RasL61S35 displayed distinct morphological changes. Most noticeably, RasL61S35 caused profound proliferation of the distal dendrites (Fig. 4C,P). As a result, the terminal tips and TDBL were significantly increased without changes in primary dendrites or soma size (Fig. 4M–O), and the dendritic caliber also did not change compared with control cells (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). In contrast, RasL61C40-expressing cells displayed morphological changes resembling those induced by CA PI3K and CA Akt (Fig. 4D,E), such as an increased soma size and dendritic caliber, as well as moderate increases in primary dendrite number, terminal tips, and TDBL (Fig. 4M–O).
Numerous studies have consistently demonstrated that PI3K–Akt signals through or cooperates with mTOR to control cell size in various cell types, including neurons (Fingar and Blenis, 2004; Hay and Sonenberg, 2004). The prominent effects of the Ras mutants CA PI3K and CA Akt on soma size led us to focus on mTOR as a central converging point for coordinately regulating a variety of aspects of dendrite formation. We therefore used rapamycin, a highly specific inhibitor of mTOR, to further dissect the downstream signaling mechanisms. Strikingly, chronic inactivation of mTOR by rapamycin uniformly reduced the dendrite and soma size as well as dendritic complexity, as evidenced by a reduction of the terminal tips and TDBL in all cases and even in the control condition (Fig. 4), suggesting a central role of mTOR signaling in both basal and the Ras–PI3K–Akt and Ras–MAPK-dependent dendrite formation. Rapamycin only partially blocked the effects of RasL61S35 and RasL61C40 on overall dendrite morphology, indicating that an rapamycin-insensitive pathway(s) is also involved. In contrast, rapamycin completely blocked the dendritic growth by CA PI3K and CA Akt, suggesting that dendrite growth through the Ras–PI3K–Akt signaling is absolutely dependent on mTOR function. The specificity and efficacy of rapamycin was confirmed by its selective inhibition on pS6 with only slight or no effect on pAkt and pMAPK (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) (for short-term treatment, see Fig. 6).
To verify the efficacy of these mutants, we used immunocytochemical staining for an antibody specific for the phosphorylated mTOR (Ser 2448). A significant increase in p-mTOR immunoreactivity was found for RasL61C40-expressing neurons compared with the empty vector-transfected control cells (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). The effect of RasL61S35 and RasL61C40 mutants on dendritic spine/filopodia morphology was next assessed. Similar to RasL61, both mutants produced prominent filopodia-like protrusions (Fig. 5B–D), suggesting that activation of either pathway is sufficient to alter spine morphology. In contrast to RasL61, RasL61S35 caused only a small but yet a significantly decreased spine density, whereas no change in spine density was seen in RasL61C40-expressing cells. Consistent with the above finding that PI3K signaling is involved in the development of both spines and filopodia in control cells (Fig. 4), chronic blockade of mTOR by rapamycin also significantly reduced both spine and filopodia density in cells transfected with empty vector. Interestingly, chronic blockade of mTOR activity significantly reduced the filopodia-like protrusions in RasL61S35-, RasL61C40-, CA PI3K-, and CA Akt-expressing cells but not in RasL61-expressing neuron (Fig. 5N). These results further support the notion that mTOR functions as a converging point downstream of Ras–PI3K–Akt and Ras–MAPK signaling to regulate spine morphology. As seen in LY294002-treated cells, however, the spine density did not show dramatic changes, with the exception of a significant reduction in RasL61-expressing cells and a small but significant increase in RasL61S35-expressing cells (Fig. 5M). Because RasL61 activates both pathways, one simple explanation for the lack of affect of rapamycin on filopodia-like protrusions on RasL61-expressing cells might be that a rapamycin-insensitive mTOR signaling (Sarbassov et al., 2005) and/or mTOR-independent MAPK signaling is sufficient to compensate for the loss of rapamycin-sensitive mTOR function to regulate spine morphology. The inconsistent changes or lack of change in spine density attributable to chronic inhibition of these pathways could be explained by an intriguing dual role of these pathways in spine formation (see below), as well as by a possible developmental compensation.

Regulation of dendritic size and shape by Ras–PI3K–Akt–mTOR signaling. A–F depict overall view of 15 DIV neurons transfected with GFP (A), RasL61 (B), RasL61S35 (C), RasL61C40 (D), CA PI3K (E), and CA Akt (F) cells on 6 DIV. G–L depict matching groups of neurons treated with rapamycin (Rap) (1μm) starting on 6 DIV. M–O show mean ± SEM for TDBL, terminal tip number, and soma size, respectively, from more than two independent experiments (n = 24 GFP, 20 RasL61, 19 RasL61S35, 33 RasL61C40, 30 CA PI3K, 35 CA Akt, 26 GFP plus rapamycin, 22 RasL61 plus rapamycin, 26 RasL61S35 plus rapamycin, 21 RasL61C40 plus rapamycin, 20 CA PI3K plus rapamycin, and 25 CA Akt plus rapamycin cells). Statistical difference within group: *p < 0.05, **p < 0.01, ***p < 0.001. Difference with control group: ###p < 0.001, one-way ANOVA (Dunnett's test). Scale bar, 50 μm. P, Sholl analysis of 15 DIV CA1/CA3 neurons to analyze detailed morphometric assessment shows that neurons overexpressing RasL61S35 have more profound effect on the distal dendrites without effects on primary dendrites, whereas RasL61C40-overexpressing neurons show a larger dendrite size and increases in both primary and distal dendrite branches. Application of rapamycin reduced the dendrite size in both the RasL61 mutants, but the increased distal dendritic branching persists in RasL61S35-expressing cells, whereas rapamycin completely blocked the primary and distal dendrite increases in RasL61C40-expressing cell. Concentric circles at 20μm spacing difference were drawn around the cell body, and the number of intersections of all dendritic branches with the circles were counted and plotted. n is same as above for all groups. Statistical difference shown is only with respect to control group: *p < 0.01, **p < 0.0001, one-way ANOVA (least significant difference).
A dual role of the PI3K–Akt–mTOR signaling in dendritic spine formation
The experiments described in the previous sections demonstrated that multiple parallel pathways operate cooperatively and independently to promote filopodia formation, and chronically elevated levels of these signaling pathways retard dendritic spine development. These novel findings raise the possibility that, although several parallel signaling pathways are involved in the dendritic filopodia formation, additional signal(s) and tightly regulated signaling pathways are required for developing fully functional dendritic spines. To further test this hypothesis, we took advantage of the well known effects of BDNF on neuronal morphogenesis.
It is well documented that BDNF simultaneously activates multiple signaling cascades, including the Ras–MAPK and Ras–PI3K–Akt pathways (McAllister et al., 1999) (Fig. 6). Numerous studies have demonstrated that chronic and short-term manipulation of BDNF level and the activity of its receptor (TrkB) produce profound morphological changes both in vivo and in vitro (McAllister et al., 1999), although the underlying signaling mechanisms are not yet well elucidated.

Regulation of spine morphology by Ras–PI3K–Akt–mTOR signaling. A–F illustrate close-up views of dendritic protrusions from 15 DIV hippocampal neurons transfected with GFP (A), RasL61 (B), RasL61S35 (C), RasL61C40 (D), CA PI3K (E), and CA Akt (F) on 6 DIV. G–L show neurons from sister coverslips treated with rapamycin (1μm) (Rap) starting on 6 DIV. The bar graphs indicate mean ± SEM for spine (M) and filopodia (N) densities. The densities were quantified from two independent set of experiments (n = 600 GFP, 350 RasL61, 375 RasL61S35, 200 RasL61C40, 450 CA PI3K, 500 CA Akt, 270 GFP plus rapamycin, 240 RasL61 plus rapamycin, 430 RasL61S35 plus rapamycin, 340 RasL61C40 plus rapamycin, 240 CA PI3K plus rapamycin, and 300 CA Akt plus rapamycin dendritic protrusions). Statistical difference within group: *p < 0.05, **p < 0.01, ***p < 0.001. Difference with control group: ###p < 0.001, one-way ANOVA (Dunnett's test). Scale bar, 10 μm.
As shown in Figure 6, a 24 h application of BDNF (20–50 ng/ml) in culture medium on 14 DIV hippocampal neurons caused profound increases in the number of filopodia concomitant with a reduction of spines (Fig. 6I,J). A similar destabilization of the dendritic spines in terms of induction of filopodia and retraction of spine also has been reported by Horch et al. (1999). These effects of BDNF on spine morphology were blocked by 200 nm K252a, a general cell-permeable protein kinase inhibitor known to block tyrosine protein kinase activity of the neurotrophin receptor (Fig. 6C). Remarkably, inhibition of PI3K by LY294002 or more prominently by wortmannin resulted in a significant rise in spines and reduction in filopodia (Fig. 6D,E). Coapplication of 50 μm LY303511, a structural analog of LY294002, did not block the effect of BDNF on spine morphology (data not shown), further supporting that these effects were specifically attributable to an inhibition of PI3K signaling. Inhibition of MEK by U0126, however, did not result in a significant increase in spine. Interestingly, the increases in filopodia by BDNF were primarily blocked by U0126 (Fig. 6F). Concurrent application of LY294002 and U0126 to block both of the PI3K and MAPK signaling completely blocked the effects of BDNF on spine morphology (Fig. 6G). In fact, both the number of spines and filopodia were reduced to a level significantly lower than those in sham controls that were only exposed to the vehicle, DMSO. Importantly, 24 h treatment of the neurons at this stage with BDNF and the drugs did not cause any detectable change in cell survival, even with concurrent application of LY294002 and U0126 (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). To examine further whether these effects are mediated by mTOR, we inhibited the mTOR signal by coapplication of BDNF with rapamycin for 24 h (Fig. 6H). A significant rise in spines and a reduction in filopodia densities were observed compared with BDNF-treated spines. In addition, the treatment of neurons with the drugs without BDNF did not cause any significant difference in the spine or filopodia density compared with the control cells (data not shown). The efficacy and specificity of the drugs for inhibiting the signaling cascades were verified by Western blots (Fig. 6K,L) for pAkt, pMAPK, and pS6. LY294002 and rapamycin primarily blocked the S6 phosphorylation, whereas U0126 alone had no effect on pS6.
Together, these results demonstrate that BDNF activates coordinated PI3K and MAPK signaling through mTOR to regulate dendritic spine morphology. However, the involvement of other signaling pathway(s) cannot be ruled out. Furthermore, these data underscore a dual role of key signaling pathways in the formation and promotion of dendritic spines and the importance of spatiotemporal downregulation of the PI3K–Akt–mTOR signaling pathways in dendritic spine development.
To further support our hypothesis for a dual role of PI3K–pAkt–mTOR signaling on spine formation, in a second series of experiments, we examined the effects of short-term blockade (24 h) of PI3K and mTOR in cells expressing the constitutively active mutants of Ras, PI3K, and Akt. As shown in Figure 7, inhibition of PI3K (E–H) or mTOR (I–L) consistently caused a significant increase in mushroom-shaped spines with a concomitant decrease in filopodia-like protrusions (M, N).
mTOR signaling regulates dendritic complexity, soma size, and spine morphology in hippocampal slice culture
To confirm the effects seen on dendrite and spine morphogenesis in a more intact system, we used organotypic hippocampal slice cultures. The neurons were cotransfected biolistically with CA Akt along with EGFP at 2 DIV and were analyzed at 10 DIV for morphology. A substantial increase in TDBL, soma area (Fig. 8D–F), terminal tips, and dendritic caliber (data not shown) was observed for the CA Akt-transfected CA1 pyramidal neurons. Furthermore, an increase in filopodia-like protrusions and a decrease in spines were noted (Fig. 8G). To verify the key role of mTOR in mediation of these effects, the slices were treated with rapamycin. Remarkably, rapamycin treatment even for 24 h caused a significant decrease in the soma size, TDBL, and terminal tip number, suggesting that a rapamycin-sensitive signaling is involved in the maintenance of the neuronal structures. In addition, the 24 h application of rapamycin in CA Akt-expressing cells was sufficient to partially restore the normal spine morphology with a concomitant reduction in filopodia-like protrusions (Fig. 8G,H).
Discussion
Here we demonstrate a plausible role of one of the common signaling pathways, PI3K–Akt–mTOR, downstream of Ras and BDNF, in dendritic arbor and dendritic spine morphogenesis. Using calcium phosphate transfection to introduce constitutively active mutants of Ras, PI3K, and Akt in hippocampal CA1/CA3 culture, along with pharmacological manipulations of the different signaling cascade, we have demonstrated that this key cell survival signaling pathway also plays a central role in the regulation of dendrite size and shape as well as dendritic spine morphology in neurons. During normal development, soma size usually positively correlates with dendrite growth. Furthermore, it has been long recognized that dendritic atrophy and hypertrophy as well as change of soma size can be mediated by innervation, deafferentation, and release or depletion of trophic factors; the signaling cascades underlying these changes, however, are not well understood. Neurotrophic factors, particularly BDNF, have been shown to be involved in regulating dendritic complexity and soma size (McAllister et al., 1999). As in a variety of other cell types, the soma size in neurons is also controlled by mTOR signaling (Fingar and Blenis, 2004). Our study confirmed these observations and further demonstrated, for the first time to our knowledge, that mTOR signaling also controls dendrite size and shape and is involved in the regulation of spine morphology. Intriguingly, the soma size is controlled by the mTOR pathway alone; however, dendritic growth and spine formation require a coordinated action of both of the MAPK and mTOR signaling and possibly other unidentified pathway(s).

BDNF promotes growth of filopodia and destabilizes dendritic spines, and short-term inhibition of PI3K–Akt–mTOR promotes spine development. The micrographs depicts close-up views of 15 DIV dendritic protrusions from hippocampal neurons transfected with GFP on 6 DIV and treated with BDNF and BDNF plus drugs for 24 h starting on 14 DIV. GFP control (A), BDNF (20 ng/ml) (B), and coapplication of BDNF with K252a (200 nm)(C), LY294002 (50μm)(D; LY), wortmannin (1μm)(E; Wort), U0126 (10μm)(F; U0), LY294002 (50μm) plus U0126 (10μm)(G), and rapamycin (1μm)(H; Rap). I and J show mean ± SEM for spine and filopodia densities, respectively. K–L, Western blot analysis to ascertain the efficacy and specificity of treatment with BDNF and pharmacological inhibitors for above morphological experiments. Sister coverslips were harvested and probed for pAkt, pMAPK, and pS6. A representative experiment is shown in K. L, Quantifications of the Western blots from three independent experiments. The quantifications for spine density were from at least three independent experiments (n = 600 GFP, 350 BDNF, 470 BDNF plus K252a, 458 BDNF plus LY294002, 487 BDNF plus wortmannin, 463 BDNF plus U0126, 460 266 BDNF plus LY294002 plus U0126, and 300 BDNF plus rapamycin dendritic protrusions). ***p < 0.0001, statistical difference with BDNF. ##p < 0.001, ###p < 0.0001, difference with respect to GFP control group, one-way ANOVA (Dunnett's test). Scale bar, 10 μm.
It is well known that Ras can activate PI3K by directly interacting with the catalytic subunit of type I PI3Ks, leading to activation of the lipid kinase as a result of its translocation and conformational changes. PI3K catalyzes the production of PI(3,4)P2 (phosphatidylinositol 3,4-biphosphate) and PI(3,4,5)P3 (phosphatidylinositol 3,4,5-triphosphate), second messengers that bind to a large number of proteins through the PH domains. The serine–threonine kinase Akt (protein kinase B) and phosphoinositide-dependent protein kinase-1 are two such effectors that have been shown to be the immediate downstream effectors of PI3K in the survival cascades. It has been shown that PI3K activity is necessary and sufficient for growth factor-dependent activation of Akt. Besides Ras, PI3K can be activated by a variety of growth signals, such as trophic factors, growth factors, cytokines, and neurotransmitters, as well as patterned neuronal activity (Rodgers and Theibert, 2002). Previous works have shown a role of PI3K and Akt in axon growth (Atwal et al., 2000; Kuruvilla et al., 2000; Markus et al., 2002; Shi et al., 2003; Jiang et al., 2005), neurite initiation in the PC12 cell line (Rodgers and Theibert, 2002; Tyson et al., 2003), and cell and organ development (Yang et al., 2004). In this study, we have extended the effect of PI3K signaling in several new directions. First, we have shown that overexpression of constitutively active PI3K and Akt is sufficient to alter spine morphology by producing filopodia-like protrusions with a concomitant loss of mushroom-shaped spines. A second new insight gained from the current study is that these constitutively active mutants also facilitate an increase in dendritic complexity and soma size. An increase in both the proximal branching and terminal tips indicated the widespread effect of this signaling. Moreover, we found that, although the cultured hippocampal neurons are critically dependent on the PI3K–Akt signaling pathway for survival during the first several days after plating, at a later stage when neurons undergo active dendrite development, the role of PI3K–Akt has switched from protecting from cell death and promoting survival into a dominant role in dendritic patterning, to control the size and shape of the dendritic trees. We think that this phenotype recapitulates the primitive stage of development during which the neurons compete for survival and growth. Once a proper connection has been established, the process of dendritic pruning and axonal refinement will take place, thus eliminating dendrites and spines nonessential to the cell function. Furthermore, changes in morphology of the dendritic tree and dendritic spines are correlated with synaptic plasticity and may relate mechanistically to its expression and stabilization.

Short-term manipulation of PI3K–Akt–mTOR signaling promotes mushroom-shaped spines. A–D depict dendritic protrusions in 15 DIV cells transfected with GFP, RasL61, CAPI3K, and CA Akt on 6 DIV. The middle and right panels shows cells from sister coverslips treated with LY294002 (50 μm)(E–H; LY) and rapamycin (1 μm)(I–L; Rap) for 24 h starting on 14 DIV. The bar graphs show mean ± SEM for spine (M) and filopodia (N) densities. Quantification for the data were done from at least three experiments (n = 600 GFP, 350 RasL61, 450 CA PI3K, 500 CA Akt, 325 GFP plus LY294002, 250 GFP plus rapamycin, 275 RasL61 plus LY294002, 275 RasL61 plus rapamycin, 450 CA PI3K plus LY294002, 350 CA PI3K plus rapamycin, 400 Ca Akt plus LY294002, and 315 CA Akt plus rapamycin dendritic protrusions). ###p < 0.0001, statistical difference with respect to GFP control. *p < 0.05, ***p < 0.0001, difference within respective groups, one-way ANOVA (Dunnett's test). Scale bar, 10 μm.
It is of interest to compare the effects of the three major signaling pathways identified so far. Selective activation of the Ras–MAPK pathway as shown here as well as overexpression of constitutively active mutants of Rac/CDC42 (Luo, 2002; Whitford et al., 2002; Van Aelst and Cline, 2004) (our unpublished results) leads to increased dendritic complexity primarily by increasing more distal dendrite dynamics without affecting primary dendrites; therefore, the overall shape of the dendritic tree is essentially not affected. In contrast, activation of the Ras–PI3K–Akt–mTOR pathway consistently increases the number of primary dendrites, often leading to a profound alteration of its overall shape and size. Dijkhuizen and Ghosh (2005) have demonstrated recently that BDNF can induce rapid formation of primary dendrite formation via activation of the PI3K and MAPK pathways. Interestingly, a constitutively active form of PI3K, but not MEK, was found to be sufficient to induce primary dendrite formation in cortical neurons in their studies (Dijkhuizen and Ghosh, 2005).
How do the Ras–PI3K–Akt–mTOR and the Ras–Raf–MEK–MAPK pathways act in parallel and coordination to regulate dendrite formation? It is conceivable that they both involve the regulation of protein synthesis by translational and transcriptional control and alteration of actin and microtubule cytoskeleton. It is well known that Ras can cross-talk with other small GTPases, including Rho/Rac/CDC42 (Bar-Sagi and Hall, 2000). PI3K, Akt, or mTOR all can potentially signal to actin dynamics (Rodgers and Theibert, 2002). In some cell lines, PI3K as well as Akt have been shown to be upstream of Rac/CDC42 or to activate PAK, the major effector for mediating Rac/CDC42 effect on actin through a Rac/CDC42-independent mechanism (for review, see Rodgers and Theibert, 2002). The involvement of synergistic activation of both the PI3K and MAPK pathway for controlling dendrite (this study) and axon (Atwal et al., 2000) development is reminiscent of their well documented role in cell transformation and tumorigenesis, underscoring a highly conservative fundamental mechanism for controlling cell growth.
By analogy to growth control in other types of cells, it is highly likely that mTOR acts mainly through its regulation of translational control (Hay and Sonenberg, 2004; Klann et al., 2004) to mediate its effects on dendrite formation and soma size. The translational initiation by mTOR is primarily through two down-stream effectors: p70S6Ks and 4EBPs. p70S6Ks requires hierarchical phosphorylation by mTOR and MAPK for optimal activation (Schmelzle and Hall, 2000). Similarly, 4EBP1 is also phosphorylated by mTOR and MAPK at multiple sites (Herbert et al., 2002). These might be partially accountable for the involvement of both PI3K and MAPK in dendrite formation. Interestingly, a role of MAPK-dependent translational control in long-term synaptic plasticity and memory formation has been revealed recently (Kelleher et al., 2004). Conversely, our biochemical results suggested that the PI3K–Akt–mTOR pathway is the major regulator for S6 activation, the downstream target of p70S6Ks. Although a simple inhibition of the MAPK pathway did not affect the basal and BDNF-induced activation of S6 in the cell culture system at the particular stage we studied, a concurrent inhibition of both of the PI3K and MAPK (MEK) pathways further reduced the S6 activity when compared with that by the inhibition of the PI3K alone (Fig. 6 and data not shown), revealing a more intriguing signaling network in the regulation of the 70S6Ks/S6 activity. This is in line with the recent finding of a redundancy between the 70S6Ks and the MAPK pathways in mediating early S6 phosphorylation in response to mitogens (Pende et al., 2004). Additional investigation into the signaling mechanism for controlling 70S6Ks and 4EBPs activation as well as a potential role of 70S6Ks and 4EBPs in dendrite development and spine formation would be of great interest.

PI3K–Akt–mTOR signaling regulates dendritic complexity, soma size, and spine morphology in slice culture. The top micrograph panels depicts neurons from 10 DIV hippocampal slice culture, transfected with GFP (A), CA Akt (B), and CA Akt (C) on 2 DIV and treated with rapamycin (1μm) for 24 h starting at 9 DIV. D–F illustrates the mean ± SEM for TDBL, primaryorder dendrite, and soma area, respectively. G depicts dendritic protrusions and their quantification (H). Quantification of the data were done from two independent experiments (n > 25 cells for each group and >1200 μm dendritic segments for quantifying protrusion density). Statistical difference with respect to control: #p < 0.005, ###p < 0.0001. Difference with respect to CA Akt: *p < 0.05, **p < 0.005, ***p < 0.0001, one-way ANOVA (Tukey's test). Scale bars: C, 50 μm; G, 10 μm.
In summary, by globally manipulating the designated signaling pathways using well established pharmacological and molecular tools, we have identified a signal transduction pathway, mediated by PI3K–Akt–mTOR downstream of BDNF and Ras, for the regulation of soma size, dendritic branching pattern, and spine morphology. The growing evidence has implicated that the presence of dendritic and synaptic localization of PI3K–Akt–mTOR-regulated translational machinery in neurons (Asaki et al., 2003; Si et al., 2003; Takei et al., 2004) may well position this key signaling pathway in a complex signaling network to regulate localized structural and functional plasticity. Whether and how local Ras–PI3K–Akt–mTOR signaling is sufficient or coordinates with a cell-wide signaling network to achieve synapse-specific changes in neurons remains one of the biggest challenges for future investigations.
Footnotes
This work was supported by United States Public Health Service Grants DA17919 (G.-Y.W.) and GM068098 (J.K.), Welch Foundation Grant Q-1536 (J.K.), and United States Army Grant W81XWH-04-1-0260 (G.-Y.W.). We thank Drs. Alex Toker and Lewis Williams for providing plasmids and members of Eric Klann's and David Sweatt's laboratories for helpful suggestions for the paper as well as technical assistance for slice culture. We also thank Jacqui Alldritt for carefully reading this manuscript and Dr. Rana A. K. Singh for technical support for gene gun transfection of the slice cultures.
Correspondence should be addressed to Gang-Yi Wu, One Baylor Plaza, Room 420B, Baylor College of Medicine, Houston, TX 77030. E-mail: gangyiw{at}bcm.tmc.edu.
DOI:10.1523/JNEUROSCI.2284-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511288-12$15.00/0




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}