

Abstract
Alzheimer’s disease (AD) is found to have striking brain inflammation characterized by clusters of reactive microglia that surround senile plaques. A recent study has shown that microglia placed in contact with isolated plaque fragments release neurotoxins. To explore further this process of immunoactivation in AD, we fractionated plaque proteins and tested for the ability to stimulate microglia. Three plaque-derived fractions, each containing full-length native Aβ1-40 or Aβ1-42 peptides, elicited neurotoxin release from microglia. Screening of various synthetic peptides (Aβ1-16, Aβ1-28, Aβ12-28, Aβ25-35, Aβ17-43, Aβ1-40, and Aβ1-42) confirmed that microglia killed neurons only after exposure to nanomolar concentrations of human Aβ1-40 or human Aβ1-42, whereas the rodent Aβ1-40 (5Arg→Gly, 10Tyr→Phe, 13His→Arg) was not active. These findings suggested that specific portions of human Aβ were necessary for microglia–plaque interactions. When coupled to microspheres, N-terminal portions of human Aβ (Aβ1-16, Aβ1-28, Aβ12-28) provided anchoring sites for microglial adherence whereas C-terminal regions did not. Although itself not toxic, the 10–16 domain of human Aβ was necessary for both microglial binding and activation. Peptide blockade of microglia–plaque interactions that occur in AD might prevent the immune-driven injury to neurons.
Alzheimer’s disease (AD) is the most frequent cause of dementia in the elderly, accounting for >15 million cases worldwide. The neuropathological hallmarks of this disorder include neuritic and core senile plaques (Selkoe, 1991; Terry et al., 1994a,b), complex aggregations of proteins composed primarily of a distinctive peptide β-amyloid (Aβ). It is generally believed that Aβ is in some way responsible for the synaptic and neuronal loss associated with dementia (Selkoe, 1991; Yankner and Mesulam, 1991; Price et al., 1992;Younkin, 1995). Aβ is found within the gray matter of AD patients as a component of neuritic and core plaques and is associated with reactive microglia, astrogliosis, and neuronal loss. A second type of Aβ accumulation found in both AD and aged normal brain consists of diffuse plaques (discrete mesh-like structures of 70–100 μm diameter, visualized by silver staining, thioflavine S, or immunohistochemistry) that are not associated with such pathological changes as dystrophic neurites or decline in cognitive function (Yamaguchi et al., 1988; Masliah et al., 1990, 1993). Finally, diffuse, amorphous deposits of Aβ, demonstrable only by immunohistochemistry, have been described in aged brain and as an early manifestation of AD-like pathology in Down’s syndrome (Giaccone et al., 1989; Verga et al., 1989). Although the mechanisms that link neuritic and core plaques to dementia remain unresolved, two principal hypotheses have been advanced: (1) that Aβ acts as a potent and direct neurotoxic agent (Yankner et al., 1990) or (2) that neuritic/core plaques elicit a cascade of cellular events leading to neuronal pathology (Davies, 1994;Giulian et al., 1995a). Support for the first hypothesis comes fromin vitro observations in which synthetic Aβ peptides appear toxic to enriched cultures of neurons (Pike et al., 1991; Cotman et al., 1992) or to various non-neuronal cell lines (Behl et al., 1994;Pollack et al., 1995). Support for the second hypothesis comes from evidence that neuritic/core plaques are not directly neurotoxic, as shown by the fact that neurons can be grown successfully atop Aβ peptides (Koo et al., 1993; Wujek et al., 1996), that neuritic/core plaques added directly to neurons do not cause neuron damage (Giulian et al., 1995a), and that Aβ peptides infused into the brain do not cause tissue injury (Games et al., 1992; Podlisny et al., 1992;Stephenson and Clemens, 1992).
One pathway for Aβ-induced neuron damage may involve inflammatory cells, for it has long been recognized that reactive microglia (activated brain mononuclear phagocytes) are closely associated with AD neuritic plaques (Bolsi, 1927; McGeer et al., 1987; Rogers et al., 1988; Giulian, 1992, 1995a; Perlmutter et al., 1992). Microglia found in normal adult brain are highly ramified, quiescent cells that retract processes and become reactive during CNS injury (Rio-Hortega, 1932). In AD, quantitative histopathology has determined that >80% of core plaques are associated with clusters of reactive microglia, whereas <2% of diffuse Aβ deposits show such an association (Giulian et al., 1995a). These observations suggest that brain inflammatory responses may be directed specifically against the constituents of neuritic and core plaques. As the principal immune effector cells of the brain, activated microglia are capable of releasing such cytotoxic agents as proteolytic enzymes, cytokines, complement proteins, reactive oxygen intermediates, NMDA-like toxins, and nitric oxide (Thery et al., 1991; Giulian, 1992; Rogers et al., 1992; Lees, 1993). To explore the immunopathology of AD, we have recently tested microglial interactions with neuritic/core plaques in vitro and found that plaque fragments stimulate reactive microglia and secretion of neurotoxins (Giulian et al., 1995a), thus linking reactive microgliosis with neuronal pathology. Here, we demonstrate that Aβ1-42 is the plaque-derived component that elicits neurotoxic responses in microglia. Importantly, the N terminus of human Aβ provides an anchoring site necessary for initiating this neurotoxic cascade. Our data point to strategies that may block activation of neurotoxic microglia and, thus, serve as therapies for AD dementia.
MATERIALS AND METHODS
Isolation of microglia. Rat microglia were isolated from newborn animals by the method of Giulian and Baker (1986) with recovery of ∼0.5 × 106 ameboid microglia per brain with >99% purity. Criteria used to identify mononuclear phagocytes included the presence of scavenger receptors as shown by binding a fluorescent probe (acetylated low-density lipoprotein labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine [DiI-ac-LDL]), the presence of CR3 complement receptor (labeling with OX-42 antibody), and characteristic spine-bearing cell-surface morphology seen by scanning EM (Giulian et al., 1995b). Human microglia were isolated from 50 to 100 gm of normal adult cortical gray matter within a 6 hr postmortem interval as described previously (Giulian et al., 1995b). We obtained cells of a high degree of purity (>98%, ∼0.5 × 106 cells/gm wet weight of tissue) that were active phagocytes and that showed the presence of CD4, spine-bearing surface morphology, scavenger receptors, and the class II marker HLA-DR (Giulian et al., 1995b).
Isolation, purification, and characterization of plaque proteins.AD brains were obtained from patients with both clinical history and pathological features to meet diagnostic criteria as defined by the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD;Mirra and Heyman, 1993). Amyloid proteins were isolated from AD cerebral cortex laden with neuritic and core plaques by using discontinuous sucrose gradients of (in m): 1.2, 1.3, 1.4, 1.7, and 2.0 (Roher et al., 1993a,b). Amyloid cores were recovered from the 1.4/1.7 m sucrose interface as discrete, dense particles (15–25 μm diameters) and found to be thioflavine S(+) and 6E10 anti-amyloid antibody(+) (from Institute of Basic Research, Staten Island, NY). These purified cores were solubilized in 80% formic acid, fractionated by Superose 12 FPLC, and dialyzed (1000 Da cutoff) against 20 mm ammonium bicarbonate containing 0.7% zwitterion betaine (Roher et al., 1993a). The protein content of each fraction before dialysis was determined by amino acid analysis (Roher et al., 1993a), and morphology was examined by transmission electron microscopy (TEM; Roher et al., 1988). The resulting five fractions obtained from pooled brain samples were highly reproducible both in content and quantity, as described in detail elsewhere (Roher et al., 1988,1993a,b). Estimates of Aβ1-40 and Aβ1-42 content in plaque fractions were based on an ELISA method similar to that described byKuo et al. (1996) using synthetic peptides as standards. In this report, the capture antibody was the murine monoclonal 468 (Senetek, Maryland Heights, MD), and the reporter rabbit polyclonal antibodies to recognize specific peptides were biotinylated S40 and S42 (a gift from M. Shoki, Gunma University, Japan). The amyloid components of the plaque fractions also were characterized by Tris-tricine PAGE and by Western blots. The presence of α-1-antichymotrypsin (ACT) and apolipoprotein E (apoE) in fractions was confirmed by immunostaining with TEM, using a colloidal gold technique (Roher et al., 1993b).
Diffuse plaque proteins were isolated from postmortem brains rich in diffuse plaques (>85% diffuse deposits as viewed by thioflavine S-stained sections) from patients with no clinical history of dementia. Diffuse plaque aggregates were recovered with the same methods for obtaining neuritic/core plaque fragments but appeared as fine, thioflavine S(+), anti-Aβ antibody(+) strands (1–5 μm diameters) from the 1.4 m/1.7 m sucrose gradient interface. Then this material was solubilized in 80% formic acid at room temperature for 30 min, centrifuged at 250,000 × g for 30 min, and sequentially dialyzed (1000 Da cutoff) against 40 mm ammonium bicarbonate, 6% betaine, pH 7.8; against 40 mm ammonium bicarbonate, 2% betaine, pH 7.8; and against 20 mm ammonium bicarbonate, 0.7% betaine, pH 7.8. Protein content before dialysis was estimated by measuring total amine content, using the fluorescamine assay on acid hydrolysates (6N HCl; 24 hr; 105°C). Solubilized proteins from diffuse or neuritic/core plaques were added to cultures at a concentration of 400 μm total amine. All synthetic peptides were purchased from California Peptide (Napa, CA) or Bachem (King of Prussia, PA).
Aβ coupling to beads and cell adherence assays. Synthetic Aβ peptides were linked to Sepharose beads (Pharmacia, Piscataway, NJ) in coupling buffer (0.1 m NaHCO3 and 0.5 m NaCl, pH 8.3) containing 10% DMSO. This solution was combined with an appropriate volume of CNBr-activated Sepharose 4B (10 mg protein/ml bead; Pharmacia protocol) and mixed overnight on a platform mixer at room temperature. Remaining active groups on the beads were blocked by 1.0 m glycine; excess uncoupled peptides were removed from this bead–peptide complex by washing with three cycles of alternating pH (0.1 m acetate buffer, pH 4.0, followed by 0.1 m Tris-HCl buffer, pH 8.0, each buffer containing 0.5 m NaCl). Coupled beads (104/ml) were placed in 35 mm culture dishes covered with 250,000 adhering microglia at 37°C in 1.5 ml N2 medium (Giulian et al., 1995a). The numbers of microglia that detached from the plate and bound to coupled beads were determined at the end of a 6 hr incubation period by inverted phase microscopy with 100 beads scored from each of three sister cultures. Glycine and bovine serum albumin (BSA)-coupled beads were used as controls.
Fluoresbrite carboxylate microspheres (YG 1.0 μm, Polysciences, Warrington, PA; 0.5 ml of a 2.5% suspension) were washed twice in 0.1 m carbonate buffer, pH 9.6, and three times in 0.02 m sodium phosphate, pH 4.5. Carbodiimide was then added dropwise to a final concentration of 1%, and the suspension was mixed for 4 hr at room temperature. After three more washes in 0.02 m sodium phosphate, the pellet was resuspended in 1.2 ml of 0.2 m borate buffer, pH 8.5, and 300 μg of Aβ (or control peptide) in 6% DMSO was added. After overnight mixing at room temperature, the microspheres were blocked by 1 m glycine, pH 8.0, for 30 min and washed in 10 mg/ml BSA in borate buffer. Isolated microglia (1000/mm2) were placed atop 16 mm glass coverslips in 24-well culture plates. Each well then received 250,000 microspheres with binding assays performed at 37°C. After 4 hr, coverslips were dipped 10 times in PBS and fixed with 4% buffered paraformaldehyde overnight. Microsphere adherence to cells was scored at 200× magnification with phase/fluorescence microscopy. Data were expressed as mean microspheres per field ± SE after subtracting out a background estimated from glycine-coupled spheres bound to sister cultures.
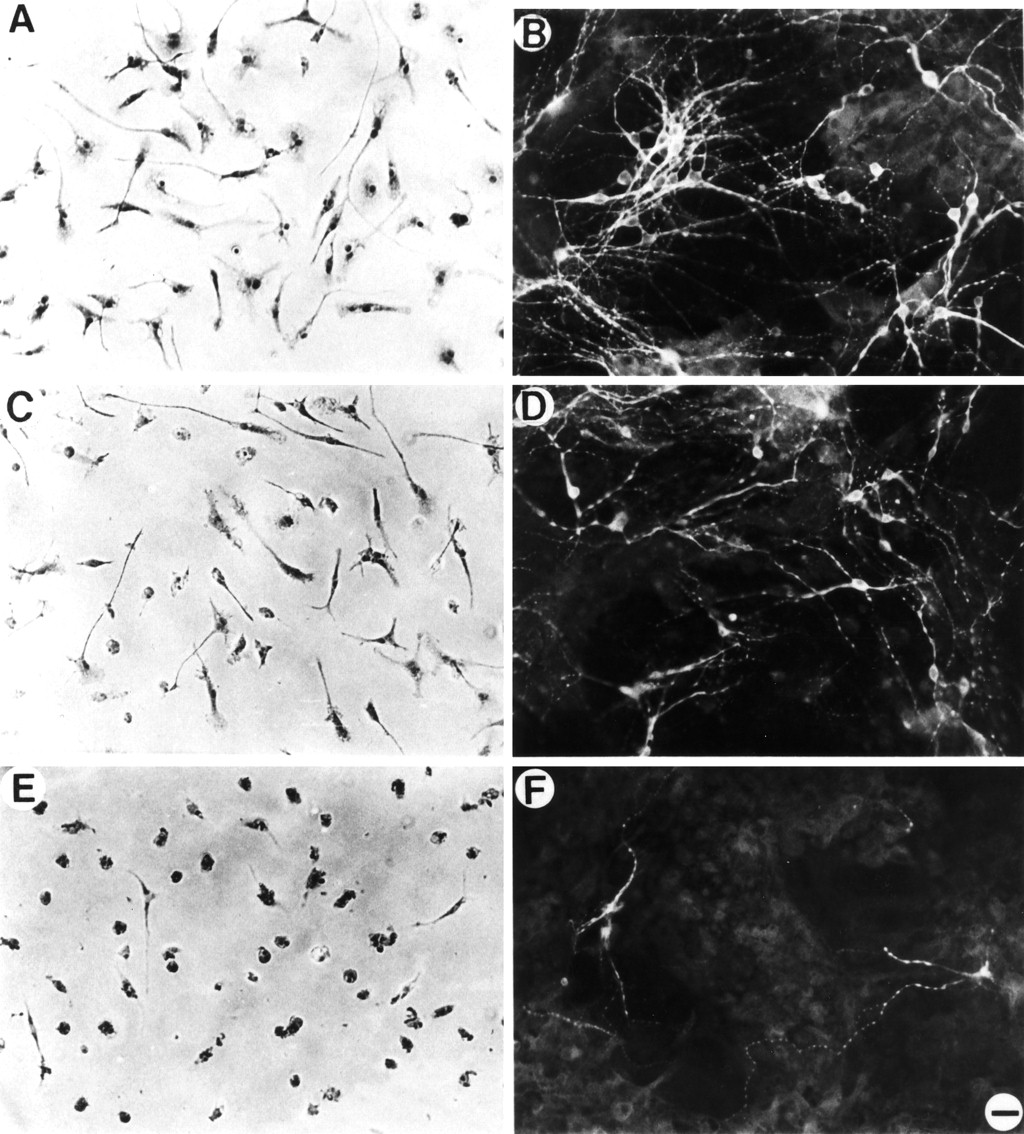
Neuron cultures and toxicity assays. Cultured neurons prepared from rat hippocampus were used to assay for neurotoxins (Giulian et al., 1995a). Briefly, hippocampal cells obtained from embryonic stage (E) 18 rat fetuses were dispersed mechanically in the presence of 0.083% trypsin and plated onto poly-l-lysine-coated glass coverslips in 24 well plates at 250,000 cells/well (resulting in adhering cell densities of ∼1250 cells/mm2) in chemically defined N2 culture media containing 5% fetal bovine serum. Gradual reduction of serum began on day 10 in vitro with daily 1:1 volume replacements with chemically defined media until a final concentration of 0.6% serum was achieved. Mature cultures consisted of process-bearing neurons (15–20% of total cell population) atop a bed of astroglia (>75%) mixed with microglia (from 3 to 8%; see Fig. 1). To eliminate microglia, cultures were exposed to saporin (sap, a ribosome-inactivating protein; Davis and Wiley, 1989) coupled to acetylated LDL (ac-LDL) at a concentration of 10 μg/ml for 12 hr. Saporin-ac-LDL selectively bound to scavenger receptors and reduced microglial numbers to <0.1% of the total population but had no effect on numbers or viability of either neurons or astroglia (see Fig. 1). Then cultures depleted of microglia were exposed to test substances in the presence or absence of exogenous microglia (100,000 cells/culture). Typically, the microglia/neuron ratio at the time of assay was ∼3:1 and the astroglia/neuron ratio ∼6:1. Controls included cultures without saporin treatment, cultures without test substances, and cultures with test substances but lacking microglia.

Selective elimination of microglia from mixed hippocampal cultures. A, C, E, Control cultures show complex neuronal networks revealed by MAP-2/NF immunostaining (A), the presence of DiI-ac-LDL(+) microglia (C), and near-confluent feeder layer of GFAP(+) astrocytes (E). B, D, F, After treatment of cultures with saporin coupled to acetylated LDL, microglia were eliminated (D) without effect on survival of either neurons (B) or astroglia (F). Scale bar, 25 μm. G, Counts of specific cell populations with and without Sap-ac-LDL treatment confirm the specific depletion of microglia. Data are expressed as mean values ± SE obtained from nine randomly selected fields from at least five independent cultures viewed at 200× magnification.
On day 14 of culture, immediately after saporin treatment, test substances were added to the hippocampal cultures. Synthetic peptides were supplied as 1 mm stock solutions in 0.1 mPBS, pH 7.2, stored for at least 1 week at 4°C before use. After 72 hr incubation, the cultures were fixed in 4% paraformaldehyde at room temperature for 18 hr and immunostained by overnight incubation with a mixture of anti-neurofilament (NF) antibodies (SMI-311, 1:600; Sternberger Monoclonals) plus anti-MAP-2 (184959, 1:600; Boehringer Mannheim, Indianapolis, IN) at 4°C in the presence of 2% horse serum and 0.3% Triton X-100 to delineate both neuronal cell bodies and neurites. Astroglia were visualized with rabbit anti-glial fibrillary acidic protein (GFAP, DAKO) as described earlier (Giulian et al., 1989). Finally, to label all cell nuclei, the coverslips were exposed to 0.01% bisbenzimide (Hoechst 33258, Sigma, St. Louis, MO) in 0.1 m phosphate buffer, pH 6.9, and rinsed in distilled water and mounted in glycerin. Immunolabeled cells per field and nuclei per field were scored at 200× magnification with fluorescence microscopy. Data were expressed as percentage of mean survival expressed in terms of parallel untreated control cultures after scoring at least 20 randomly selected fields for each of three coverslips.
Neuron-free cultures were prepared from postnatal day 7 rat optic nerve. After 3 d in N2 culture media supplemented with 5% fetal bovine serum, cells were maintained for an additional 4 d in serum-free N2 medium. Then cultures were fixed and immunostained for neuronal and astroglial markers, as described for hippocampal cells. Additionally, oligodendroglia containing galactocerebroside were immunostained with the monoclonal antibody O1 (Sommer and Schachner, 1981; Bansal et al., 1989; provided by Pamela Knapp, University of Kentucky). Immunostaining of neuron-specific enolase was performed in a 1:2000 dilution of rabbit anti-rat polyclonal antibody (Polysciences) overnight at 4°C after rinsing in 50 mm glycine, 0.3% Triton X-100, and 10% horse serum.
Ciliary neurons from E9 chick embryos were plated onto poly-l-lysine-coated coverslips in 24 well plates at two ganglia per well in N2 media (diluted to 90%) and supplemented with 30 mm KCl plus 0.6% horse serum (modified from Giulian et al., 1993b). Cultures consisted of ∼50% NF(+) neurons mixed with Schwann cells and were free of mononuclear phagocytes and astroglia. Ciliary neurons were sensitive to the toxic effects of NMDA and quinolinic acid (Giulian et al., 1993a,b). Neurotoxic activity was measured after 48 hr as described in detail previously (Giulian et al., 1993a). The percentage of neuron kill score was calculated as [1-(neurons per field in treated group/neurons per field in the untreated control group)] × 100%. Data were expressed as mean values ± SE, with each value obtained from 18 fields per coverslip, using at least six coverslips per group.
Biochemical studies of toxic agents. Purification of neuron-killing activity from culture media conditioned by activated microglia has been described previously (Giulian et al., 1995a) and involved ultrafiltration through YM-30 membrane followed by YM-1 membrane. Then the ultrafiltrates were washed with equal volumes of ethyl acetate under acidic conditions, pH 4.0. and extracted into ethyl acetate under alkaline conditions, pH 10.5. All neurotoxic activity was recovered into this basic organic phase. Material was reextracted into an acidic aqueous phase, pH 2.0, dried under vacuum, flushed with nitrogen gas, and subjected to acid hydrolysis (in 6N HCl for 24 hr at 105°C). Then hydrolysates were extracted into basic ethyl acetate and eluted twice from C18 RP-HPLC (3.9 × 150 mm Nova-Pak, Waters, Milford, MA) with a 0–20% acetonitrile gradient developed over 35 min (solvent A, 0.1% trifluoroacetic acid in dH2O; solvent B, 0.1% trifluoroacetic acid in dH2O/acetonitrile 5:95, vol/vol). Purification of neurotoxic activity from AD neocortex involved an aqueous extract (10 vol sterile distilled water per tissue weight) from 1 kg of minced gray matter of frozen human brain that was subjected to an identical fractionation as described above by ultrafiltration, organic extraction, acid hydrolysis, and RP-HPLC (Giulian et al., 1995a). Phenolic and amine contents were used to estimate concentrations of neurotoxin found within highly purified HPLC fractions. Assigning a UVmax of 265 nm (0.1% trifluoroacetic acid in 14% acetonitrile in dH2O), peaks of activity eluted from C18-HPLC were compared with a standard curve of tyramine eluted under identical conditions measured with a multiple wavelength detector (Rainin Dynamax UV-M). Amine content was determined by the fluorescamine method with tyramine as a standard. These detection methods gave similar values for a given toxin preparation; the estimates of toxin concentration assumed one amine and one phenolic ring per molecule (Giulian et al., 1995a).
Acid-catalyzed esterification of neurotoxin was performed with 3N HCl in n-butanol (Regis Chemical, Morton Grove, IL) for 60 min at 80°C and short acetylation in acetic anhydride in methanol (1:3 vol/vol; Sigma) for 1 min at 25°C; the reaction was terminated by addition of excess glycine at room temperature. Neurotoxin was also modified by excess pentafluoropropionic anhydride (PFPA; Fluka Chemie AG, Switzerland) at 60°C for 60 min, with 100 U/ml plasma amine oxidase (amine:oxygenoxidoreductase; 1.4.3.6; Worthington Biochemical, Freehold, NJ) at 25°C in 1 ml of 10 mm PBS, pH 7.0, for 4 hr or with 390 units of polyphenol oxidase (monophenol, dihydroxyphenylalanine: oxygenoxidoreductase; 1.14.18.1, Worthington) at 25°C in 2 ml of 10 mm PBS, pH 7.0, for 2 hr. In all cases, enzymatic reactions were terminated by boiling for 15 min. Inactivated enzyme controls were prepared by boiling before incubation with neurotoxins.
Nitrites and nitrates, stable byproducts of nitric oxide synthetase (NOS) served as markers for nitric oxide (NO) synthesis (Ignarro, 1990). Nitrite/nitrate concentrations in media conditioned by isolated human microglia (106 cells/ml; for 24–72 hr incubated in the presence of neuritic/core plaques or Aβ peptides) were measured by the Griess method (Beckman et al., 1990) against a standard curve ranging from 0.1 to 50.0 μm nitrate.
RESULTS
Establishing cultures to study microglia–neuron interactions
Reactive microglia in AD brain cluster around neuritic and core plaques but do not interact with diffuse ones (Perlmutter et al., 1992;Giulian et al., 1995a). Recent studies have indicated that such reactive microglia are a source of neurotoxic factors and may injure neurons in a variety of disorders (Giulian, 1992, 1995a). Study of such putative glia–neuron interactions requires suitable in vitro methods. Current models of AD pathogenesis have relied heavily on brain cell cultures, particularly those prepared from dissociated embryonic rat hippocampus. To examine microglia–neuron interactions, it was necessary to develop long-term in vitrosystems that both contained robust hippocampal neurons and allowed control of microglial populations. We found that dissociated E18 hippocampal cells grew well at >1000 cells/mm2 in N2 media supplemented with 5% fetal bovine serum. To approach more closely a chemically defined media for study of cellular interactions, we reduced the serum supplement to a minimal level by serial dilution. Although neurons anchored themselves atop a feeder layer of astroglia within 5 d, rapid reductions in serum concentrations caused astroglia to form thin processes that, in turn, dislodged adhering neurons. To preserve the neuron–astroglia relationships, sera levels were reduced gradually beginning on day 10 in vitro by partial media changes (see Materials and Methods). Under such conditions, hippocampal populations contained ∼15% NF(+) MAP2(+) neurons (Fig. 1A), ∼5% scavenger receptor(+) microglia (Fig. 1C), and >75% GFAP(+) astroglia (Fig. 1E). The microglia present in these cultures have been shown previously to be a significant source of cytokines and cytotoxins (Giulian et al., 1989, 1994, 1995a). To eliminate microglia from these hippocampal preparations, we coupled saporin, a ribosomal inactivating protein, to ac-LDL. As shown in Figure 1D, saporin-ac-LDL (10 μg/ml for 12 hr) essentially destroyed all scavenger receptor(+) microglia while sparing neurons (Fig. 1B) and astroglia (Fig.1F). Monitoring each of the cell populations (Fig.1G) confirms that this treatment brought about a selective elimination of microglia in embryonic hippocampal cultures. Thus, saporin-ac-LDL provided a selective agent to deplete microglia; alternatively, the addition of isolated microglia (>99% homogeneity) to the neuron/astroglia cultures offered a means to selectively reestablish this glial population. By controlling mononuclear phagocyte populations, it was possible to determine whether AD plaque proteins influenced microglia–neuron interactions.
Senile plaques and microglial killing of neurons
As reported earlier, cultured microglia incubated with neuritic/core plaque fragments released neuron-killing factors (Giulian et al., 1995a). To investigate the specificity of this plaque–microglia interaction, neuritic/core plaques from AD brains and diffuse plaques from normal, aged brains were isolated, solubilized, and applied to cultured hippocampal neurons in the presence or absence of microglia. As shown in Figure 2A, solubilized neuritic/core plaque proteins stimulated microglial release of neurotoxins, whereas the solubilized constituents of diffuse plaques did not. To elucidate the signaling mechanism, solubilized neuritic/core plaque material was next fractionated into five major peaks by sizing chromatography (Fig. 2B), as described previously by Roher et al. (1993a,b). Dominant constituents found in these plaque fractions included glycoproteins and ACT in peak S1, apoE in S2, and significant amounts of Aβ-amyloid (predominately Aβ1-42) in peaks S3, S4, and S5, as trimers, dimers, and monomers, respectively (Table 1). Plaque fractions S3, S4, or S5 added to microglial cultures led to dramatic retraction of cellular processes and engulfment of amyloid aggregates (Fig. 3B,D), whereas fractions S1 and S2 had little effect on microglial behavior (Figs. 2C,3A,C). The addition of plaque fractions S3, S4, and S5 to hippocampal cultures led to a severe loss of neurons, but only in the presence of microglia (Fig. 2D). These data suggested that plaque-derived fractions S3, S4, and S5 contained factors capable of inducing neurotoxic microglia. As shown in Table 1, Aβ1-40 or Aβ1-42 peptides were common to these three fractions and were, therefore, likely candidates as microglial activators.

Constituents of solubilized native senile plaques elicit neuron killing. A, Neuritic/core or diffuse plaques were isolated from cortical gray matter, solubilized in formic acid, and dialyzed against a betaine buffer. Equal amounts of plaque protein (normalized to total amine content at 400 μm) were added to neuronal cultures in the presence (100,000 cells/culture) or absence of rat microglia. As shown, solubilized neuritic/core plaque proteins (Neuritic/Core Plaque) led to significant killing of neurons, but only in the presence of microglia. Neither solubilized diffuse plaque proteins (Diffuse Plaque) nor the betaine buffer (Buffer Control) elicited neurotoxic activity. B, Size exclusion chromatography of neuritic/core plaque proteins using two Superose 12 columns in tandem (300 mm × 10 mm × 2; beads, 10 μm diameters). The chromatogram was developed with 80% glass-distilled formic acid at a flow rate of 0.3 ml/min and monitored at 280 nm. Approximate molecular masses of the fractions included the following: S1, 200; S2, 45; S3, 15; S4, 10; and S5, 5 kDa (see Table 1 for fraction composition).C, Histogram shows that exposure to peaks S3, S4, and S5 all elicit significant increases in the percentage of reactive microglia as defined by morphological criteria (see Materials and Methods), whereas peaks S1 and S2 do not. D, Fractions of solubilized neuritic/core plaques were applied to hippocampal cultures in the presence or absence of microglia. No neuron killing was detected in cultures free of microglia. Neuron loss appeared, however, in microglia-containing cultures exposed to peaks S3, S4, and S5, all of which contain Aβ (see Table 1).
Characterization of protein fractions derived from neuritic/ core plaques

Soluble fractions of native plaques induce microglial reactivity. Bright-field photomicrographs of rat microglia cultures exposed to peak S1 (A) or peak S5 (B) and immunostained for the presence of Aβ. As shown, aggregates of Aβ are found throughout the cultures incubated with peak S5. Scale bar, 25 μm. Phase photomicrographs show cultured microglia as process-bearing cells with spinous surfaces typical of nonreactive cells, despite exposure to peak S1 (C). In contrast, microglia exposed to peak S5 retract processes and take on a reactive cell morphology similar to that found in AD brain (D). Scale bar, 5 μm.
β-Amyloid peptides and microglial killing of neurons
To test whether Aβ alone could drive neurotoxic microglia, next we examined the actions of synthetic peptides (Table2). Generally, Aβ peptides applied in μm concentrations had no damaging effects on neurons grown in microglia-free cultures (Fig.4A,C). When, however, microglia were added to this culture system and incubated with either human Aβ1-40 or Aβ1-42, there was widespread neuronal loss (Figs. 4C,5). In the presence of 1 μm Aβ1-42, the microglial density required for maximum neuron killing was ∼150 cells/mm2 (microglia/neuron ratio of 0.8:1; Fig.4D), although we noted neuron killing in the presence of <50 microglia/mm2. Without saporin-ac-LDL pretreatment, the endogenous microglia population normally found in these cultures ranged from 40 to 80 cells/mm2. The levels of neuron killing in preparations containing endogenous microglia (incubated with 1 μm Aβ1-42) were above values predicted by cell-density curves constructed from the addition of exogenous glia (Fig. 4D), indicating that native microglia seeded with the original E18 cultures were particularly efficient as neuron-killing cells. Clearly, depletion of microglia from mixed neuron–glia cultures was necessary to demonstrate killing through inflammatory cells activated by Aβ.
Properties of Aβ peptides

Toxic actions of synthetic Aβ peptides on neurons. A, B, High concentrations of most Aβ peptides placed in hippocampal cultures containing neurons and astroglia (but depleted of microglia) show little effect. There is, however, a generalized cytotoxic action by Aβ25-35 at ≥30 μm on both neurons (A) and astroglia (B). In the absence of microglia, none of the Aβ peptides (at 1 μm) produces destruction of neurons. When rat microglia are added to neuronal cultures, however, only Aβ1-40 and Aβ1-42 elicit neuron killing (C). D, Addition of increasing numbers of microglia shows a saturated neuron-killing response at a density of 150 microglia/mm2 when incubated with 1 μm Aβ1-42; microglia found within the E18 culture at the time of plating (Endogenous Microglia) also show an efficient killing capacity in the presence of Aβ. These observations point to the need to deplete neuron cultures of microglia when mechanisms of Aβ toxicity are assessed. E, Dose–response curves reveal Aβ1-42 to be the most potent microglial stimulus, with an estimated ED50 of 10 nm, as compared with 80 nm for Aβ1-40 (500 microglia/mm2).

Cellular responses on exposure to synthetic Aβ peptides. Phase microscopy shows that cultured rat microglia undergo morphological changes with retraction of processes when exposed to 1 μm Aβ1-42 (E); in contrast, 1 μm Aβ17-43 (C) does not alter microglial morphology, which appears identical to untreated cells grown under control conditions (A). Fluorescence microscopy of neuron plus microglia cultures shows robust NF(+) MAP2(+) hippocampal neurons (B) that are undamaged after addition of conditioned media (10% v/v) from microglia incubated with 1 μm Aβ17-43 (D). Significant neuron loss occurs, however, if hippocampal cultures are exposed to conditioned media from microglia incubated with 1 μm Aβ1-42 (F). Scale bar, 25 μm.
When 500 microglia/mm2 was added to neuron cultures, Aβ1-42 and Aβ1-40 showed an ED50 of ∼10 and 80 nm, respectively (Fig. 4E). The amount of Aβ1-42, therefore, found in plaque fractions S3, S4, and S5 (Table1), as well as the amounts of Aβ1-42 estimated to exist in AD brain (in the range of ng/gm tissue; Kuo et al., 1996), would be sufficient to elicit neurotoxic glia. For the most part, small Aβ peptides (Aβ1-16, Aβ1-28, Aβ17-43; Table 2) did not produce neurotoxicity in the presence or absence of microglia. An exception, however, was Aβ25-35, which was generally cytotoxic at high concentrations (≥30 μm) with destruction of nearly 90% of both neurons and glia when given at 100 μm(Fig. 4A,B). This lack of neuronal specificity for high concentrations of Aβ25-35 was consistent with its damaging effects noted by others on such non-neuronal cell lines as HeLa (Pollack et al., 1995) or on primary cultures of astrocytes (Harris et al., 1995). However, because Aβ25-35 is not a naturally occurring biological product, it is unlikely to participate in the pathology of AD (Roher et al., 1993a,b).
A number of reports have suggested that the neurotoxic capacity of Aβ is associated with specific structural features, such as aggregation or fibril formation (Pike et al., 1991, 1993). For example, Pike et al. (1991) described greater neuron loss after Aβ1-42 was stored at 37°C for several days to increase the appearance of aggregates in distilled water. We found, however, that both fresh and “aged” Aβ1-42 peptides prepared as described by Pike et al. (1993) to be equally effective in stimulating neurotoxic microglia (75.1 ± 5.8% vs 77.5 ± 2.6% neuron kill, respectively). It has been suggested that fibril formation occurring with Aβ1-42 in solution was a critical feature for its neurotoxicity (Lorenzo and Yankner, 1994; Simmons et al., 1994; Howlett et al., 1995). To determine whether conformational states such as aggregation were pertinent to microglia-dependent neuron killing, we covalently coupled various Aβ forms to 1 μm fluorescent microspheres. Aβ1-42-coupled microspheres were readily taken up by microglia in E18 cultures (Fig. 6A,B), similar to the rapid cell recognition of neuritic/core plaque fragments (Giulian et al., 1995a) and native Aβ aggregates (Fig. 3). In contrast, β17-43-coupled microspheres were not engulfed by microglia (Fig. 6C,D). Importantly, microsphere-coupled Aβ1-42 and Aβ1-40, as well as unbound forms in solution, activated neurotoxic microglia, whereas Aβ1-16 or Aβ17-43 was not effective either when tested in solution or linked to microspheres (Fig.6E). These observations suggested that the primary structure of the Aβ1-42 peptide, and not such complex features as aggregation or β-sheet formation, was sufficient to induce neurotoxic glia.

Aβ activates microglia after coupling to microspheres. Fluorescently labeled microspheres were covalently coupled to Aβ1-42 and placed in hippocampal cultures containing rat microglia (500 cells/mm2). After 72 hr, Aβ1-42 spheres (A) were localized specifically within DiI-ac-LDL(+) microglia (B; colocalization noted byarrows). In contrast, Aβ17-43 microspheres (C) showed no consistent association with microglia (D). Scale bar, 20 μm. E, Comparison of capacity of Aβ in solution or coupled to microspheres (Bead-Bound) to elicit neurotoxic microglia (250,000 microspheres/culture; 100,000 microglia/culture; 72 hr incubation). Neuronal loss was similar whether Aβ peptides were in solution or bound to beads, indicating that fibril formation or other changes in tertiary structure were not necessary to stimulate neurotoxic microglia.
How potent is Aβ as a direct neurotoxin?
As noted above, Aβ1-42 did not kill neurons directly when applied at 100 μm in cultures free of microglia (Fig. 7B), whereas 100 nm Aβ in the presence of microglia brought about significant neuron loss (Fig.7C). Because previous reports (Pike et al., 1991, 1993;Cotman et al., 1992) described direct effects of Aβ1-42 on neuronsin vitro, we sought to identify culture conditions, other than the presence of microglia, which might lead to neuron killing. Because astroglia might participate in AD mechanisms of neuronal injury, next we examined whether astroglia-free cultures of ciliary ganglia (∼50% neurons and 50% Schwann cells) were sensitive to Aβ. Direct application of Aβ1-42 again had no apparent effect on survival of ciliary cells, whereas Aβ1-42 stimulated microglia-secreted toxin to destroy ciliary neurons (Fig.7H). Similarly, plaque-stimulated microglia have been found to destroy ciliary neurons (Giulian et al., 1995a).

Fluorescent photomicrographs of hippocampal cultures after exposure to Aβ1-42. A, Control cultures show complex networks of NF(+) MAP-2(+) neurons. B, Exposure of cultures to 100 μm Aβ1-42 in the absence of microglia has no effect on neuron number, whereas (C) addition of 100 nm Aβ1-42 in the presence of rat microglia (500 cells/mm2) destroyed nearly all neurons.D–G, Immunostaining for neuron-specific enolase (NSE) is not specific to neurons in CNS cultures, as shown by immunofluorescent visualization of glia in cultures of neuron-free optic nerve, including galactocerebroside(+) oligodendroglia (D) and GFAP(+) astrocytes (F), which are both NSE(+) (E and G, respectively). Scale bar, 10 μm. H, Ciliary neuron cultures showed that Aβ1-42 is not toxic to neurons in the absence of brain glia (Aβ1-42 only) after 48 hr exposure. Conditioned media from Aβ1-42-stimulated microglia (Microglia+Aβ1-42) did, however, kill neurons, indicating that astrocytes are not necessary for microglial neurotoxicity.
As an alternative strategy to assess the role of astroglia in Aβ interactions with microglia, we sought to eliminate glia from dissociated E18 hippocampal cells by using mitotic inhibitors, as described by Koh et al. (1990) and Pike et al. (1993). We found, however, that such culture systems did not provide a reliable assay for monitoring neuron killing, because these cell preparations were inherently unstable, with neuron survival dropping to <80% within 48 hr. Moreover, the use of chemically defined media or mitotic inhibitors did not actually eliminate non-neuronal cells [such as glial precursors, scavenger receptor(+) microglia, or GFAP(+) astroglia] but simply slowed glial differentiation and the degree of antigen expression. In addition, astrocytes grown under such marginal culture conditions take on a reactive morphology with long, thin processes similar to neurons. Despite reports by others (Whitson et al., 1989;Koh et al., 1990; Pike et al., 1991, 1993; Cotman et al., 1992), we were unable to obtain a reliable neuron count in such preparations by phase microscopy. Introduction of fetal calf sera to poisoned cultures stimulated cells with neuron-like morphology to develop into GFAP(+) astroglia. Although NF(+) MAP2(+) neurons made up <20% of the hippocampal cell population, >90% of the cells were found to be neuron-specific enolase(+). However, neuron-free cultures of developing optic nerve also contained >85% neuron-specific enolase(+) cells, including process-bearing galactocerebroside(+) oligodendroglia (Fig.7D,E) and GFAP(+) astroglia (Fig. 7F,G). Overall, we could neither establish healthy cultures of highly enriched hippocampal neurons using methods described by other laboratories nor reliably interpret Aβ toxicity within such preparations. We were unable, therefore, to assess the direct action of Aβ on primary cultures of glia-free brain neurons.
Specific plaque proteins activate human microglia
Responses of human microglia to Aβ, as well as to other stimulants, might differ from the responses of rodent microglia. Activated rodent macrophages, for example, are richly supplied with inducible nitric oxide synthetase (iNOS) and produce cytotoxic levels of NO (Lees, 1993). We find in the hippocampal culture system used here that lipopolysaccharide (≥100 ng/ml) induces rat microglial iNOS, which resulted in NO-dependent killing of neurons. In this culture system there was a dose-dependent relation of nitrate/nitrite levels of the media and neuronal loss, with loss apparent at 15 μmnitrate/nitrite and above (data not shown). Human macrophages, on the other hand, are thought to contain little iNOS and produce negligible amounts of NO (Denis, 1994). For this reason, iNOS involvement in AD pathology, as recently proposed by Meda et al. (1995), remains uncertain. To compare responses of human cells to those of rat, we isolated human microglia from normal adult brains recovered rapidly at autopsy (Giulian et al., 1995a,b). These human brain mononuclear cells behaved as did the rat microglia, engulfing neuritic/core plaques and retracting processes (Giulian et al., 1995a). Both the synthetic Aβ peptides and native plaque fractions S3, S4, and S5 induced human microglia to become neurotoxic (Fig.8A,B) in patterns identical to those of rat microglia. Although Aβ1-40 and Aβ1-42 were very potent inducers of neurotoxic human microglia, these peptides did not bring about release of nitrate or nitrites (Fig. 8C,D). Neither Aβ exposure to rat microglia (data not shown) nor LPS exposure to human microglia elicited nitrate or nitrite levels above 1.5 μm. Such observations argue against involvement of microglial iNOS in the neuronal pathology of AD. Overall, human and rat microglia responded identically to Aβ, both exhibiting neurotoxicity when in culture with intact human Aβ1-42 or Aβ1-40 peptide (Fig. 8E).

Human microglia and neuron killing.A, Only Aβ-containing fractions from solubilized neuritic/core plaques [peaks S3 (54 nm), S4 (220 nm), and S5 (250 nm)] elicit human microglia to engage in neurotoxic behaviors. B, When tested at 1 μm concentrations, synthetic Aβ1-40 and Aβ1-42 also stimulated release of neurotoxin from human microglia, whereas smaller Aβ fragments had no effect. Despite neuron killing, there is no evidence of increased production of nitrate or nitrite by human cells stimulated with either native (C) or synthetic (D) Aβ. E, Neuron killing could be induced by human or rat microglia exposed to 1 μm of the human forms of either Aβ1-42 or Aβ1-40. The rodent form of Aβ1-40, however, was inactive, as were fragments of human Aβ, including 1-28, 12-28, and 17-43.
Aβ as an indirect neurotoxin
A number of cytotoxic factors have been reported to participate in Aβ neurotoxicity, including free radicals (Behl et al., 1994), NO (Meda et al., 1995), cytokines (Mrak et al., 1995), and NMDA-like molecules (Giulian et al., 1995a). To determine whether short-lived factors play a role in Aβ-induced neuron killing by microglia, we compared neuronal loss when microglia were either mixed among neurons (contact) or separated from neurons by placement in filter-bottomed Millex cell chambers (no contact). Aβ1-40 and 1–42, as well as the native plaque fractions, stimulated microglia to destroy neurons despite segregation of microglia and neurons (data not shown). These observations rule out involvement of short-lived free radical intermediates, because such agents required close proximity between secretory and target cells. Moreover, there was no reduction of microglia-mediated neuron killing after exposure of either human or rodent cells to Aβ on incubation with such free-radical scavengers as vitamin E, catalase, or glutathione or with such potent inhibitors of iNOS as diphenyl- eneiodium (DPI) orl-N-5-(1-iminoethyl)ornithine hydrochloride (l-NIO; Fig. 9A). Although glutamate antagonists acting on non-NMDA receptor sites did not protect neurons, NMDA receptor antagonists (Fig. 9B), including AP5, AP7, MK-801, and ifenprodil, prevented neuronal loss when applied at low concentrations (10 μm).

Drug blockade of Aβ-induced neuron killing by rat and human microglia. To investigate mechanisms of cell killing, we stimulated rat microglia with 1 μm Aβ1-42 (Rat/Aβ1-42) and human cells with fraction S5 (containing 250 nm of native Aβ1-42) from solubilized neuritic/core plaques (Human/S5 Peak). A, Agents that act as free radical scavengers (vitamin E, 100 μm; catalase, 25 U/ml; glutathione, 100 μm) did not block microglial killing of neurons. No protective effects were observed with the nitric oxide synthetase inhibitorsl-N-5-(1-iminoethyl)ornithine hydrochloride (l-NIO; 10 μm) or diphenyl iodonium (DPI; 300 nm), although the NMDA antagonist AP5 prevented neuron death. B, Other NMDA antagonists acting at the receptor site (AP7), at the polyamine regulatory site (Ifenprodil), or at the ion channel (MK801) all blocked neuron death, whereas the non-NMDA glutamate antagonists (GAMS, BNQX) did not. All drugs were applied at 10 μm.C, Isolation of neurotoxin from culture media conditioned by Aβ-stimulated rat microglia (Aβ1-42/Microglia) or from frozen AD gray matter (AD Brain) involved extractions in ethyl acetate, pH 10.5, acid hydrolysis, and sequential gradient RP-HPLC (C18 column using a 0–20% acetonitrile gradient in dH2O with 0.1% trifluoroacetic acid). Neurotoxin activities from microglial-conditioned media copurify with that from AD brain tissue via a coelution using RP-HPLC at ∼14% acetonitrile. Neurotoxicity was not found within control brain extracts or from unstimulated microglial culture media (data not shown).
The neurotoxin recovered from Aβ1-40 or Aβ1-42-stimulated human microglia withstood boiling, showed a low molecular mass (<1000 Da), extracted into ethyl acetate at pH 10.5, and bound to cationic exchangers such as SP-Sephadex C25, as described for plaque-exposed microglial toxin in previous work (Giulian et al., 1995a). Each of these properties is shared by the neurotoxic phenolic amine, which can be extracted from AD brain. Inactivation of both the microglia-derived and brain-derived neurotoxin by PFPA, fluorescamine, and plasma amine oxidase suggested the presence of a terminal amine group at the active site, whereas insensitivity to acidified butanol esterification indicated a lack of carboxyl groups (Giulian et al., 1995a). Overall, the active principal derived from Aβ-stimulated microglia exhibited properties identical to those of the neurotoxin recovered from AD gray matter or from culture media of plaque-stimulated microglia (Giulian et al., 1995a). Protease insensitivity and resistance to acid hydrolysis (6N HCl, 105°C, 24 hr) of the neurotoxin ruled out peptide factors, including cytokines and Aβ itself. Coelution on tandem ion exchange columns confirmed the identical character of microglia-derived and AD brain-derived lipophilic killing factor. As shown in Figure9C, a single peak of biological activity from Aβ-stimulated microglia coeluted with the toxin extracted from AD brain by RP-HPLC. Previous study has shown that this purified agent was an effective toxin against hippocampal neurons in vitro orin vivo in the picomolar range (Giulian et al., 1995a).
Specific Aβ domains bind to microglia
We next turned to the issue of how Aβ peptides activate microglia. Because adherence to plaques might serve as an important first step in the recruitment of reactive glia, it was reasonable to consider Aβ as a potential anchoring site for microglia on plaque surfaces. To test this hypothesis, synthetic Aβ peptides or native plaque-derived proteins were covalently coupled to 90 μm Sepharose beads. Then these beads were floated atop cultured microglia adherent to plastic culture dishes. Within 30 min, microglia began to detach from the culture dish and anchored to beads that were covalently coupled to native plaque proteins or synthetic Aβ1-42 (Fig. 10A,B). Within 6 hr, the number of microglia adhering to plaque protein-coated beads had increased by fivefold when compared with cells adhering to control beads coupled to glycine or BSA (Fig. 10C). Interestingly, Aβ peptides that contained N-terminal residues such as Aβ1-28 also promoted cell binding, whereas the C-terminal portion (Aβ17-43) did not (Fig.10C).

Aβ domains and interactions with microglia.A, Phase photomicrograph of rat microglial cell adhering to Sepharose bead coupled to human Aβ1-42 peptides. B, Fluorescence photomicrograph of the same bead showing adherent cell labeled by the fluorescent microglial marker DiI-ac-LDL. Scale bar, 20 μm. C, Rat microglial adherence to Sepharose-coupled beads after 6 hr. Plaque proteins derived from neuritic/core plaques provided an anchoring site for microglia, as did Aβ1-42. Importantly, Aβ1-28 also promoted bead binding, whereas Aβ17-43 did not. Controls included beads coupled to glycine (Control-glycine) and to bovine serum albumin (Control-BSA). Data shown are expressed as the numbers of adhering cells per 100 randomly selected beads ± SE after 6 hr incubation at 37°C.
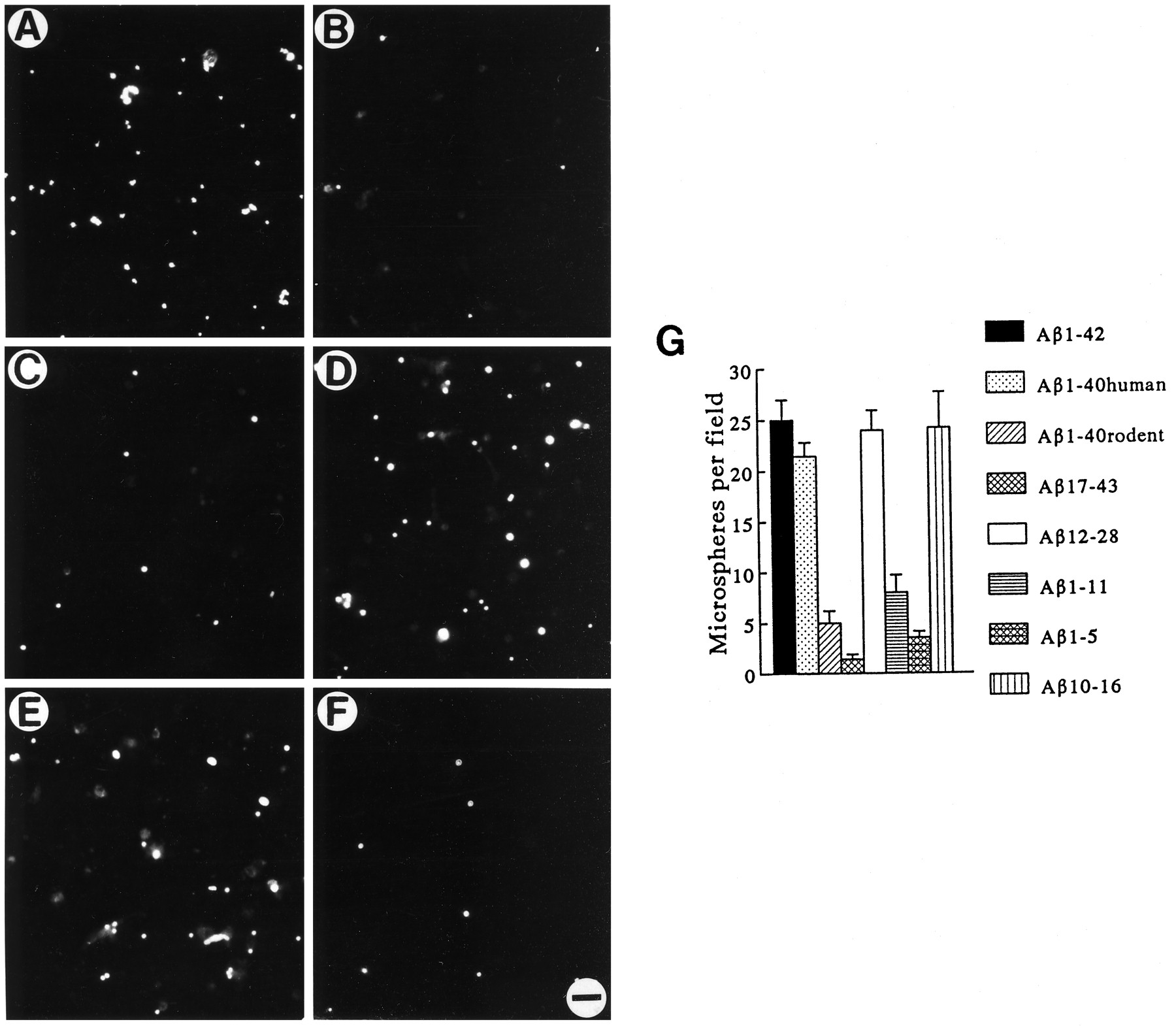
To delineate further which portions of the Aβ peptide served as a microglial binding site, we next compared a variety of synthetic peptides coupled to 1-μm-diameter microspheres. We found that, within 4 hr of incubation, marked glial binding to microspheres coupled to Aβ1-42, Aβ1-40, Aβ1-16, or Aβ12-28 occurred (Fig. 11A–F), with little cell binding of spheres coupled to Aβ17-43, Aβ25-35, Aβ36-42, or to rodent Aβ1-40 (5Arg→Gly, 10Tyr→Phe, 13His→Arg). Because structural differences between the human and rodent forms of Aβ occur between residues 5 and 13 of the N terminus, we next focused on properties of this specific domain. As shown in Figure 11G, Aβ12-28 microspheres provided an anchoring substrate for cells, whereas Aβ1-11 did not. Examination of a heptapeptide confirmed a microglial binding domain between residues 10 and 16 (Fig. 11G).

Aβ cell-binding domain is required for activation of neurotoxic microglia. Fluorescent photomicrographs showing microsphere binding to enriched cultures of rat microglia (500/mm2) after 4 hr incubation at 37°C. Coupling of Aβ peptides to fluorescent microspheres showed that Aβ1-42 (A), Aβ12-28 (D), and Aβ10-16 (E) readily bind, whereas peptides Aβ17-43 (B), Aβ1-11 (C), and Aβ1-5 (F) did not. Quantitations of binding pattern (G) indicated that regions of the N terminus containing amino acid residues 10–16 were necessary for Aβ binding to microglia. Data are expressed as mean values ± SE when viewed at 200× magnification.
The importance of the N-terminal cell-binding domain for Aβ-microglial interactions was supported by the fact that neither Aβ17-43 nor Aβ1-40rodent induced neurotoxic cells despite test concentrations 100-fold above that required for human Aβ1-40 (Figs. 4E, 8E). The patterns of binding and toxicity predicted that the 10–16 binding region would be a necessary component for activation of neuron-killing microglia. To test this hypothesis, we next incubated Aβ10-42 at increasing concentrations and found it to be nearly as potent as Aβ1-42 in eliciting microglia-dependent killing of cells (Figs.4E, 12A). However, the Aβ10-16 binding domain or the 17–43 region by themselves did not injure neurons (Fig. 12A). Thus, the N terminus of human Aβ (particularly residues 10–16) was necessary, although not sufficient, for eliciting neurotoxic microglia (Fig.12B).

Comparison of Aβ effects on microglia.A, Dose–response curves show that, although it is able to bind to microglia, Aβ10-16 did not elicit neurotoxic microglia. The addition of this microglial-binding domain to Aβ17-42 (which neither binds to microglia nor elicits toxicity) created a peptide, Aβ10-42, which both bound to microglia and stimulated microglia to kill neurons. B, Diagram comparing the structures and functions of synthetic peptides under study. Shaded areaillustrates the N-terminal portion of Aβ that differs between human and rat forms and is necessary for microglial adherence.
DISCUSSION
Since the description by Bolsi (1927) of reactive microglia near plaques in AD brain, it has been uncertain whether these reactive non-neuronal cells actually contribute to the disease process or merely reflect ongoing pathology. Recently, however, it has become clear that reactive microglia surround only certain types of amyloid deposits in the brain (the neuritic and core plaques) while ignoring nearby deposits of other types, including diffuse plaques (Perlmutter et al., 1992; Giulian et al., 1995a). Such selectivity in the distribution of reactive glia suggests that specific signals within neuritic and core plaques drive brain inflammation. With the increasing recognition that reactive microglia can mediate neuronal injury via release of cytotoxic factors (Banati et al., 1993; Giulian et al., 1993a), speculation on the involvement of microglia in AD has encompassed the release of complement proteins (Rogers et al., 1988,1992), cytokines (Meda et al., 1995; Mrak et al., 1995), NMDA-like toxins (Piani et al., 1991; Giulian et al., 1993a,b), and free radicals (Thery et al., 1991; Hensley et al., 1994). Recently, our laboratory has shown that cultured human microglia exposed to neuritic/core plaque fragments secreted a highly neurotoxic phenolic amine (Giulian et al., 1995a). This phenolic amine seems to exert its toxic effects via direct interactions with the NMDA receptor complex, as its effects are blocked by NMDA receptor antagonists. Moreover, this microglia-derived neurotoxin was also isolated from plaque-laden AD gray matter. The strong correlation between tissue concentrations of the neurotoxin and the tissue densities of reactive microglial clusters (Giulian et al., 1995a) provides further evidence for a link between plaque–microglia interactions and neuronal pathology.
As described here, the primary signal for plaque induction of microglial neurotoxicity is the Aβ peptide. Plaque fragments from AD brain induce microglia to become neurotoxic; however, only those solubilized plaque fractions that contained Aβ1-42 (or Aβ1-40) stimulated both rat and human microglia to take on reactive morphologies and become neurotoxic. Testing with synthetic peptides confirmed that the full-length 1–42 peptide was a potent inducer of neuron-killing microglia and the truncated Aβ1-40 slightly less so. Other forms of Aβ, including the peptides Aβ1-28, Aβ12-28, and Aβ17-43, were inactive. Several lines of evidence suggest that secondary or tertiary structures of β-amyloid are not critical for activation of microglia. For example, rodent Aβ1-42, which does not induce toxicity, also forms β-pleated sheets, as found with human peptide (Fraser et al., 1992). In addition, Aβ17-42, which is even more prone to aggregation than is Aβ1-42 (Gowing et al., 1994; Table2), is also unable to induce toxicity. In fact, testing a variety of peptide fragments, we have shown that the N-terminal and C-terminal regions seem to play separate and necessary roles in microglial activation. The interactions of microglia with peptide-coupled beads reveal that the N-terminal region is necessary for anchoring of the peptide to the cells. This finding may account for the inability of rodent Aβ to induce neurotoxicity, because the first 16 amino acids of rodent Aβ are unlike the human Aβ domain. Interestingly, residues 1–16 compose the hydrophilic portion of the molecule and thus may be accessible for microglial attachment to the plaque. Without this attachment domain, Aβ is unable to induce toxicity, as shown by the inability of Aβ17-42 to activate microglia. The C-terminal portion of Aβ remains necessary to toxicity induction, however, because the N terminus (1–16) alone was unable to induce microglial neurotoxicity. Overall, the residues neighboring 13His play an essential role in directing Aβ interactions with microglia (Giulian et al., 1996).
It is important to note that the Aβ effects on microglial neurotoxicity reported here are distinct from the directneuron-killing effects of Aβ described by other laboratories (Yankner, 1990; Pike et al., 1991, 1993; Cotman et al., 1992). Most laboratories exploring a direct toxicity carefully describe those specific cell-culture conditions, or particular protocols for Aβ peptide preparation, which have been essential to create an environment for cell killing (Pike et al., 1991, 1993; Mattson et al., 1992;Howlett et al., 1995; Pollack et al., 1995). For example, low cell numbers seem to be necessary to demonstrate direct killing by Aβ, with cell densities typically <100/mm2 (Mattson and Rydel, 1992). In addition, some groups report that toxic effects are seen only if cultures are exposed to the peptide after a defined period of incubation in vitro (Yankner et al., 1990), only if glia are poisoned (Pike et al., 1995), only if batch-to-batch variability among synthetic peptides is considered (May et al., 1992), only if synthetic peptides are “aged” (Pike et al., 1991, 1993), or only if glutamate or other glutamate receptor agonists are present (Koh et al., 1990;Mattson et al., 1992). Unfortunately, specific labeling for microglia (that may compose 5% to 10% of cells in embryonic rat hippocampal cultures) is seldom used, so the contribution of neurotoxic microglia to these other culture systems cannot be assessed.
Our in vitro systems were optimized to maintain healthy and long-lived neuron/astroglia cultures and were controlled to examine microglial interactions with neurons. These cultures differed in several important ways from assays described by other investigators. First, our neurotoxicity assays use high-density cultures, an order of magnitude greater than the low-density systems cited above. Under this condition we find, in agreement with Mattson and Rydel (1992), no directly toxic effects of Aβ. Second, the culture system used here is very supportive of neuronal growth, as shown by extensive neuritic projections and viability for several weeks beyond the test period. In culture preparations poisoned with mitotic inhibitors or in very low-density cultures, neuronal survival drops by at least 50% spontaneously (Mattson and Rydel, 1992), making it very difficult to monitor and interpret the effects of any cytotoxic agent. Third, microglial content of the culture system must be demonstrated clearly, because the endogenous population of microglia present in primary hippocampal cultures is fully neurotoxic when exposed to Aβ1-42. Finally, we use a combination of neuron-specific markers (MAP-2 and NF) to allow accurate monitoring of neurons among a mixed population of cells. In contrast, phase microscopy (which is very difficult to interpret in developing brain cell cultures), release of lactate dehydrogenase into culture media (which occurs after any cell damage), or a decline in neuron-specific enolase(+) cells (which includes both neurons and glia in embryonic cultures) cannot differentiate the survival of neuronal and non-neuronal cells. Clearly, in vitro models must be interpreted with caution, for they represent only approximations of AD pathology, and all are prone to artifacts of cell culture. Although both the direct and indirect neurotoxic effects of Aβ may play roles in the neuronal pathology of AD, the striking potency of Aβ to induce neurotoxic microglia suggests that indirect, immune-mediated pathways may be substantial.
The observations described here point to strategies for intervention in the pathology resulting from neurotoxic microglia in AD; these include (1) suppression of signaling steps as neuritic/core plaques turn quiescent microglia into reactive ones, (2) inhibition of microglial synthesis and secretion of neurotoxins, and (3) the blockade of neurotoxin attack on neurons. In pursuit of the first of these strategies, experiments are in progress to identify and manipulate the specific domains of Aβ responsible for the various steps in the Aβ-induced cascade of cellular response leading to neurotoxic microglia. Because the cell attachment domain in the N-terminal portion of Aβ is not itself toxic, it may be possible to block induction of neurotoxic microglia selectively by competition with small Aβ peptides. Treatments encompassing the second strategy are reflected in several retrospective studies that have implicated anti-inflammatory drugs as beneficial for AD, and treatment trials have been suggested on the basis of these data (Breitner et al., 1990; McGeer et al., 1990;Schnabel, 1993; Eikelenboom et al., 1994; Lucca et al., 1994). However, because commonly used immunosuppressants (including glucocorticoids) do not reduce neurotoxic activities of brain mononuclear phagocytes (Giulian, 1992), further investigation of such microglial suppressants as chloroquine is indicated (Giulian et al., 1989; Giulian and Robertson, 1990). Finally, as shown in Figure 9B, the neurotoxin secreted by plaque-activated microglia can be blocked by antagonists of the NMDA receptor. Perhaps NMDA receptor antagonists now in clinical trials for stroke, trauma, and epilepsy might offer benefit to the AD patient. We suggest that suppression of neurotoxic microglia offers a number of therapeutic strategies, which may slow neuronal loss in Alzheimer’s disease.
Footnotes
This work was supported by National Institutes of Health Grants AG12548, NS35972, NS52637, and AG11925; the Methodist Hospital Foundation; and the Alzheimer’s Disease Research Center at Baylor College of Medicine. We thank Dr. Janos Varga of California Peptide for preparation of synthetic peptides.
Correspondence should be addressed to Dr. Dana Giulian at the above address.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}