

Abstract
In postnatally developing and adult brains, interneurons of the olfactory bulb (OB) are continuously generated at the subventricular zone of the forebrain. The newborn neuroblasts migrate tangentially to the OB through a well defined pathway, the rostral migratory stream (RMS), where the neuroblasts undergo collective migration termed “chain migration.” The cell-intrinsic regulatory mechanism of neuroblast chain migration, however, has not been uncovered. Here we show that mice lacking the actin-binding Akt substrate Girdin (a protein that interacts with Disrupted-In-Schizophrenia 1 to regulate neurogenesis in the dentate gyrus) have profound defects in neuroblast chain migration along the RMS. Analysis of two gene knock-in mice harboring Girdin mutants identified unique amino acid residues in Girdin's C-terminal domain that are responsible for the regulation of neuroblast chain migration but revealed no apparent requirement of Girdin phosphorylation by Akt. Electron microscopic analyses demonstrated the involvement of Girdin in neuroblast cell–cell interactions. These findings suggest that Girdin is an important intrinsic factor that specifically governs neuroblast chain migration along the RMS.
Introduction
In the rodent brain, neural stem cells and the generation of neurons are sustained throughout the postnatal period and adulthood at two locations: the subventricular zone (SVZ) near the lateral ventricles and the subgranular zone (SGZ) of the dentate gyrus (DG) (Alvarez-Buylla and García-Verdugo, 2002; Lledo et al., 2006; Zhao et al., 2008). In the SVZ, neuroblasts are generated from dividing astrocytic stem cells and migrate over a long distance to the olfactory bulb (OB) through the rostral migratory stream (RMS) (Doetsch et al., 1999; Alvarez-Buylla and García-Verdugo, 2002). When the neuroblasts arrive at the OB, they detach from the RMS and become interneurons, and this developmental pathway is essential for neuronal replacement in the OB (Heck et al., 2002; Lledo et al., 2006; Imayoshi et al., 2008).
A fascinating feature of the migration of SVZ neuroblasts is that they undergo collective migration termed “chain migration,” in which the neuroblasts form a homotypic chain-like organization (Alvarez-Buylla and García-Verdugo, 2002). However, limited information is available about how the neuroblasts interact with each other to coordinate their chain migration along the RMS. One candidate protein for intrinsic control of neuroblast migration is neural cell adhesion molecule (NCAM) (Cremer et al., 1994; Rutishauser, 2008). NCAM-deficient mice exhibit a severe OB size reduction due to migration defects of SVZ neuroblasts. A subsequent study, however, revealed that NCAM is not essential for the formation of chains by the neuroblasts; rather, it is required for linking the neuroblasts to neighboring astrocytes and promoting cell survival (Chazal et al., 2000; Gascon et al., 2007). Other studies have identified that chain migration of neuroblasts is regulated by intracellular signaling mediated by the ErbB4 receptor and integrin β1 (Anton et al., 2004; Belvindrah et al., 2007). However, it remains poorly understood how the neuroblasts use locally acting extracellular cues and intrinsic molecular machinery to coordinate their proliferation, migration, and terminal differentiation.
We recently reported that the actin-binding Akt substrate Girdin (girders of actin filaments) is a component of a protein complex with Disrupted-In-Schizophrenia 1 (DISC1), a susceptibility gene for schizophrenia and bipolar disorder, in newborn neuroblasts in the DG (Enomoto et al., 2009; Kim et al., 2009). Girdin-deficient (Girdin−/−) mice appear developmentally normal, but our close examination revealed severe defects in the migration and positioning of SGZ neuroblasts in the DG (Enomoto et al., 2009).
Considering previous findings that SGZ and SVZ neuroblasts share common mechanisms for their generation, migration, and differentiation (Duan et al., 2008; Zhao et al., 2008), it is intriguing to ask whether Girdin has a role in postnatal and adult neurogenesis in the SVZ and development of the OB. In the present study, we show that Girdin is expressed in SVZ neuroblasts and regulates their migration to the OB. Along with a single-cell genetic approach using oncoretroviruses, examination of Girdin−/− and two newly generated sets of mutant mice demonstrated that Girdin is essential for the chain migration of neuroblasts, which is, unexpectedly, independent of its phosphorylation by Akt and interaction with DISC1.
Materials and Methods
Knock-out and knock-in mice.
Construction of the Girdin gene-targeting vector and generation of Girdin−/− mice were described previously (Kitamura et al., 2008). Conventional gene-targeting techniques were used to generate the SA (serine-1417 was replaced with alanine) and Basic-mut knock-in mice. The 5′ 2.4 kb pairs and 3′ 6.3 kb fragments of the mouse girdin gene (ccdc88a), which contain exons 24 and 25 (5′ arm) and 26–28 (3′ arm), respectively, were isolated by PCR and cloned into the targeting vector. Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene) to introduce the SA and the Basic-mut mutations. The final targeting construct contains the SA/Basic-mut carrying exon 25 with a LoxP-flanked PGK-neo-resistant gene cloned downstream of exon 25. The diphtheria toxin-A chain gene was used for negative selection. The linearized targeting vector was electroporated into CSL3 ES cells (a gift from Dr. C.-S. Lin, Columbia University, New York, NY), and homologous recombinants were selected by growth in G418. Targeted ES cell clones identified by Southern blot analysis using an external PCR probe were injected into C57BL/6 blastocysts for generation of chimeric mice. Chimeras that were consistently germline transmitting were mated with β-actin promoter/Cre transgenic mice to excise the neo-resistant gene and evaluate its possible interference. The presence of mutations was verified by genomic DNA sequencing. Heterozygous 129Sv background mice were crossed with each other to generate homozygous mice or were mated with C57BL/6 mice to generate mice on a C57BL/6 genetic background. We confirmed that the phenotypes of the Girdin mutant mice do not depend on the genetic backgrounds (data not shown). All animal protocols were approved by the Animal Care and Use Committee of Nagoya University Graduate School of Medicine.
Immunohistochemistry and immunofluorescence studies.
Brains of wild-type, Girdin−/−, and SA/Basic-mut mice were perfused with 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer, postfixed in the same fixative overnight, cut into 50–60 μm sections on a microslicer (VT1200S; Leica), and immunolabeled for light and fluorescence microscopy. For GABA immunohistochemistry, the fixative contained 4% PFA and 0.25% glutaraldehyde in 0.1 m phosphate buffer. Endogenous peroxidase activity was blocked with 1% hydrogen peroxide in PBS. Free-floating sections were then incubated in a blocking buffer [included in tyramide signal amplification (TSA) kit 15; Invitrogen] containing Triton X-100 (0.1%) for 30 min at 25°C. Primary antibodies were diluted in the blocking buffer and applied to sections overnight at 4°C. After washes with PBS with 0.05% Tween 20 (PBS-T), sections were incubated with horseradish peroxidase-conjugated secondary antibodies and diluted in the blocking buffer for 1 h at 25°C. After washes with PBS-T, immunostaining was detected using a TSA system (CSAII; Dako). Alternatively, biotin-conjugated AffiniPure donkey anti-goat and anti-rabbit IgG secondary antibodies (Jackson ImmunoResearch Laboratories) were used to increase sensitivity. Sections were examined and imaged with a light microscope (BX50; Olympus) equipped with a digital camera (DP70; Olympus).
For immunofluorescence studies, sections were incubated with primary antibodies diluted in PBS containing Triton X-100 (0.1–0.2%) and applied to sections overnight at 4°C. After washes with PBS, sections were incubated with Alexa Fluor 488/594-conjugated secondary antibodies (Invitrogen) and diluted in PBS for 3 h at 25°C. The sections were then mounted with ProLong Gold antifade reagent containing DAPI (Invitrogen), and fluorescence was examined using a confocal laser-scanning microscope (Fluoview FV500; Olympus).
Antibodies.
Rabbit anti-Girdin polyclonal antibodies were developed against the 19 C-terminal amino acids of Girdin and affinity purified with the immunized peptide (Enomoto et al., 2005). Other antibodies and dilutions used in this study include rabbit anti-Girdin polyclonal antibody (1:100; Santa Cruz Biotechnology), anti-V5 monoclonal antibody (1:200; Invitrogen), anti-GFP polyclonal antibody (1:100; MBL International), anti-doublecortin (Dcx) polyclonal antibody (1:100; Santa Cruz Biotechnology), anti-ZsGreen polyclonal antibody (1:100; Clontech), anti-glial fibrillary acidic protein (GFAP) antibody (1:1000; Sigma), anti-NeuN antibody (1:100; Millipore), anti-MASH1 (1:100; BD Biosciences), anti-C-Caspases-3 (1:100; Cell Signaling Technology), anti-Ki67 antibody (1:5000; Novocastra), anti-β-galactosidase antibody (1:5000; Abcam), anti-Reelin monoclonal antibody (G10, 1:1000; Calbiochem), anti-Reelin antibody (CR-50, 1:1000) (Ogawa et al., 1995), anti-calretinin polyclonal antibody (1:500; Millipore), anti-parvalbumin monoclonal antibody (1:2000; Sigma), anti-Calbindin-D-28K monoclonal antibody (1:2000; Sigma), anti-Ctip2 rat monoclonal antibody (1:500; Abcam), and anti-GABA polyclonal antibody (1:5000; Sigma).
Cresyl violet (Nissl) staining.
Paraffin-embedded brain tissue sections were deparaffinized and placed in 0.5% cresyl violet in distilled water at 37°C for 10 min. The sections were rinsed briefly in 90% ethanol twice and dipped in 100% ethanol three times before being dehydrated in xylene three times for 5 min. Sections were covered with glass coverslips adhering to Permount and examined under a light microscope.
Immunoelectron microscopic analysis.
Brains of wild-type, Girdin−/−, and Basic-mut mice were perfused with 4% PFA in 0.1 m phosphate buffer for 1 d at 4°C and cut into 50–60 μm sections on a microslicer (VT1200S; Leica). For immunohistochemistry, floating sections were stained with rabbit anti-Girdin polyclonal antibody (1:100; Santa Cruz Biotechnology) for 2 d at 4°C and washed with PBS-T, followed by incubation with biotin-conjugated anti-rabbit IgG antibody either for 2 d at 4°C or for 2 h at room temperature. Immunostaining was detected by DAB reaction for 10 min. After washing, the sections were postfixed for 20 min in 2% osmium tetroxide at 4°C, dehydrated with alcohols, and embedded in Epon. Ultrathin sections (90 nm thick) including the RMS were cut on an Ultracut S (Reichert-Jung), counterstained with uranyl acetate, and analyzed by an electron microscope (JEM-1400EX; JEOL).
Fluorescent labeling of neuroblasts on acute brain slices.
Sagittal brain sections through the SVZ, RMS, and OB of postnatal day 6 (P6) mice were cut into 350-μm-thick slices with a McIlwain tissue chopper. The slices were dissected out and dissociated in small tissue fragments using needles in L-15 medium (Invitrogen) on a 35 mm dish. A small crystal of DiI was placed onto the SVZ using a microneedle, followed by incubation at 37°C for 12 h under 5% CO2. Images were acquired using a confocal laser-scanning microscope FV500 (Olympus).
Explant cultures and chemoattraction assays.
Brains isolated from P6 mice were dissected in ice-cold Leibovitz's L-15 medium (+) (Invitrogen) and sectioned coronally through the RMS into 350-μm-thick slices with a tissue chopper. Then the RMS slices were cut into square pieces of 100–200 μm in diameter, embedded in a mixture of Matrigel and L-15 medium (1:1), and cultured with Neurobasal medium (Invitrogen) in the presence of 2% B-27 supplement (Invitrogen), 10% FBS, 100 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) on glass-based dishes (Iwaki). The RMS explants were allowed to congeal in a culture dish and were cultured at 37°C under 5% CO2. After 2 d in culture, the explants were fixed in 4% PFA for 30 min, followed by immunofluorescent staining.
For chemoattraction assays, RMS explants were embedded in a mixture of Matrigel and L-15 medium (1:1) together with agarose beads (Sigma) soaked in BSA (control condition), glial cell line-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF), or vascular endothelial growth factor (VEGF) and cultured in Neurobasal medium for 2 d. The explants were fixed in 4% PFA for 30 min and stained with Dcx antibody.
Stereotaxic injection of engineered retroviruses.
Oncoretroviruses that express shRNAs, ZsGreen, and the Girdin N-terminal domain specifically in proliferating cells and their progeny were engineered as described previously (Duan et al., 2007; Enomoto et al., 2009; Kim et al., 2009). To label and deliver exogenous shRNAs or genes in dividing neuroblasts and their progeny in the neonatal SVZ, P5 wild-type or Girdin−/− mice were anesthetized on ice, and 1 μl of the shRNAs/ZsGreen-expressing retroviruses was stereotactically injected into the bilateral SVZ (coordinates from bregma: anterior, 0.4 mm; lateral, 1.0 mm; ventral, 1.0 mm). Three to seven days after retroviral injection, mice were anesthetized with sodium pentobarbital and transcardially perfused with 0.01 m PBS, followed by 4% PFA in 0.1 m phosphate buffer, pH 7.4, at room temperature. Brains were postfixed overnight in 4% PFA. Brains were sectioned with a cryostat (Leica) into 50-μm-thick sections in a plane perpendicular to the septo-temporal axis of the hippocampal formation. Immunostaining was essentially performed as described previously (Enomoto et al., 2009). Fluorescent signals were detected using a confocal laser-scanning microscope (FV500; Olympus). A minimum of 5–10 neurons of randomly picked sections from each animal and at least five animals was analyzed for migration under each experimental condition.
β-Galactosidase staining of mouse sections.
Brains of wild-type and Girdin−/+ mice were perfused with 4% PFA in 0.1 m phosphate buffer, embedded in O.C.T. compound, and cryosectioned into 15-μm-thick sections. The sections were then fixed in LacZ fixative (0.1% glutaraldehyde and 2 mm MgCl2 in PBS) for 10 min on ice, washed twice in LacZ washing buffer (2 mm MgCl2, 0.01% sodiumdeoxycholate, and 1% Nonidet P-40 in PBS) for 10 min, and stained in X-Gal staining solution (1 mg/ml X-Gal, 5 mm potassiumferrocyanide, and 5 mm potassiumferricyanide in LacZ washing buffer) for 1–3 d at 37°C. After color development, sections were washed in PBS, counterstained with Nuclear Fast Red (Vector Laboratories), and mounted. Images were acquired using a light microscope (BX-50; Olympus).
Data analyses.
Data are presented as the mean ± SEM. Statistical significance was evaluated with Student's t tests.
Results
Severe morphological defects in OB development in Girdin−/− mice
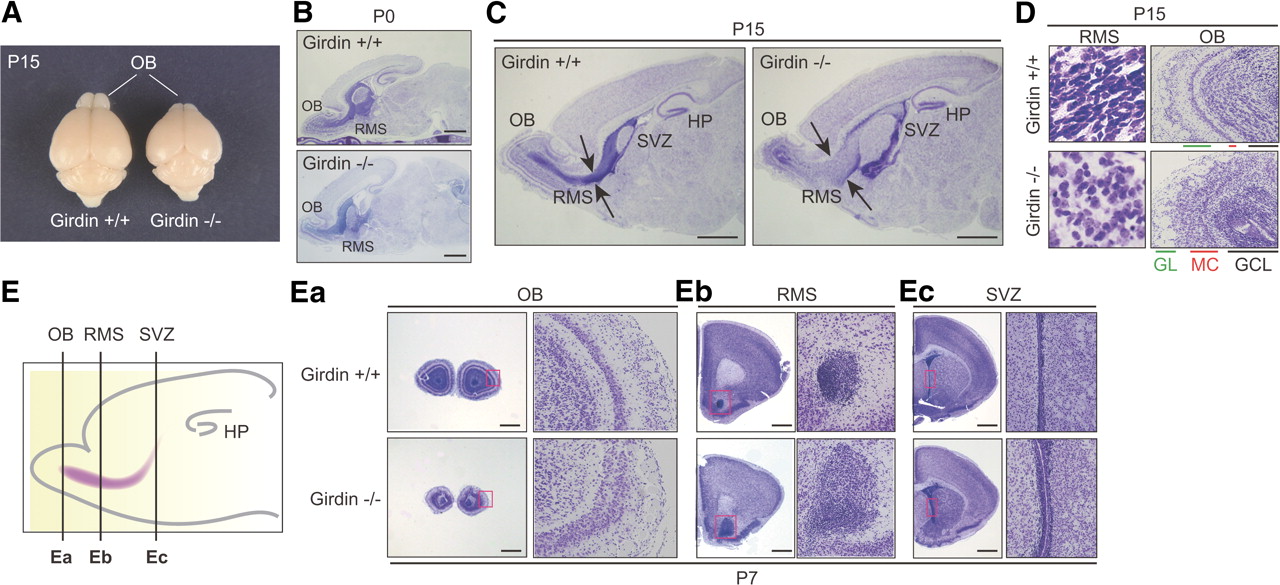
We previously generated Girdin−/− mice to study the function of Girdin and its interaction with DISC1 in postnatal development of the DG (Kitamura et al., 2008; Enomoto et al., 2009; Kim et al., 2009). Following this line of study, we also found severe defects in the development of the OB in Girdin−/− mice (Fig. 1). Grossly, at P15, the brains of Girdin−/− mice exhibited much smaller OBs than wild-type mice although they also showed a mild reduction in overall brain size compared with wild-type mice (Fig. 1A). Examination of the brain structure using Nissl staining revealed an almost normal brain cytoarchitecture in Girdin−/− mice at birth (P0), whereas a reduction in OB size became apparent postnatally compared with wild-type mice (Fig. 1B–E). Sagittal and coronal sections through the brain of Girdin−/− P15 mice suggested migratory defects of SVZ neuroblasts toward the OB, leading to their accumulation at the SVZ and the halfway point of the RMS (Fig. 1C,E). At high magnification, the shape and distribution of SVZ and RMS neuroblasts were distinct between wild-type and Girdin−/− mice (Fig. 1D,E). These data imply that Girdin is involved in directional migration of SVZ neuroblasts toward the OB and that the loss of this signal causes a reduction of OB size. Accordingly, the cytoarchitecture of the granular cell layer (GCL), mitral cell layer, and glomerular layer are all severely disorganized in Girdin−/− mice compared with wild-type mice (Fig. 1D,Ea).

Defects in the development of the RMS and OB in Girdin−/− mice. A, Gross view of the brains of wild-type (left) and Girdin−/− (right) P15 mice, showing the decrease in OB size in Girdin−/− mice. B, C, Nissl-stained brain sections of P0 (B) and P15 (C) wild-type and Girdin−/− mice show significant deficits in the development of the RMS and the OB in Girdin−/− mice. Arrows indicate the width of the RMS. HP, Hippocampus. Scale bars, 1 mm. D, High-magnification views of the RMS (left) and the OB (right) of wild-type and Girdin−/− mice. GL, Glomerular layer; MC, mitral cell layer. E, Coronal sections through the OB (Ea), the RMS (Eb), and the SVZ (Ec) of wild-type (top) and Girdin−/− (bottom) P7 mice. The regions within the red boxes are shown at a higher magnification in adjacent panels. The far left panel shows schematic presentations of the cutting levels of coronal brain sections throughout this study. Scale bars, 200 μm.
Expression of Girdin in type A SVZ neuroblasts migrating to the OB
Supporting the defects in neuronal cytoarchitecture in Girdin−/− mice, immunohistochemical studies using anti-Girdin antibodies showed prominent expression of Girdin in interneurons that populate the GCL of the OB and neuroblasts in the RMS and the SVZ (in addition to the DG and the CA1 and CA3 regions of the hippocampus as described previously), in agreement with in situ hybridization data (Fig. 2A) (Lein et al., 2007; Enomoto et al., 2009). Girdin expression patterns were also confirmed by β-galactosidase staining of brain sections from mice heterozygous for a knock-in mutation that places the LacZ gene containing a nuclear localization signal (LacZ-NLS) in frame with the initiator methionine sequence of the girdin gene (Fig. 2B) (Kitamura et al., 2008). As expected, the expression of the LacZ-NLS was detected in a similar pattern to the immunohistochemical data, except that some sets of cells were positive for the LacZ-NLS in the cortex and other brain regions, which could be attributable to higher sensitivity of β-galactosidase staining procedures.

Expression of Girdin in Dcx-positive migrating neuroblasts. A, Brain sections from wild-type (right) and Girdin−/− (far left) P15 mice were stained for Girdin. Brown staining denotes a positive Girdin signal. The regions within the black boxes (A and B) are shown at a higher magnification in adjacent panels. LV, Lateral ventricle; IHC, immunohistochemistry. Scale bars, 1 mm. B, X-gal staining of brain cryostat sections from wild-type (far left) and Girdin−/+ (right) P7 mice. The regions within the black boxes [the RMS and the hippocampus (HP)] are shown at a higher magnification in adjacent panels. Scale bars, 1 mm. C, Expression of Girdin in Dcx-positive type A neuroblasts but neither GFAP-positive astrocytes nor MASH1-positive transit amplifying cells in the SVZ. SVZ sagittal sections from wild-type and Girdin−/− P16 mice were double stained with the indicated antibodies for neuronal differentiation markers and Girdin antibodies. Enlarged images are shown in adjacent panels. D, Expression of Girdin in migrating neuroblasts in the RMS. RMS sagittal sections from wild-type P15 mice were double stained with anti-Girdin and indicated antibodies. Enlarged images are shown in insets. Scale bars, 50 μm.
To determine which neuroblast developmental stages express Girdin, the sections were double stained with Girdin and several markers for neuronal development (Fig. 2C,D). Girdin expression was detected only in SVZ neuroblasts positive for Dcx, a marker for dividing type A neuroblasts (type A cells), but not in either neural stem cells (type B cells) positive for GFAP or transient amplifying cells positive for the basic helix-loop-helix transcription factor MASH1 (Fig. 2C) (Duan et al., 2008; Zhao et al., 2008). In line with this finding, Dcx (but not other markers) was expressed in Girdin-positive cells in the RMS (Fig. 2D). Together, these data support the notion that Girdin is specifically expressed in type A neuroblasts, the majority of which migrate to the OB.
Girdin deficiency impairs chain migration of neuroblasts in the RMS
Supporting the findings of the Nissl staining experiments (Fig. 1), the involvement of Girdin in the formation and maintenance of chain migration of SVZ neuroblasts became more apparent when sections of Girdin−/− and wild-type mice were stained for Dcx (Fig. 3A). In wild-type mice, the Dcx-positive neuroblasts form densely packed, chain-like structures as they migrate in groups within the RMS, whereas, in Girdin−/− mice, the neuroblasts migrate individually and sometimes perpendicular to each other along the direction of the stream (Fig. 3A). One interpretation of this phenomenon is that cell–cell interactions and cell polarity of the neuroblasts may be compromised in Girdin−/− mice, which results in a widely dispersed RMS. Additionally, we observed that in coronal sections through the SVZ of Girdin−/− mice, some neuroblasts seemed to be mismigrating toward the striatum, further implicating a role for Girdin in determining cell directionality, the loss of which may lead to a dysregulated commitment of neuroblasts into other migratory pathways (Fig. 3B) (Inta et al., 2008). Neuroblasts were quantified by counting the Dcx-positive cells through coronal sections, and this analysis revealed that the defects in the migration of Dcx-positive neuroblasts result in their accumulation in the RMS and the SVZ in Girdin−/− mice (Fig. 3C).

Girdin deficiency impairs chain migration of RMS neuroblasts. A, P15 sagittal sections through the RMS were stained with anti-Dcx antibody, followed by DAB detection. The regions of the RMS within black boxes (a–d) are shown at a higher magnification on the right. In Girdin−/− mice, most of the Dcx-positive neuroblasts are migrating individually, and their leading processes are sometimes perpendicular to the direction of the stream (arrowheads). Scale bars: far left, 1 mm; a–d, 20 μm. B, P15 coronal sections through the OB, RMS, and SVZ were stained with anti-Dcx antibodies, showing dispersed RMS in Girdin−/− mice. RMS regions are indicated by dashed lines. Note that some neuroblasts were mismigrating toward the striatum in Girdin−/− mice (arrowheads). St., Striatum. Scale bars, 100 μm. C, Quantification of the distribution of Dcx-positive neuroblasts in wild-type and Girdin−/− P15 mice. The number of neuroblasts per high power field was counted and quantified. Five sections of the OB, RMS, and SVZ from three brains each of wild-type and Girdin−/− mice were evaluated. Data are expressed as the mean ± SEM, and comparison between wild-type and Girdin−/− mice was done by Student's t test. Asterisks indicate significant difference (p < 0.01) between wild-type and Girdin−/− mice. D, Gliosis of the RMS of Girdin−/− mice. Sagittal sections through the RMS (far left) and coronal sections through the OB (right) of wild-type and Girdin−/− P15 mice were stained with anti-Dcx (red) and anti-GFAP (green) antibodies. GFAP-positive cells (arrowheads) were frequently found in the RMS and intermixed with Dcx-positive neuroblasts in Girdin−/− mice. Scale bars, 100 μm. IHC, Immunohistochemistry.
We next investigated whether the increased number of SVZ and RMS neuroblasts was caused by a decrease in their apoptosis and/or an increase in their proliferation. Neuroblast apoptosis was evaluated by immunohistochemical staining of brain sections prepared from P15 wild-type and Girdin−/− mice with an antibody that recognizes only activated and cleaved caspase-3 (Srinivasan et al., 1998). In Girdin−/− mice, apoptosis was significantly increased in the SVZ but not in the RMS (Fig. 4A,B). In contrast, no significant change in the number of Ki-67-positive cells was observed in either the SVZ or RMS between wild-type and Girdin−/− mice (Fig. 4C,D). These data indicate that neuroblast migration defects in Girdin−/− mice resulted in increased neuroblast apoptosis in the SVZ.

Effects of Girdin deficiency on the apoptosis and the proliferation of SVZ neuroblasts. A, P15 coronal sections through the SVZ and RMS were stained with anti-activated Caspase-3 antibody, followed by DAB detection. Activated Caspase-3-positive cells (brown, highlighted by arrowheads) in the SVZ were numerous in Girdin−/− mice (right) compared with wild-type mice (left). B, Activated Caspase-3-positive cells were counted in six sections of the RMS or SVZ. Data are expressed as the mean ± SEM, and a comparison between wild-type and Girdin−/− mice was done by Student's t test. An asterisk indicates a significant difference (p < 0.01) between wild-type and Girdin−/− mice. C, P15 coronal sections through the SVZ and the RMS were stained with anti-Ki-67 antibody, followed by DAB detection. Ki-67-positive cells that progressed through the cell cycle (brown, highlighted by arrowheads) in these regions were similar between Girdin−/− mice (right) and wild-type mice (left). D, Ki-67-positive cells were counted in six sections of the RMS or SVZ. Data are expressed as the mean ± SEM, and a comparison between wild-type and Girdin−/− mice was done by Student's t test. No apparent significant difference between wild-type and Girdin−/− mice was found. IHC, Immunohistochemistry.
In the SVZ, newborn neuroblasts are arranged in chains surrounded by astrocyte-like stem cells, and they subsequently migrate tangentially along the RMS within a tunnel of astrocytes and extracellular matrices (Alvarez-Buylla and García-Verdugo, 2002). To examine the role of Girdin in the interactions between neuroblasts and surrounding astrocytes, sagittal and coronal sections from wild-type and Girdin−/− mice were double stained for Dcx and GFAP (Fig. 3D). In Girdin−/− mice, the Dcx-positive neuroblasts were significantly intermixed with GFAP-positive astrocytes, resulting in extensive gliosis in the RMS and OB, as was also reported in NCAM- and Slit1-deficient mice (Chazal et al., 2000; Kaneko et al., 2010). Thus, Girdin seems to be fundamental not only for directional migration but also for the segregation of neuroblasts and astrocytes, both of which are required for the neuroblasts to move in chains through the RMS.
Girdin is essential for neuroblast chain migration along the RMS
To precisely track the fate and distribution of postnatally generated neuroblasts, we used a retroviral-mediated approach (Ming and Song, 2005; Duan et al., 2007; Enomoto et al., 2009; Kim et al., 2009) to label only newly generated neuroblasts in wild-type and Girdin−/− neonatal mice (Fig. 5A). A retrovirus engineered to express Zoanthus green fluorescent protein (ZsGreen) was stereotactically injected into the SVZ of P5 mouse brain to infect only dividing stem cells or transient amplifying cells in vivo. In wild-type mice, at 3 d after injection, the majority of ZsGreen-expressing neuroblasts had migrated along the RMS with proper directionality, seemed to be closely apposed to each other to form chains, and reached the OB at 7 d after injection (Fig. 5A). In contrast, in Girdin−/− mice, ZsGreen-expressing neurons were randomly oriented with leading processes crossing each other at different angles and never reached the OB within the experimental period.

Girdin is essential for neuroblast chain migration in the RMS. A, Severe defects in the migration of newborn neuroblasts in Girdin−/− mice. A retrovirus bearing the ZsGreen cDNA was stereotactically injected into the SVZ of wild-type (top) and Girdin−/− (bottom) neonatal (P5) mice. Shown are sample confocal images of immunostaining with ZsGreen (green) and Dcx (red) antibodies. Chromosomal DNA was visualized with DAPI staining. RMS regions within white boxes are shown at a higher magnification in adjacent panels. At 3 and 7 d after the injection, the leading processes of the ZsGreen-positive neuroblasts are perpendicular to each other, leading to defects in neuroblast migration in Girdin−/− mice. In Girdin−/− mice, the majority of neuroblasts fail to reach the OB 7 d after retroviral injection (far right). Scale bars: far left, 1 mm; middle, 20 μm. B–D, Analysis of tangential neuroblast migration in brain slices. SVZ neuroblasts in acute brain slices isolated from wild-type and Girdin−/− P6 mice were labeled with DiI crystals placed in the SVZ (asterisks) and cultured for 12 h. The number of DiI-labeled migrating neuroblasts (C) and their migration distance (D) were quantified. In brain slices isolated from wild-type mice, the DiI-labeled neuroblasts tangentially migrated from the SVZ toward the OB in contrast to brain slices isolated from Girdin−/− mice, where the neuroblasts stayed within the SVZ and poorly migrated out from the SVZ. Asterisks indicate a significant difference (p < 0.0001) between wild-type and Girdin−/− mice. HP, Hippocampus. E, Cell-intrinsic function of Girdin in the migration of neuroblasts. Neuroblasts in the SVZ isolated from wild-type P6 mice were dissociated, labeled with DiI, and transplanted onto acute brain slices prepared from Girdin−/− mice (top). In a reciprocal experiment, SVZ neuroblasts from Girdin−/− mice were transplanted onto wild-type brain slices (bottom). Twenty-four hours after transplantation, fluorescent images show that Girdin−/− mice-derived neuroblasts failed to migrate toward the OB on the wild-type brain slices. Yellow arrowheads denote migrating neuroblasts.
To further confirm the role of Girdin in neuroblast migration along the RMS, solubilized carbocyanine dye DiI (1,1′-dioctadecyl-3,3,3c3′-tetramethylindocar-bocyanine perchlorate) was administered to the SVZ of acute brain slices prepared from wild-type and Girdin−/− P6 mice, and the slices were cultured for 12 h to label SVZ neuroblasts (Fig. 5B). The number of migrating neuroblasts and their migration distance were quantified and analyzed (Fig. 5C,D). The DiI-labeled neuroblasts in wild-type slices, but not Girdin−/− slices, had successfully migrated toward the OB, suggesting that Girdin is required for neuroblasts in brain slices to respond to guidance cues provided under this experimental condition.
A more important finding was that dissociated neuroblasts from the SVZ of P5 Girdin−/− mice were defective in migrating toward the OB through the RMS when they were transplanted into the SVZ of wild-type slices (Fig. 5E). In a reciprocal experiment, in which the neuroblasts from the SVZ of wild-type mice were transplanted into the SVZ of Girdin−/− slices, the neuroblasts retained the ability to migrate toward the OB. Together, our findings demonstrate that Girdin regulates neuroblast migration downstream of extrinsic growth factors provided by their environment, an effect that seems to be maintained in acute slices.
Girdin is required for chain migration of cultured neuroblasts
From the data described above, it was concluded that impaired migration of newborn neuroblasts is the responsible mechanism for OB developmental defects in Girdin−/− mice. To test in detail the effects of Girdin deficiency on the migratory mode of individual neuroblasts, and to ask whether Girdin regulates their chain migration in a cell-autonomous fashion independent of extracellular matrix and surrounding astrocytes, we next used in vitro cultures of isolated neuroblasts or acute RMS explants (Fig. 6) (Wu et al., 1999; Murase et al., 2008). First, when acute RMS explants obtained from wild-type P6 mice (Girdin+/+ RMS) were embedded in Matrigel and cultured for short periods (<24 h), Dcx-positive neuroblasts started to migrate out around the explant (Fig. 6A). After additional culture of Girdin+/+ RMS explants (>48 h), compact chains of Dcx-positive migrating neuroblasts were symmetrically distributed around the explant (Fig. 6B,C). In contrast to the above findings, when acute RMS explants obtained from Girdin−/− P6 mice (Girdin−/− RMS) were cultured under the same conditions, most of them failed to translocate their soma and nuclei forward and were motionless, although the neuroblasts started to extend their leading processes (Fig. 6B,C). A few neuroblasts were able to migrate out of Girdin−/− RMS explants, but they failed to form chains and migrated at a lower velocity than neuroblasts from Girdin+/+ RMS explants (Fig. 6B,D). These data indicate that Girdin is an important intrinsic protein that determines the migratory mode of neuroblasts in in vitro cultures.

Girdin is essential for neuroblast chain migration and chemoattraction. A, Expression of Girdin in Dcx-positive cultured neuroblasts. RMS explants from wild-type P6 mice were cultured in Matrigel for 24 h. The explants and emigrating neuroblasts were stained with anti-Girdin (green) and anti-Dcx (red) antibodies. Scale bars, 100 μm. B, RMS explants from wild-type (left) and Girdin−/− (right) P6 mice were cultured in Matrigel for 2 d. The explants and emigrating neuroblasts were stained with anti-GFAP (green) and anti-Dcx (red) antibodies. The transmitted differential interference contrast (DIC) images show that the neuroblasts migrate out from the explant as chains (yellow arrowheads) in wild-type mice, whereas cells in Girdin−/− mice failed to migrate out from the explants (red arrowheads) with only the extension of leading processes (white arrowheads). C, D, Time-lapse observation of neuroblasts emigrating from wild-type and Girdin−/− RMS explants cultured in Matrigel. The morphology of individual neuroblasts was observed in vitro by taking sequential DIC images of neuroblasts at 30 min intervals. Arrows and arrowheads denote cell bodies and leading processes of migrating neuroblasts, respectively. In neuroblasts from Girdin−/− RMS explants, the nuclei seem to stay in the proximity of the explant with only an extension of leading processes. Presented are representative images of two independent experiments. D, The velocity of neuroblasts migrating from Girdin+/+ and Girdin−/− RMS explants was quantified and analyzed. An asterisk indicates a significant difference (p < 0.0001) between wild-type and Girdin−/− mice. E, Girdin is essential for chemoattractive migration of cultured neuroblasts. RMS explants from wild-type (top) and Girdin−/− (bottom) P6 mice were cocultured with agarose beads (arrowheads) coated with the indicated growth factors for 2 d. The explants and emigrating neuroblasts were stained with anti-Dcx antibodies (magenta). F, G, Quantification of the effects of growth factors on neuroblast migration after Dcx immunostaining. F, Schematic illustration of the scoring principle: the number of cells within quadrant regions being proximal and distal against the beads were counted and quantified as shown in G (left). G, Right, Areas of migrating neuroblasts, which was defined as the sum of the pixels of the areas with positive Dcx staining, were evaluated and quantified. For each condition, a total of 10–15 explants were evaluated in three independent experiments. Data are expressed as the mean ± SEM, and comparisons between proximal and distal quadrants were analyzed by Student's t test. Asterisks indicate a significant difference (p < 0.01) between proximal and distal quadrants.
Next, Girdin+/+ and Girdin−/− RMS explants were cultured in close proximity with agarose beads coated with growth factors including GDNF, BDNF, and VEGF, which have been shown to induce the migration of neuroblasts (Paratcha et al., 2006; Chiaramello et al., 2007; Wittko et al., 2009). We found that the neuroblasts in Girdin−/− RMS explants displayed an impaired ability to migrate out in response to the chemogradients provided by the growth factors, suggesting that Girdin acts downstream of their cognate receptors to activate intracellular migratory machinery (Fig. 6E–G).
The basic amino acid residue-rich region in the C-terminal domain of Girdin is responsible for the regulation of neuroblast migration
Possible mechanisms that control neuroblast migration include the phosphorylation of serine at residue 1417 (Ser-1417) in the C-terminal (CT) domain of Girdin by Akt downstream of growth factor stimulation (Enomoto et al., 2005; Jiang et al., 2008; Kitamura et al., 2008; Weng et al., 2010). Another possibility concerns a basic amino acid residue-rich (BR) region (amino acids 1399–1408) located adjacent to the Akt phosphorylation site (Fig. 7A). The mutation of two basic residues (lysine and arginine) to alanines within this site attenuates the localization of the CT1 domain of Girdin to the plasma membrane (Fig. 7A) (Enomoto et al., 2005). To investigate whether one or both of these putative mechanisms are necessary for neuroblast migration in the RMS, we generated knock-in mice in which we substituted either (1) Ser-1417 with an alanine (termed ”SA”) or (2) the six basic amino acid residues (1399, 1401, 1402, 1405, 1407, 1408) with alanines (termed “Basic-mut”) by replacing exon 25 of the mouse girdin gene with the mutated exon (Fig. 7A–C). Western blot analysis using brain lysates from mice with homozygous mutations (SA/SA and Basic-mut/Basic-mut, used in subsequent experiments) showed that the expression levels of Girdin in these mutant mice are comparable to those observed in wild-type mice (Fig. 7C). As expected, serine-1417 phosphorylation of Girdin was undetectable in SA/SA mice (Fig. 7C). Interestingly, phosphorylation of this site was also markedly decreased in Basic-mut/Basic-mut mice.

The BR region in the CT domain of Girdin, but not its phosphorylation by Akt, is responsible for regulation of neuroblast migration. A, Proposed domain structures of mouse Girdin. Girdin is composed of three domains: an N-terminal domain (NT) that binds to DISC1, a central coiled-coil domain, and a CT domain that binds to actin filaments and the Gα protein family (Enomoto et al., 2005; Ghosh et al., 2008; Jiang et al., 2008; Kitamura et al., 2008; Weng et al., 2010). An Akt phosphorylation site and a BR region are contained in the CT1 domain located in the first half of the CT domain. Shown in red characters are amino acid mutations introduced in the mutant mice generated in this study. B, Generation of SA and Basic-mut knock-in mutant mice. The mouse girdin gene consists of 33 coding exons, and exons 23–29 are shown here. Homologous recombination of the gene-targeting vector at the girdin locus (wild-type allele; top) was designed to insert a PGK-neo cassette into intron 25 and introduce indicated amino acid mutations that are encoded by exon 25. The structure of the targeted girdin allele (bottom) is shown. The position of the PCR-amplified genomic DNA probe used to screen ES cell colonies by Southern blotting is shown in the red box. Restriction enzyme sites for Southern blotting and predicted size of bands on Southern blotting are also shown. All data pertaining to mutant mice in this study were prepared in mutant mice without the neo gene. DTa, diphtheria toxin-A. C, Western blot analysis of brain lysates from wild-type and indicated mutant mice using anti-Girdin, anti-phospho-Girdin (P-Girdin), and anti-β-actin antibodies. WB, Western blot. D, Body weights and mortality rates in offspring from wild-type and Basic-mut mice. E, P15 sagittal brain sections through the OB, RMS, and hippocampus of SA and Basic-mut mice were Nissl stained (far left) and immunostained with Dcx antibodies (right). Subregions of the RMS and the DG within the black boxes (a–d) are shown at a high magnification in adjacent panels. In Basic-mut mice, but not SA mice, the migration of Dcx-positive neuroblasts was dysregulated both in the RMS and the DG, which seems to recapitulate the phenotype of Girdin−/− mice. IHC, Immunohistochemistry; HP, hippocampus. F, The BR region of Girdin is responsible for the regulation of migration in cultured neuroblasts. RMS explants from the SA and the Basic-mut P6 mice were cultured in Matrigel for 2 d. The explants and emigrating neuroblasts were stained with Dcx antibodies (magenta). Nuclei were visualized with DAPI staining (white).
Similar to what we observed with Girdin−/− mice (Kitamura et al., 2008; Enomoto et al., 2009), both the SA and the Basic-mut mice survive embryogenesis and are born without apparent gross abnormalities (data not shown). It is of interest that the Basic-mut mice displayed significant weight loss after P7–P8 and the survival rate declined over time to about 40% at P52 (Fig. 7D). These mice do survive longer than Girdin−/− mice, which have been reported to die by P25 (Kitamura et al., 2008). We did not observe any defects in the growth and survival rate of the SA mice (data not shown), suggesting that Akt-mediated phosphorylation of Girdin is dispensable for embryonic and early postnatal life.
We next examined the cytoarchitecture of the RMS and the OB by Nissl staining and Dcx immunohistochemistry (Fig. 7E). In the Basic-mut mice, but not the SA mice, neuroblasts accumulated along the way within the RMS, and the development of the OB was significantly impaired because of these defects in neuroblast migration, reminiscent of the phenotype of Girdin−/− mice (Fig. 7E). Also, in the DG of the hippocampus of the Basic-mut mice (but not the SA mice), we observed an overmigration and an altered positioning of newborn dentate granule cells (DGCs) in the GCL, suggesting that the BR region of Girdin is important for the development of both the OB and the DG (Fig. 7E).
Importantly, in vitro culture of RMS explants also supports this notion. Most of the neuroblasts in RMS explants isolated from the Basic-mut mice did not migrate out of the explants, in contrast to those taken from the SA mice (Fig. 7F).
Involvement of Girdin in neuroblast cell–cell interactions
The defects in chain migration in Girdin−/− mice may be attributed to the loss of appropriate cell–cell adhesion of neuroblasts. In agreement with the Dcx immunostaining data described above, an electron microscopic analysis showed zonula adherens-like cell–cell contact zones between neuroblasts (Doetsch et al., 1997), which were observed at the ultrastructural level on coronal sections of the RMS in wild-type mice but were not evident in Girdin−/− mice (Fig. 8A). It is of note that, in Basic-mut mice, the contact zones between closely apposed neuroblasts seem to be immature and persist for a shorter distance compared with those observed in wild-type mice, which continuously extend up to 0.5–1 μm in length without interruption.

Involvement of Girdin in neuroblast cell–cell interactions. A, Defects in cell–cell interactions of neuroblasts in Girdin−/− and Basic-mut mice. Selected coronal sections through the RMS from indicated mice were analyzed by electron microscopy. The regions in white boxes are shown at a higher magnification in adjacent panels. In wild-type mice (left), zonula adherens-like contact zones appear between closely apposed neuroblasts over a long distance (arrowheads), which seem to be absent or immature in Girdin−/− (middle) or Basic-mut (right, arrowheads) mice, respectively. N, Nuclei. B, C, Electron immunocytochemistry for the detection of Girdin localization in the RMS of wild-type (left), Girdin−/− (middle), and Basic-mut (right) mice. In wild-type mice, Girdin preferentially localizes to small zonula adherens-like contacts between neuroblasts (arrowheads). In contrast, in Basic-mut mice, Girdin seems to be localized to the cytoplasm apart from the plasma membrane (white arrowheads). C, Quantification of the percentage of membrane-localized immunoreactivity for Girdin in the neuroblasts of wild-type and Basic-mut mice. Girdin subcellular localization was classified into three different categories (both sides of cell–cell contacts; one side of cell–cell contacts; cytoplasm) based on the observation of Girdin immunoreactivity.
We also performed immunoelectron microscopic analysis of brain sections through the RMS of wild-type and Basic-mut mice (Fig. 8B,C). Girdin−/− mice were also examined as a control for staining. In the wild-type mice, Girdin preferentially localizes at cell–cell contact sites in neuroblasts, whereas in the Basic-mut mice, the Girdin mutant protein tends to be localized in the cytoplasm without associating with the plasma membrane (Fig. 8B,C). These results suggest that Girdin is involved in neuroblast cell–cell interactions through the function of the BR region in its CT domain.
No apparent involvement of DISC1 in neuroblast migration in the RMS
We recently showed that Girdin interacts with DISC1 in immature DGCs, where the Girdin/DISC1 protein complex has a critical role in both the migration and proper positioning of the DGCs within the granule cell layer (Enomoto et al., 2009; Kim et al., 2009; Porteous and Millar, 2009). Given that the DG and the SVZ, two major late-developing structures of the brain, share common mechanisms for neurogenesis, it is feasible to consider a possible involvement of the Girdin/DISC1 protein complex in neuroblast migration along the RMS. Injection of an oncoretrovirus engineered to knock down endogenous DISC1 (Fig. 9A) (Duan et al., 2007; Kim et al., 2009) into the SVZ, however, resulted in no apparent delay in neuroblast migration compared with the control condition at 4 d after injection (Fig. 9B,C), indicating that neuroblast migration is most likely independent of DISC1 function in the RMS. This is also supported by an additional time course experiment, which showed that the numbers of neuroblasts reaching the OB are comparable between control and DISC1 knockdown groups at 7 d after injection (Fig. 9D,E). These findings suggest that, in SVZ neuroblasts, DISC1 may regulate subtle alterations in cellular morphology and function but not the migration of neurons, leading to the hypothesis that distinct mechanisms exist for neuroblast migration in the DG versus the SVZ.

No apparent involvement of DISC1 and its interaction with Girdin in neuroblast migration in the RMS. A, Schematic diagram of the retroviral vector (pUEG) harboring control or DISC1 shRNAs used for in vitro birth-dating and genetic manipulation. LTR, Long terminal repeat. B–E, No apparent effects of DISC1 knockdown on the migration of newborn neuroblasts. Control or DISC1 shRNA-expressing retroviruses were injected into the SVZ of neonatal (P4) mice. Four (B, C) and seven (D, E) days after the retrovirus injection, the OB and RMS sagittal sections were prepared and immunostained (IS) with anti-GFP antibodies. Chromosomal DNA was visualized with DAPI staining. Shown in C and D is a quantification of the numbers of neuroblasts that reach the OB at the experimental time points. N.S., Not significant.
Cytoarchitectonic structure of the neocortex in Girdin−/− mice
Based on the above understanding of the phenotype of Girdin−/− mice, one of the goals of the present study was to determine how Girdin depletion affects the migration and differentiation of other neuronal populations in the forebrain. Immunostaining of brain sections of wild-type and Girdin−/− P15 mice for the calcium-binding protein Calbindin-D-28K and Ctip2 (COUP-TF-interacting protein 2), which are markers for cortical layers II/III and V, respectively, showed no evident disruption of cortical layers or heterotopic displacement of neurons (Fig. 10A). Consistent with this finding was the complementary results obtained from the immunostaining of brain sections of embryonic day 14 (E14) mice for Reelin, a marker of Cajal-Retzius (CR) cells that regulates cortical plate lamination during embryogenesis (D'Arcangelo et al., 1995), showing that the number and the distribution of CR cells were comparable between wild-type and Girdin−/− E14 embryos (Fig. 10B).

Cytoarchitectonic structure of the neocortex in Girdin−/− mice. A, Cortical layering and the distribution of Calbindin/Ctip2-positive interneuron in Girdin−/− mice. P15 sagittal sections of wild-type and Girdin−/− mice were stained with anti-Calbindin (left) and Ctip2 (right) antibodies, followed by DAB detection. On the right of each panel, the number of Calbinding/Ctip2-positive neurons in each layer was counted and quantified. An asterisk indicates a significant difference (p < 0.01) between wild-type and Girdin−/− mice. B, Distribution of Cajal-Retzius (CR) cells in Girdin−/− mice. E15 coronal sections of wild-type and Girdin−/− mice were stained with anti-Reelin antibody (left), and the number of Reelin-positive cells in the marginal zone in each hemisphere was counted and quantified (right), showing that Girdin−/− mice show a comparable distribution/positioning of CR cells as observed in wild-type mice. Arrowheads indicate Reelin expression in CR cells. C, Distribution of Reelin-positive neurons in Girdin−/− postnatal mice. P15 sagittal sections were stained with anti-Reelin antibody. Right, The cortex area of each section was roughly divided into three layers, and the number of Reelin-positive neurons in each layer was counted and quantified, showing no evident deficits in the distribution of Reelin-positive neurons in Girdin−/− mice. D, Defects in the development of cortical interneurons in Girdin−/− mice. P15 sagittal sections were stained with anti-GABA (far left), anti-parvalbumin (middle), and anti-Calretinin (far right) antibodies. On the right of each panel, the number of immunoreactive neurons was counted and quantified, showing impaired migration and/or differentiation of GABAergic cortical interneurons in Girdin−/− mice. Asterisks indicate a significant difference (p < 0.001) between wild-type and Girdin−/− mice. N.S., Not significant.
In contrast, subsequent immunostaining for GABA, a marker for inhibitory cortical interneurons, showed that the number of GABAergic interneurons is significantly decreased in Girdin−/− mice compared with wild-type mice (Fig. 10D). In agreement with this was the observation that cells positive for parvalbumin and calretinin, markers of differentiated interneuron subtypes, are also significantly decreased in Girdin−/− mice (Fig. 10D). In addition, we observed a significant decrease in Calbindin-positive interneurons in cortical layers IV–VI of Girdin−/− mice (Fig. 10A). The data suggest that Girdin appears to be required for either cortical interneuron migration from their birthplace (the ganglionic eminence of the basal telencephalon) toward the developing neocortex (Wonders and Anderson, 2006) or for the acquisition of a GABAergic phenotype at later times. Given that SVZ neuroblasts also give rise to interneurons in the OB, this observation raises the possibility that Girdin serves as a common regulator for the development of cortical interneuron subgroups from the ventral telencephalon. However, this may be an overly simplistic hypothesis. In our subsequent experiments, the number of neurons expressing Reelin, which is known to be expressed in some subsets of interneurons in the neocortex of postnatal and adult brains (Alcántara et al., 1998), was similar between wild-type and Girdin−/− P15 mice (Fig. 10C). Additional studies are needed to discern how Girdin is involved in the development and fate determination of interneurons and neuronal architecture in the neocortex.
Discussion
To date, a multitude of proteins including growth factors and their receptors, transcription factors, and adhesion molecules have been determined to be important for neuroblast migration in the RMS (Ming and Song, 2005; Lledo et al., 2006; Duan et al., 2008; Zhao et al., 2008). To our knowledge, only a few proteins whose whole-body knockout in mice causes severe defects in the development of the RMS have been identified, including the cell–cell adhesion molecule NCAM (Cremer et al., 1994; Ono et al., 1994; Rutishauser, 2008) and the extracellular signaling molecule Reelin (Hack et al., 2002). Our current study identifies Girdin as another specific regulator of neuroblast migration in the postnatal development of the RMS and neurogenesis in the SVZ. The phenotype of Girdin−/− mice was analogous to that of NCAM-deficient mice in that both mice exhibit severe defects in the development of the OB. Our study, however, revealed unique features of Girdin−/− mice that are distinct from those of NCAM-deficient mice. First, the defect in neuroblast migration and resulting traffic jam in the RMS seem to be more evident in Girdin−/− mice than in NCAM-deficient mice. Second, NCAM-deficient mice appear healthy for up to 4 months of age (and fertile) in contrast to the shortened life span of Girdin−/− mice (Cremer et al., 1994; Kitamura et al., 2008). Third and most importantly, in in vitro cultures of RMS explants isolated from Girdin−/− mice, the neuroblasts failed to form chains, which stands in contrast to previous reports that NCAM is not essential for chain migration but is required for the interaction between neuroblasts and surrounding astrocytes and neuroblast intracellular signal transduction (Chazal et al., 2000; Gascon et al., 2007). At present, the precise mechanisms for how Girdin regulates neuroblast chain migration are unknown, and there remains a possibility that the phenotype found in Girdin−/− mice could be attributable to indirect developmental alterations resulting from the defects in cell migration. Insights into these issues come from our immunohistochemical and electron microscopic analyses, which revealed that cell–cell interactions and localization of Girdin on cell–cell contact sites were severely compromised in Girdin−/− and Basic-mut mice. Given that Girdin is an actin-binding protein (Enomoto et al., 2005), our data suggest possible previously unknown functions for Girdin at the interface between the plasma membrane and intracellular cytoskeleton.
One of the remarkable findings of the present study is that the Basic-mut mice exhibited a phenotype indistinguishable from that of Girdin−/− mice, with severe cytoarchitectural abnormalities in the OB and RMS as well as the DG (Fig. 7). We previously reported from in vitro protein–lipid overlay assays the possibility that the BR region in the CT domain of Girdin associates with the plasma membrane via binding to phosphoinositides (Enomoto et al., 2005). Although no direct evidence has been presented demonstrating that the BR region directly interacts with the plasma membrane in cells or in vivo, the deletion of the BR region leads to an attenuation of EGF- or insulin-like growth factor 1-induced directional migration of fibroblasts and breast cancer cells, indicating a biological importance of the BR region in cell migration (Enomoto et al., 2005; Jiang et al., 2008). It seems that the phenotype observed in the Basic-mut mice can be attributed to defects in neuroblast migration along the RMS, suggesting that the BR region of Girdin is an essential site that transmits extrinsic signals to intracellular migratory mechanisms. In this regard, what is interesting are the findings from our in vitro experiments showing that most of the neuroblasts in acute RMS explants isolated from Girdin−/− and the Basic-mut mice totally failed to translocate their soma forward and were motionless (Figs. 6, 7). This is somewhat inconsistent with the cytoarchitecture of the RMS of Girdin−/− and Basic mut mice in which SVZ neuroblasts migrate very slowly but steadily toward the OB (Figs. 1, 7). Therefore, what should be noted here is the seeming importance of synergistic interactions between neuroblasts and surrounding astrocytes that create a niche for the development of the RMS and the OB.
Our previous data have shown that Akt-mediated phosphorylation of Ser-1417 has an important role in the motility of breast cancer cells and endothelial cells (Jiang et al., 2008; Kitamura et al., 2008; Weng et al., 2010). Much to our surprise, analysis of SA mice revealed that Akt-mediated phosphorylation of Ser-1417 is dispensable for neuroblast migration in the RMS, thus providing a new view that there are distinct mechanisms of action for Girdin between neural cells and non-neural cells. We propose that Girdin is an essential regulator of the migration of SVZ neuroblasts, whereas its phosphorylation by Akt is dispensable, which is in contrast to postnatal angiogenesis and cancer cell migration mechanisms. Nonetheless, there remains the possibility that these regulatory mechanisms fine-tune the function of Girdin to evoke subtle alterations of cellular morphology and function in a tissue- or context-dependent manner.
Despite an extensive array of studies on DISC1, the expression of DISC1 and its role in SVZ neurogenesis remain elusive, in part because of a lack of DISC1-deficient mice and a significant difficulty in studying DISC1 expression and function. We showed that retroviral-mediated knockdown of DISC1 in newborn neuroblasts in the SVZ had no apparent effects on neuroblast migration (Fig. 9), which is in contrast to previous reports demonstrating a pivotal role for DISC1 in determining the migration and the positioning of newborn neurons in the DG (Duan et al., 2007, 2008). Although it is unclear whether DISC1 is distinctly expressed in the neural stem cells or the neuroblasts born in the SVZ, these findings implicate distinct mechanisms for neurogenesis between the DG and the SVZ. Also, it should be noted that our present study investigated the role of Girdin in postnatal development of the OB and the RMS, leaving the function of Girdin and DISC1 in adult SVZ neurogenesis unexplored. Additional studies are required to demonstrate whether neuroblasts born in postnatal and adult SVZ constitute a functionally homogeneous neuronal population and share common mechanisms regulating their migration to the OB.
An important, but unanswered question concerns the upstream event activating Girdin during neuroblast migration in the RMS. The most likely candidates for this include various receptor tyrosine kinases for growth factors including neuregulins, GDNF, BDNF, etc., and the extracellular matrix receptor integrins, which have proven to be crucial for neuroblast migration in the RMS (Anton et al., 2004; Paratcha et al., 2006; Belvindrah et al., 2007; Chiaramello et al., 2007; Garzotto et al. 2008; Murase et al., 2008; Wittko et al., 2009). It is worth noting that ErbB4 (activated by neuregulin 1) plays a central role in neuroblast migration, the loss of which results in changes in chain morphology and organization (Anton et al., 2004). It seems not to be the case that Akt (which is generally activated downstream of these receptors) transmits extracellular signals to the intracellular migratory machinery via Girdin. Additional studies will be needed to identify the precise mechanisms connecting extracellular signals and the function of Girdin in neuroblast migration.
Footnotes
This work was supported by a Grant-in-Aid for Global Center of Excellence Research, Scientific Research (A), a Grant-in-Aid for Scientific Research on Innovative Areas (to M.T.), the Program for Improvement of Research Environment for Young Researchers, Special Coordination Funds for Promoting Science and Technology (to A.E.), and a Grant-in-Aid for Scientific Research (C) (to N.A.) commissioned by the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We gratefully acknowledge Chyuan-Sheng Lin and Frank Costantini (Columbia University, New York, NY) for their considerable support in generating Girdin mutant mice. We thank Daisuke Tsuboi (Nagoya University, Nagoya, Japan), Akira Sakakibara (Nagoya University), Takeshi Kawauchi (Keio University Graduate School of Medicine, Minato, Tokyo), and Kazunori Nakajima (Keio University Graduate School of Medicine) for helpful discussions and Kaori Ushida for technical assistance.
- Correspondence should be addressed to Masahide Takahashi, Department of Pathology, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya 466-8550, Japan. mtakaha{at}med.nagoya-u.ac.jp




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}