

Abstract
The nucleolus represents an essential stress sensor for the cell. However, the molecular consequences of nucleolar damage and their possible link with neurodegenerative diseases remain to be elucidated. Here, we show that nucleolar damage is present in both genders in Parkinson's disease (PD) and in the pharmacological PD model induced by the neurotoxin 1,2,3,6-tetrahydro-1-methyl-4-phenylpyridine hydrochloride (MPTP). Mouse mutants with nucleolar disruption restricted to dopaminergic (DA) neurons show phenotypic alterations that resemble PD, such as progressive and differential loss of DA neurons and locomotor abnormalities. At the molecular level, nucleolar disruption results in increased p53 levels and downregulation of mammalian target of rapamycin (mTOR) activity, leading to mitochondrial dysfunction and increased oxidative stress, similar to PD. In turn, increased oxidative stress induced by MPTP causes mTOR and ribosomal RNA synthesis inhibition. Collectively, these observations suggest that the interplay between nucleolar dysfunction and increased oxidative stress, involving p53 and mTOR signaling, may constitute a destructive axis in experimental and sporadic PD.
Introduction
The synthesis of ribosomal RNA (rRNA), the rate-limiting step in ribosome synthesis, is intricately regulated to be responsive to metabolism and specific environmental challenges (Grummt, 2003). Therefore, transcription of rRNA genes and their maturation play a central role in the complex network that controls cell growth and proliferation. Several stress stimuli, like DNA damage, hypoxia, and nutrient deprivation, inhibit rRNA synthesis, causing nucleolar disruption and release of proteins from the nucleolus. The aberrant accumulation of nucleolar proteins in the nucleoplasm interferes with the MDM2–p53 degradation complex, leading to elevated p53 levels and apoptosis (Lohrum et al., 2003; Kurki et al., 2004; Ofir-Rosenfeld et al., 2008), making the nucleolus a decisive checkpoint of cellular well being (Rubbi and Milner, 2003).
The transcriptional control of rRNA genes is mediated by the transcription initiation factor IA (TIF-IA), which regulates the activity of RNA polymerase I in response to extracellular signals such as growth factors, nutrients, and environmental stress like hypoxia or reactive oxygen species (ROS) (Mayer and Grummt, 2005; Moss et al., 2007). In line with this, genetic ablation of TIF-IA leads to nucleolar disruption followed by p53-dependent cell death (Yuan et al., 2005).
Nucleolar malfunction has been reported to contribute to the pathology of several genetic disorders, such as Werner's syndrome, Bloom's syndrome, and Treacher Collins' syndrome. More recently, decreased rRNA synthesis has been reported in neurodegenerative diseases like Alzheimer's disease and Huntington's disease (Boisvert et al., 2007). However, despite the increasing appreciation of the role of the nucleolus (and rRNA synthesis) in the regulation of cellular growth and apoptosis, the molecular mechanisms that link ribosome biosynthesis and nucleolar structure to cell survival and neurodegenerative diseases are still poorly understood.
We have recently shown that TIF-IA loss in mature neurons has protracted effects on p53 stability and cell survival, reproducing the chronic nature of neurodegenerative processes (Parlato et al., 2008). Here, we show that nucleolar disruption induced by TIF-IA deletion selectively in dopaminergic (DA) neurons in the mouse leads to a Parkinson-like state, characterized by increased oxidative damage and progressive loss of substantia nigra neurons accompanied by marked deficiencies in motor performance. In addition, we show that increased oxidative stress in DA neurons, induced by the neurotoxic compound 1,2,3,6-tetrahydro-1-methyl-4-phenylpyridine hydrochloride (MPTP), leads to nucleolar dysfunction. Finally, we identify p53 and mammalian target of rapamycin (mTOR) as key players in this destructive interplay between nucleolar dysfunction and oxidative stress, which we indicate is operant also in sporadic Parkinson's disease (PD).
Materials and Methods
Mice.
Mice were maintained in C57BL/6 background on a 12 h light/dark cycle with water/food ad libitum. TIF-IADATCre mice were supplied with liquid food to extend their life span. TIF-IADATCre and TIF-IADATCreERT2 mice were produced by mating homozygous TIF-IAflox/flox mice (Yuan et al., 2005) with DATCre (Parlato et al., 2006) and DATCreERT2 cloned in frame with the ATG of the dopamine transporter gene contained in a bacterial artificial chromosome (BAC). The modified BAC was used for the injection into pronuclei of C57BL/6 oocytes. Induction of the inducible Cre recombinase was achieved by injection of 1 mg of tamoxifen (Sigma-Aldrich) intraperitoneally, twice daily for 5 consecutive days. As control, we used littermates harboring only the floxed alleles. Recombination pattern of DATCreERT2 was assayed by crossing with ROSA26lacZ reporter mice (Soriano, 1999). All experimental procedures were approved by the Committee on Animal Care and Use (Regierungspräsidium Karlsruhe) and performed in accordance with the local Animal Welfare Act and the European Communities Council Directive of 24 November 1986 (86/609/EEC). Both male and female mice were used for the phenotypic analysis.
Pharmacological treatment and behavior.
MPTP intoxication was performed by three intraperitoneal injections at 24 h intervals of MPTP (Sigma-Aldrich; three times 20 mg/kg body weight) and treated mice were killed 1 d after the last injection as described previously (Schober et al., 2007). Treatment of mice with l-3,4-dihydroxyphenylalanine (l-DOPA) was performed as described previously (Szczypka et al., 2001). Mice were intraperitoneally injected daily with 50 mg/kg l-DOPA for 3 weeks. For long-term experiments, l-DOPA were implanted with the constant release amount of 1 mg/kg (Innovated Research of America). One week after tamoxifen, TIF-IADATCreERT2 and control littermates were injected with pifithrin-α (Alexis Biochemicals) (2.2 mg/kg body weight) in PBS for 6 weeks. Motor coordination measurements were done on the rotating drum with accelerated speed (accelerator: Rotarod; Jones & Roberts; for mice 7650, TSE). After 10 min adaptation at 2.5 rpm, the time the animal spent on the accelerating rod was recorded.
Immunohistochemistry, in situ hybridization, and cytochrome c oxidase assay.
Mice were killed with CO2 and treated as described previously (Parlato et al., 2006). The following antibodies were used: tyrosine hydroxylase (TH) (Millipore), Cre recombinase (Parlato et al., 2006), p53 (Novocastra), nucleophosmin (NPM) (Millipore), phospho-S6 (235–236) (Cell Signaling), S6 (Cell Signaling), dopamine D1 receptor (Sigma-Aldrich), nitrosylated tyrosine (NITT) (Millipore), neuroketals (NK) (Millipore), and 8-hydroxydeoxyguanosine (8-OHdG) (Millipore). The histological assay to test the activity of cytochrome c oxidase (COX) was performed on unfixed frozen brain sections as previously described (Kraytsberg et al., 2006). For immunofluorescence, as secondary antibodies anti-sheep Alexa 594 and anti-mouse Alexa 488 (Invitrogen) were used to detect TH and NPM or S6, respectively. Formalin-fixed, paraffin-embedded sections of the midbrain autopsies from four PD and four control cases were obtained from the German Brain Bank “Brain-Net.” Immunohistochemistry (IHC) was performed as for the mouse samples. β-Galactosidase (X-gal) staining was performed as previously described (Parlato et al., 2006).
HPLC–electrochemical detection.
For measurements of striatal dopamine content, HPLC–electrochemical detection (HPLC-ED) was performed as described previously (Schober et al., 2007).
RNA expression analyses.
Substantia nigra (SN) and ventral tegmental area (VTA) were punched out of 50-μm-thick vibratome sections immersed in RNALater using a puncher with 4 mm diameter. Total RNA was prepared with the RNeasy Mini Kit (QIAGEN). Analysis of gene expression was performed with quantitative PCR (qPCR) using fluorescent TaqMan beacons (Applied Biosystems) according to the manufacturer's instructions. Briefly, total RNA was isolated and then reverse transcribed with TaqMan Reverse Transcription kit (Applied Biosystems). The abundance of the following transcripts was measured: YY1, UCP-2, TH. To verify the equal amount of input cDNA in each reaction, we have also measured the abundance of two housekeeping gene transcripts, Hprt1 and B2m.
Quantitative analysis of DA neurons.
The total number of TH-positive neurons in SN and VTA, identified according to established anatomical landmarks, were counted on vibratome sections, immunostained with a specific TH antibody. For the oxidative damage, every fourth paraffin section was used for cell counting, spanning the entire midbrain region containing dopaminergic neurons. For each experimental group, four to eight mice were used. The ImageJ program (http://rsb.info.nih.gov/ij/) was used to determine the optical density of TH immunoreactivity in the striata of control and mutant mice. Statistical analysis was performed by using the GraphPad Prism program (GraphPad). Statistical significance was assessed by Student's t test or one-way ANOVA. Values of p are expressed compared with respective control (*p < 0.05; **p < 0.01; ***p < 0.001). Nucleolar integrity in TH-positive neurons was quantified on postmortem midbrain paraffin sections from age-matched control (n = 7), progressive supranuclear palsy (PSP) (n = 4), and PD patients (n = 4) immunostained with anti-NPM (1:1000; Millipore MAB4500) and anti-TH (1:500; Millipore AB1542) antibodies. For each subject, the number of TH-positive neurons with clearly visible or disrupted nucleoli was counted on four midbrain sections by the investigator, blind to the experimental conditions. Statistical significance was calculated by one-way ANOVA followed by Tukey's post hoc test using GraphPad Prism software (GraphPad).
Results
Nucleolar damage leads to progressive loss of DA neurons and parkinsonism
To analyze whether nucleolar damage is present in PD, we examined in postmortem human brain sections from four different PD patients and matched controls (supplemental Table 1A, available at www.jneurosci.org as supplemental material) the distribution of NPM in DA neurons, by IHC with TH and NPM (Fig. 1A,B). Interestingly, we found a significant decrease of nucleolar integrity in DA neurons from these PD patients (Fig. 1C). To assess whether nucleolar damage is also present in other parkinsonian disorder, we analyzed brain sections from patients affected by PSP (supplemental Fig. 1, supplemental Table 1B, available at www.jneurosci.org as supplemental material). Also, for this disease there is a significant higher level of nucleolar disruption than in age-matched controls. Although these findings do not prove that the nucleolar damage initiates the neurodegenerative process, they indicate that nucleolar integrity is lost during neurodegeneration in PD and may be a contributing factor to the cell death.

Nucleolar disruption in PD. A, B, Analysis of nucleolar integrity by NPM immunostaining (brown) in DA neurons (TH positive; blue) in age-matched controls and PD patients. C, Quantification of visible nucleoli in four PD samples at different pathology progression show a dramatic decrease of visible nucleoli in TH+ neurons compared with controls. Scale bar: A, B, 20 μm. Error bars represent SEM.
To analyze the consequences of nucleolar impairment in DA neurons, we ablated the TIF-IA gene using the Cre/loxP system in DA neurons by crossing TIF-IAfl/fl mice, homozygous for the TIF-IA floxed allele (Yuan et al., 2005) with transgenic mice expressing the Cre recombinase exclusively in DA neurons (DATCre) (Parlato et al., 2006). In TH-immunoreactive DA neurons of control embryonic day 18.5 (E18.5) embryos, the nucleoli were visible as distinct punctuate structures immunolabeled by NPM-specific antibodies. In TIF-IAfl/fl; DATCre mutants (TIF-IADATCre) mice, NPM was mainly nucleoplasmatic, consequently to nucleolar disruption (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). At the same stage, we observed increased p53 protein level and apoptotic cell death (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Until day 15, TIF-IADATCre mice were indistinguishable from control littermates. Thereafter, they showed a progressive reduction in weight gain (Fig. 2A). At 4 weeks, mutant mice exhibited abnormal behavior, starting with slowness of movements, gait and posture disturbances, and finally culminating in twitching similar to PD tremor (supplemental Movie S1, available at www.jneurosci.org as supplemental material). By IHC with TH-specific antibodies (Fig. 2B), we found a dramatic decrease in the number of DA neurons in TIF-IADATCre mice. Intriguingly, DA neurons in the SN were more rapidly and severely affected than those in the VTA (Fig. 2C,D) despite similar levels of recombination and p53 induction (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
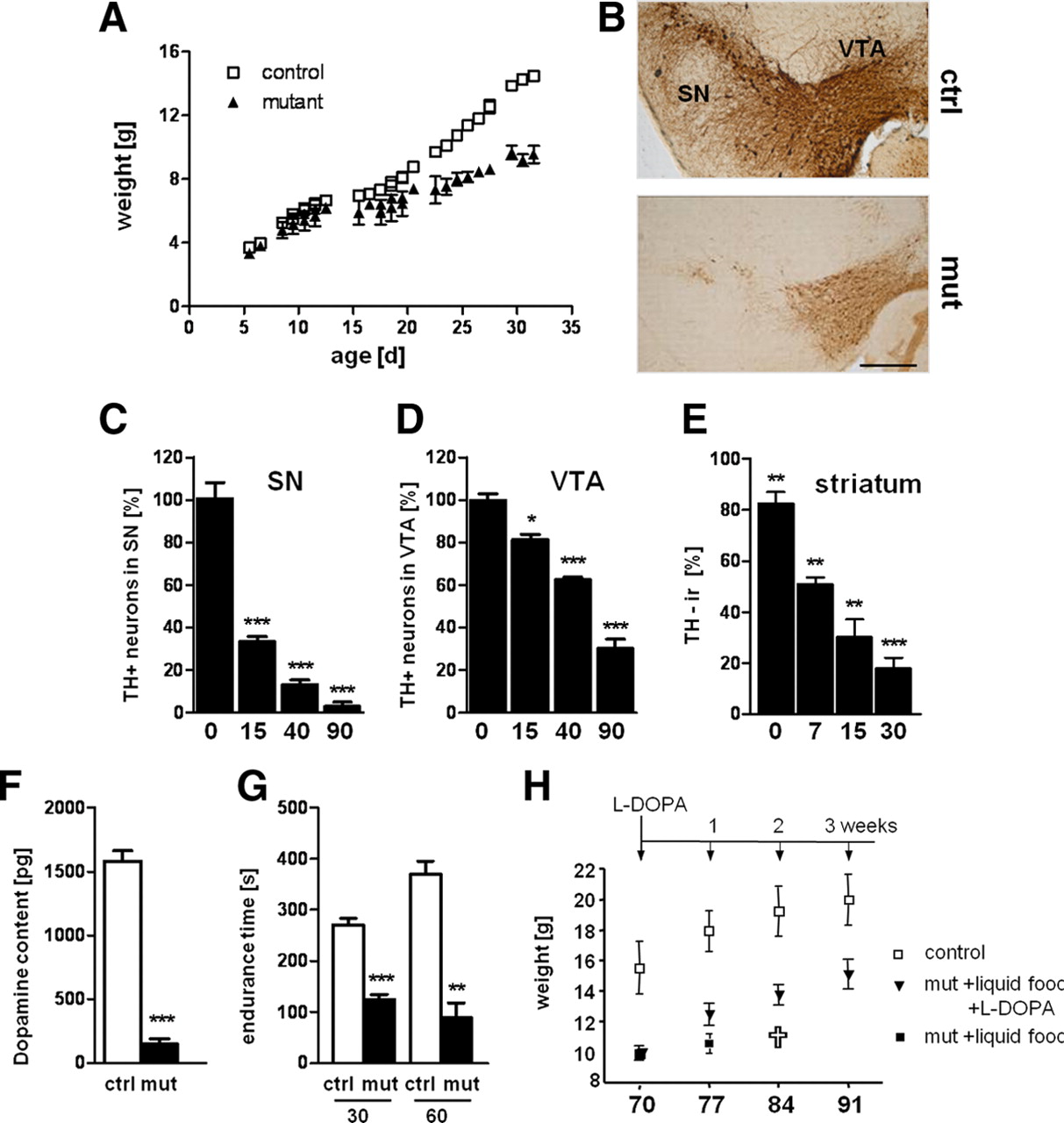
Ablation of TIF-IA in DA neurons leads to parkinsonism in mice. A, Weight curves show growth differences between mutant and control littermates starting at P15 (n = 8). B, Loss of DA neurons in mutant mice at P30, visualized by immunostaining using antibodies against TH. C, D, Quantification of DA neurons at different postnatal stages (P0, P15, P40, and P90), normalized to control littermates and plotted as a histogram for each time point (n = 5), revealed that mutant and control mice did not show any significant differences in number of DA neurons at birth, whereas they are progressively lost at later stages. SN neurons (C) are more susceptible to the loss of TIF-IA than VTA neurons (D). E, Levels of TH immunoreactivity in striata at P7, P15, and P30 were normalized to control littermates and plotted as a histogram for each time point (n = 4). F, The striatal dopamine content measured by HPLC-ED shows a 95% reduction compared with the control group at P40 (n = 5). G, Locomotor deficits of TIF-IADATCre mice determined by the accelerating rotarod assay. At P30, mutants show a locomotor deficit of 55% and at P60 of 76% compared with control mice (n = 5). H, Rescue of TIF-IADATCre mice by l-DOPA injection. Mutant mice (>7 weeks of age) treated by daily injecting l-DOPA for 3 weeks gained weight similarly to control mice, whereas untreated mutant mice died without gaining weight despite supplementation with liquid food (n = 4). Scale bar, 150 μm. *p < 0.05; **p < 0.01; ***p < 0.001. Error bars represent SEM.
TH immunoreactivity in DA terminals in the striatum was decreased already at postnatal day 0 (P0) (Fig. 2E), before any loss of DA neurons (Fig. 2C,D). Measurement of the dopamine content by HPLC in TIF-IADATCre mice at 5 weeks of age showed a 95% reduction of dopamine levels compared with control mice (Fig. 2F). At 4 weeks of age, we observed a ∼55% reduction in motor performance of mutant mice on the accelerating rotarod. This decrease reached a ∼76% decline at 8 weeks of age (Fig. 2G). To establish the responsiveness to the dopamine precursor l-DOPA, we gave l-DOPA (50 mg/kg, i.p.) to mice that already showed a rather severe phenotype. Whereas mutants without treatment died within 1 week, the treated mice survived and gained weight during the 3 weeks analyzed (Fig. 2H). Moreover, increased growth of TIF-IADATCre mice was achieved 6 weeks after continuous treatment by subcutaneous l-DOPA pellets (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Together, these analyses demonstrate that ablation of TIF-IA leads to progressive and differential loss of DA neurons in the SN and VTA, reduction of the striatal dopamine content, and severe impairment of motor performance.
Inducible ablation of TIF-IA in adult DA neurons
To dissect the sequence of events that link TIF-IA ablation with loss of DA neurons and avoid possible developmental effects in TIF-IADATCre mutants, we generated mice in which loss of TIF-IA is induced in adulthood using an inducible Cre recombinase under the control of the dopamine transporter gene (Engblom et al., 2008). To this end, we used a modified Cre-recombinase (CreERT2) that is inactive under normal conditions but can be activated by applying tamoxifen. The line expressing the CreERT2, referred to as DATCreERT2, was crossed with the ROSA26 reporter line (Soriano, 1999) to monitor recombinase activity. DATCreERT2 activity was induced in SN and VTA on tamoxifen treatment (Fig. 3A,B) and recombination was restricted to DA neurons as shown by colocalization of LacZ activity (blue) and TH immunoreactivity (brown) (Fig. 3C).

Generation of the inducible DATCreERT2 transgenic mouse line. A, B, Brain sections of the transgenic reporter line ROSA26 expressing the inducible Cre-recombinase injected with either oil or tamoxifen. Whereas in mice injected only with oil, no activity of the reporter gene is detected (A), mice injected with tamoxifen show specific activity of the reporter in the mesencephalic region (B). C, D, The activity of the Cre recombinase is restricted to DA neurons, as determined by X-gal staining (blue) in combination with TH immunohistochemistry (brown). The inset (D) shows a higher magnification of double-stained neurons (arrows). Scale bars: A, B, 150 μm; C, 50 μm.
TIF-IAfl/fl; DATCreERT2 mice (TIF-IADATCreERT2) were generated, and at the age of 2 months, mutant and control littermates were injected with tamoxifen and analyzed after 7 weeks (Fig. 4A,B), 13 weeks (Fig. 4C,D), and 21 weeks (Fig. 4E,F). Interestingly, Cre-induced inactivation of TIF-IA in adult mice leads to a similar spatiotemporal sequence of the neurodegenerative process as observed in TIF-IADATCre mice, including early decline of TH immunoreactivity and dopamine content in the striatum (Fig. 4G,J) followed by progressive differential loss of DA neurons in SN and VTA (Fig. 4H,I) and motor dysfunction (Fig. 4K).

Progressive loss of DA neurons in TIF-IADATCreERT2 adult mice. A–F, Loss of TH immunoreactivity in striata is visible in TIF-IADATCreERT2 mutants (B) 7 weeks after tamoxifen injection compared with control (A), whereas TH+ neurons in SN/VTA are minimally affected. Thirteen weeks after injection, TH+ fibers in striata disappear almost completely (D), and severe loss of DA in SN is visible accompanied by partial loss of VTA TH+ neurons. Loss of DA fibers and neurons further advances 21 weeks after induction with tamoxifen (F). No changes are visible in the respective controls at 7, 13, and 21 weeks (A, C, E). G, Quantification of the TH+ immunoreactivity in TIF-IADATCreERT2 mice in the striatum compared with respective control mice (n = 5). H, I, Percentage of remaining DA neurons in SN (H) and VTA (I) of TIF-IADATCreERT2 mutants compared with control littermates (n = 4). J, Measurement of the dopamine content in striata by HPLC-ED reveals in TIF-IADATCreERT2 mice 7 weeks after tamoxifen a ∼45% reduction, reaching ∼80% 30 weeks after tamoxifen (n = 5). K, Locomotor deficits of TIF-IADATCreERT2 mice determined by the accelerating rotarod assay are detected 13 weeks after induction and worsen over time (n = 5). L, Analysis of TH+ neurons in control and TIF-IA; p53DATCreERT2 mice 7 weeks after tamoxifen injection in control (n = 7) and double mutants (DM) (n = 7). Error bars represent SEM. Scale bars: A–F, top panels, 400 μm; bottom panels, 150 μm. *p < 0.05; **p < 0.01; ***p < 0.001.
Together, these analyses demonstrate that ablation of TIF-IA in adult mice leads to similar PD-like characteristics as shown for the TIF-IADATCre mutant.
Nucleolar disruption induced by TIF-IA loss leads to elevation of p53 levels and p53-dependent apoptosis (Yuan et al., 2005; Parlato et al., 2008), raising the possibility to prevent neuronal degeneration by blocking p53 function. We therefore used pifithrin-α, a chemical inhibitor of p53 (Komarov et al., 1999). One week after tamoxifen induction, TIF-IADATCreERT2 mice were given pifithrin-α once daily for 6 weeks intraperitoneally. Mock-injected TIF-IADATCreERT2 mice showed a 25% reduction of DA neurons, but this was prevented by pifithrin-α treatment (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These data are also supported by the genetic ablation of p53 in DA neurons on nucleolar damage analyzed 7 weeks after induction of the mutation by tamoxifen injection (Fig. 4L).
Mitochondrial dysfunction inhibits mTOR and nucleolar activity in DA neurons
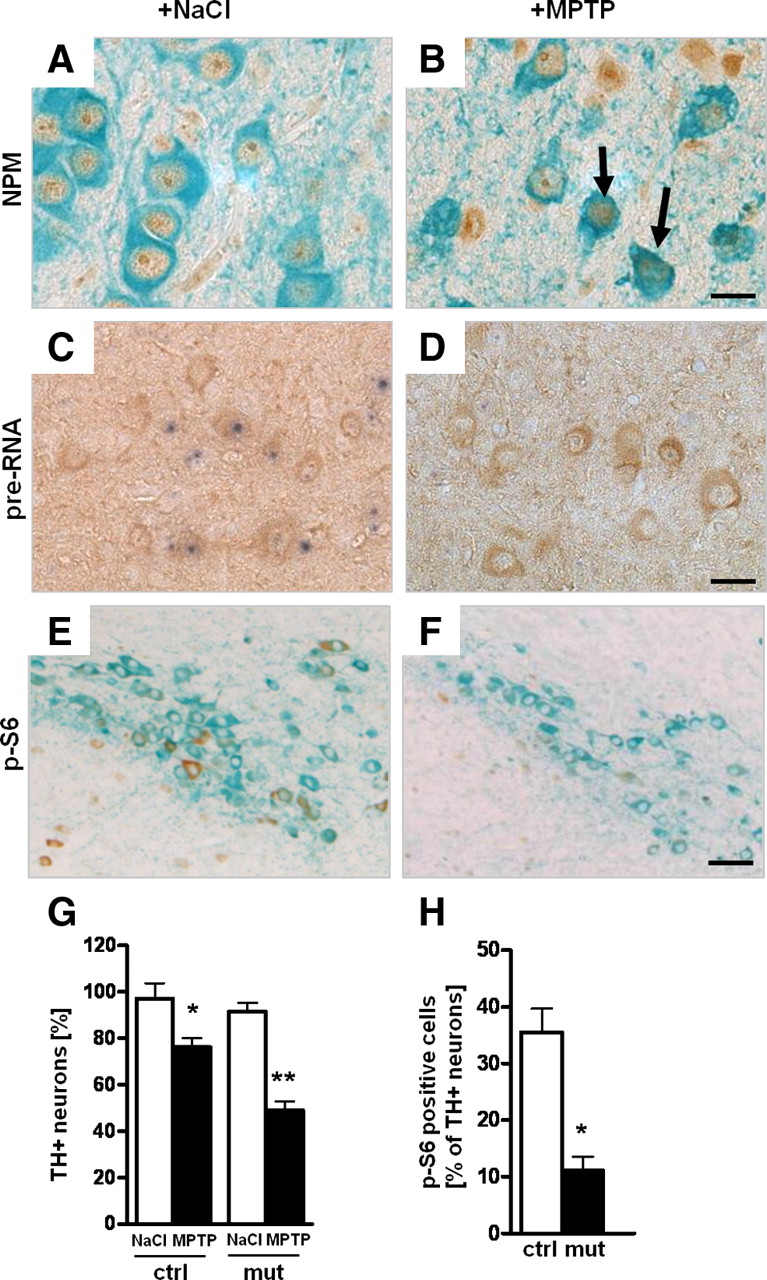
DA neurons are known to suffer from higher oxidative stress most likely because of their high rate of oxygen metabolism, low levels of antioxidants, and high iron content (Abou-Sleiman et al., 2006). Increased oxidative stress has been shown to reduce rRNA synthesis (Mayer et al., 2004). To investigate whether nucleolar activity is affected by increased oxidative stress in DA neurons, we injected the neurotoxin MPTP (20 mg/kg body weight) for 3 d into wild-type mice and analyzed them 1 d after the last injection. Whereas in animals treated with NaCl, the protein NPM is mostly localized in the nucleoli (Fig. 5A), NPM in DA neurons shows a more than threefold higher release into the nucleoplasm after injection of MPTP (Fig. 5B) (5.33 ± 1.38 vs 17 ± 1.41%; p < 0.01). In situ hybridization using a probe for the 5′-external transcribed spacer (ETS) to detect pre-rRNA synthesis revealed a strong reduction of transcriptional activity in DA neurons of mice treated with MPTP (Fig. 5D,E) (74.54 ± 5.76 vs 46.98 ± 3.74%; p < 0.01). No significant change of pre-rRNA synthesis was observed in the hippocampus that is not targeted by the toxin (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These analyses show that increased oxidative stress leads to reduced nucleolar activity in DA neurons.

Oxidative stress after MPTP treatment affects nucleolar function. A, B, Analysis of nucleolar integrity in DA neurons by immunohistochemistry using antibodies against NPM (brown) and TH (green) in 2-month-old wild-type mice injected for 3 d with either MPTP or NaCl and analyzed 1 d after the last injection (n = 5). C, D, Effect on 47S pre-rRNA synthesis in DA neurons after NaCl (C) or MPTP injection (D) analyzed by in situ hybridization with a riboprobe for the 5′-ETS of the 47S pre-rRNA (blue) in combination with TH immunohistochemistry (brown) to identify DA neurons. E, F, IHC using antibodies against p-S6 (brown) and TH (light blue) shows a decrease of phosho-S6 (p-S6) in MPTP-treated wild-type mice. G, Effect of treatment with the neurotoxin MPTP on the number of TH+ neurons in control and TIF-IADATCreERT2 mutant mice 2 weeks after tamoxifen (n = 5). Control mice show moderate loss of DA neurons on MPTP treatment, whereas TIF-IADATCreERT2 mutants treated with the neurotoxin show a more severe reduction of TH+ neurons. H, Quantification of the number of p-S6-positive DA neurons in TIF-IADATCreERT2 and control littermates 4 weeks after tamoxifen. Scale bars: A, B, 20 μm; C, D, 25 μm; E, F, 50 μm. *p < 0.05; **p < 0.01. Error bars represent SEM.
A major regulator of cell growth and metabolism in response to environmental cues by promoting rRNA and protein synthesis is the mTOR (Wullschleger et al., 2006). To determine whether nucleolar dysfunction might be triggered by a stress-induced reduction in mTOR activity, we analyzed the effects of MPTP on the phosphorylation level of the ribosomal protein S6 (p-S6), a major target of mTOR (Gingras et al., 2001) (Fig. 5E,F). We saw a significant decrease of p-S6-positive DA cells after MPTP treatment (4.29 ± 1.63 vs 31.33 ± 3.03%; p < 0.01). Although we cannot exclude other mechanisms independent of mTOR, the fact that mTOR is a major regulator of TIF-IA activity and rRNA synthesis (Mayer et al., 2004) and its activity is strongly downregulated on MPTP treatment suggest that decreased rRNA synthesis in the MPTP model could be ascribed to decreased mTOR activity.
To elucidate whether nucleolar disruption makes DA neurons more vulnerable to the mitochondrial damage induced by MPTP, we treated TIF-IADATCreERT2 and control mice with MPTP 2 weeks after tamoxifen. As expected, at this stage TIF-IADATCreERT2 mutants treated with NaCl showed no significant difference in the number of TH+ neurons compared with control mice. Moreover, whereas control mice show a 15% reduction in the number of TH+ neurons, TIF-IADATCreERT2 mice treated with MPTP displayed a considerably higher loss of DA neurons (∼40%) (Fig. 5G). This analysis shows that DA neurons are more vulnerable to the oxidative damage induced by MPTP if this insult is combined with nucleolar damage.
Because increased p53 levels are an early consequence of nucleolar damage, and p53 may set a negative feedback on mTOR signaling (Ellisen et al., 2002; Budanov and Karin, 2008; DeYoung et al., 2008), we asked whether mTOR signaling itself is impaired in TIF-IADATCreERT2 mice. Indeed, we observed a significant decrease in p-S6-positive DA neurons of TIF-IADATCreERT2 mutant mice (Fig. 5H; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This observation not only suggests a positive-feedback loop active on mTOR after nucleolar damage, probably p53 dependent, but also that other mTOR-dependent functions might be influenced by the loss of TIF-IA.
TIF-IA ablation leads to increased oxidative stress in DA neurons
The mTOR pathway regulates not only cell growth by controlling protein synthesis and stress responses (Wullschleger et al., 2006) but also energy metabolism by controlling mitochondrial function (Schieke et al., 2006). The transcription factor yin-yang 1 (YY1) increases mitochondrial gene transcription in response to mTOR activity (Cunningham et al., 2007). To further characterize the impact of mTOR downregulation observed in TIF-IADATCreERT2 mutant mice, we analyzed the expression of YY1 by qPCR on RNA isolated from SN/VTA at early stages after tamoxifen injection. We found ∼40% reduction of YY1 transcripts in TIF-IADATCreERT2 already 2 weeks after TIF-IA ablation (Fig. 6A). Among the genes transcriptionally regulated by YY1, the uncoupling protein 2 (UCP-2) transcripts were downregulated in TIF-IADATCreERT2 mice (Fig. 6B).

Perturbation of rRNA synthesis leads to mitochondrial impairment followed by increased oxidative stress. A, The bars represent abundance of YY1 transcripts by qPCR normalized to HPRT in control and TIF-IADATCreERT2 mice 2 weeks after tamoxifen (n = 3). B, The graph shows UCP-2 gene expression by qPCR. The bars represent abundance of UCP-2 transcripts normalized to the levels HPRT 2 and 3 weeks after tamoxifen treatment (n = 3). C, Two weeks after tamoxifen injection, reduced COX activity in TIF-IADATCreERT2 mutants is measured as optical density compared with control mice (n = 3). D–I, Brain sections through the ventral midbrain were analyzed for neuroketals (D, E) as marker for ROS-induced lipid damage (brown), for nitrosylated proteins (NITT) (F, G), for 8-hydroxydeoxyguanosine (8-OHdG) (H, I), as marker for ROS-induced DNA damage (brown), in combination with TH staining (light blue) to identify DA. The insets show higher magnification of immunostained cells. Scale bars, 50 μm. J, Quantification of DA neurons positive for oxidative stress markers NITT, neuroketals, and 8-OHdG in control and mutant mice 2 and 4 weeks after injection with tamoxifen. Differences are expressed as fold change compared with the mean of the controls at different stages. Higher levels of oxidative stress markers were observed in the mutants (n = 4). *p < 0.05; **p < 0.01. Error bars represent SEM. K, Diagram showing the sequence of events triggered by nucleolar damage. The decrease and increase of the parameters indicated (left) are depicted in green or red, respectively. The bars indicate the time points (weeks) analyzed after injection of tamoxifen to induce TIF-IA loss in adult mice. L, Schematic representation showing the molecular mechanisms shared between TIF-IA mutation and MPTP pharmacological models. Nucleolar damage as a consequence of TIF-IA mutation induces p53 and inhibits mTOR, causing mitochondrial dysfunction and increased oxidative damage. In the pharmacological model, mitochondrial dysfunction caused by MPTP leads to increased oxidative stress. This increase inhibits mTOR and rRNA synthesis, probably by downregulation of TIF-IA activity, causing nucleolar damage and additional consequences on cell survival.
To assay mitochondrial function in tissue sections, we measured COX activity by histochemical staining and densitometric analysis (Ekstrand et al., 2007). A decrease of COX activity by ∼40% was observed 2 weeks after tamoxifen treatment in TIF-IADATCreERT2 mutants (Fig. 6C), indicating that the mitochondrial damage represents an early consequence of the nucleolar damage. A profound effect on mitochondrial activity is also present in the constitutive mutants as early as E19 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The impairment of mitochondrial activity increases ROS levels and causes oxidative damage to proteins, lipids, and DNA (Finkel, 2005). To monitor the oxidative damage in TIF-IADATCreERT2 mutants, we determined the number of TH+ neurons positive for markers of oxidative stress, like nitrosylated proteins (NITT), NK, and 8-OHdG. Significantly higher levels of NK (Fig. 6D,E), NITT (Fig. 6F,G), and 8-OHdG (Fig. 6H,I) were found within DA neurons of TIF-IADATCreERT2 mice 4 weeks after tamoxifen injection (Fig. 6J). Together, these data identify nucleolar disruption as a potent trigger of oxidative stress and thus indicate a novel role for the nucleolus in neurodegeneration.
Discussion
We have recently shown that, in contrast to dividing neural progenitor cells, the consequences of nucleolar damage triggered by ablation of TIF-IA in differentiated neurons are protracted over time and involve the activation of an endogenous suicide response (Parlato et al., 2008). Here, we have exploited the possibility to use this mutation to study the impact of nucleolar damage and its molecular consequences for neurodegenerative diseases. In particular, we have restricted the nucleolar damage to DA neurons. Thus, we were able to reproduce essential characteristics of PD at a functional and molecular level, as summarized in Figure 6K. We found that nucleolar damage leads to p53 increase and mTOR inhibition followed by mitochondrial damage and increased oxidative stress. The decreased striatal dopamine precedes the loss of DA neurons, whereas a significant motor impairment is evident at later stages. Increased oxidative damage, before cell death—certainly one of the most relevant aspects of parkinsonism—provides a novel mechanism for neurodegeneration triggered by nucleolar damage. Animal models based on genetic mutations found in familial forms of PD (i.e., PARK1-7) have not been able to reproduce all features of the disease, such as the preferential loss of substantia nigra DA neurons (Farrer, 2006). The observation made in TIF-IA mutants, which show such differential vulnerability, reveals an intriguing specificity for the effects of nucleolar disruption in DA neuron survival, indicating a possible differential role of nucleolar-dependent functions in both DA neuron subtypes.
Although our study does not prove that the nucleolar damage is a primary cause of neurodegeneration, it shows that the impaired nucleolus is an essential alarm bell for DA neurons with consequences extending to other organelles, such as the mitochondria. Moreover, the molecular consequences of TIF-IA loss share similarities with critical signaling for growth and survival involved in neurodegenerative diseases. Attenuation of mTOR signaling has been reported in several neurodegenerative diseases, including Parkinson's disease (Inoki et al., 2005). Neurotoxins, such as 6-OHDA, MPTP, and rotenone, block the activation of mTOR signaling in neuronal cultures and in animals (Malagelada et al., 2006). In fact, we show here that mitochondrial damage induced by MPTP treatment results in a significant decrease of mTOR activity and inhibition of rRNA transcription. mTOR signaling is also decreased in TIF-IA mutants, although we cannot exclude that this is attributable to decreased mTOR protein levels. mTOR is one of the main regulators of TIF-IA activity and rRNA synthesis (Mayer et al., 2004; Grewal et al., 2007). In combination with previous data, our findings could be fit into the following model, integrating nucleolar disruption and oxidative stress and indicating mechanistic similarities between TIF-IA and MPTP models (Fig. 6L). In the pharmacological MPTP model based on induction of mitochondrial impairment and ROS accumulation, we found severe nucleolar damage, probably mediated by inhibition of mTOR function and consequent downregulation of TIF-IA and rRNA synthesis (Mayer et al., 2004; Grewal et al., 2007). In TIF-IA mutants, one of the earliest events triggered by nucleolar damage is the increased stabilization of the transcription factor p53, and we show that inhibition of p53 abrogated the cellular loss. Interestingly, the importance of p53 for neurodegeneration has been shown in Huntington's, Alzheimer's, and Parkinson's diseases (Bae et al., 2005; Alves da Costa et al., 2006; Nair et al., 2006). The question remains how p53 can relay its devastating function in neurons. In this context, it has been shown that in the presence of stress signals such as low levels of ribosome biogenesis, hypoxia, or DNA damage, p53 may set a negative feedback on mTOR signaling by activating AMP-activated protein kinase and/or by the synthesis of the protein REDD1 (Ellisen et al., 2002; Budanov and Karin, 2008; DeYoung et al., 2008). TIF-IADATCreERT2 mutants are characterized by both increased p53 levels and mTOR downregulation leading to increased oxidative stress and final neuronal demise. We assume that the stabilization of p53 after TIF-IA loss causes the negative feedback on mTOR (Levine et al., 2006). mTOR inhibition may in turn cause mitochondrial impairment and oxidative damage by deregulation of the mitochondrial metabolism via interaction with YY1 and PCG-1 (Schieke et al., 2006; Cunningham et al., 2007).
Here, we could show that TIF-IA ablation leads to a strong reduction of the transcription factor YY1, responsible for the transcription of mitochondrial genes, like UCP-2. Together, these observations suggest that interplay between nucleolar dysfunction and increased oxidative stress, involving p53 and mTOR signaling, leads to a destructive axis in TIF-IA mutants as well as in PD models (Fig. 6L).
Although pifithrin-α inhibits p53 transcriptional activity, based on the experiments presented here, we cannot exclude that other mechanisms may also contribute to the oxidative damage in TIF-IADATCreERT2 mutants [e.g., p53 may interfere directly with mitochondrial function (Vaseva and Moll, 2009)]. Upregulation of p53 can lead to translocation of Bax to mitochondria (Yuan et al., 2005) followed by permeabilization of the mitochondrial membrane, release of cytochrome c, and activation of the apoptotic program (Chipuk and Green, 2006). To assess the specific contribution of nontranscriptional p53 activity to cell survival, specific inhibition of p53 interaction with mitochondrial proteins by using the pharmacological inhibitor pifithrin-μ (Strom et al., 2006) would be a valid approach.
Another possibility involves imbalanced synthesis of mtDNA-encoded proteins leading to ROS accumulation (Bonawitz et al., 2007). A mutant mouse lacking the mitochondrial transcription factor A (Tfam) in dopaminergic neurons shows that dysfunction of the respiratory chain might be important in the pathogenesis of parkinsonism (Ekstrand et al., 2007). Consequences of altered TIF-IA function on mitochondrial gene transcription mediated by Tfam have to be considered, in light of the observation that p53 physically interacts with Tfam (Yoshida et al., 2003).
The observation that nucleolar damage is present in PD and PSP patients and after MPTP treatment encourages the next series of studies to unravel in which precise way structural and functional perturbations of the nucleolus are involved in the development of PD and other neurodegenerative disorders. As of yet, only a relatively small group of these diseases can be ascribed to simple genetic defects and it is likely that environmental factors influence their onset and progression. Enormous efforts are invested in the identification of the molecular mechanisms behind PD (Lesage and Brice, 2009). Future studies addressing these mechanisms will be important to develop procedures aiming to alleviate the consequences of this and possibly other neurodegenerative diseases. The present study indicates that cross talk between nucleolar dysfunction and oxidative stress is an interesting mechanism in this context.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft through Collaborative Research Centers Sonderforschungsbereich (SFB) 488 and SFB 636, by Fonds der Chemischen Industrie, the European Union through Grant LSHM-CT-2005-018652 (CRESCENDO), Bundesministerium für Bildung und Forschung (BMBF) through NGFNplus Grants FZK 01GS08153 and 01GS08142 and Project 0313074C (HepatoSys), Helmholtz Gemeinschaft Deutscher Forschungszentren through Initiative CoReNe and Alliance HelMA, and Deutsche Krebshilfe through Project 108567. The German Brain Bank “Brain-Net” is supported by BMBF Grant 01GI0505. D.E. was supported by the Parkinson Foundation at Linköping University. We thank R. Hertel for HPLC-ED and P. Gass for providing us with the rotarod equipment. Furthermore, we thank U. Moll and B. Liss for discussions and critical reading of this manuscript.
- Correspondence should be addressed to Günther Schütz, Molecular Biology of the Cell I, INF 280 German Cancer Research Center, D-69120 Heidelberg, Germany. g.schuetz{at}dkfz.de




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}